
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium storage with high plateau capacity in nitrogen doped carbon derived from melamine–terephthalaldehyde polymers†
Ivan K.
Ilic
 *a,
Konstantin
Schutjajew
a,
Wuyong
Zhang
a and
Martin
Oschatz
*ab
*a,
Konstantin
Schutjajew
a,
Wuyong
Zhang
a and
Martin
Oschatz
*ab
aDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: ivan.ilic@mpikg.mpg.de; martin.oschatz@mpikg.mpg.de
bFriedrich-Schiller-University Jena, Institute for Technical Chemistry and Environmental Chemistry, Center for Energy and Environmental Chemistry Jena (CEEC Jena), Philosophenweg 7a, 07743, Jena, Germany
First published on 26th February 2021
Abstract
Sodium, one of the most widespread metals in the earth's crust, can be mined virtually everywhere and holds great promise as an electroactive material batteries to compete with commercial lithium-ion batteries in terms of price and availability. However, sodium, unlike lithium, cannot be intercalated in graphite and therefore there is a need to develop sodium anodes from alternative abundant and low-cost materials. Here we show that one-step carbonisation of melamine–terephthalaldehyde mixtures, both low-cost chemicals available on an industrial scale, yields carbons with promising properties as anodes for sodium-ion batteries. The produced materials apparently contain a large number of small pores, unreachable to gas molecules, which can store sodium (in a quasi-metallic state) as Na(0). The carbon shows plateau behaviour above and below 0 V vs. Na/Na+, with a capacity of up to 170 mA h g−1 until bulk sodium plating occurs. Additionally, for 60 cycles the carbon shows high stability, exhibiting low degradation and efficiencies close to 100%. This report is a first step towards the use of nitrogen-doped carbon materials from low-cost, industrially widespread precursors as anodes for sodium-ion batteries.
1. Introduction
In the last few decades, portable electronic devices such as mobile phones, laptops, or tablets became widespread and affordable to the public. At the same time, these devices became more and more powerful causing the surge of their energy consumption. Partially, this progress was made possible by the development of lithium-ion battery technology that allowed high energy density at relatively low prices.1 Testimony to their importance is The Nobel Prize in Chemistry 2019 awarded to Goodenough, Whittingham and Yoshino “for the development of lithium-ion batteries”. However, the natural scarcity of lithium, as compared to other metals, inspired scientists to develop alternative metal-ion batteries. A metal that holds great promise is sodium – one of the most abundant metals on Earth which can be mined virtually everywhere.Just like lithium, metallic sodium is unfortunately not a suitable anode material for batteries as such, due to limited cyclability. Unlike lithiation, the sodiation of graphite is not yet an attractive option as an electrochemical process on the anode side of sodium batteries due to weaker covalent interactions between sodium and graphite.2 This led to the exploration of alternative anode materials, especially non-graphitisable carbons, so-called “hard carbons”.3 The structure of these carbons can be described by the “Falling Cards Model”,4 where cards represent graphitic sheets that upon “falling” do not perfectly stack on top of each other. This random orientation can lead to the formation of very small pore systems that remain inaccessible for most fluids. This restricted access can have a strong influence on most of the conventional experiments such as gas physisorption or electrochemical measurements. Some common examples of hard carbons are porogen-free cellulose-derived carbons. These carbons are characterised by a low surface area formed during carbonisation. Upon increasing the carbonisation temperature to 1000 °C or higher, the surface area as determined by gas sorption experiments tends to decrease even further as pores close and become inaccessible to the probing gases.5–7
With decreasing potential versus Na/Na+, the sodiation of hard carbons usually occurs gradually in different steps that can be distinguished by the mechanism of sodium storage.8–10 At higher potential, sodium ions are first bonded to high energy stabilization sites followed by storage in small pores and slightly expanded graphite-like layers. This mechanism causes constantly changing potential because full reduction of sodium ions does not take place, and the repulsive forces between the electrons localized in the carbon and the positively charged sodium ions are constantly increasing. For this reason, this region of the sodiation curve is known as the “sloping region”. A subsequent step may occur as a result of adsorption and reduction of sodium in possibly present very small, “closed” pores (i.e. pores that remain undetected by classical gas physisorption techniques). During this process, zero-valent sodium as atoms and clusters can be formed.11 This step occurs close to 0 V and is characterised by only a weak change in potential, making this step of sodium storage most suitable for battery applications. The plateau, coupled with a cathode with a defined plateau, can set the base for a battery with high capacity at a stable potential. The chemical state of sodium ions during sloping was proved by in situ12 and ex situ13 powder X-ray diffraction, and the formation of quasi-metallic clusters in the plateau region was proved by in situ SAXS11 and in operando NMR.14 Nevertheless, it is worth mentioning that there is an ongoing scientific debate whether closed pores are responsible for low-voltage plateaus observed in hard carbons.15,16
The particular role of nitrogen heteroatoms in sodium storage is intensely investigated as well.17 For instance, an almost 40% improvement of sodium storage capacity was reported after nitrogen-doping of hard carbon nanospheres. This could be explained by an increase of the number of active storage sites due to enhanced defects and porosity, with the increase of electric conductivity and with the reduction of volume expansion during sodiation.18 Furthermore, nitrogen atoms can affect sodium transport and also influence the formation of the solid-electrolyte interphase (SEI) and by that the irreversible capacity loss in the 1st cycle.19 The particular effect of heteroatom doping on the electrochemical properties is still difficult to understand and depends to a large extent on the structure of the chemical bonds between the heteroatoms and the carbon framework. For that reason, the investigation of sodium storage in defined model materials is a suitable method to get valuable insights into the underlying mechanisms under experimental conditions.20–23 For practical applications, however, such compounds are often unsuitable because they are mainly derived from expensive precursors with multi-step and complicated synthesis protocols.
A simple way for the synthesis of nitrogen-doped carbon materials is presented here, namely the fabrication of melamine–terephthalaldehyde polymer networks, followed by subsequent carbonization. Melamine and terephthalaldehyde are both inexpensive materials.24,25 It has been shown that such carbons have hard-carbon-like properties, that is, they close the pores at high carbonization temperatures but their performance as anodes for sodium storage was not previously assessed.26
Melamine-based microporous polymer networks were firstly prepared solvothermally from melamine and terephthalaldehyde in dimethyl sulfoxide. This simple catalyst-free reaction generates microporous polymers with surface areas higher than 1000 m2 g−1. Initially, the condensation of amine and aldehyde groups to imines was proposed as a scaffold-forming mechanism but no imine could be detected in the formed microporous polymer network.27 A related class of materials, melamine-based conjugated polymers, were produced from the same reactants without the addition of external solvent by heating melamine and terephthalaldehyde above the melting point of terephthalaldehyde of around 115 °C. Non-porous, conjugated polymers with aromatic centres connected by imine bonds were obtained.28 Melamine-based microporous polymer networks and conjugated polymers both proved to be interesting precursors for carbons, forming materials with excellent properties as supercapacitors,26,29–32 electrocatalysts33 and anodes for lithium-ion batteries.34
In this work, the synthesis and subsequent carbonization of a melamine-based conjugated polymer are described. The characterization of the carbons by nitrogen/carbon dioxide physisorption and small-angle X-ray scattering indicates that the variation of the carbonisation temperature leads to a of different porosities. It can be observed that the internal volume that remains inaccessible for the physisorption of nitrogen but may still be accessible for sodium storage (in the following referred to as “closed pores”) increases at higher temperature. A clear correlation between the presence of closed pores and the properties of the materials in sodium storage is observed. The carbon formed at 1000 °C, with the largest number of closed pores among all samples, shows the most attractive properties as an anode material with the charging plateau capacity around 84 mA h g−1 at C/20 current density.
2. Materials and methods
2.1 Materials
Melamine (99%), terephthalaldehyde (99%), ethylene carbonate (99%), diethyl carbonate (99+, %), metallic sodium (99.9%), absolute ethanol (99.8+, %) and polytetrafluoroethylene preparation (60 wt% in water) were bought from Sigma Aldrich. N-Methyl-2-pyrrolidone (99+, %), copper foil (annealed, uncoated 99.8%, thickness 0.025 mm) and sodium hexafluorophosphate (99+, %) were bought from Alfa Aesar and polyvinylidene fluoride (PVDF, 99.5+, %) was bought from MPI Corporation. All the chemicals were used without further modification.2.2 Synthesis of MT-X
Melamine (2.0 g, 15.9 mmol) and terephthalaldehyde (3.2 g, 23.9 mmol) were mixed in an agate mortar with a pestle until a homogeneous powder was obtained. The powder was transferred to a porcelain boat and placed in a quartz glass tube in a tubular oven. The tube was purged with an argon flow and heated (4 K min−1) firstly to 150 °C for 3 h and subsequently (4 K min−1) to temperature X (600, 800 or 1000 °C) for 1 h. After the oven cooled down, the sample was removed, ground with a mortar and pestle, and activated in a vacuum oven at 150 °C for 4 h. The obtained samples are labelled MT-X. The sample MT-150 was prepared by heating at 150 °C for 3 h, without subsequent heating or activation.2.3 Characterization
Thermogravimetric analysis (TGA) measurements were performed by using a NETZSCH TG 209F1 Libra machine under a N2 atmosphere at a heating rate of 10 K min−1. Combustive elemental analysis was performed by using an Avario Micro CHNOS machine. Powder X-ray diffraction (PXRD) measurements were performed on a Bruker D8 diffractometer with a Cu-Kα (λ = 0.154 nm) X-ray source equipped with a scintillation counter-Scinti-Detector. Diffraction data were recorded in the 2θ range between 3 and 70° with an angular resolution of 0.03°. The morphology of the carbon and electrodes was investigated by scanning electron microscopy (SEM) on a Zeiss Leo Gemini 1550. An acceleration voltage of 3.00 kV was used. The atomic distribution of C, O, and N was mapped by energy-dispersive X-ray spectroscopy (EDX, X-Max, Oxford instruments). Transmission electron microscopy (TEM) images were acquired using a Zeiss TEM 912 Omega with an acceleration voltage of 120 kV. The electrode that was investigated by TEM after cycling was firstly immersed in absolute ethanol for two days (ethanol was exchanged after a day) and dried in a vacuum oven at 120 °C for 2 h. Gas physisorption measurements were carried out on a Vapor200C gas sorption analyzer (3P Instruments). N2 at −196 °C and CO2 at 0 °C were used as probe gases. The samples were degassed in a vacuum at 150 °C for at least 20 h before each measurement. The specific surface areas (SSAs) of the materials are calculated by using the multipoint Brunauer–Emmett–Teller (BET) model in the relative pressure range 0.05–0.2. The total pore volumes for nitrogen physisorption (Vt) were determined at p p0−1 = 0.99. Small-angle X-ray scattering (SAXS) experiments were performed on a Bruker Nanostar 2 device using Cu-Kα radiation in transmission geometry with a 278 mm distance between the sample and detector. Fourier-transform infrared spectroscopy was performed by putting a small amount of carbon powder on a Nicolet iS 5FTIR spectrometer (Thermo Fisher Scientific). Raman spectroscopy was performed by putting a small amount of carbon sample on a Confocal Raman Microscope Alpha 300 and measuring it at 0.5 mW with an integral time of 1 s 60 times.2.4 Conductivity measurements
The samples were prepared by mixing 50 mg of the respective carbon with 10 μL of PTFE dispersion (60 wt% in water). It was homogenised using a mortar and pestle. The pallet was prepared using a manual hydraulic press P/N. 15.011 (Graseby Specac) by holding the formed mixture at 5 tons for one minute. The formed pallet was dried for 2 h in a vacuum oven (70 °C) before it was cut in a circle of 1 cm diameter. The leftover of the pallet was used for the thickness measurements with the following results: MT-600 – 0.46 mm, MT-800 – 0.35 mm and MT-1000 – 0.34 mm. The conductivity was measured on an Ossila's Four-Point Probe System.2.5 Electrochemical measurements
All electrochemical measurements were carried out using an MPG2 potentiostat/galvanostat (BioLogic Instruments). Anodes were prepared by grinding the prepared carbon (40 mg) and super P conductive additive (5 mg) in an agate mortar with a pestle. Subsequently, the mixture was dispersed in PVDF containing NMP (1 mL; 5 mg mL−1). The formed slurry was coated on circular copper foil (11 mm), which was priorly roughened with sandpaper, cleaned with acetone and ethanol, and dried at 60 °C. Swagelok-type cells were used for all the electrochemical experiments. Sodium metal was used as the anode (a circular piece with a diameter of 12 mm), the synthesized material served as the cathode, and 200 μL of 1.0 M NaPF6 dissolved in 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 (v/v) ethylene carbonate/diethyl carbonate (EC/DEC) was used as the electrolyte. A circular piece of a Cytiva Whatman Grade GF/C Glass Microfibre filter with a diameter of 13 mm was used as a separator. Electrochemical impedance spectroscopy (EIS) spectra were recorded at an open-circuit voltage from 10 mHz to 20 kHz. Series resistance (Rs) and charge transfer resistance (Rct) were evaluated based on the Nyquist plot. Error bars indicate the standard deviation of every experiment as calculated from 3 measurements. Whenever the C value is used, 1C is defined as 372 mA g−1. Efficiency is defined as the ratio of desodiation to sodiation capacity within one and the same cycle.
:60 (v/v) ethylene carbonate/diethyl carbonate (EC/DEC) was used as the electrolyte. A circular piece of a Cytiva Whatman Grade GF/C Glass Microfibre filter with a diameter of 13 mm was used as a separator. Electrochemical impedance spectroscopy (EIS) spectra were recorded at an open-circuit voltage from 10 mHz to 20 kHz. Series resistance (Rs) and charge transfer resistance (Rct) were evaluated based on the Nyquist plot. Error bars indicate the standard deviation of every experiment as calculated from 3 measurements. Whenever the C value is used, 1C is defined as 372 mA g−1. Efficiency is defined as the ratio of desodiation to sodiation capacity within one and the same cycle.
3. Results and discussion
Melamine-based conjugated polymers with terephthalaldehyde and isophthalaldehyde were previously carbonised in tubular ovens. However, the condensation step needed for the formation of a conjugated polymer was performed at 250 °C. This is above the boiling point of both terephthalaldehyde and isophthalaldehyde,31,35 which raises the concern that the aldehydes might not react fast enough with melamine and they might evaporate as the tubular oven is an open system. Therefore, in this work, the condensation step was performed at lower temperature, namely 150 °C (Scheme 1). Previous reports have confirmed partial condensation at this temperature.28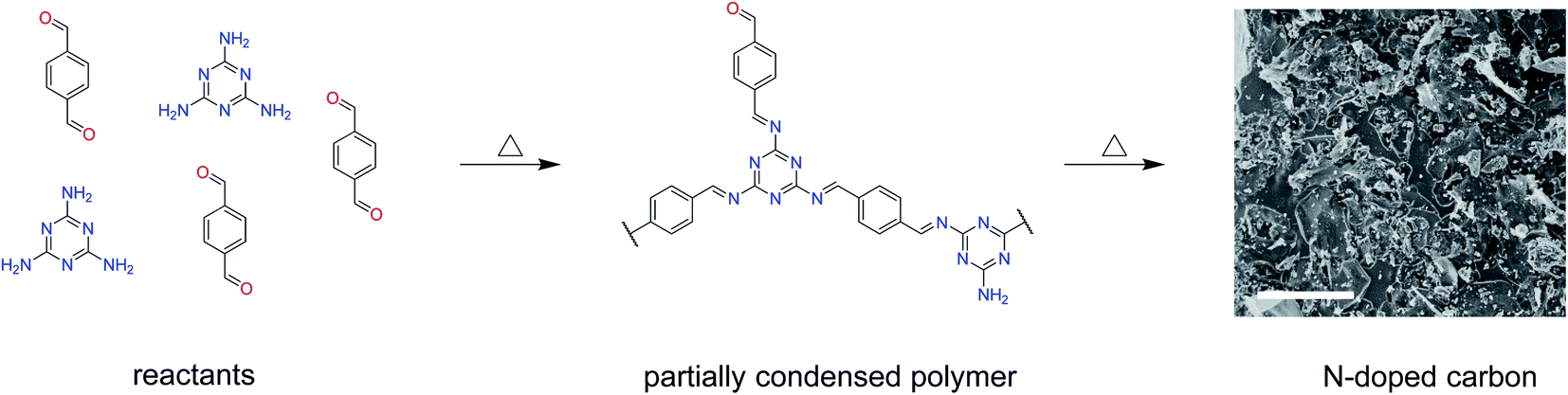 | ||
| Scheme 1 Synthesis of nitrogen-doped carbons by the carbonization of a partially condensed polymer from melamine and terephthaldehyde. N-doped carbon is represented by a SEM image of MT-1000 with the scale bar corresponding to 10 μm. | ||
The carbonization of such melamine-based networks in the chosen temperature range typically leads to nitrogen-rich carbon materials.26 Elemental analysis gives insights into the condensation process (Fig. 1a and Table S1†). MT-150 has a higher nitrogen content than the fully condensed MT polymer and a lower carbon content than the pure MT mixture. This leads to the conclusion that the argon flow facilitates the evaporation of melted terephthalaldehyde resulting in a partial removal of terephthalaldehyde from the mixture. However, excess melamine gets condensed at higher temperatures forming first a polymer and a nitrogen-rich carbon compound after subsequent heating that eventually decomposes at temperatures above 720 °C.36 The high nitrogen content remains upon carbonisation at 600 °C but drops significantly at 800 °C and even more at 1000 °C. This behaviour, along with the rising carbon contents, confirms the progression of carbonisation with the temperature. An almost constant oxygen level in all the carbonised samples indifferent of the temperature points to the introduction of oxygen upon exposing carbonised samples to air rather than from the precursors.
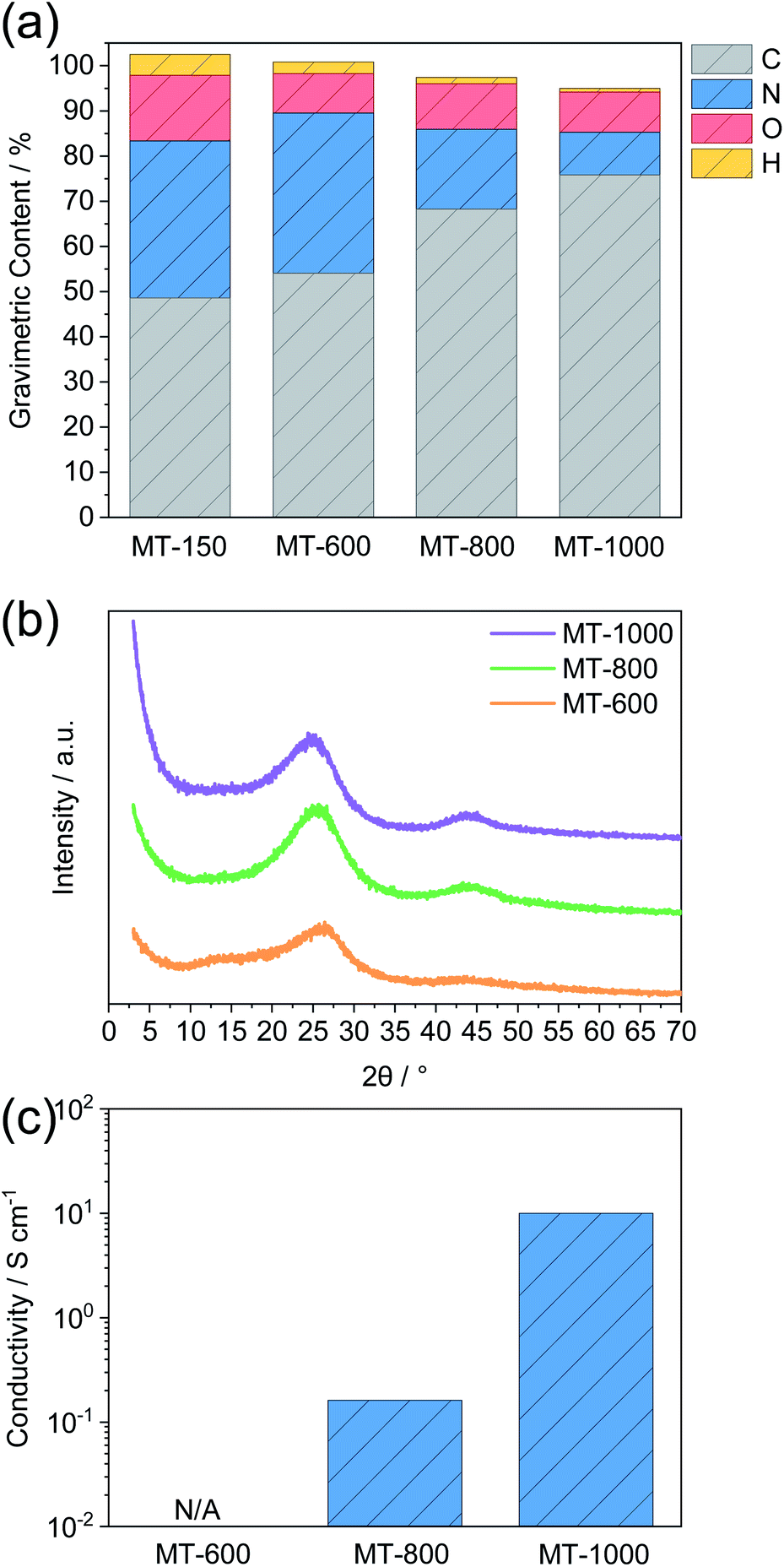 | ||
| Fig. 1 (a) Elemental analysis, (b) powder X-ray diffraction (PXRD) and (c) conductivity measurements of MT-X materials. The dotted line in (b) represents the intensity maximum of MT-800 at 25.5° 2 theta. | ||
The progress of the carbonisation with the temperature is also confirmed by PXRD (Fig. 1b). All samples show PXRD patterns that are typical of predominantly amorphous materials without a long-range order. M-600 shows several broad reflections. Two reflections around 13° and 27° 2 theta can be ascribed to the ordered stacking of units formed during condensation.37 The one at a lower angle is typical of ordered channel-like structures as typically present in graphitic carbon nitrides.37 It is of low intensity and disappears for MT-800 and MT-1000 suggesting the expected disappearance of such structural motifs after heating at higher temperature where such nitrogen-rich materials become unstable. The reflection around 26° 2 theta corresponds to the stacking of graphene- or nitrogen-rich carbon layers.38 It increases in intensity and decreases in width for MT-800 and MT-1000 as compared to MT-600 with the maximum shifting to lower values, indicating the expansion of graphitic sheets with temperature. Such a trend was also observed for comparable highly N-doped carbons,39,40 in contrast to the compression of graphitic sheets as observed for cellulose derived hard carbons.41 Together with the appearance of a peak at 44° and the elemental analysis, this indicates a higher concentration of nano-sized graphitic domains with increasing temperature, each of them representing one “Card” if the formation of hard carbons is modelled with the above-mentioned “Falling Cards Model”. The high intensity of inelastic scattering for MT-1000 at low angle points to the abundance of closed pores, as will be discussed later.
Conductivity measurements, carried out via a four-probe device, further confirm progressive graphitisation with increasing temperature (Fig. 1c). MT-600 has a conductivity even below the detection limit of the instrument. Furthermore, the conductivity of MT-1000 is approximately two orders of magnitude higher than the conductivity of MT-800. This can be ascribed to the much lower nitrogen content of the former. Furthermore, after heating at such high temperature, nitrogen is typically bonded to the carbon framework in a rather oxidized state, i.e. as graphitic nitrogen which increases the electron density in the surrounding carbon atoms.42–44 In contrast, doping with a larger amount of electron-rich nitrogen like as typically present after treatment at lower temperatures creates defects in graphene sheets and decreases the electron density of the surrounding carbon atoms, which reduces conductivity. The general trend that a small amount of nitrogen doping increases conductivity but high amounts tend to decrease it has been reported several times.45 These significant differences are also indirectly showing that a potential gradient throughout the electrode can be expected for the less conductive MT-600, whereas a more homogeneous electric potential close to the one applied to the current collector is expected for MT-1000. We would like to mention here that the expression “doping” in our case includes the substitution of carbon atoms from graphene-like domains by nitrogen atoms or the introduction of terminal nitrogen-containing groups at edges and defects. Nitrogen contents as high as those in the materials described here are far higher as in typical doping experiments in crystalline inorganic materials. In further contrast to the general understanding of “doping” examples as known from inorganic chemistry, the nitrogen is here inserted by the precursor already and not inserted post-synthetically.
The morphology of the samples investigated via scanning electron microscopy (SEM) is of similar appearance for all the samples (Fig. 2a–c). The samples consist predominantly of carbon particles with the size range of several micrometers along with smaller particles with the sizes of several hundred nanometers. Energy-dispersive X-ray spectroscopy (EDX) shows a similar C/N ratio to elemental analysis (Table S1 and Fig. S1†). This confirms that nitrogen atoms in the samples are indeed covalently bonded to the carbon framework instead of being just physically adsorbed. The significant difference in the oxygen content determined by both methods can be explained by the fact that EDX is performed under high vacuum, which leads to the desorption of oxygen-containing impurities from the carbon surface such as water, carbon dioxide and oxygen gas. Therefore, the high oxygen content in the elemental analysis may be caused by species physically adsorbed on the surface of the carbon instead of covalently bonded oxygen. Indeed, thermogravimetric analysis (Fig. S2†) confirms this as there is a significant mass loss at temperatures below 100 °C. The highest water content can be ascribed to MT-800, followed by MT-600 and MT-1000, which is in agreement with the differences between the oxygen contents of these samples determined by combustive elemental analysis and EDX. The possibility that physisorbed water can make a significant contribution to the oxygen content was recently reported for other nitrogen-rich carbon materials.46 Additionally, the presence of adsorbed water is indicated by infrared spectroscopy (Fig. S3†), which shows absorption above 3000 cm−1, characteristic of O–H groups as present in water molecules. However, there is no possibility to determine if this signal comes from water molecules or surface oxygen functionalities or to make any quantitative analysis from this measurement. Transmission electron microscopy (TEM) reveals that all three carbons are predominantly amorphous without any significant long-range order (Fig. 2d–f), confirming the previous observations from powder XRD. The Raman spectra of the samples (Fig. S4†) confirm the typical disordered character of the materials without sharply separated D- and G-bands. The fitting of the spectra with a four-band model shows ID/IG ratios in the range from 1.09 to 1.44 as widely observed for amorphous carbon materials without distinct graphitization.
 | ||
| Fig. 2 (a–c) SEM and (d–f) TEM images of (a and d) MT-600, (b and e) MT-800, and (c and f) MT-1000. The scale bar represents 5 μm (a–c) and 50 nm (d–f). | ||
The porosity of the prepared carbons was assessed using gas physisorption and small-angle X-ray scattering (SAXS). Nitrogen physisorption isotherms at 77 K (Fig. 3a) show modest nitrogen uptakes for all three carbons and therefore all samples are characterised by rather low porosity when determined with nitrogen as the probe gas at 77 K. From this method, it is suggested that MT-600 exhibits the lowest specific surface area and pore volume (Table S2†). Upon carbonisation at 800 °C, a larger volume of accessible pores is present while further increase of carbonisation temperature to 1000 °C reduces the number of pores that can be detected by nitrogen gas. Carbon dioxide physisorption isotherms at 273 K (Fig. 3b) do not follow the same trend. Although the highest uptake of carbon dioxide is observed for MT-800 it is followed by MT-600 while MT-1000 has the lowest uptake of this probe gas. This difference can be ascribed to the higher quadrupole moment exhibited by the carbon dioxide, its smaller kinetic diameter and the higher measurement temperature (i.e., faster diffusion of the probe gas). These differences can lead to the detection of pores with carbon dioxide that remain inaccessible for nitrogen but can still be suitable for sodium storage. This is of particular importance for materials with highly polar surfaces and very narrow pores, such as those discussed here.47 Especially the not fully equilibrated isotherm of MT-1000 exhibits hysteresis suggesting restricted access of CO2 to part of the internal surface area of this sample thus indicating closed pores.
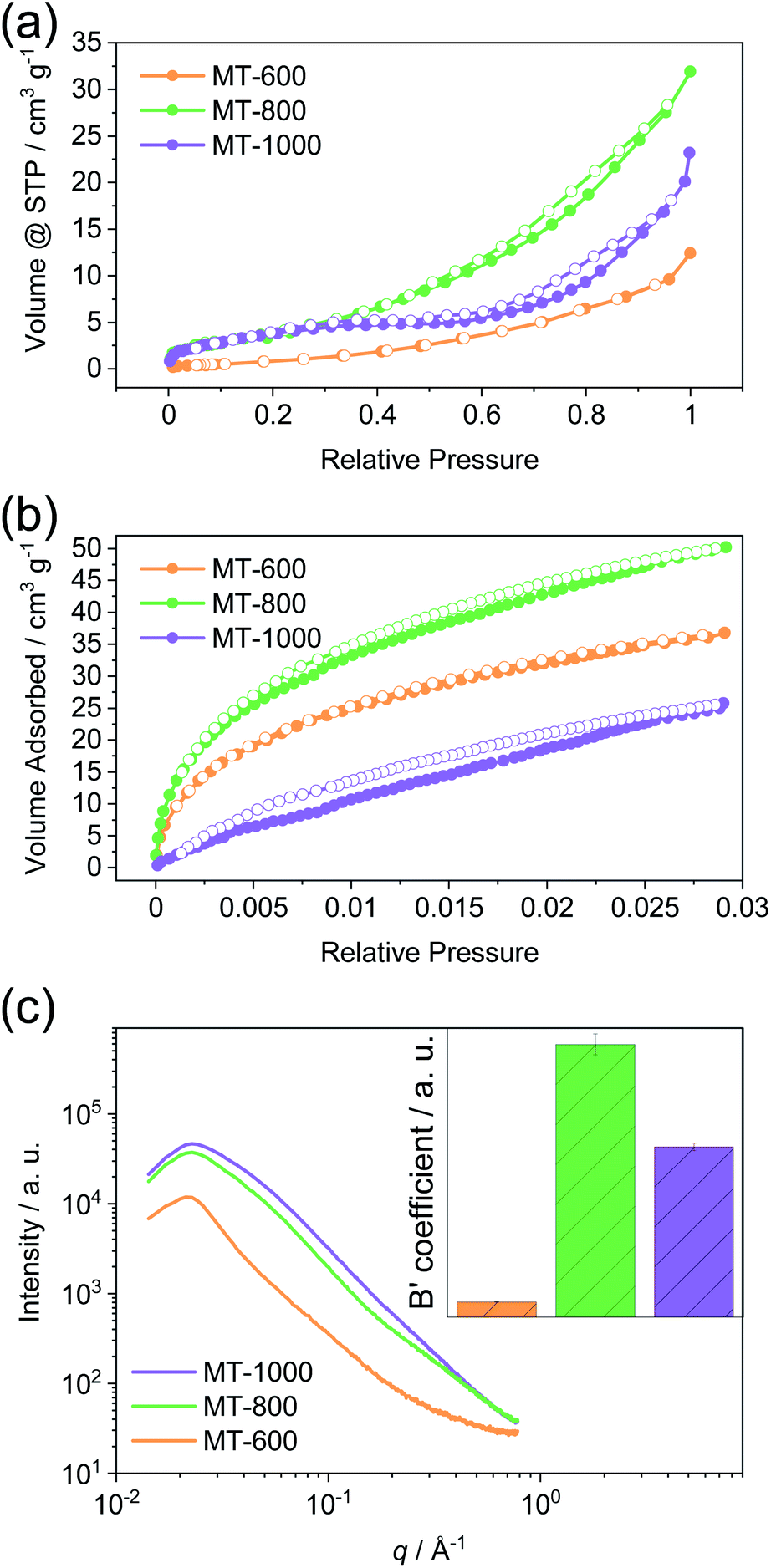 | ||
| Fig. 3 (a) Nitrogen physisorption isotherms at 77 K, (b) carbon dioxide physisorption isotherms at 273 K, and (c) small-angle X-ray scattering (SAXS) curves with the calculated B′ coefficient of MT-600, MT-800, and MT-1000. | ||
Small-angle X-ray scattering (SAXS) is able to detect inhomogeneities in the electron local density in the samples on a scale that is relevant for nanopores, as a result of a different scattering power of the empty pore and the pore walls. Previously, this method was successfully applied to study the porosity of different carbons.11,48 Unlike gas physisorption techniques, SAXS is not limited to open pores as no fluid needs to reach the interior of the pore enabling SAXS to detect the closed pores, along with the open pores and helps the full assessment of the porosity of the material. SAXS curves can be described by pore-matrix approximation represented by a function:
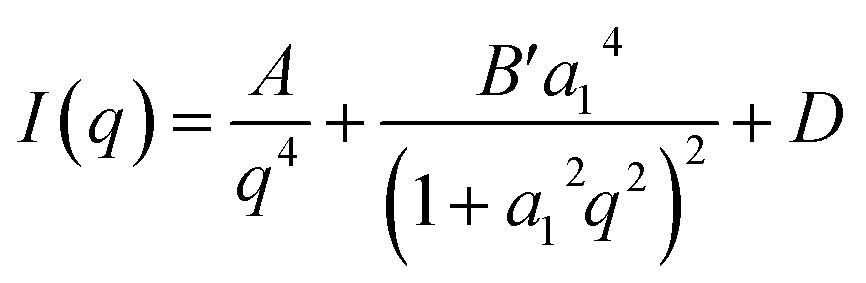
MT-800 has the largest number of pores, in agreement with data obtained from gas physisorption. However, while MT-1000 has somewhat lower porosity, MT-600 has a significantly lower porosity than both. This conclusion could not be drawn from the gas physisorption data, which can only assess open pores. Therefore, MT-600 apparently has a very low contribution of closed porosity. However, MT-800 has an overall high porosity, probably due to the high amount of gases that are formed and leave the sample upon carbonisation at this temperature. SAXS and carbon dioxide measurements agree on this point. Further heating decreases the number of open pores and MT-1000 is finally characterised by a relatively large number of closed pores.
Galvanostatic charge–discharge measurements at a current density of C/20 were used to compare the carbons electrochemically (Fig. 4). All the carbons exhibit high first sodiation and significantly lower desodiation capacity (Fig. 4a and b). This occurs due to the irreversible reactions of the electrolyte with reactive sites (such as nucleophilic nitrogen atoms or defect sites on the edges of graphitic sheets) on the carbon surface during the first sodiation forming a solid-electrolyte interphase (SEI). Therefore, efficiency, here defined as the ratio of the second and the first capacity of sodiation, decreases with the increase in the surface area.7,41 Indeed, MT-800 has the lowest efficiency, followed by MT-600, while MT-1000 exhibits the highest efficiency of 40%. Such values are also typically observed for heteroatom-rich carbon materials with a high volume of open pores. It can thus be concluded that defect sites and heteroatoms contribute to a decrease of efficiency even when they are located into "closed pores". The notably higher efficiency of MT-1000, as compared to MT-600, can be ascribed to the smaller number of heteroatoms, fewer defects and possibly also a certain volume of closed pores that might not come into contact with the electrolyte solvent and therefore no irreversible reactions may take in the closed pores. Notably, even the initial efficiency of MT-1000 does not still reach a commercially relevant level. It could be further increased by the reduction of open porosity and by the use of artificial SEIs or solvent-free electrolytes.49,50 In the context of this and many other studies, it must be stressed out that the irreversible processes occurring during SEI formation are likely to have a significant impact on the pore structure and chemical properties of the electrode materials. It is therefore more reasonable to conclude directly on the structure–property relationships of a material for SEI formation in the first cycle rather than on the electrochemical signature in the subsequent cycles. Cyclic voltammograms (CVs) recorded during the sodiation in the first cycle (Fig. S6†) all have a rather capacitor-like shape at high voltages vs. Na/Na+. Subsequently, an irreversible peak is observed around 0.4 V vs. Na/Na+ in all three materials which is caused by irreversible processes during SEI formation. Another corresponding peak is visible at 2.2 V vs. Na/Na+ for MT-800, which is shifted to a much lower potential for MT-600. All three samples exhibit the beginning of a peak approaching 2 mV and the desodiation peak is better visible around 0.2 V for all three samples in both the first and the second cycle (Fig. 4f). This peak system is well described for other materials that exhibit a plateau due to closed pores.6 In accordance to its highest first cycle efficiency in charge–discharge measurements, MT-1000 still shows a notable but overall the smallest contribution of irreversible electrochemical processes in CV measurements.
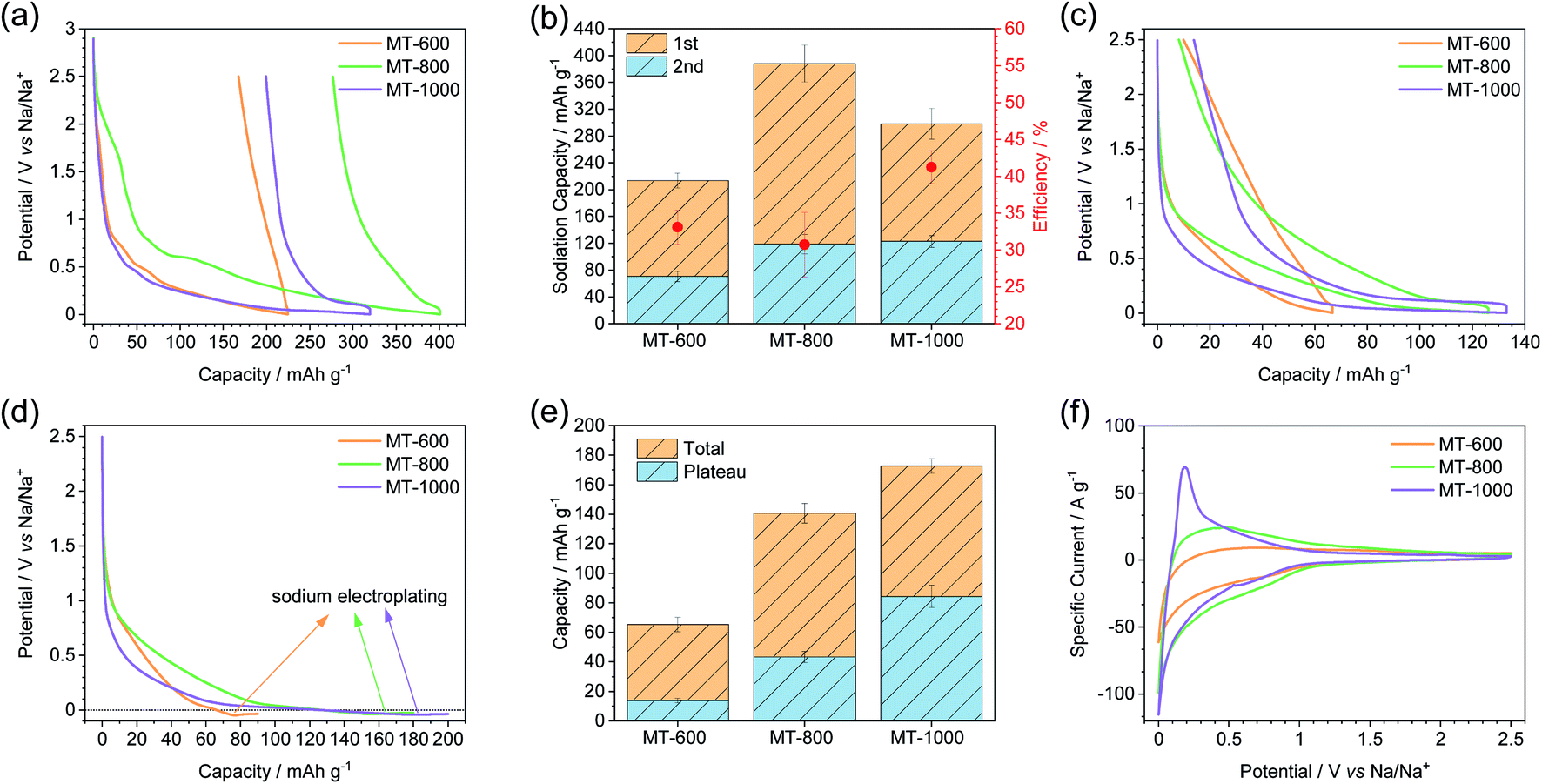 | ||
| Fig. 4 (a) First sodiation and desodiation curves, (b) first two sodiation capacities along with calculated efficiencies, (c) 5th sodiation and desodiation curves, (d) 6th sodiation curves with subsequent electroplating, (e) total capacity of the carbons along with the plateau capacity as calculated from deriving the curve, and (f) the second cycle of cyclic voltammetry of MT-600, MT-800, and MT-1000. All galvanostatic charging–discharging experiments (a–e) were performed at C/20 current density while the cyclic voltammetry experiments (f) were performed at a scan rate of 0.1 mV s−1. | ||
The charge–discharge curves of MT-600 show an insignificant plateau, while MT-800 shows a much shorter plateau than MT-1000 indicating that the closing of the pores occurs to some degree already at 800 °C but to a much higher degree at 1000 °C and therefore the sample MT-1000 shows the largest plateau below 2 mV (Fig. 4c). Sodiation up to 2 mV can underestimate possible additional sodium storage capacity within the plateau of the material.51,52 Therefore, sodiation with capacity instead of the voltage limit was performed to determine the additional sodiation capacity of the samples below 2 mV (Fig. 4d, S7 and S8†) and it is indeed shown that the capacity of all three carbons was underestimated as they all show a significant additional capacity before sodium plating occurs. The plateau is defined as a part of the sodiation curves with a constant slope, between the sloping and onset of electroplating. The slope of the curve as a function of capacity was determined from the numerical derivation of the sodiation curves (Fig. S8†). As total capacity, the plateau capacity also rises with the carbonisation temperature (Fig. 4e). Additionally, the ratio of the plateau to total capacity also increases with temperature. Therefore, MT-1000 is the material with the highest plateau capacity, which can be ascribed to the significant volume of closed pores. Similar tendencies have recently been observed for glucose-derived hard carbon materials.53
The sample with the highest plateau capacity, MT-1000, was investigated at different current densities (Fig. 5). To ensure comparable SEIs, all the samples were cycled at C/20 for 5 sodiation–desodiation cycles before sodiation at different current densities. The measurement at each current density was performed with a separate triplet of cells. As expected, the highest capacity is achieved at the lowest current density of C/20, while the capacity fades with increasing current density (Fig. 5a, S9†). The potential at which electroplating occurs varies depending on the current density, getting reduced at higher current densities, as overpotential increases at higher currents. The plateau capacity (Fig. S10†) also varies, decreasing with increasing current density (Fig. 5b).
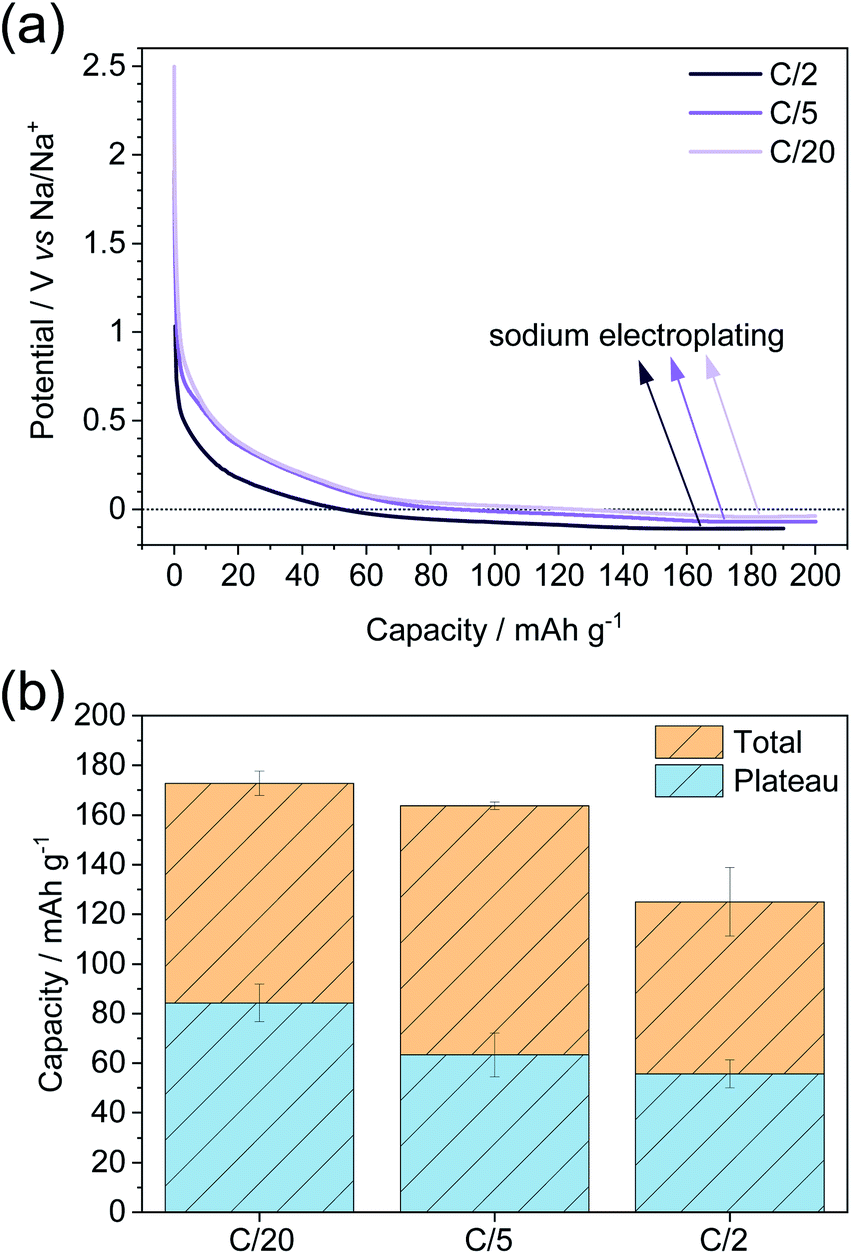 | ||
| Fig. 5 (a) Sodiation curves at different current densities and subsequent electroplating and (b) total capacity along with plateau capacity at different current densities as calculated from the 1st derivation of the curve. Before sodiation at any current density, the cells were cycled for 5 sodiation–desodiation cycles at a current density of C/20. | ||
The stability of MT-1000 upon cycling was assessed at a current density of C/5 for 60 cycles (Fig. 6a). The cells were sodiated up to 139 mA h g−1 (85% of the total capacity of MT-1000 as determined during dipping) and subsequently desodiated up to 2.5 V. Desodiation capacity is rather stable with few deviations, where efficiency goes above 100% can be ascribed to the unstable SEI and therefore further efforts should be made to stabilise the SEI. During all 60 cycles the efficiency stays close to 100%, confirming that full desodiation occurs and possible sodium electroplating that would inevitably form dendrites and therefore full desodiation would be minimized. Comparable findings have recently been published for lithium anodes and first examples for sodium also exist.54,55
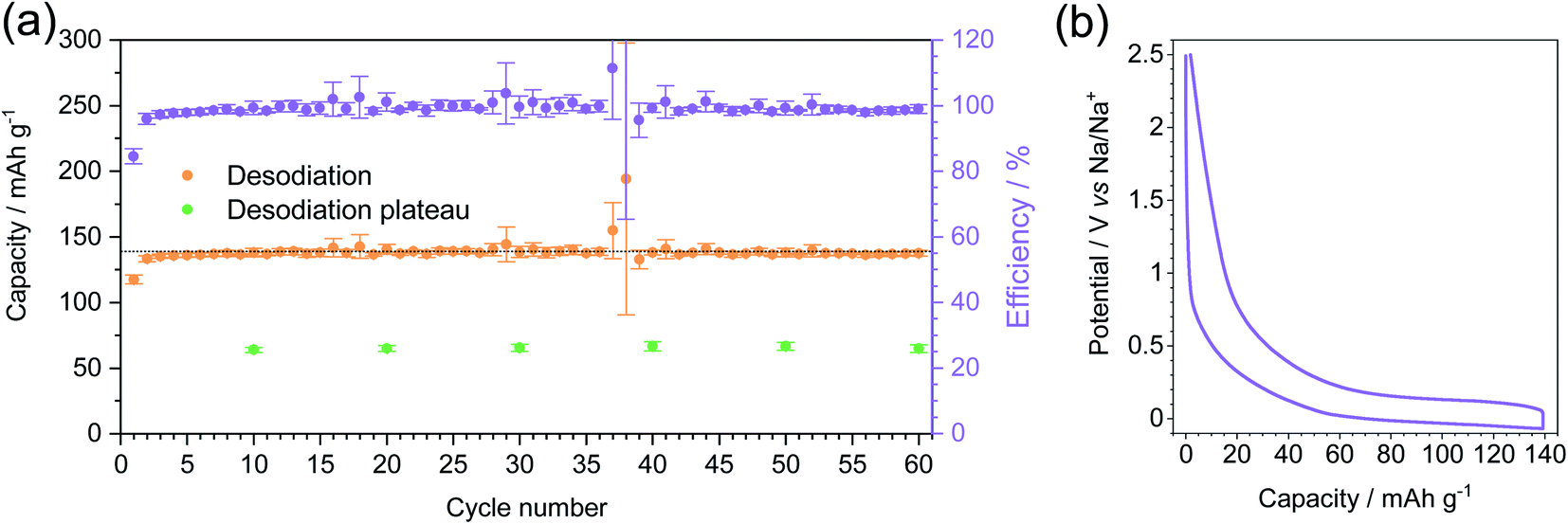 | ||
| Fig. 6 Long-term cycling of MT-1000 at a current density of C/5 with a sodiation limit of 139 mA h g−1 and desodiation limit of 2.5 V. Before sodiation at any current density the cells were cycled for 5 sodiation–desodiation cycles at a current density of C/20. (a) Desodiation capacity, desodiation plateau in selected cycles and efficiency. The dotted line represents 139 mA h g−1 sodiation capacity. (b) 5th sodiation–desodiation cycle. | ||
The plateau of desodiation was determined in selected cycles (Fig. S11†) and it exhibits rather stable values with a capacity of around 65 mA h g−1. The shape of the 5th sodiation–desodiation cycle (Fig. 6b) confirms that the plateau forms a large part of both sodiation and desodiation curves and that it is more reasonable to choose a capacity limit than a voltage limit for cycling because then the significant contribution of reversible sodium storage capacity below 0 V can be utilized.
EIS was employed to characterise the cells used for long term cycling. The Nyquist plot in Fig. S12† shows a classic semicircle and an upward line. The diameter of the semicircle is proportional to the resistivity of the cell, while the slope of the line correlates with the kinetics of the redox reaction. Before cycling the cell shows a large semicircle that gets significantly reduced after 5 cycles at a current density of C/20 indicating the stabilisation of the cell upon cycling. Prolonged cycling for 60 cycles at a current density of C/5 does not change the shape or diameter of the semicircle indicating no degradation of the cell during further cycling. All the upward lines seem to have a comparable slope, indicating no significant change in the kinetics of the battery during cycling. Unlike the last line, the line after 5 cycles at a current density of C/20 shows rather scattered points at the end indicating that prolonged cycling contributes to the stabilization of the cell.
The morphology of the electrode before and after long-term cycling was assessed by SEM (Fig. S13†). Before cycling, the electrode consists of a grainy material (MT-1000) along with bigger pieces of carbon with sizes around 10 μm, which are likely the conductive additive. After cycling there are no significant changes in the morphology confirming the stability of the electrodes. The fibers found on the top of the electrode probably come from the separator that has a fibrous structure.
4. Conclusions
A series of nitrogen-doped carbons were prepared by the carbonisation of a melamine–terephthalaldehyde mixture at various temperatures. The porosity of the prepared carbons was assessed with a combination of physisorption experiments and small-angle X-ray scattering. The carbons exhibit different heteroatom contents and porosities with variable contributions of open and closed pores due to the differences in the carbonisation temperature. During carbonisation, open pores are formed up to a point where pores close due to the rearrangement of the carbon walls, making them unreachable for fluids established for pore characterization. Although electrolyte solvent might not reach some of the closed pores, sodium ions do and upon reduction at low potential they fill the closed pores with quasi-metallic sodium, making materials rich in closed pores interesting for use as sodium anodes. Indeed, the carbon with the highest volume of closed pores proved as an interesting candidate for sodium anodes, reaching capacities higher than 170 mA h g−1, with storage in closed pores contributing to around 80 mA h g−1 at a current density of C/20. Additionally, the carbon can be cycled without significant degradation for 60 cycles. This is not a cycle number relevant for real battery applications but shows that capacity-limited sodium storage can be reversible even below 0 V vs. Na/Na+. Promising electrochemical behaviour, along with the low-priced precursors, makes the synthesised carbon a promising material for commercial sodium anodes. There is still room for optimization regarding the materials design for sodium storage under such unconventional conditions and the cycling stability along with reasons for possible capacity loss needs to be investigated in more detail. Finally, future studies will investigate the influences of the structural changes of the materials at carbonization temperatures even higher than 1000 °C.Funding
The work was supported by the Max Planck Society within the Fraunhofer-Max Planck cooperation project [CLUSTERBATT].Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge help in the laboratory by Ines Below-Lutz. We furthermore thank Heike Runge for SEM and TEM measurements, Antje Völkel for elemental analysis and Daniel Werner for SAXS. The authors thank Diana Piankova for help with SEM and TEM analysis and Jonas Pampel for fruitful discussions. Open Access funding was provided by the Max Planck Society.References
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- D. Saurel, B. Orayech, B. Xiao, D. Carriazo, X. Li and T. Rojo, Adv. Energy Mater., 2018, 8, 1703268 CrossRef.
- B. Xiao, T. Rojo and X. Li, ChemSusChem, 2019, 12, 133–144 CrossRef CAS PubMed.
- J. R. Dahn, W. Xing and Y. Gao, Carbon, 1997, 35, 825–830 CrossRef CAS.
- E. R. Buiel, A. E. George and J. R. Dahn, Carbon, 1999, 37, 1399–1407 CrossRef CAS.
- C. Bommier, W. Luo, W. Y. Gao, A. Greaney, S. Ma and X. Ji, Carbon, 2014, 76, 165–174 CrossRef CAS.
- C. Matei Ghimbeu, J. Górka, V. Simone, L. Simonin, S. Martinet and C. Vix-Guterl, Insights on the Na+ ion storage mechanism in hard carbon: discrimination between the porosity, surface functional groups and defects, Elsevier Ltd, 2018, vol. 44 Search PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- R. R. Gaddam, E. Jiang, N. Amiralian, P. K. Annamalai, D. J. Martin, N. A. Kumar and X. S. Zhao, Sustainable Energy Fuels, 2017, 1, 1090–1097 RSC.
- R. R. Gaddam, A. H. Farokh Niaei, M. Hankel, D. J. Searles, N. A. Kumar and X. S. Zhao, J. Mater. Chem. A, 2017, 5, 22186–22192 RSC.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 4428–4431 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Chem. Commun., 2016, 52, 12430–12433 RSC.
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- A. Gomez-Martin, J. Martinez-Fernandez, M. Ruttert, M. Winter, T. Placke and J. Ramirez-Rico, Chem. Mater., 2019, 31, 7288–7299 CrossRef CAS.
- N. A. Kumar, R. R. Gaddam, M. Suresh, S. R. Varanasi, D. Yang, S. K. Bhatia and X. S. Zhao, J. Mater. Chem. A, 2017, 5, 13204–13211 RSC.
- A. Agrawal, S. Janakiraman, K. Biswas, A. Venimadhav, S. K. Srivastava and S. Ghosh, Electrochim. Acta, 2019, 317, 164–172 CrossRef CAS.
- L. Chen, R. Yan, M. Oschatz, L. Jiang, M. Antonietti and K. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9067–9073 CrossRef CAS PubMed.
- Z. Wang, Y. Li and X. J. Lv, RSC Adv., 2014, 4, 62673–62677 RSC.
- S. Wang, L. Xia, L. Yu, L. Zhang, H. Wang and X. W. Lou, Adv. Energy Mater., 2016, 6, 1502217 CrossRef.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- R. Yan, K. Leus, J. P. Hofmann, M. Antonietti and M. Oschatz, Nano Energy, 2020, 67, 104240 CrossRef CAS.
- M. Volanti, D. Cespi, F. Passarini, E. Neri, F. Cavani, P. Mizsey and D. Fozer, Green Chem., 2019, 21, 885–896 RSC.
- K. Darvishi, K. Amani and M. Rezaei, Appl. Organomet. Chem., 2018, 32, e4323 CrossRef.
- E. Fiset, T. E. Rufford, M. Seredych, T. J. Bandosz and D. Hulicova-Jurcakova, Carbon, 2015, 81, 239–250 CrossRef CAS.
- M. G. Schwab, B. Fassbender, H. W. Spiess and A. Thomas, J. Am. Chem. Soc., 2009, 131, 7216–7217 CrossRef CAS PubMed.
- H. Zheng, W. Dong, X. Huang, G. Wang and Z. Wu, Green Chem., 2017, 20, 664–670 Search PubMed.
- X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081–3086 CrossRef CAS PubMed.
- X. Yang, X. Zhuang, Y. Huang, J. Jiang, H. Tian, D. Wu, F. Zhang, Y. Mai and X. Feng, Polym. Chem., 2015, 6, 1088–1095 RSC.
- N. Díez, R. Mysyk, W. Zhang, E. Goikolea and D. Carriazo, J. Mater. Chem. A, 2017, 5, 14619–14629 RSC.
- D. Xue, D. Zhu, M. Liu, H. Duan, L. Li, X. Chai, Z. Wang, Y. Lv, W. Xiong and L. Gan, ACS Appl. Nano Mater., 2018, 1, 4998–5007 CrossRef CAS.
- W. H. Lee, D. W. Lee and H. Kim, J. Electrochem. Soc., 2015, 162, F744–F749 CrossRef CAS.
- Z. Xiao, Q. Song, R. Guo, D. Kong, S. Zhou, X. Huang, R. Iqbal and L. Zhi, Small, 2018, 14, 1703569 CrossRef PubMed.
- A. Rehman and S. J. Park, Sci. Rep., 2018, 8, 6092 CrossRef PubMed.
- Y. Cui, J. Zhang, G. Zhang, J. Huang, P. Liu, M. Antonietti and X. Wang, J. Mater. Chem., 2011, 21, 13032–13039 RSC.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- M. B. Vázquez-Santos, E. Geissler, K. László, J. N. Rouzaud, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2012, 116, 257–268 CrossRef.
- R. Walczak, A. Savateev, J. Heske, N. V. Tarakina, S. Sahoo, J. D. Epping, T. D. Kühne, B. Kurpil, M. Antonietti and M. Oschatz, Sustainable Energy Fuels, 2019, 3, 2819–2827 RSC.
- R. Yan, E. Josef, H. Huang, K. Leus, M. Niederberger, J. P. Hofmann, R. Walczak, M. Antonietti and M. Oschatz, Adv. Funct. Mater., 2019, 29, 1902858 CrossRef.
- V. Simone, A. Boulineau, A. de Geyer, D. Rouchon, L. Simonin and S. Martinet, J. Energy Chem., 2016, 25, 761–768 CrossRef.
- M. Perovic, Q. Qin and M. Oschatz, Adv. Funct. Mater., 2020, 30, 1908371 CrossRef CAS.
- R. Walczak, B. Kurpil, A. Savateev, T. Heil, J. Schmidt, Q. Qin, M. Antonietti and M. Oschatz, Angew. Chem., Int. Ed., 2018, 57, 10765–10770 CrossRef CAS PubMed.
- N. Fechler, N. P. Zussblatt, R. Rothe, R. Schlögl, M. G. Willinger, B. F. Chmelka and M. Antonietti, Adv. Mater., 2016, 28, 1287–1294 CrossRef CAS PubMed.
- Z. R. Ismagilov, A. E. Shalagina, O. Y. Podyacheva, A. V. Ischenko, L. S. Kibis, A. I. Boronin, Y. A. Chesalov, D. I. Kochubey, A. I. Romanenko, O. B. Anikeeva, T. I. Buryakov and E. N. Tkachev, Carbon, 2009, 47, 1922–1929 CrossRef CAS.
- S. Youk, J. P. Hofmann, B. Badamdorj, A. Völkel, M. Antonietti and M. Oschatz, J. Mater. Chem. A, 2020, 8, 21680–21689 RSC.
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC.
- T. Zheng, W. Xing and J. R. Dahn, Carbon, 1996, 34, 1501–1507 CrossRef CAS.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- U. Subramanya, C. Chua, V. G. He Leong, R. Robinson, G. A. Cruz Cabiltes, P. Singh, B. Yip, A. Bokare, F. Erogbogbo and D. Oh, RSC Adv., 2019, 10, 674–681 RSC.
- Y. Zheng, Y. Lu, X. Qi, Y. Wang, L. Mu, Y. Li, Q. Ma, J. Li and Y. S. Hu, Energy Storage Mater., 2019, 18, 269–279 CrossRef.
- Z. Li, Z. Jian, X. Wang, I. A. Rodríguez-Pérez, C. Bommier and X. Ji, Chem. Commun., 2017, 53, 2610–2613 RSC.
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O'Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M. M. Titirici, Energy Environ. Sci., 2020, 13, 3469–3479 RSC.
- W. Ye, F. Pei, X. Lan, Y. Cheng, X. Fang, Q. Zhang, N. Zheng, D. L. Peng and M. S. Wang, Adv. Energy Mater., 2020, 10, 1902956 CrossRef CAS.
- K. Schutjajew, J. Pampel, W. Zhang, M. Antonietti and M. Oschatz, Small, 2021 DOI:10.1002/smll.202006767.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10960f |
| This journal is © The Royal Society of Chemistry 2021 |