A phosphite-based layered framework as a novel positive electrode material for Na-ion batteries†
Received
28th October 2020
, Accepted 12th January 2021
First published on 13th January 2021
Abstract
Na-ion batteries (SIBs), perceived as the most promising alternative energy storage technology, are attractive for large-scale stationary applications due to the cost effectiveness and global abundance of sodium. One of the formidable challenges in the way of their extensive commercialization is the development of suitable low-cost positive electrode materials with high energy density and long cycle life. In this study, a phosphite-based layered polyanionic material with the formula, Na2[(VOHPO3)2(C2O4)]·2H2O, has been investigated as a novel positive electrode for SIBs. The material was synthesized through a room temperature precipitation method and undergoes reversible Na+-ion insertion at an average discharge voltage of 3.65 V which is higher than that of the same V4+/V5+ redox couple of NaVOPO4 (3.4 V) in a non-aqueous Na cell. The material exhibits a high discharge capacity of ∼101 mA h g−1 at 0.1C rate in Na half-cells. Capacity fading encountered by the pristine material was overcome with the help of ball-milling with carbon. The layered material facilitates the migration of large Na+ ions, resulting in a superior rate performance (∼80 mA h g−1 at 10C rate). In addition, a long-term cycling stability over 1000 cycles was demonstrated at 2C rate with 62% capacity retention. Operando XRD studies reveal that the reversible Na+-ion insertion in the framework happens via a bi-phasic mechanism. The feasibility of full cells was demonstrated using NaTi2(PO4)3 as the negative electrode and the full cell exhibited a reversible capacity of 71 mA h g−1 at 0.1C rate and 65 mA h g−1 at 1C rate with good capacity retention.
Introduction
In recent years, the sales of electric vehicles have soared thanks to the high energy densities offered by Li-ion batteries (LIBs). The electrification of vehicles is also projected to rise exponentially in the next decades. Another large-scale application of LIBs is the stationary electrical energy storage (EES) in smart grids to store renewable energy, such as wind and solar, an important step to alleviate the problems of global warming. Since the size of stationary storage in grids is of several MW h, such large-scale projects using LIBs would rapidly increase the demand for Li.1 This may disrupt the supply chain of Li, given the low abundance and the associated geopolitical issues in the production of lithium, which in turn may trigger a tremendous escalation of Li cost. Hence, the focus on alternative energy storage technologies using other alkali ions as charge carriers has increased recently. In particular, Na-ion batteries (SIBs) are actively studied owing to the global uniform distribution and high abundance of sodium resources, and an energy storage mechanism similar to that of LIBs has helped in a rapid progress in the field of SIBs.2–4
Exploration and development of suitable positive and negative electrode materials for the reversible Na storage with high energy density and at low-cost are vital for the extensive commercialization of SIBs. Hard carbon, as a promising negative electrode material for Na-ion batteries, delivers a high capacity of >300 mA h g−1.5,6 Thus, the energy density of practical SIBs depends on the performance of positive electrodes. Of the various prospective candidates such as layered transition metal oxides7,8 and polyanionic frameworks,9 vanadium containing phosphates and fluorophosphates such as Na3V2(PO4)3,10 NaVOPO4,11–14 NaVPO4F (ref. 15 and 16) and Na3V2(PO4)2F3 (ref. 17 and 18) have attracted huge attention due to their satisfactory capacity, high redox potentials and ultra-long cycle life. To understand the structural differences in the frameworks, various polymorphs of NaVOPO4 have been studied and compared by Aparicio et al. They reported that layered αI-NaVOPO4 with tunnels in the layer exhibits high ionic conductivity due to the high mobility of Na+ ions and 3D Na diffusion pathways with low activation energy which facilitate better Na-storage properties.12 For the structural similarity, hybrid inorganic–organic open framework materials such as Na2[(VOHPOx)2(C2O4)]·2H2O (x = 3, 4) are interesting candidates for SIB positive electrodes since they have a layered structure with tunnels in the layer and suitable interplanar distance to host the large Na+ ions.19–21 These metal–organic phosphate/phosphite open frameworks (MOPOFs) were reported earlier as reversible Li-ion and K-ion hosts with excellent electrochemical properties.21–24 For example, K-half cells with the rGO/K2[(VOHPO4)2(C2O4)] composite as a positive electrode delivered a reversible capacity of >100 mA h g−1 at an average voltage of ∼3.85 V and a superior rate performance.22 In addition, an oxalate sulphate framework material, Na2Fe(C2O4)SO4·H2O, demonstrated 3 V operation as a positive electrode for SIBs.25 With two distinct channels of 5.907 and 4.126 Å, the material demonstrates facile Na migration and delivered a capacity close to its theoretical value of 88 mA h g−1 and good cycle stability over 500 cycles.
Herein, we propose a phosphite based layered open framework, Na2[(VOHPO3)2(C2O4)]·2H2O (NVPox), as a novel positive electrode material for Na-ion batteries. The material was prepared by a facile room temperature precipitation synthesis and is scalable. Owing to the combined inductive effects of the oxalate26–28 and phosphite ligands on vanadium, the material results in an average discharge voltage of 3.65 V which is higher than the 3.4 V of NaVOPO4 for the same V4+/V5+ redox couple in non-aqueous Na cells. A high discharge capacity of 101 mA h g−1 at 0.1C rate and a superior rate performance of 80 mA h g−1 at 10C were delivered by the material. The material demonstrated long-term cycling stability for 1000 cycles with 62% capacity retention at 2C rate.
Experimental
Synthesis
Na2[(VOHPO3)2(C2O4)]·2H2O was prepared in an earlier study by a hydrothermal synthesis at 120 °C for 3 days using V2O5, oxalic acid, NaOH, and H3PO3.21 In this study, the material has been prepared at room temperature as reported for another MOPOF material, K2[(VOHPO4)2(C2O4)]·6H2O.23 10 mmol of V2O5, 20 mmol of tartaric acid, 20 mmol of NaOH and 100 mmol of H3PO3 were dissolved in deionized water by sonication and the resulting blue solution was stirred at room temperature for 12 h. After slow evaporation of the solvent from the reaction mixture under ambient conditions, a green precipitate of NVPox was obtained. The suspension was filtered and the residue of the green NVPox was washed with deionized water and dried in air at room temperature. To improve the crystallinity, the material was reheated solvothermally in isopropyl alcohol at 150 °C for 2 h and thus obtained NVPox is referred from here on as p-NVPox. The pristine material was ball-milled with acetylene black using ZrO2 balls at 300 rpm for 6 h resulting in a black powder which is referred from here on as bm-NVPox.
Characterization
Structure of the prepared samples was analysed by powder X-ray diffraction (PXRD) using a Rigaku SmartLab diffractometer with Ni-filtered Cu-Kα radiation. The obtained XRD pattern was fitted by the Pawley method using TOPAS software. Morphology of the materials was studied using a scanning electron microscope (FE-SEM, JEOL, 4500 FE). The thermal behaviour of as-synthesized NVPox was tested by thermogravimetric analysis using a DTG-60, Shimadzu Corporation, Japan. The electronic conductivity of the samples was determined using a Loresta-GX MCP-T700 resistivity meter. In this study, pellets of the material with a uniform surface were prepared without any binder. The thickness and diameter of the pellets were ∼1.5 mm and 10 mm, respectively. The instrument uses a 4-pin probe which directly measures the electronic conductivity. The particle size of the samples was measured using a LA-350 laser diffraction particle size distribution analyser, Horiba, Japan. Samples were dispersed in water by ultrasonication for 2 min and used for the particle size measurement.
Electrochemical measurements were carried out with NVPox working electrodes fabricated from an aqueous slurry containing 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio of the active material, acetylene black and sodium polyacrylate (PANa) binder. The amount of conducting carbon in the electrodes of p-NVPox and bm-NVPox was the same. However, acetylene black and the active material were ball-milled in the case of bm-NVPox prior to electrode preparation. Coin cells (R 2032) were assembled in an Ar-filled glove box with the prepared electrodes, sodium metal foil as the counter electrode, 1 M NaPF6 dissolved in EC:PC (1:1 v/v) as the electrolyte and a glass fiber filter (GB-100R, ADVANTEC) as the separator. Similarly, Li-cells and K-cells were fabricated using lithium and potassium metal foils, respectively, as the counter electrodes. 1 M LiPF6 and 1 M KPF6 dissolved in EC:DEC (1:1 v/v) were used as electrolytes for Li- and K-cells, respectively. Na-ion full cells were fabricated as coin cells with bm-NVPox as the positive electrode and carbon coated NaTi2(PO4)3 (NTP) as the counter (negative) electrode with a capacity ratio of 0.8:1 (positive:negative). Structural changes of NVPox during the charge/discharge tests were examined using an operando XRD technique with a two-electrode cell (Rigaku Co., Ltd.). Electrochemical impedance spectroscopy measurements were carried out in a frequency range of 200 kHz to 0.01 Hz using a VMP3 potentiostat (Biologic). The Nyquist plots were fitted with an equivalent circuit using ZView software (Scribner).
:2:1 ratio of the active material, acetylene black and sodium polyacrylate (PANa) binder. The amount of conducting carbon in the electrodes of p-NVPox and bm-NVPox was the same. However, acetylene black and the active material were ball-milled in the case of bm-NVPox prior to electrode preparation. Coin cells (R 2032) were assembled in an Ar-filled glove box with the prepared electrodes, sodium metal foil as the counter electrode, 1 M NaPF6 dissolved in EC:PC (1:1 v/v) as the electrolyte and a glass fiber filter (GB-100R, ADVANTEC) as the separator. Similarly, Li-cells and K-cells were fabricated using lithium and potassium metal foils, respectively, as the counter electrodes. 1 M LiPF6 and 1 M KPF6 dissolved in EC:DEC (1:1 v/v) were used as electrolytes for Li- and K-cells, respectively. Na-ion full cells were fabricated as coin cells with bm-NVPox as the positive electrode and carbon coated NaTi2(PO4)3 (NTP) as the counter (negative) electrode with a capacity ratio of 0.8:1 (positive:negative). Structural changes of NVPox during the charge/discharge tests were examined using an operando XRD technique with a two-electrode cell (Rigaku Co., Ltd.). Electrochemical impedance spectroscopy measurements were carried out in a frequency range of 200 kHz to 0.01 Hz using a VMP3 potentiostat (Biologic). The Nyquist plots were fitted with an equivalent circuit using ZView software (Scribner).
Results and discussion
Crystal structure
X-Ray powder diffraction pattern of p-NVPox is shown in Fig. 1a. The XRD data show that the structure is similar to that of the hydrothermally obtained Na2[(VOHPO3)2(C2O4)]·2H2O, reported earlier21 and without any impurity phases. Whole powder pattern fitting of the peaks by the Pawley method indicates that it can be well indexed to a triclinic lattice of the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The lattice parameters were calculated as a = 6.3257(10) Å, b = 6.7612(10) Å, c = 8.0855(16) Å, α = 104.997(8)°, β = 101.636(16)°, and γ = 98.975(11)°, which are close to the previously reported values. The material has a layered open-framework structure consisting of two-dimensional anionic sheets of [(VOHPO3)(C2O4)], stacked along the b-axis with Na+ ions and H2O in the interlayer space (shown in Fig. 1c). The anionic sheet (shown in Fig. 1d) parallel to the (01−1) plane is composed of the one-dimensional chains of VOHPO3 along the a-axis, which are interconnected by oxalate ligands to form the 2D open frameworks having a large tunnel in the sheet. In the VOHPO3 chains, VO6 octahedra are corner-shared with three different (HPO3)2− tetrahedra. The interlayer space was calculated to be 5.8 Å from the peak position of 01−1 reflection in Fig. 1a. Further details on the crystal structure of Na2[(VOHPO3)2(C2O4)]·2H2O can be found in a previous paper.21 TGA of the material (shown in Fig. S1†) infers that the dehydration occurs in two steps with formation of an intermediate monohydrate compound. While the first dehydration occurs at 80–125 °C, the subsequent dehydration happens at 170–220 °C. The TGA also infers that the oxalate group decomposes at temperatures of >360 °C, indicating its good thermal stability. The SEM image of p-NVPox (Fig. 1b) demonstrates that the material contains primary particles with plate-like morphology which aggregate to form secondary particles with a size of several microns.
space group. The lattice parameters were calculated as a = 6.3257(10) Å, b = 6.7612(10) Å, c = 8.0855(16) Å, α = 104.997(8)°, β = 101.636(16)°, and γ = 98.975(11)°, which are close to the previously reported values. The material has a layered open-framework structure consisting of two-dimensional anionic sheets of [(VOHPO3)(C2O4)], stacked along the b-axis with Na+ ions and H2O in the interlayer space (shown in Fig. 1c). The anionic sheet (shown in Fig. 1d) parallel to the (01−1) plane is composed of the one-dimensional chains of VOHPO3 along the a-axis, which are interconnected by oxalate ligands to form the 2D open frameworks having a large tunnel in the sheet. In the VOHPO3 chains, VO6 octahedra are corner-shared with three different (HPO3)2− tetrahedra. The interlayer space was calculated to be 5.8 Å from the peak position of 01−1 reflection in Fig. 1a. Further details on the crystal structure of Na2[(VOHPO3)2(C2O4)]·2H2O can be found in a previous paper.21 TGA of the material (shown in Fig. S1†) infers that the dehydration occurs in two steps with formation of an intermediate monohydrate compound. While the first dehydration occurs at 80–125 °C, the subsequent dehydration happens at 170–220 °C. The TGA also infers that the oxalate group decomposes at temperatures of >360 °C, indicating its good thermal stability. The SEM image of p-NVPox (Fig. 1b) demonstrates that the material contains primary particles with plate-like morphology which aggregate to form secondary particles with a size of several microns.
 |
| | Fig. 1 (a) Whole pattern fitting results (Pawley method) of the XRD pattern of p-NVPox. Red circles represent the experimental values while the calculated pattern is shown as blue lines. The black line represents the difference between the calculated and observed intensities. The vertical bars correspond to the positions of Bragg reflections. (b) The SEM image of p-NVPox. (c and d) Schematic illustrations of crystal structures for Na2[(VOHPO3)2(C2O4)]·2H2O; (c) the layered structure with Na+ ions in the interlayer space (H2O in the interlayers is not shown for clarity) and (d) the intralayer structure of the [(VOHPO3)2(C2O4)] sheet parallel to the (01−1) plane. | |
Electrochemical Na extraction/insertion
The Na extraction/insertion performance of the layered NVPox was investigated using Na half-cells at a current density of 11 mA g−1 (∼0.1C) in the potential range of 2.0–4.3 V vs. Na. Charge–discharge profiles of p-NVPox for selected cycles are shown in Fig. 2a. The charge processes, during which Na+ ion extraction and the simultaneous oxidation of V4+ to V5+ should occur, are characterized by three distinct plateaus at 3.58, 3.82 and 3.97 V in Fig. 2a and from the peaks in the corresponding dQ/dV plot (Fig. 3a). However, the discharge processes involve the insertion of Na into the framework in two steps at 3.73 V and 3.49 V (Fig. 2a). The difference in the number of charge and discharge plateaus can be inferred as the difference in the number of intermediate compositions formed during the charge/discharge. A similar phenomenon was reported for the NaFePO4 phase in Na cells, in which an intermediate composition of Na0.7FePO4 was formed during the charge processes while the discharge processes happened without any intermediate composition.29,30 The initial discharge capacity of p-NVPox was 101 mA h g−1, which is reasonably close to the theoretical capacity of 115 mA h g−1. However, the initial charge capacity of 177 mA h g−1 is higher than the theoretical capacity. The excess oxidative capacity may be attributed to the side reactions of the material with the electrolyte.31 As shown in the dQ/dV plots of charge curves (Fig. S2†), the intensity of the 3.97 V peak decreases from the 1st cycle to the 4th cycle which can be attributed to the side reaction with the electrolyte.
 |
| | Fig. 2 (a) Charge–discharge profiles for selected cycles of p-NVPox at 0.1C current rate in the potential range of 2.0–4.3 V and (b) comparison of the cycling stability of p-NVPox and bm-NVPox at 0.1C rate. | |
 |
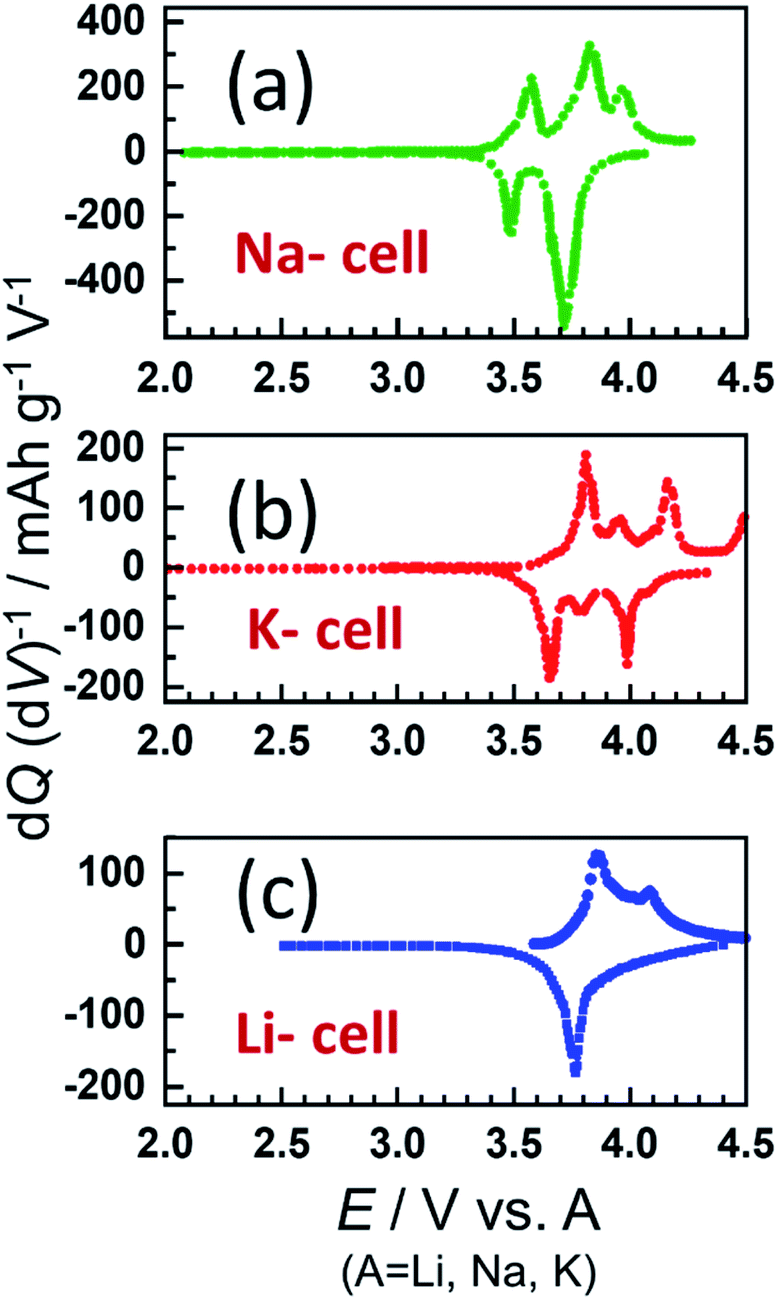
| | Fig. 3 The dQ/dV plots of p-NVPox in (a) Na, (b) K and (c) Li cells for the 10th cycle. | |
To compare the Li- and K-insertion properties of the material, Li//p-NVPox and K//p-NVPox half-cells were cycled at 0.1C. During the initial charge, Na+ ions are removed from the framework while the initial discharge predominantly involves the insertion of Li and K in the Li- and K-cells, respectively. Charge/discharge curves of the material in the three half-cells and the dQ/dV plots of the 10th cycle are compared in Fig. S3† and 3a–c, respectively. The dQ/dV plots indicate that the Li insertion/extraction into/from the framework happens at a higher potential than in Na cells. Similarly, a higher potential was observed for the same redox couple during K insertion/extraction. Average discharge voltages of 3.76 V and 3.75 V were exhibited by the Li- and K- half cells which is ∼0.12 V higher than that of Na-half cells. However, the discharge capacities of Na cells were higher than those of the Li- and K- cells (Fig. 2a and S4†). From the dQ/dV curves in Fig. 3a–c, it is found that the number of plateaus in the discharge curves increases in the order of Li < Na < K and a similar trend is reported for layered transition metal oxide cathode materials.32
During the initial cycles of p-NVPox, high capacities of ∼100 mA h g−1 were demonstrated which gradually fades during the cycling. At the end of 100 cycles, a discharge capacity of only 53 mA h g−1 was delivered by the p-NVPox electrodes as shown in Fig. 2b. The inferior cycling stability may be linked to the poor electronic conductivity of the material (2 × 10−7 S cm−1). In earlier reports of MOPOF materials in Li and K cells, improvement in the cycling stability was achieved through enhancement of electronic conduction in the electrode by compositing with reduced graphene oxide (rGO).22,23 Since NVPox was prepared in this study by a slow precipitation method, compositing with rGO would result in an inhomogeneous mixture of NVPox and rGO. Therefore, an alternative process was adopted to improve the electronic conductivity of NVPox. The pristine material was ball-milled with acetylene black at 300 rpm for 6 h. The bm-NVPox exhibits a high electronic conductivity of 2.05 × 10−1 S cm−1. This ball-milling process did not induce any phase change as confirmed by the XRD pattern of bm-NVPox (Fig. S5†). However, broadening of the peaks infers that the crystallite size was mechanically reduced due to the milling. This is further supported by the SEM micrographs (Fig. S6†) and particle size distribution measurements (Fig. S7†) indicating a decrease in the size of aggregated particles. As shown in Fig. 2b and S8,† bm-NVPox exhibited better cycling stability. The discharge capacity of bm-NVPox for the initial cycles was ∼93 mA h g−1 and after 100 cycles, a discharge capacity of 81 mA h g−1 was retained. In comparison, the p-NVPox electrodes demonstrated a poor capacity retention of only 53 mA h g−1 after 100 cycles at the same current density. The improvement in the capacity of bm-NVPox can be attributed to the following two factors. The average particle size of Na2[(VOHPO3)2(C2O4)]·2H2O is smaller in bm-NVPox as observed from the particle size measurement and SEM micrographs. In addition, the ball-milling results in improved contact of NVPox and acetylene black carbon due to the smaller particle size of NVPox, enhancing the electronic conductivity.
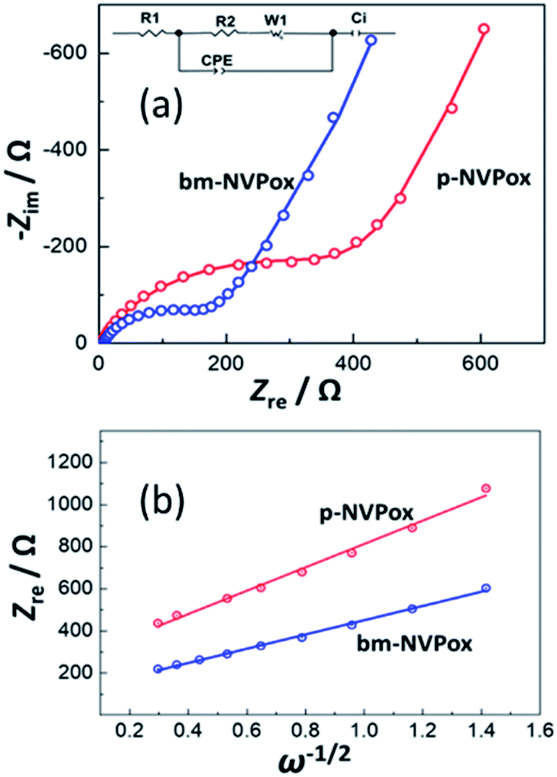
To further investigate the improved performance of bm-NVPox, EIS measurements of p-NVPox and bm-NVPox were carried out. Nyquist plots (Zrevs. Zim) of the two electrodes are compared in Fig. 4a. Fitting of these Nyquist plots with an equivalent circuit (show in the inset of Fig. 4a) reveals that the charge transfer resistances (Rct) of p-NVPox and bm-NVPox electrodes were 475 and 203 Ω, respectively. The significant decrease in the resistance after the ball-milling shows the improved electronic conductivity of the material. The bm-NVPox electrodes also exhibited lower Rct than p-NVPox at the charged state (4.3 V) and the discharged state (2.0 V) as shown in Fig. S9.† The Na+-ion diffusion coefficient in the material was calculated by determining the Warburg coefficients (σ) from the low-angular frequencies of the impedance spectra. The slope of a linear plot of Zreversus the reciprocal root-square of the angular frequencies (ω−1/2) gives the Warburg coefficient. Fig. 4b compares the Zrevs. ω−1/2 plots of p-NVPox and bm-NVPox electrodes. Using the calculated slope (σ) values, the diffusion coefficient (DNa) can be deduced from eqn (1).
| |  | (1) |
where
R is the gas constant,
T is the absolute temperature,
F is the Faraday's constant,
A is the geometrical electrode area,
n is the number of electrons per molecule, and
C is the molar concentration of Na
+ ions. The
DNa of bm-NVPox (3.14 × 10
−14 cm
2 s
−1) was nearly twice that of p-NVPox (1.7 × 10
−14 cm
2 s
−1). The downsizing and modifying crystallinity of NVPox particles by ball-milling could result in a faster Na
+-ion diffusivity in the bm-NVPox. Thus, the bm-NVPox electrodes exhibit lower charge transfer resistance and higher diffusion coefficients in the framework.
 |
| | Fig. 4 (a) Nyquist plots of p-NVPox and bm-NVPox before cycling, and (b) Zrevs. ω−1/2 plots of p-NVPox and bm-NVPox before cycling. | |
Rate capability
The rate performance of the bm-NVPox electrodes in Na cells was examined at different current rates ranging from 0.2C to 10C (1C corresponds to 115 mA g−1) on discharge. The charge current was kept constant at 11 mA g−1 throughout the cycling. Fig. 5a shows the comparison of discharge curves of the bm-NVPox electrode at these rates. The ball-milled sample demonstrated a better rate capability than the p-NVPox electrodes (Fig. 5b). The bm-NVPox electrode delivered higher reversible capacities of 91, 88, 86, 84, 82 and 80 mA h g−1 at 0.2, 0.5, 1, 2, 5, and 10C rates, respectively, compared to 90, 83, 74, 64, 57 and 34 mA h g−1 of the p-NVPox electrode (Fig. S10† and 5b). When the rate was reduced to 0.2C, both bm-NVPox and p-NVPox delivered 86 mA h g−1 which corresponds to ∼95% of initial discharge capacity. The excellent capacity retention of the bm-NVPox electrodes, at high current rates of up to 10C, highlights the facile Na-ion migration in the framework.
 |
| | Fig. 5 Rate capability and long-term cycling of NVPox: (a) discharge profiles of bm-NVPox at current rates of 0.2, 0.5, 1, 2, 5 and 10C; (b) comparison of capacity retention of p-NVPox and bm-NVPox at these current rates; and (c) capacity retention plots of bm-NVPox at 2C rate for 1000 cycles. The inset figure in (c) shows the dQ/dV plots of discharge curves of selected cycles. | |
Long-term cycling
The long-term cyclability of the material was evaluated at a current rate of 2C. Both the charge and the discharge processes were carried out at 2C rate with a constant voltage charging step at 4.3 V for 15 min. As shown in Fig. 5c, the capacity of the bm-NVPox electrode for initial cycles was ∼80 mA h g−1. The capacity gradually decreases during the cycling and a capacity retention of 62% was observed after 1000 cycles. Throughout the cycling of bm-NVPox, the average discharge voltage remained at 3.6 V (as shown in the inset of Fig. 5c). In contrast, p-NVPox demonstrated a poor cycling stability at 2C rate as shown in Fig. S11.† The capacity fading of p-NVPox is more pronounced at 2C rate than at 0.1C rate, delivering only 17 mA h g−1 after 200 cycles. The excellent voltage stability and high capacity retention of bm-NVPox demonstrate the stability of the layered open framework structure during the long-term cycling.
Structural evolution
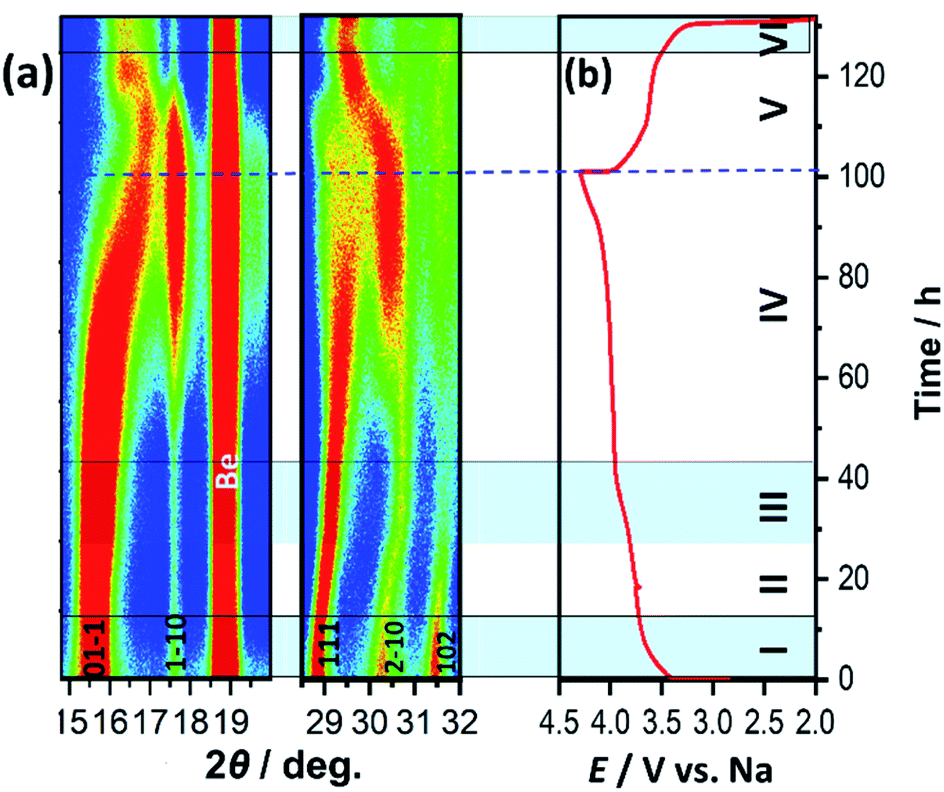
Structural evolution of NVPox during the sodiation/de-sodiation was studied by operando XRD using an in situ XRD cell assembled in the Na//p-NVPox cell configuration. Contour maps of the XRD patterns of the first cycle for selected 2θ regions are shown in Fig. 6a and the corresponding charge–discharge profile is shown in Fig. 6b. Using differential voltage (dV/dQ) analysis of the first charge curve (Fig. S12†), it is estimated that a single-phase domain exists in region I (Fig. 6b). In this region, Na extraction from the framework results in a solid solution and 111, 2−10 and 102 Bragg peaks initially observed at 2θ = 28.9°, 30.1°, and 31.2°, respectively, shift to higher angles. It is followed by a biphasic reaction (region II) and a monophasic reaction (region III) to form intermediate materials, Na2(1−x)[(VOHPO3)2(C2O4)]·2H2O. Since the peak positions are closer to the initial phase, it is estimated that the structure of the intermediate phases is similar to the initial phase. Upon further Na extraction, another biphasic reaction proceeds in region IV. The region is characterized by increasing intensities of peaks at 16.9, 17.6, 19.6, 30.4, and 31.6°, which can be assigned to the de-sodiated phase, [(VOHPO3)2(C2O4)]·2H2O. The decrease in the interplanar distance upon charging is inferred by the higher-angle shift of the 01−1 peak. The side reaction of the material with the electrolyte, which results in a large irreversible capacity, overlaps in region IV. As seen in Fig. S12 and S13,† the capacity in the plateau region at 3.97 V is larger during the operando XRD measurement than in coin cells. The fact indicates that the side reactions with the electrolyte are more pronounced in the in situ cell configuration compared to coin cells due to the more complicated internal structure of the in situ cell and very slow in situ operation at a low current density. During the first discharge, the intensity of the peaks at 17.6 and 19.6° assigned to the [(VOHPO3)2(C2O4)]·2H2O phase decreases with increasing amount of Na. This is indicative of a biphasic reaction in region V, and the de-sodiated phase coexists with an intermediate phase Na2y[(VOHPO3)2(C2O4)]·2H2O. Followed by this two-phase domain, there is a monophasic region until the lower cut-off voltage (region VI). At the end of discharge, the peak position of the main diffraction peaks is slightly different from that of the initial phase. However, an ex situ XRD pattern at the discharged state (Fig. S14†) displays almost the same pattern as that of the pristine electrode. The fact indicates that the structural changes during Na extraction and insertion are completely reversible. The different peak position at the end of the first discharge in the operando XRD pattern may be explained by incomplete Na-insertion as only the higher-voltage plateau region was observed in the initial discharge curve during the operando measurement. During the second cycle (Fig. S15†), the charged state has the same interplanar distance as the first charge as inferred from the peak position at 17.6° for both cycles. However, due to the significantly low intensities of the peaks and closely related structures of the intermediate phases, it is difficult to understand the complete mechanism of Na storage in the material with the current data. Measurements with synchrotron XRD or neutron diffraction may provide further insight into the mechanism.
 |
| | Fig. 6 Structural evolution during Na extraction/insertion from/into p-NVPox: (a) contour maps of operando XRD during the first cycle for selected 2θ regions and (b) Charge–discharge profile during the operando XRD measurement. | |
Full cells
To evaluate the feasibility of the material for application in Na-ion full cells, coin cells were fabricated using bm-NVPox as the positive electrode and carbon coated NaTi2(PO4)3 (NTP) as the negative electrode. Fig. S16† shows the charge–discharge profiles of the NTP electrode in Na half-cells. The half-cells delivered a reversible capacity of ∼104 mA h g−1 at 2.1 V vs. Na with good capacity retention (shown in Fig. S17†) indicating its suitability as a negative electrode for full cell studies. Voltage profiles of the full cell NTP//bm-NVPox cycled at a current rate of 0.1C are shown in Fig. 7a. In the first cycle, a discharge capacity of 71 mA h g−1 was obtained. Charge capacities of the initial few cycles were higher as noticed in the half-cells. The current rate was increased to 1C after 5 cycles and the discharge capacity decreased slightly to 65 mA h g−1. The full cell manifests a good capacity retention as shown in Fig. 7b, delivering 61 mA h g−1 after 50 cycles at 1C rate.
 |
| | Fig. 7 Demonstration of full cells of NVPox: (a) Charge–discharge profiles for the first three cycles of the NTP//bm-NVPox cell at a current rate of 0.1C; (b) capacity retention plot at 1C rate (the capacity is calculated based on the weight of the positive electrode). | |
Conclusions
A vanadium based hybrid inorganic–organic open framework material, Na2[(VOHPO3)2(C2O4)]·2H2O, with a layered structure was investigated as a Na insertion positive electrode for the first time. It was prepared via a room temperature precipitation method which enables scaling-up. A high discharge capacity of 101 mA h g−1 was delivered by Na-half cells at an average discharge voltage of 3.65 V which is slightly higher than the working potential of the pure inorganic material NaVOPO4 (∼3.4 V) functioning on the same V4+/V5+ redox couple. Since the pristine material (p-NVPox) suffered from capacity decay due to its poor electronic conductivity, ball-milling was employed. The bm-NVPox exhibited higher capacities at high current rates up to 10C (80 mA h g−1) while p-NVPox delivered only 32 mA h g−1 at 10C. In particular, the material can be cycled over 1000 cycles with a satisfactory retention rate of 62% at a high rate of 2C. The enhanced performance of bm-NVPox electrodes can be attributed to the enhanced electronic conductivity and faster Na diffusion due to smaller particles and modified crystallinity. The feasibility of the full cell was also demonstrated using NaTi2(PO4)3 as the counter electrode. The NTP//bm-NVPox full cells exhibited discharge capacities of 71 and 65 mA h g−1 at current rates of 0.1C and 1C, respectively, with good capacity retention for 50 cycles.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was partly funded by the MEXT program “ESICB” (No. JPMXP0112101003), the Japan Science and Technology Agency (JST) through A-STEP program (No. JPMJTS1611), and JSPS KAKENHI Grant Numbers JP16K14103, JP16H04225, JP18K14327, and JP20K05690.
References
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy Mater., 2020, 10, 2001310 CrossRef CAS.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Chem. Rec., 2018, 18, 459–479 CrossRef CAS.
- A. Kamiyama, K. Kubota, T. Nakano, S. Fujimura, S. Shiraishi, H. Tsukada and S. Komaba, ACS Appl. Energy Mater., 2020, 3, 135–140 CrossRef CAS.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- K. Kubota, S. Kumakura, Y. Yoda, K. Kuroki and S. Komaba, Adv. Energy Mater., 2018, 8, 1703415 CrossRef.
- C. Delmas, D. Carlier and M. Guignard, Adv. Energy Mater., 2020, 2001201 Search PubMed.
- P. Barpanda, L. Lander, S.-i. Nishimura and A. Yamada, Adv. Energy Mater., 2018, 8, 1703055 CrossRef.
- W. Duan, Z. Zhu, H. Li, Z. Hu, K. Zhang, F. Cheng and J. Chen, J. Mater. Chem. A, 2014, 2, 8668–8675 RSC.
- Y. Fang, Q. Liu, L. Xiao, Y. Rong, Y. Liu, Z. Chen, X. Ai, Y. Cao, H. Yang, J. Xie, C. Sun, X. Zhang, B. Aoun, X. Xing, X. Xiao and Y. Ren, Chem, 2018, 4, 1167–1180 CAS.
- P. A. Aparicio, J. A. Dawson, M. S. Islam and N. H. de Leeuw, J. Phys. Chem. C, 2018, 122, 25829–25836 CrossRef CAS.
- G. He, A. Huq, W. H. Kan and A. Manthiram, Chem. Mater., 2016, 28, 1503–1512 CrossRef CAS.
- J. Song, M. Xu, L. Wang and J. B. Goodenough, Chem. Commun., 2013, 49, 5280–5282 RSC.
- J. Barker, M. Y. Saidi and J. L. Swoyer, Electrochem. Solid-State Lett., 2003, 6, A1–A4 CrossRef CAS.
- M. Law and P. Balaya, Energy Storage Materials, 2018, 10, 102–113 CrossRef.
- L. Zhu, H. Wang, D. Sun, Y. Tang and H.-Y. Wang, J. Mater. Chem. A, 2020, 8, 21387–21407 RSC.
- C. Zhu, C. Wu, C.-C. Chen, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Chem. Mater., 2017, 29, 5207–5215 CrossRef CAS.
- J. F. Colin, T. Bataille, S. E. Ashbrook, N. Audebrand, L. Le Pollès, J. Y. Pivan and E. Le Fur, Inorg. Chem., 2006, 45, 6034–6040 CrossRef CAS.
- M. Nagarathinam, K. Saravanan, E. J. H. Phua, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, Angew. Chem.,
Int. Ed., 2012, 51, 5866–5870 CrossRef CAS.
- A. S. Hameed, M. V. Reddy, N. Sarkar, B. V. R. Chowdari and J. J. Vittal, RSC Adv., 2015, 5, 60630–60637 RSC.
- A. S. Hameed, A. Katogi, K. Kubota and S. Komaba, Adv. Energy Mater., 2019, 9, 1902528 CrossRef CAS.
- A. S. Hameed, M. V. Reddy, M. Nagarathinam, T. Runčevski, R. E. Dinnebier, S. Adams, B. V. R. Chowdari and J. J. Vittal, Sci. Rep., 2015, 5, 16270 CrossRef CAS.
- A. Shahul Hameed, M. Nagarathinam, M. Schreyer, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, J. Mater. Chem. A, 2013, 1, 5721–5726 RSC.
- T. Song, W. Yao, P. Kiadkhunthod, Y. Zheng, N. Wu, X. Zhou, S. Tunmee, S. Sattayaporn and Y. Tang, Angew. Chem., Int. Ed., 2020, 59, 740–745 CrossRef CAS.
- W. Yao, A. R. Armstrong, X. Zhou, M.-T. Sougrati, P. Kidkhunthod, S. Tunmee, C. Sun, S. Sattayaporn, P. Lightfoot, B. Ji, C. Jiang, N. Wu, Y. Tang and H.-M. Cheng, Nat. Commun., 2019, 10, 3483 CrossRef.
- X. Wang, R. Kurono, S.-I. Nishimura, M. Okubo and A. Yamada, Chem.–Eur. J., 2015, 21, 1096–1101 CrossRef CAS.
- W. Yao, M.-T. Sougrati, K. Hoang, J. Hui, P. Lightfoot and A. R. Armstrong, Chem. Mater., 2017, 29, 2167–2172 CrossRef CAS.
- W. Tang, X. Song, Y. Du, C. Peng, M. Lin, S. Xi, B. Tian, J. Zheng, Y. Wu, F. Pan and K. P. Loh, J. Mater. Chem. A, 2016, 4, 4882–4892 RSC.
- P. Moreau, D. Guyomard, J. Gaubicher and F. Boucher, Chem. Mater., 2010, 22, 4126–4128 CrossRef CAS.
- X. Bie, K. Kubota, T. Hosaka, K. Chihara and S. Komaba, J. Mater. Chem. A, 2017, 5, 4325–4330 RSC.
- Y. Hironaka, K. Kubota and S. Komaba, Chem. Commun., 2017, 53, 3693–3696 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10517a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Mirai
Ohara
a,
Kei
Kubota
ab,
Mirai
Ohara
a,
Kei
Kubota