
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar-driven valorisation of glycerol on BiVO4 photoanodes: effect of co-catalyst and reaction media on reaction selectivity†
Yi-Hsuan
Wu
 a,
Denis A.
Kuznetsov
*a,
Nicholas C.
Pflug
b,
Alexey
Fedorov
a and
Christoph R.
Müller
*a
a,
Denis A.
Kuznetsov
*a,
Nicholas C.
Pflug
b,
Alexey
Fedorov
a and
Christoph R.
Müller
*a
aDepartment of Mechanical and Process Engineering, ETH Zürich, Leonhardstrasse 21, 8092 Zürich, Switzerland. E-mail: denisk@ethz.ch; muelchri@ethz.ch
bDepartment of Environmental Systems Science, ETH Zürich, Universitätstrasse 16, 8092 Zurich, Switzerland
First published on 4th February 2021
Abstract
The electrochemical valorization of glycerol, a by-product from biodiesel production, has received significant attention, yet systems for the efficient reforming of glycerol that are based on non-precious metals have rarely been reported. Here, we introduce tungsten-doped bismuth vanadate (W:BiVO4) electrodes combined with an atomic-layer-deposited nickel (oxy)hydroxide (NiOx(OH)y) co-catalyst, as a promising photoanode material for the photoelectrochemical (PEC) oxidation of glycerol. To reveal trends in the reaction kinetics and selectivities, glycerol oxidation reaction (GOR) was investigated in varying electrolytes and at different applied biases. The photoanode developed in our study provides a rare example of the efficient production of the high value-added products, dihydroxyacetone (DHA), glyceraldehyde (GALD), and glycolaldehyde (GCALD), in the absence of precious metal catalysts. Under optimized conditions, W:BiVO4 with a NiOx(OH)y co-catalyst features oxidation currents and onset potentials for glycerol/water oxidation that are on par with state-of-the-art transition-metal-oxide photoanodes employed for the reforming of organic species, which marks an important step towards affordable solar-driven electrolyzers and direct alcohol fuel cells.
Introduction
Molecular hydrogen H2 is a promising energy carrier, which can be used directly in emission-free fuel cell devices,1–3 or in hydrogenation reactions to produce high-energy-density chemicals such as ammonia4,5 or methanol.6–8 Conventionally, hydrogen is produced by the reforming of fossil fuels thus contributing appreciably to industrial CO2 emissions.9 Electrolysis of water10 offers a potentially sustainable alternative way for H2 synthesis, if coupled with renewable electricity sources such as wind, hydropower or sunlight-driven photovoltaics.11,12 Complementing water splitting, the (photo)electrochemical oxidation of cheap and abundant organics or biomass feedstocks (e.g. glycerol) is an emerging field with enormous potential for the production of hydrogen and value added chemicals.13,14 While the splitting of water into H2 and O2 is highly endothermic (ΔG° = 237 kJ mol−1), requiring a minimum cell voltage of ΔE° = 1.23 V,12 supplemented by the reaction overpotential (typically >0.5 V to achieve industrially relevant current densities), the dehydrogenative reforming of abundant carbohydrates or alcohols requires a relatively low external energy input.15 Moreover, the products of the partial oxidation of organic feedstocks can be of high market value (for instance, dihydroxyacetone is used in the cosmetics industry).16 Yet the highly efficient catalysts composed of abundant elements for the reforming of organic species are not developed, in part as a result of a limited fundamental understanding of the reaction pathways of the organics valorisation to selectively produce H2 and valuable oxygenates.The use of semiconductor electrodes in a photoelectrochemical (PEC) setup allows to reduce considerably the reaction overpotentials, paving the way towards unbiased solar-driven photocatalysis.17,18 Bismuth vanadate (BiVO4) has been identified as one of the most promising photoanodes, exploited initially for water splitting.19 In the monoclinic phase, BiVO4 is the photoactive n-type semiconductor with a bandgap of 2.4 eV,20,21i.e. it absorbs in the visible light range. However, unmodified BiVO4 features low catalytic activity for photoelectrochemical water oxidation. To enhance kinetics, improve charge transport characteristics and reduce surface recombination of the photo-generated charge carriers in BiVO4, different strategies have been employed, i.e. aliovalent doping or deposition of co-catalysts for the oxygen evolution reaction.22 As a result, the range of application for BiVO4-based photocatalysts expanded from water oxidation20 to other photo(electro)catalytic transformations, such as the oxidation of 5-hydroxymethylfurfural, degradation of organic dyes or CO2 conversion.23–25 Despite these advances, BiVO4 remains underutilized for the reforming of abundant organic feedstock molecules to H2 and partially-oxidized value-added organic chemicals.
In the present study, we evaluate the performance of BiVO4-based photoanodes for the photoelectrocatalytic reforming of glycerol, C3H5(OH)3. Glycerol, a side product of biodiesel production, is a highly abundant and inexpensive chemical26 formed during the transesterification of triglycerides with methanol (one mole of glycerol is generated per three moles of biodiesel,27 which corresponds to ca. 100 kg of glycerol produced per ton of biodiesel). Oxidation, or the partial dehydrogenative reforming of glycerol, can in principle generate a plethora of the high value-added chemicals, e.g. 1,3-dihydroxyacetone, glyceraldehyde, tartronic acid, glycolic acid or hydroxypyruvic acid (Scheme 1).28 On the other hand, the complete dehydrogenative decomposition of one mole of glycerol can generate up to four moles of hydrogen at a relatively low energy input (ΔG = 3.9 kJ molglycerol−1).16 Therefore, glycerol has a potential to be used in both H2 generating electrolyzers and direct alcohol fuel cells.29,30
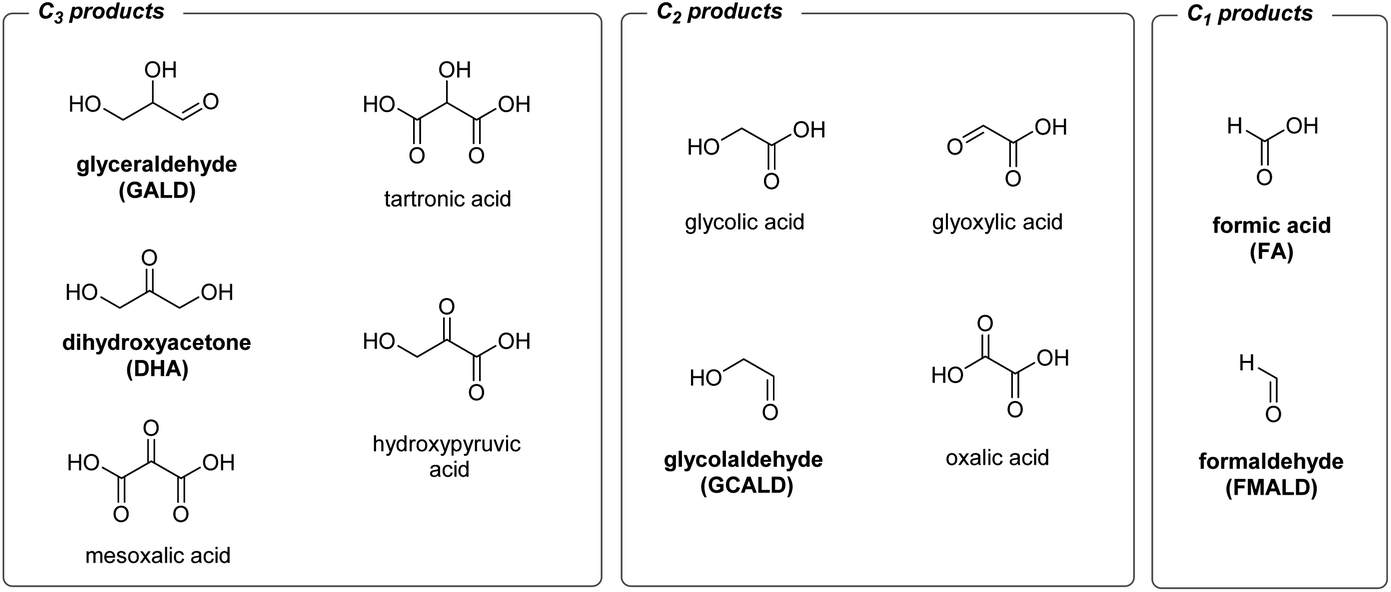 | ||
| Scheme 1 Products of electrochemical glycerol reforming identified in the literature. Products found in this study are shown in bold. | ||
Here, we present the study of the photoelectrochemical glycerol oxidation on BiVO4-based photoanodes in different pH environments. Our work demonstrates that tungsten doping in the BiVO4 structure, aided by the deposition of a nickel oxyhydroxide (denoted in the text as NiOx(OH)y, see discussion below) co-catalyst layer, notably improves the kinetics of glycerol oxidation on the photoanode. We focused on the development and optimization of the nickel-based coatings as the latter demonstrated promising electro- and photoelectrocatalytic activity and stability in a related reaction of water oxidation31,32 as well as in electrocatalytic glycerol oxidation.33 In addition, the pH of the reaction media substantially affects the distribution of products, e.g. formation of glyceraldehyde (GALD) is only observed in acidic conditions. Glycolaldehyde (GCALD), the simplest α-hydroxycarbonyl synthon for sugar formation, to the best of our knowledge, was for the first time observed in PEC glycerol oxidation. Our study clarifies the effects of different reaction variables (e.g. pH, applied bias, presence of co-catalysts) on the kinetics of PEC glycerol reforming. Thus, tungsten-doped bismuth vanadate (W:BiVO4) with an atomic-layer-deposited NiOx(OH)y co-catalyst achieved a photocurrent of 4.2 mA cm−2 at 1.2 VRHE (0.5 M Na2SO4), which is on par with the most active photocatalysts employed to date for the decomposition of organic substrates and biomass derivatives.34 Overall, our approach represents an effective, scalable strategy for producing solar hydrogen and offers a pathway to valorise abundant low cost glycerol.
Results and discussion
Materials synthesis and characterization
Electrodes containing W-doped BiVO4 films on fluorine doped tin oxide (FTO) coated glass substrates were fabricated by a general electrodeposition-calcination method,34–36 modified slightly in this work (details are given in ESI†). The tungsten dopant was introduced into BiVO4 during a calcination step in order to enhance the bulk charge transfer efficiency of the material37 (denoted W:BiVO4; W content is 0.5 at% according to ICP, Fig. S1†). Nickel-based coating was deposited onto W:BiVO4-based electrodes via atomic layer deposition (ALD)38 using pulses of nickelocene (Ni(η5-C5H5)2, NiCp2) and H2O at 250 °C (see ESI† for detailed experimental procedures). Nickel-coated W:BiVO4 electrodes (Ni content of 0.65 at%) were used for PEC measurements without further thermal treatment.X-ray diffraction (XRD) pattern of as-synthesized W:BiVO4 matches that of a monoclinic scheelite reference (no. 01-083-1699, ICDD database, Fig. 1a). W:BiVO4 has a porous structure according to scanning electron microscopy imaging (SEM, Fig. S2†). No additional peaks were observed in the diffraction pattern of W:BiVO4, after atomic-layer-deposition of nickel coating (Fig. 1a).
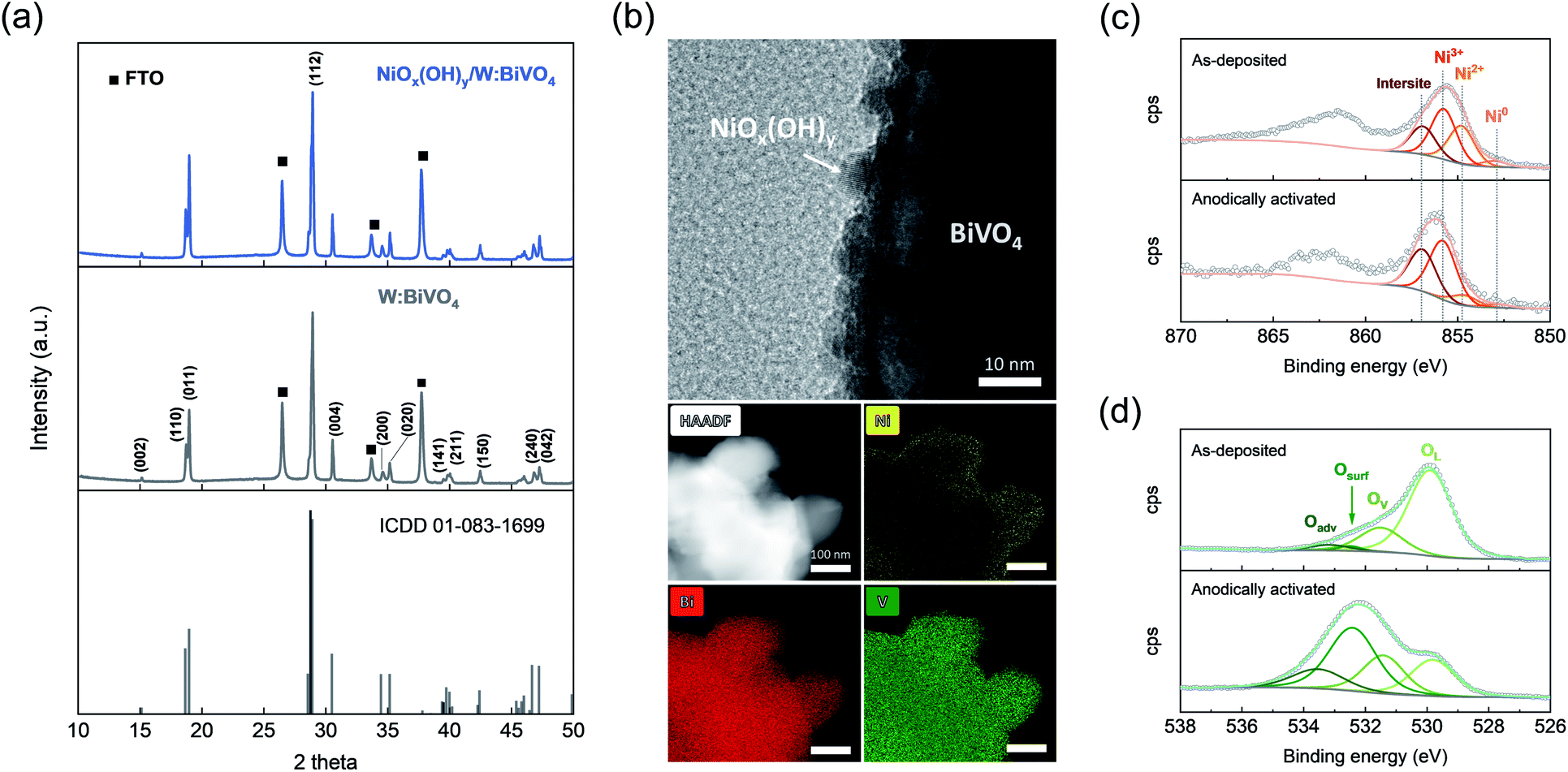 | ||
| Fig. 1 (a) XRD patterns of BiVO4 (ICDD no. 01-083-1699), W:BiVO4 and as-deposited NiOx(OH)y/W:BiVO4. The peaks corresponding to the FTO substrate are indicated by squares. (b) HRTEM, HAADF-STEM, and EDX elemental mapping images of electrochemically activated NiOx(OH)y/W:BiVO4 electrodes. Overlay of the (c) Ni 2p and (d) O 1s core level XPS signals of as-deposited and electrochemically activated NiOx(OH)y/W:BiVO4. The feature denoted as intersite (856.9 eV) is due to defects in the structure, as discussed elsewhere.39 | ||
Nickel-coated W:BiVO4 electrodes were subjected to five anodic scans, from −0.05 to 1.50 VRHE in 0.5 M potassium borate buffer solution (KBi, pH = 9.3) (Fig. S3a†), and then used for glycerol oxidation. Such electrochemical activation yields NiOx(OH)y species in the form of uniformly distributed crystalline nanoparticles on the surface of W:BiVO4, according to high-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDX) elemental mapping (Fig. 1b). XRD of anodically-activated NiOx(OH)y/W:BiVO4 electrodes is identical to that of the as prepared NiOx(OH)y/W:BiVO4 (Fig. S3b†). A negligible difference in the absorption spectra of NiOx(OH)y/W:BiVO4 and W:BiVO4 indicates that ALD-deposited NiOx(OH)y does not alter noticeably light absorption properties of the photoanode (Fig. S3c†).
The surface composition and valence states of as-deposited and anodically activated NiOx(OH)y/W:BiVO4 were probed by X-ray photoelectron spectroscopy (XPS). The Ni 2p core level XPS spectrum of as-deposited NiOx(OH)y/W:BiVO4 reveals features ascribed to metallic Ni, Ni2+ and Ni3+ at 852.9, 854.7 and 855.7 eV, respectively, while an additional feature at higher binding energy (denoted as intersite, 856.9 eV) can be ascribed to the presence of defects in the nickel (oxy)hydroxide structure (Fig. 1c).39 As expected, anodic activation increased the relative amount of oxidized nickel species (NiOx(OH)y), evidenced by the much enhanced intensity of the feature at 855.7 eV.
The O 1s core level spectrum (Fig. 1d) of as-deposited and activated NiOx(OH)y/W:BiVO4 films can be deconvoluted into four components: lattice oxygen (OL, 529.5 eV), lattice oxygen sites located in the vicinity of oxygen vacancies/defects (OV, 531 eV), surface oxygen species (Osurf, 532 eV), and adventitious species (weakly bound adsorbed water/CO2, Oadv).40,41 Comparison of the spectra of the as-deposited and activated films reveals a distinct increase in the concentration of surface oxygen groups for the electrochemically activated films, i.e. an increase from 2 to 42%, consistent with the formation of layered nickel oxyhydroxide species with a highly exposed surface (Table S1†).42
Photoelectrochemical oxidation of glycerol
To investigate the performance of the electrochemically activated NiOx(OH)y/W:BiVO4 electrodes (see above) for PEC glycerol oxidation, photoelectrochemical studies were carried out using a custom-made, two-compartment cell with an anion-exchange membrane. Glycerol oxidation was studied in mild alkaline media (0.5 M potassium borate buffer, KBi, pH = 9.3) and in non-buffered electrolyte (0.5 M Na2SO4, pH = 7.0), which gradually acidifies during the PEC reaction. In the following sections, we discuss the photoelectrochemical activity of the fabricated materials for glycerol oxidation, their long-term stability for GOR, and the distribution of the reaction products as a function of the reaction conditions (electrolyte, applied bias).Fig. 2a and b show the linear sweep voltammetry (LSV) profiles under dark and illumination conditions for W:BiVO4 and NiOx(OH)y/W:BiVO4 in 0.5 M KBi and 0.5 M Na2SO4 electrolytes. In Na2SO4 solution, the PEC performance of W:BiVO4 is characterized by an onset potential of ca. 0.43 VRHE and a photocurrent for water oxidation of 2.3 mA cm−2 at 1.2 VRHE (in the absence of glycerol). The NiOx(OH)y co-catalyst enhances the photocurrent to 3.3 mA cm−2 (1.2 VRHE), leading to a ca. 50 mV cathodic shift of the onset potential. In the presence of glycerol (0.1 M), the photocurrent increases in the entire potential range, reaching 4.2 mA cm−2 at 1.2 VRHE.
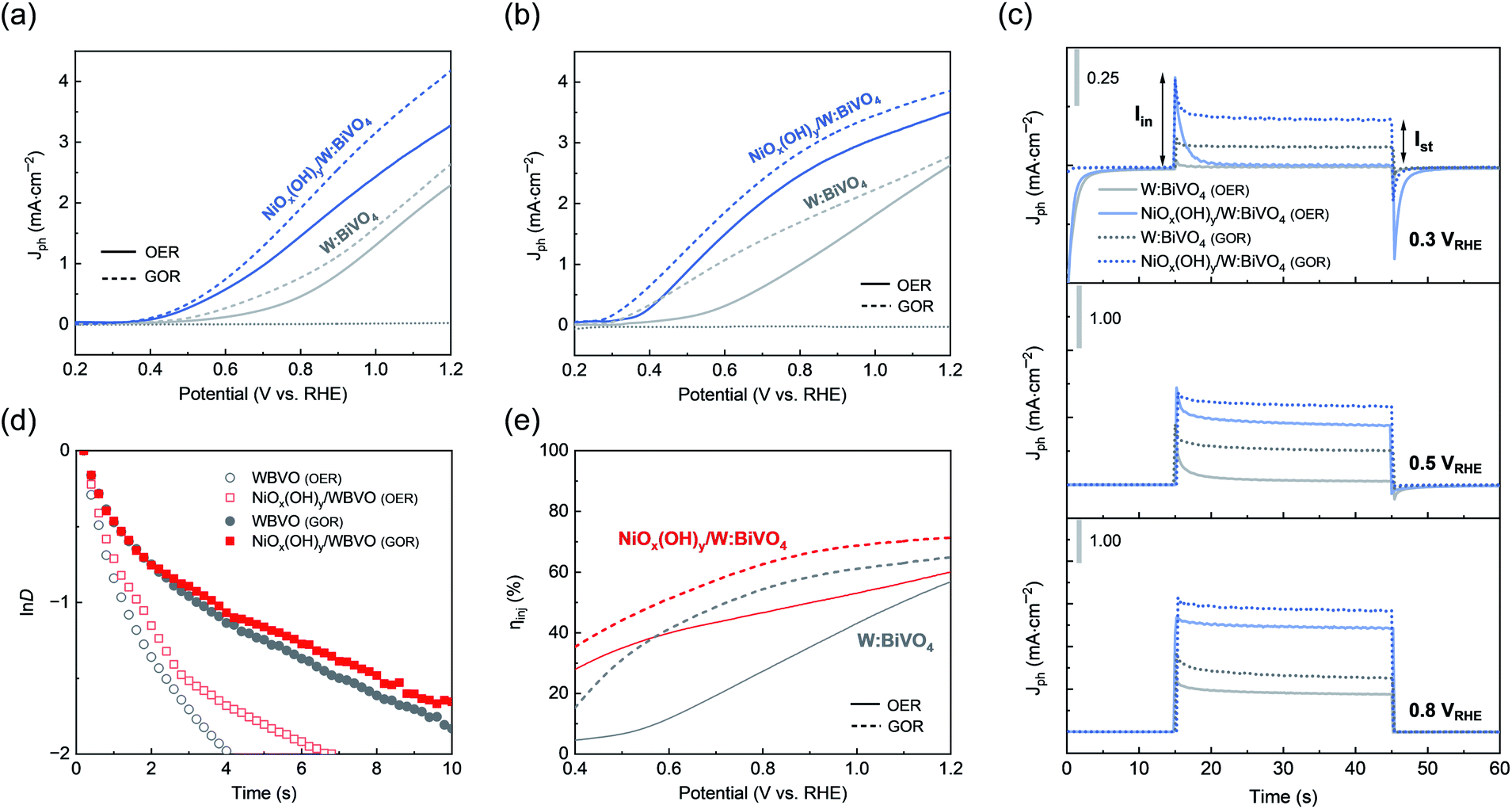 | ||
Fig. 2 Linear sweep voltammetry of W:BiVO4 and NiOx(OH)y/W:BiVO4 in (a) 0.5 M Na2SO4 and (b) 0.5 M KBi with and without the addition of 0.1 M glycerol under dark and AM 1.5 G, 100 mW cm−2 illumination; scan rate = 10 mV s−1. (c) Chopped chronoamperometry plots of W:BiVO4 and NiOx(OH)y/W:BiVO4 in 0.5 M KBi with (dotted lines) and without (solid lines) 0.1 M glycerol at 0.3 VRHE, 0.5 VRHE, and 0.8 VRHE. The color code is the same as on panels a and b. (d) Plots of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D as a function of time of photoanodes for water oxidation and glycerol oxidation (E = 0.5 VRHE). (e) Hole injection efficiency vs. applied bias for glycerol oxidation in 0.5 M KBi; the corresponding linear sweep voltammetry of sulfite oxidation is presented in the ESI.† D as a function of time of photoanodes for water oxidation and glycerol oxidation (E = 0.5 VRHE). (e) Hole injection efficiency vs. applied bias for glycerol oxidation in 0.5 M KBi; the corresponding linear sweep voltammetry of sulfite oxidation is presented in the ESI.† | ||
In mild-alkaline conditions (0.5 M KBi), W:BiVO4 and NiOx(OH)y/W:BiVO4 exhibit ca. 50 mV cathodic shift of the onset potential on the RHE scale relative to that in the Na2SO4 electrolyte (Fig. 2b). The photocurrent for glycerol oxidation on NiOx(OH)y/W:BiVO4 electrodes reaches 2.5 mA cm−2 (0.8 VRHE) and increases to 3.5 mA cm−2 at 1.2 VRHE, thus matching or exceeding the performance of some transition-metal-oxide-based PEC systems (e.g. TiO2) employed for the oxidation of organic alcohols (methanol, ethanol or glycerol).34 Control experiments show a high reproducibility of the data collected on W:BiVO4 and NiOx(OH)y/W:BiVO4 photoelectrodes (Fig. S3d†). Overall, the enhancement of the catalytic activity of NiOx(OH)y/W:BiVO4 for water and glycerol oxidation in mildly alkaline conditions relative to neutral conditions may originate from the pH-dependence of the electrocatalytic activity of the NiOx(OH)y co-catalyst layer, which is a typically observed phenomenon for nickel or cobalt-based oxides and (oxy)hydroxides.43,44
The charge-transfer dynamics at the semiconductor–electrolyte interface (SEI) was analysed by the transient photocurrent profile at a constant potential. Here, we used alkaline 0.5 M KBi conditions and relatively low biases, i.e. 0.3 VRHE, 0.5 VRHE, and 0.8 VRHE (Fig. 2c). The initial transient photocurrent, Iin, is caused by the separation of the photogenerated hole–electron pairs, where trapping or recombination of charge carriers takes place by surface states or reduced species in an electrolyte.45 The continuous decay of the photocurrent indicates ongoing recombination until a steady state, Ist, is reached. When the light is off, back-reaction of electrons at the conduction band with accumulated holes results in a negative current spike.
In the absence of glycerol, the steady-state photocurrent at 0.3 VRHE (i.e. close to the onset potential) is nearly zero for W:BiVO4 and NiOx(OH)y/W:BiVO4 electrodes, coinciding with the onset potential observed in the LSV profile. In the presence of glycerol, a distinct non-zero photocurrent is detected reaching 80 μA cm−2 (0.3 VRHE), 0.53 mA cm−2 (0.5 VRHE) and 1.22 mA cm−2 (0.8 VRHE) for the W:BiVO4 electrodes (Fig. 2c). In addition, the back-reaction current was significantly suppressed at 0.3 VRHE, likely indicating a reduced accumulation of holes at the material surface. In the presence of glycerol, the photocurrent of the NiOx(OH)y/W:BiVO4 electrodes increased to 0.2 mA cm−2 (0.3 VRHE), 1.2 mA cm−2 (0.5 VRHE) and 2.9 mA cm−2 (0.8 VRHE). Note that all the studied potentials are below the thermodynamic potential of the water oxidation, 1.23 VRHE.
The photocurrent relaxation dynamics were also analysed using the transient decay time τ, expressed by a logarithmic plot of the parameter D (eqn (1)) versus time, where It is the current at time t.45
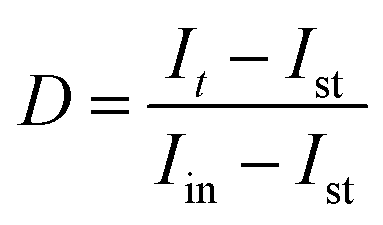 | (1) |
The transient decay time is defined as time at which lnD = −1. The value of τ reflects the electron lifetime within bulk semiconductor. BiVO4 is known to undergo facile recombination of charge carriers due to sluggish charge-transfer kinetics at the SEI.46 Since the accumulated holes at the surface facilitate electron–hole recombination leading to a reduced lifetime of the separated charges, a facilitated hole transfer to the reagents in the electrolyte will prolong the lifetime of the separated electrons. At 0.5 VRHE, W:BiVO4 suffers from severe surface recombination, resulting in τ of ca. 1.3 s. Upon addition of glycerol acting as a hole acceptor, an enhanced charge transport from the near-surface region to the electrolyte gives rise to a near three-fold enhancement of τ reaching 4.0 s (Fig. 2d). Overall, these observations of an increased photocurrent and a longer transient decay time imply faster kinetics of the photoelectrochemical glycerol oxidation reaction as compared to the oxygen evolution reaction (OER) in water.
The analysis of the hole-injection efficiency (ηinj, see ESI and Fig. S4 for details†) for glycerol oxidation on the studied photoanodes further corroborates this conclusion. The sulfite anion (SO32−) is a typically used hole scavenger, whose ηinj can be taken as a reference value (100%).47 The hole injection efficiency for water oxidation reaches ca. 55% and 60% on W:BiVO4 and NiOx(OH)y/W:BiVO4 electrodes, respectively, at 1.2 VRHE (Fig. 2e). In the presence of glycerol (0.1 M), NiOx(OH)y/W:BiVO4 photoelectrode exhibits a superior hole transfer to glycerol than W:BiVO4 throughout the whole potential window, reaching an efficiency of 72% at 1.2 VRHE, that confirms the enhancement of the kinetics of glycerol oxidation with respect to OER.
Long-term photoelectrochemical oxidation of glycerol
Two static potentials, 0.8 and 1.2 VRHE, were used to investigate the effect of the applied bias on the kinetics of glycerol reforming under constant potential electrolysis conditions. Fig. S5† demonstrates 10 h chronoamperometry tests under illumination at 1.2 VRHE in 0.5 M Na2SO4 or 0.5 M KBi, both in the presence of 0.1 M glycerol or in a glycerol-free electrolyte. Anodic photocorrosion, i.e. oxidation during OER under illumination, was reported previously for BiVO4 (ref. 48) and indeed, a rapid decay of the photocurrent in 0.5 M Na2SO4 and 0.5 M KBi is observed for W:BiVO4 electrodes (Fig. S5†). After 5 h of electrolysis, merely 26% and 20% of the photoactivity is retained in 0.5 M Na2SO4 and 0.5 M KBi, respectively. In contrast, in the presence of glycerol, an excellent stability along with a marked increase of the current was observed, likely indicative of the preferential oxidation of glycerol over water on the W:BiVO4 photoanode (Fig. S5†). Noteworthy, for the 0.5 M Na2SO4 electrolyte, a decrease in the pH in the anodic compartment to <3 was detected, whereas the pH remains unchanged in a 0.5 M KBi buffer solution.While the excellent stability of the W:BiVO4 electrodes in the presence of glycerol is observed at both low (0.8 VRHE) and high (1.2 VRHE) biases, the degradation of NiOx(OH)y/W:BiVO4 electrodes in 0.5 M KBi takes place, as the loss of 21% (0.8 VRHE) and 33% (1.2 VRHE) in the photocurrent after 10 h is observed (Fig. S6†). We attribute this decrease to a gradual dissolution of the nickel catalyst, however the poisoning of the surface by adsorbed reaction intermediates or by-products can also take place.49,50 Notably, in the absence of glycerol, the photoelectrochemical stability of NiOx(OH)y/W:BiVO4 in 0.5 M KBi is substantially higher (Fig. S7†). Inductively coupled plasma optical emission spectrometry (ICP-OES) was used to analyse the electrolyte composition after the reaction under various conditions (Fig. S8†). In the presence of glycerol, we observed a negligible dissolution of Bi and V into the electrolyte (≤0.6% of the total loading) from W:BiVO4 electrodes in 0.5 M KBi, while somewhat higher values (e.g. ≥6% loss of Bi and V after continuous GOR at 1.2 VRHE) are observed in 0.5 M Na2SO4. In contrast to bismuth vanadate, the NiOx(OH)y overlayer proved to be unstable under the reaction conditions, e.g. 10 h of continuous electrolysis in 0.5 M KBi resulted in a loss of >90% of the nickel content. However, in 0.5 M Na2SO4, the rate of nickel dissolution decreases significantly, i.e. to 35% (0.8 VRHE) and 24% (1.2 VRHE) after 10 h of electrolysis. XPS analysis on NiOx(OH)y/BiVO4 electrode revealed absence of the noticeable changes of the chemical states of nickel after GOR in Na2SO4 (Fig. S9†).
HRTEM imaging of the electrodes after 10 h of glycerol oxidation in 0.5 M KBi (Fig. S10†) confirms that BiVO4 maintains its original morphology. Consistent with ICP data, the used NiOx(OH)y/W:BiVO4 electrodes reveal no detectable amounts of nickel on the surface after the PEC test. Thus, the dissolution of the nickel co-catalyst layer coincides with a decrease of the photocurrent during constant potential electrolysis, serving as an indirect evidence for the crucial role of nickel surface species in catalysing glycerol oxidation on the photoanode (Fig. S6†).
To summarize, atomic-layer-deposited NiOx(OH)y enhances the OER kinetics of W:BiVO4 as reflected by an increased photocurrent and transient decay time. In the presence of glycerol, both W:BiVO4 and NiOx(OH)y/W:BiVO4 exhibit a high activity towards glycerol oxidation. NiOx(OH)y/W:BiVO4 electrodes demonstrate appreciable photoelectrochemical stability under PEC glycerol oxidation conditions, although the gradual dissolution of the nickel cocatalyst was observed in the presence of glycerol that resulted in a continuous decay of the catalytic currents for NiOx(OH)y/W:BiVO4 electrodes over time in alkaline condition. An increase of the current despite the dissolution of the NiOx(OH)y layer during photoelectrolysis in a Na2SO4 solution can be attributed to the gradual acidification of the medium, which can have a positive effect on the reaction kinetics.
Product analysis: influence of electrolyte, applied bias, and NiOx(OH)y co-catalyst on the chemoselectivity of GOR
Next, the C1–C3 products of glycerol oxidation during long-term GOR conditions (10 h) over W:BiVO4 and NiOx(OH)y/W:BiVO4 electrodes were identified and quantified by 1H and 13C NMR spectroscopy and high performance liquid chromatography (HPLC).1H and 13C NMR spectroscopy
Previous reports have identified 1,3-dihydroxyacetone (DHA), glyceraldehyde (GALD), glyceric acid (GLA), hydroxypyruvic acid (HyP), tartronic acid (TA), glycolic acid (GLC), glyoxylic acid (GLX), oxalic acid (OA), formic acid (FA), and carbonate, CO32−, as possible products of the (photo)electrochemical oxidation of glycerol (Scheme 1).28,511H and 13C NMR data acquired in our study under different PEC conditions are shown in Fig. S11, S12 and Table S2.† Note that to account for the possible photochemical degradation, the NMR spectra of glycerol discussed here are recorded from a glycerol solution exposed to AM 1.5 G, 100 mW cm−2 illumination for 10 h without applied bias.0.5 M Na2SO4 electrolyte
In 0.5 M Na2SO4, GALD, GCALD, DHA, FA, and FMALD (predominantly hydrated) were detected by 1H and 13C NMR by comparing with standards and literature data52 (Fig. S11, S13a and S14a†). Noteworthy, illumination of a glycerol solution (0.5 M Na2SO4) without applying a bias did not lead to formation of the new species, as evidenced by NMR analysis, which is indicative of a high stability of glycerol towards spontaneous photo-oxidation in Na2SO4 electrolyte (Fig. S11a†).1H and 13C spectra collected for the NiOx(OH)y/W:BiVO4 electrode at 1.2 VRHE in 0.5 M Na2SO4 exhibit complex patterns (δH ∼3.5–4.5 ppm and δC ∼60–100 ppm) (Fig. S11a and b†), in which new signatures in addition to the previously identified peaks of GCALD, GALD, and DHA are found. Presumably, these signatures are due to oxygenated species that we are currently unable to identify. Previous studies have shown that GCALD, GALD, and DHA form complex equilibria of monomers and oligomers in aqueous solutions.53–55 Analysis of the 1H NMR spectrum of the product mixture obtained for the NiOx(OH)y/W:BiVO4 photoanode at 1.2 VRHE assumes a presence of GCALD, GALD, and DHA (by matching with the signals of the respective reference chemicals, Fig. S14†), however 13C NMR of the same solutions (Fig. S11b†) showed that GCALD, GALD, and DHA do not account for all of the signals. A 1H–1H Correlation Spectroscopy (COSY) analysis for this mixture showed many overlapping signals making its interpretation difficult, and heteronuclear single quantum coherence spectroscopy (HSQC) (Fig. S15†) and heteronuclear multiple bond correlation (HMBC) analysis failed to show correlations for the 1H signals of interest. Thus, further work is necessary to identify the as yet undetermined specie(s).
0.5 M KBi electrolyte
Unlike with 0.5 M Na2SO4 electrolyte, as yet unidentified products of spontaneous glycerol decomposition under illumination were observed in a 0.5 M KBi buffer solution, as evidenced by the appearance of noise level signals at δH = 2.09, 2.72, 2.91 and 3.07 ppm in the 1H NMR spectra (Fig. S12a†). The product mixture contains GCALD, along with FA and FMALD, and each of these species are observed for all electrode materials tested (NiOx(OH)y/W:BiVO4 and W:BiVO4) and at both electrolysis potentials studied. The presence of GALD cannot, however, be ruled out, as the GALD signals overlap with those of GCALD. Notably, DHA was found in the product mixture for NiOx(OH)y/W:BiVO4 electrodes, but not on bare W:BiVO4 without a nickel co-catalyst (Fig. S12a, S14b and S16†). Note that no carbonate was detected in the reaction mixture as followed from the absence of the corresponding signals in 13C NMR spectra (δ = 166–168 ppm).56Quantitative analysis and mechanistic hypothesis
Following the identification of the key products by NMR spectroscopy, HPLC analysis of the reaction mixture was carried out to obtain quantitative information about the products formed after 5 h and 10 h of continuous photoelectrolysis under varying conditions (Fig. S17†). In general, the analysis of the products by HPLC is consistent with results of NMR spectroscopy. In 0.5 M Na2SO4, GALD and GCALD are identified as products by the direct comparison of retention times with commercial references measured in the same electrolyte (Fig. S17a†). In the case of glycerol oxidation in a 0.5 M KBi buffer (pH = 9.3), GCALD is found under all experimental conditions (Fig. S17b†). Further analysis of the HPLC data is complicated by the overlap of the glycerol signal with the signals of some of the reaction products. In both electrolytes, due to the overlap of the chromatogram peaks of FMALD with those of glycerol and the overlap of the FMALD 1H NMR chemical shifts with that of water, quantification of FMALD could not be performed, therefore FMALD is excluded from the discussion below.The effect of the NiOx(OH)y co-catalyst layer and the applied bias on the production rate (R) and faradaic efficiency (FE) of the detected products of the glycerol oxidation is illustrated in Fig. 3, based on 1H NMR spectra and chromatography results. The distribution of the products of the electrolysis reaction depends on the applied potential, type of electrolyte and the electrode material (Table 1 and S19†). Specifically, in the 0.5 M Na2SO4, the average production rate of DHA, GALD, GCALD and FA increases with increasing potential when using the W:BiVO4 electrode (Fig. 3a). At 0.8 VRHE, FA is a primary product of glycerol oxidation on W:BiVO4 electrodes with a FE of over 70% and a production rate of ca. 54 mmol h−1 m−2, while at the higher potential of 1.2 VRHE, a four-fold and five-fold enhancement of the production rate of DHA (70 mmol h−1 m−2) and GCALD (ca. 60 mmol h−1 m−2), corresponding to FE's of 19% and 11%, is observed.
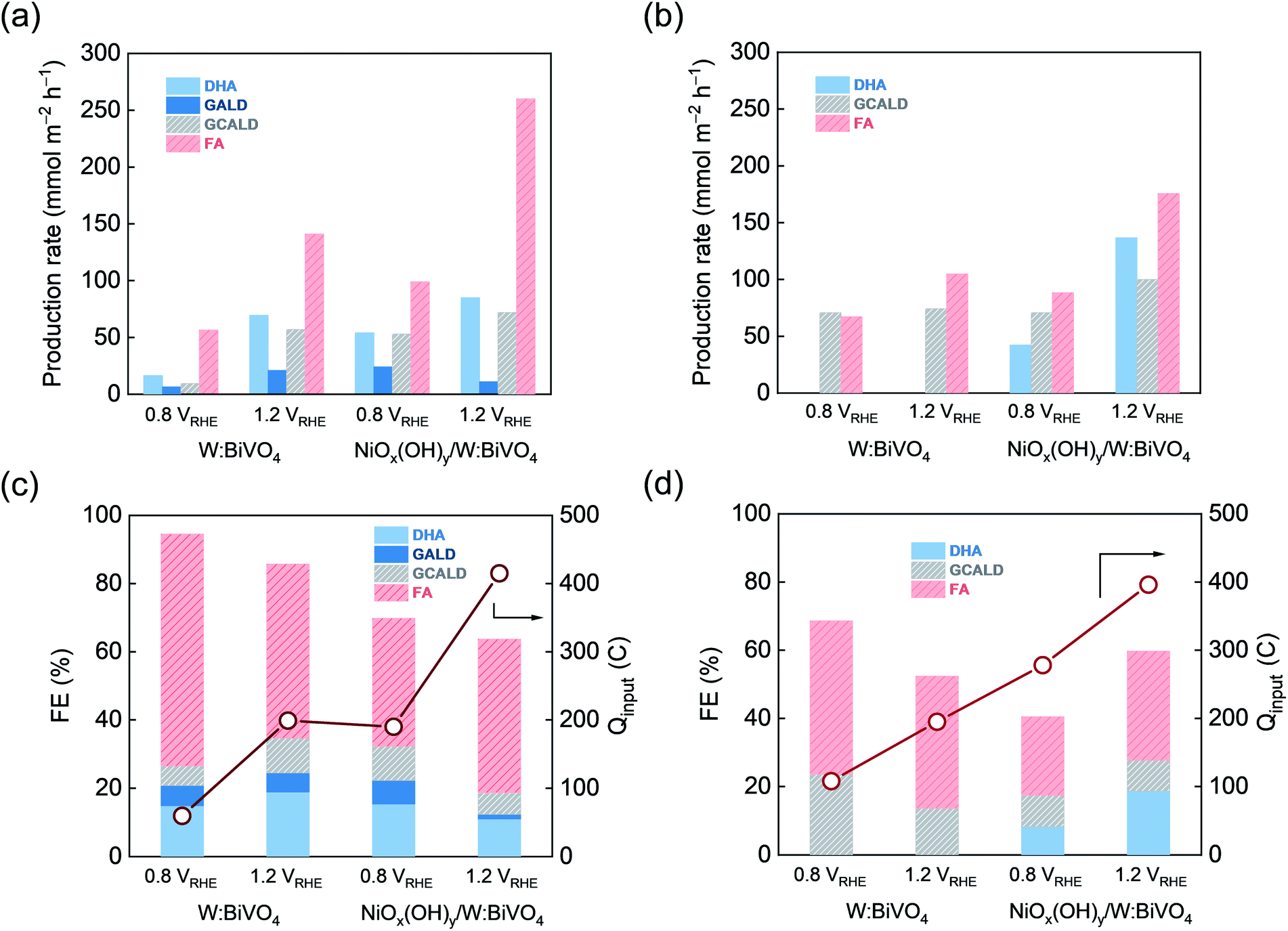 | ||
| Fig. 3 Quantitative product analysis for different electrolytes after 10 h of continuous photoelectrolysis of 0.1 M glycerol solution over W:BiVO4 and NiOx(OH)y/W:BiVO4 electrodes at different applied voltages. Production rate (R, mmol m−2 h−1) determined in (a) 0.5 M Na2SO4(aq) and (b) 0.5 M KBi; faradaic efficiency per mole of specific compound (FE, %; bars) and total charge transfer (Qinput, C; line) determined in (c) 0.5 M Na2SO4(aq) and (d) 0.5 M KBi using HPLC and 1H NMR (DHA: dihydroxyacetone, GALD: glyceraldehyde, GCALD: glycolaldehyde dimers, and FA: formic acid or formate). | ||
| Electrolyte | Electrode | Applied bias | Identified productsa | Enhanced rate and FE of specific products observed with increased potential |
|---|---|---|---|---|
| a Reaction conditions: 0.1 M glycerol, electrode area 2.8 cm2, volume of anode part 33 ml, temperature at 25 °C (DHA: dihydroxyacetone, GALD: glyceraldehyde, GCALD: glycolaldehyde dimers, FA: formate, and FMALD: formaldehyde). b Product that is identified by 1H NMR only. c Product that is identified by 13C NMR only, hence the quantitative analysis is not available. d Product that is identified by 1H NMR; however, its chemical shifts overlap with GCALD. | ||||
| Na2SO4 | W:BiVO4 | 0.8 VRHE | GALD, DHA, GCALD, FA, FMALD | R DHA, RGALD, RGCALD, RFA, FEDHA, FEGALD, FEGCALD |
| 1.2 VRHE | GALD, DHA, GCALD, FA, FMALD | |||
| NiOx(OH)y/W:BiVO4 | 0.8 VRHE | GALD, DHA, GCALD, FA, FMALD | R DHA, RGCALD, RFA | |
| 1.2 VRHE | GALD, DHA, GCALD, FA, FMALD | |||
| KBi | W:BiVO4 | 0.8 VRHE | GALDb,d, GCALD, FAb, FMALDc | R FA |
| 1.2 VRHE | GALDb,d, GCALD, FAb, FMALDc | |||
| NiOx(OH)y/W:BiVO4 | 0.8 VRHE | GALDb,d, GCALD, DHAb, FAb, FMALDc | R DHA, RGCALD, RFA, FEDHA, FEFA | |
| 1.2 VRHE | GALDb,d, GCALD, DHAb, FAb, FMALDc | |||
For both 0.5 M Na2SO4 and 0.5 M KBi electrolytes, the deposition of a NiOx(OH)y co-catalyst layer results in an enhanced catalytic glycerol oxidation reflected in the higher photocurrents (higher total charge transfer) and product yields. For instance, at an electrolysis potential of 1.2 VRHE in 0.5 M Na2SO4, ca. two-fold increase of the photocurrent was achieved upon deposition of a NiOx(OH)y co-catalyst. In addition, an increase in the applied bias increases the production rate of the value-added DHA in the presence of NiOx(OH)y. Specifically, in 0.5 M KBi, a three-fold enhancement of production rate of DHA (138 mmol h−1 m−2) was achieved at 1.2 VRHE (compared to 0.8 VRHE) along with an improved FE of 19% (Fig. 3b and d).
Product analysis and identification enabled the proposal of reaction path(s) for the photoelectrocatalytic glycerol decomposition using W:BiVO4 and NiOx(OH)y/W:BiVO4 electrodes. It is proposed that DHA and GALD isomers are the first stable species to form upon 2e− oxidation of one of the hydroxyl moieties of glycerol.51,57 A higher yield of DHA compared to GALD (Fig. 3 and S19†) corroborates with DFT calculations that have identified DHA as the thermodynamically more stable product than GALD.58 GCALD is proposed to form from isomeric DHA/GALD species through C–C bond cleavage, releasing one equivalent of FA. Subsequent C–C bond cleavage to form one more equivalent of FA and FMALD from GCALD is proposed as one of the further steps. The presence of hydrated GCALD was confirmed by HPLC and NMR spectroscopy (identification of its exact structure(s) in the solution, i.e. hydrated monomers, dimers or oligomers,59 is beyond the scope of this work).
In 0.5 M Na2SO4, PEC glycerol oxidation using a W:BiVO4 electrode leads to higher amounts of GALD and GCALD at increased applied bias (1.2 VRHEvs. 0.8 VRHE). In contrast, NiOx(OH)y/W:BiVO4 electrodes preferentially produce GALD and GCALD at a lower potential, 0.8 VRHE, presumably due to their overoxidation at higher applied voltage. Although the deposition of a NiOx(OH)y co-catalyst does not appear to alter the general reaction pathway, the presence of a nickel co-catalyst strongly influences the product distribution (which is also affected by the applied bias and reaction media). For instance, NiOx(OH)y/W:BiVO4 electrodes show an increased selectivity toward DHA formation over GCALD at 1.2 VRHE in alkaline KBi buffer. In contrast to a recent report, in which it was argued that the selective formation of DHA from glycerol is feasible,60 our findings indicate that multiple products inevitably form upon glycerol photoelectrooxidation, emphasizing in turn the importance of the use of complementary analytical tools (e.g. HPLC and NMR spectroscopy) for an accurate quantitative analysis of the complex product mixture (see also Fig. S27†).
Conclusions
This study reveals key trends in the product distribution of the photoelectrochemical glycerol oxidation over BiVO4-based electrodes in the presence/absence of a NiOx(OH)y co-catalyst, at varying electrolyte environments. A quantitative product analysis was achieved by combining HPLC and 1H NMR spectroscopy. We have found that GALD, DHA, GCALD, FA and FMALD are the primary products of glycerol oxidation. To the best of our knowledge, in this study GCALD was detected as a product of PEC glycerol oxidation for the first time. Importantly, we identify the conditions favouring the formation of the high value-added chemicals, DHA and GALD, thus providing a rare example of the system enabling production of valuable C3 products from glycerol without the use of precious metal catalysts. We believe that the reported development of a system for the photoelectrochemical glycerol reforming relying on earth-abundant elements is an important step in the advancement of affordable solar-driven electrolyzers and direct alcohol fuel cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Giovanni Bovone and Prof. Dr Mark Tibbitt for providing access to HPLC. We thank Prof. Dr Kristopher McNeill for providing access to the NMR spectrometer and for helpful discussions. Dr Sung Min Kim is acknowledged for assistance in the atomic layer deposition of the NiOx(OH)y coatings. Dr Agnieszka Kierzkowska and Dr Felix Donat are thanked for ICP analysis. The work was supported by the Swiss Office of Energy (BFE, Grant Agreement No. SI/501598-01).Notes and references
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M. G. Willinger, A. Fedorov and C. R. Muller, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Norskov, Nat. Mater., 2017, 16, 70–81 CrossRef.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- C. Mondelli, G. Gözaydın, N. Yan and J. Pérez-Ramírez, Chem. Soc. Rev., 2020, 49, 3764–3782 RSC.
- P. R. d. l. Piscina and N. Homs, Chem. Soc. Rev., 2008, 37, 2459–2467 RSC.
- M. Simões, S. Baranton and C. J. C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229–230 CrossRef CAS.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef CAS.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- L. Zhang, D. Chen and X. Jiao, J. Phys. Chem. B, 2006, 110, 2668–2673 CrossRef CAS.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng and B. Ye, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328 CrossRef CAS.
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- L. C. Meher, D. V. Sagar and S. N. Naik, Renewable Sustainable Energy Rev., 2006, 10, 248–268 CrossRef CAS.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- K. Zakaria, M. McKay, R. Thimmappa, M. Hasan, M. Mamlouk and K. Scott, ChemElectroChem, 2019, 6, 2578–2585 CrossRef CAS.
- Z. Zhang, L. Xin, J. Qi, D. J. Chadderdon and W. Li, Appl. Catal., B, 2013, 136–137, 29–39 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS.
- D. K. Bediako, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 3662–3674 CrossRef CAS.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, J. Electroanal. Chem., 2013, 703, 56–62 CrossRef CAS.
- X. Lu, S. Xie, H. Yang, Y. Tong and H. Ji, Chem. Soc. Rev., 2014, 43, 7581–7593 RSC.
- K. J. McDonald and K.-S. Choi, Energy Environ. Sci., 2012, 5, 8553–8557 RSC.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- H. S. Park, K. E. Kweon, H. Ye, E. Paek, G. S. Hwang and A. J. Bard, J. Phys. Chem. C, 2011, 115, 17870–17879 CrossRef CAS.
- A. S. Asundi, J. A. Raiford and S. F. Bent, ACS Energy Lett., 2019, 4, 908–925 CrossRef CAS.
- E. L. Ratcliff, J. Meyer, K. X. Steirer, A. Garcia, J. J. Berry, D. S. Ginley, D. C. Olson, A. Kahn and N. R. Armstrong, Chem. Mater., 2011, 23, 4988–5000 CrossRef CAS.
- K. A. Stoerzinger, W. T. Hong, X. R. Wang, R. R. Rao, S. B. Subramanyam, C. J. Li, Ariando, T. Venkatesan, Q. Liu, E. J. Crumlin, K. K. Varanasi and Y. Shao-Horn, Chem. Mater., 2017, 29, 9990–9997 CrossRef CAS.
- D. A. Kuznetsov, M. A. Naeem, P. V. Kumar, P. M. Abdala, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2020, 142, 7883–7888 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef.
- D. A. Kuznetsov, J. Peng, L. Giordano, Y. Román-Leshkov and Y. Shao-Horn, J. Phys. Chem. C, 2020, 124, 6562–6570 CrossRef CAS.
- A. Hagfeldt, H. Lindström, S. Södergren and S.-E. Lindquist, J. Electroanal. Chem., 1995, 381, 39–46 CrossRef.
- C. Zachäus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS.
- F. M. Toma, J. K. Cooper, V. Kunzelmann, M. T. McDowell, J. Yu, D. M. Larson, N. J. Borys, C. Abelyan, J. W. Beeman, K. M. Yu, J. Yang, L. Chen, M. R. Shaner, J. Spurgeon, F. A. Houle, K. A. Persson and I. D. Sharp, Nat. Commun., 2016, 7, 12012 CrossRef.
- N. S. Porter, H. Wu, Z. Quan and J. Fang, Acc. Chem. Res., 2013, 46, 1867–1877 CrossRef CAS.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS.
- C. Dai, L. Sun, H. Liao, B. Khezri, R. D. Webster, A. C. Fisher and Z. J. Xu, J. Catal., 2017, 356, 14–21 CrossRef CAS.
- T. Chatterjee, E. Boutin and M. Robert, Dalton Trans., 2020, 49, 4257–4265 RSC.
- J. Kua, M. M. Galloway, K. D. Millage, J. E. Avila and D. O. De Haan, J. Phys. Chem. A, 2013, 117, 2997–3008 CrossRef CAS.
- V. Waagen, T. K. Barua, H. W. Anthonsen, L. K. Hansen, D.-J. Fossli, E. Hough and T. Anthonsen, Tetrahedron, 1994, 50, 10055–10060 CrossRef CAS.
- A. C. Garcia, Y. Y. Birdja, G. Tremiliosi-Filho and M. T. M. Koper, J. Catal., 2017, 346, 117–124 CrossRef CAS.
- S. Leukel, M. Mondeshki and W. Tremel, Inorg. Chem., 2018, 57, 11289–11298 CrossRef CAS.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354–362 CrossRef.
- L. Cheng, C. Doubleday and R. Breslow, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4218–4220 CrossRef CAS.
- V. A. Yaylayan, S. Harty-Majors and A. A. Ismail, Carbohydr. Res., 1998, 309, 31–38 CrossRef CAS.
- D. Liu, J. C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10480a |
| This journal is © The Royal Society of Chemistry 2021 |