
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis of novel rutile-type high entropy fluorides for electrocatalysis†
Parvathy Anitha
Sukkurji‡
a,
Yanyan
Cui‡
a,
Seunghwa
Lee
b,
Kai
Wang
a,
Raheleh
Azmi
c,
Abhishek
Sarkar
 ad,
Sylvio
Indris
c,
Subramshu S.
Bhattacharya
e,
Robert
Kruk
a,
Horst
Hahn
ad,
Qingsong
Wang
*a,
Miriam
Botros
*a and
Ben
Breitung
*a
ad,
Sylvio
Indris
c,
Subramshu S.
Bhattacharya
e,
Robert
Kruk
a,
Horst
Hahn
ad,
Qingsong
Wang
*a,
Miriam
Botros
*a and
Ben
Breitung
*a
aInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein Leopoldshafen, Germany. E-mail: qingsong.wang@kit.edu; miriam.botros@kit.edu; ben.breitung@kit.edu
bLaboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), ISIC-LSCI, 1015 Lausanne, Switzerland
cInstitute for Applied Materials–Energy Storage Systems, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen 76344, Germany
dKIT-TUD Joint Research Laboratory Nanomaterials Institute of Materials Science, Technische Universität Darmstadt (TUD), 64206 Darmstadt, Hessen, Germany
eDepartment of Metallurgical and Materials Engineering, Nano Functional Materials Technology Centre (NFMTC), Indian Institute of Technology Madras, Chennai, 600036, India
First published on 8th February 2021
Abstract
Multicomponent rutile (P42/mnm) structured fluorides, containing 4 to 7 transition metals (Co, Cu, Mg, Ni, Zn, Mn, and Fe) in equiatomic ratios, were synthesized using a simple mechanochemical approach. The high entropy fluorides were characterized using different techniques, all of which indicate that the high entropy fluorides tend to crystallize into a homogeneously mixed solid solution and single-phase structure. These high entropy fluorides represent an additional class of high entropy ceramics, which have recently attracted attention especially due to the development of high entropy oxides. With the introduction of these novel high entropy fluorides, similar interest could be generated due to the variety of different applications for fluoride materials and the improvements the high entropy concept might bring. Here we present an in-depth characterization study and the potential application of high entropy fluorides as a catalyst for the oxygen evolution reaction, in which the high entropy fluorides do show increased performance compared to a state-of-the-art catalyst for the oxygen evolution reaction, IrO2, despite eliminating noble metal constituents.
Introduction
In recent years, high entropy materials (HEMs) have gathered growing interest due to their remarkable and often unexpected properties, and a whole class of promising materials for future applications has been discovered.1,2 The high entropy concept is based on the idea of utilizing high configurational entropy (or disorder) to induce manifold interactions between the incorporated elements, or sometimes even to stabilize the crystal structure according to the Gibbs–Helmholtz equation.3 Additionally, these materials offer great versatility and the possibility of tailoring the properties, since with the exchange of one single element, or with a varied stoichiometry, the complete inter-elemental interactions and therefore the properties, can be changed. The origin of HEMs is based on the well-known high entropy alloys (HEAs), which consist of a solid solution of a large number of metals.4,5 Similarly, this concept of using high configurational entropy to achieve desired properties (controversial discussion if many high entropy materials are really entropy-stabilized or “just” showing high configurational entropy is ongoing) can be applied to ceramic compounds, wherein multiple cations or anions are occupying the same sublattice, thereby giving rise to configurational entropy.6,7 In 2015 Rost et al. reported entropy stabilized oxides ((CoCuMgNiZn)O) as a special group of high entropy ceramics, which resulted in a boost of related activities.8 This material crystallizes into a rock-salt structure and shows a reversible mixing/demixing behavior upon heating and cooling, respectively, which indicates entropy stabilization. Following this, Bérardan et al. examined the electrical properties of (CoCuMgNiZn)O and investigated the effect of dopants such as Li+, In3+, Ga3+, and Ti4+.9 Additionally, co-doping with Li+ and Ga3+ led to phase pure solid solutions (CoCuMgNiZn)1−2xLixGaxO.10 Numerous reports detail such ceramic compounds, including silicides,11 borides,12 nitrides,13 carbides,14etc. These materials exhibit a wide range of applications, including thermoelectric, dielectric, and optic applications.15–17One important materials class in materials science is metal fluorides due to their special properties and manifold applications. Inorganic metal fluorides find applications in catalysis, as cathode active materials for battery cells, in superconducting devices, as corrosion protection materials etc.18–21 Non-binary compounds, like Co doped MgF2, can show additional properties, for example in the mentioned case, the modification of surface acidity properties whilst maintaining the rutile phase.22 Ternary metal fluorides for instance, AgCuF3,23 CuxFe1−xF2 or Li3MF6 (M = V, Cr, Mn, Co, Fe) demonstrate reversible conversion reactions with Li-ions,24–26 rendering them potential energy storage materials. It is also found that multi-element incorporation described by the high entropy concept is beneficial for electrochemical storage in sodium ion batteries (SIB).27 This includes well-designed open framework strategies and doping with conversion type materials28 such as ReO3-type FeF3 (ref. 29 and 30) yielding high performance SIB cathodes.
Taken together, the transfer of the high entropy concept to transition metal fluorides in general, seems to be a promising approach for novel catalytic and electrochemical applications.31,32 First reports on rare-earth-based fluorite and perovskite structured high entropy fluorides and rock-salt structured oxyfluorides have shown promising properties for energy storage, and optical and catalytic properties.6,16,18 Wang et al. reported that the existence of weak bonds between transition metal (TM)-fluoride species, intrinsic structural defects and distortion of the HEM lattice due to different TM constituents can provide enhanced oxygen evolution reaction (OER) activity.18 Due to their special electronic configuration and surface complexity, HEMs are in general expected to provide desirable electrocatalytic properties.33,34
In the present work, we detail the synthesis and in-depth characterization of multi-cation (with 4 to 7 cations) composed rutile (P42/mnm) structured medium- (4 cations) and high-entropy (5–7 cations) fluorides (MEF and HEFs, respectively). Considering a solid solution state, the configurational entropy for a 7-cation containing compound can be calculated to be 1.95R (based on eqn (S1), where R is the universal gas constant). These compounds were synthesized using a straightforward and scalable mechanochemical milling process, therefore avoiding more complex synthesis procedures as reported in previous publications on HEFs.18 The characterization of the HEFs is demonstrated to support the assumption of a solid solution structure of the compounds, which is imperative for confirming the high configurational entropy. By using various measurement techniques, stretching of M–F bonds at the surface of the nanocrystalline particles was detected, resulting in different chemical environments, as similarly described for nanosized FeF2 particles by Ramasamy et al.35 Additionally, the compounds were tested with respect to their electrocatalytic performance for the OER. It could be shown that the Tafel slope was the lowest for all reported high entropy ceramics, including IrO2, which was taken as a state-of-the-art reference material. Additionally, the overpotential was decreased, despite the exclusion of noble metal elements. These results show the promising catalytic features of these easily synthesized high entropy fluorides.
Results and discussion
In this work, a novel class of high entropy materials, rutile structured high entropy fluorides (HEFs) are presented. Namely (CuNiFeZn)F2, (CuNiFeCoZn)F2, (CuNiFeCoMn)F2, (CuNiFeCoZnMn)F2 and (CuNiFeCoZnMnMg)F2 are synthesized and characterized, hereinafter labeled MEF4, HEF5_Zn, HEF5_Mn, HEF6, and HEF7, respectively, based on the number of incorporated cations. Configurational entropy calculations are performed and shown in Table S1† and the entropy value ranges from 1.39R for the 4-cation system to a maximum of 1.95R in the case of the 7-cation system. Materials crystallizing in a rutile structure, lie in the tetragonal P42/mnm space group. The rutile structure (Fig. 1a) is composed of cations, which are coordinated by anions in a distorted octahedral symmetry. The MF6 octahedra share two opposing edges with the adjacent octahedra and therefore form a chain-like structure. These chains are connected to each other by the corners of the octahedra. The rutile structure is commonly known for oxide and fluoride compounds, e.g. TiO2, FeF2, ZnF2, etc. The respective cations for the MF2 HEFs (M = Co, Cu, Mg, Ni, Zn, Mn, Fe) are selected by adhering to the Hume–Rothery and Pauling's rules, which propose similarity in ionic radii, coordination numbers and crystal structures to successfully achieve a solid solution.36 Table S2† presents the space group and lattice constant values of the starting binary fluorides utilized as precursors for this study. A similar range of lattice constant values is exhibited by all binary fluorides. ZnF2 (a = b = 0.4705 nm and c = 0.3134 nm, as given by ICSD #9169) was used as the reference for refining the X-ray diffraction (XRD) patterns of the MEF and HEFs. Table S3† presents the structural parameters of all samples obtained from Rietveld analysis. For more understanding of the phase purity of the materials, log(I) vs. 2θ plots are given in Fig. S1.†Fig. 1b indicates that all high entropy materials show a single-phase rutile structure and the Miller indices hkl are labeled at the top diffractogram of HEF7, while a minor unidentifiable impurity phase is present in the medium entropy system MEF4. A representative Rietveld refinement result of the most complex HEF7 is presented in Fig. 1c. The single phase rutile structure is confirmed with lattice parameters values, a = b = 0.47134(0) nm, c = 0.31725(9) nm and an average crystallite size of 8.7(5) nm.37,38 Since slightly different sized ions are incorporated into one lattice, the shift of the (110) reflection can be expected. In MEF4 an average metal ion radius of 0.88 Å is calculated based on the assumption of equimolar proportions of the metal ions and the ionic radius values given by Shannon et al.; further details about the average metal ion radius of MEF4 and calculation can be found in the ESI.†39 Additionally, the high-spin configuration of the metal ions (where applicable) was assumed, since F− as a ligand leads to only a small splitting (weak ligand in the spectrochemical series for octahedrally coordinated ions) of the d-orbitals into t2g and eg states, therefore favoring high-spin configuration of the ions. For MEF4, the average metal ion radius is 0.88 Å, and adding another metal ion such as Co2+ which also has a similar ionic radius of 0.88 Å resulted in no significant shifts in the XRD patterns. Replacing Zn2+ (radius 0.89 Å) with Mn2+ (HEF5_Mn, radius Mn2+ 0.96 Å) leads to smaller lattice parameters, despite the larger cationic radius. The incorporation of both Zn2+ and Mn2+ seems to widen the unit cell in HEF6 compared to that in the HEF5 compounds. Identifying the reason for these lattice changes still requires further investigation. Adding slightly smaller Mg2+ (HEF7, radius Mg2+ 0.86 Å) reduces the size of the unit cell again. | ||
| Fig. 1 (a) Representative structural model of pristine HEF7 nanoparticles. The colored balls correspond to the positions of the different metals; the orange balls correspond to F (b) XRD patterns of MEF and HEF samples containing 4, 5, 6 and 7 cations. (c) Rietveld refinement of the XRD pattern of HEF7 confirming its phase purity. | ||
Transmission electron microscopy (TEM) analysis was carried out using HEF7 as an example, since it contains the highest number of different cations. The morphology of the as-synthesized HEF7 indicated very fine polycrystalline nanoparticles with crystallite sizes around 10 nm. This is in accordance with the Rietveld results. The sizes of the particles, composed of many crystallites, range up to hundreds of nm (Fig. 2a). The inset in Fig. 2a shows the results of selected-area electron diffraction (SAED) carried out on a small area at the edge of a HEF7 nanoparticle. The nanoparticles possess high crystallinity with a rutile-structure and the SAED annular pattern confirms that the material is obtained without any secondary phases or impurities. Fig. 2b illustrates a high-resolution TEM micrograph of the lattice planes from HEF7, with a d-spacing corresponding to the 110 planes (as confirmed from the XRD pattern).
 | ||
| Fig. 2 (a) TEM micrograph of HEF7 and the inset shows the corresponding SAED pattern. These SAED indices comply with ZnF2 (P42/mnm, 136) (b) HR-TEM image showing lattice planes (with the inset depicting a magnified area) with a d-spacing value corresponding to the (110) plane. | ||
Fig. 3 shows the results from energy-dispersive X-ray spectroscopy (EDS) analysis of the HEF7 nanopowders. The mapping indicates a solid solution with all 7 metallic cations and fluorine being homogeneously distributed at the nanometer level. From the homogeneous elemental distribution in EDS mapping, the TEM and the XRD results, it is assumed that the synthesized powders were single-phase solid solution materials. The stoichiometries of all the HEF based compounds with chemical formula MF2 were calculated based on ICP-OES results (Fig. S2†). The elemental mapping spectrum of HEF7 showing the uniform distribution of the metals is given in Fig. S3.†
 | ||
| Fig. 3 STEM-based elemental maps of HEF7, showing a homogeneous distribution. | ||
In order to further confirm the solid solution state of HEF7, 57Fe Mössbauer Spectroscopy (MS) investigation was carried out to identify the chemical environment and the oxidation state of Fe in HEF7. Fig. 4 shows the Mössbauer spectrum of HEF7. The spectrum was fitted using two quadrupole doublet subspectra, both corresponding to Fe2+ environments (doublet I and II) with nearly identical isomer shifts (IS, I = 1.34 mm s−1, II = 1.30 mm s−1) but distinct quadrupole splits (QS, I = 2.67 mm s−1, II = 1.96 mm s−1). In addition, a small fraction of Fe3+ was evident (IS = 0.40 mm s−1, QS = 0.48 mm s−1). The quantification, based on the relative area fraction, yielded the values of Fe3+, Fe2+ (doublet I) and Fe2+ (doublet II) as 19%, 50% and 31%, respectively. However, this result is in contrast to the other obtained results from SAED, XRD, TEM mapping and NMR, since it suggests a different chemical environment and even a mixed-valence state of the incorporated Fe. Nevertheless, this contrasting behavior is coherent with the conclusions drawn from the Mössbauer studies of FeF2 nanoparticles, reported by Ramasamy S. et al. They reported a similar finding, wherein MS peak splitting arose from local defects due to a large amount of grain boundaries.35 Additionally, they referred to a slightly different Fe–F bond stretching at the surface (due to minor topological hydration), which could lead to a different chemical environment and therefore requires different fitting parameters from bulk FeF2. Hence, we presume that the origin of broadening in the MS spectrum of HEF7 might be due to the vast amount of local defects and grain boundaries formed during the ball milling procedure.40
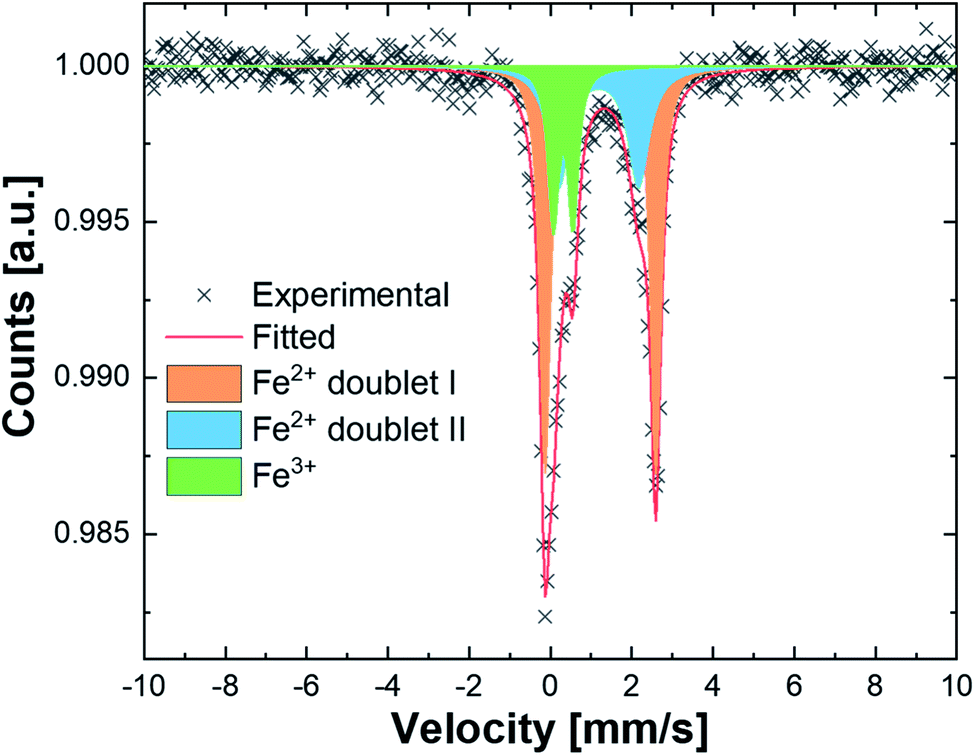 | ||
| Fig. 4 Mössbauer spectra of pristine HEF7 nanoparticles showing two different oxidation states and two different chemical environments. | ||
To elucidate the presence of Fe2+/3+ at the surface of the particles, electron energy loss spectroscopy (EELS) mapping was performed on the particle edges to identify the oxidation states of Fe, Co, Ni, Cu and Mn. Zn and Mg were assumed to be in the 2+ oxidation state. The sample was transported to a vacuum transfer TEM holder to exclude oxidation. Fig. 5 shows the EELS investigation results of HEF7 with five different areas on the edges of different particles being measured. Co, Cu, and Ni could only be found in the 2+ state and did not show any indications that point towards a mixture of different valence states. Fig. 5a shows the L3 and L2 edges of Mn, exhibiting a maximum peak shift between the different areas of about 0.6 eV. The L3/L2 ratio shows some minor differences between the areas, but the ratios for Mn3+ and Mn4+ would be much lower, so Mn in the 3+ and 4+ states can be excluded (see Fig. S2a†). Therefore, the Mn state in all the areas is measured to be around 2+. The Fe L3 and L2 edge positions are displayed in Fig. 5b. Two different L3 edges are detected, which can be indexed to Fe2+ and Fe3+ as indicated by Mössbauer spectroscopy results (more details in Fig. S2b†). Different measured areas show different valence states, which implies that an inhomogeneous change in the oxidation state takes place. We attribute this to the local defects resulting from ball-milling, as explained in the Mössbauer section of this article. Fig. 5c shows one of the measured areas and an EELS mapping, depicting that Fe2+ and Fe3+ appear mixed, and not separated into areas with only Fe2+ or Fe3+. However, it should be noted that no secondary phases were observed, either from XRD or HR-TEM, which indicates that these differences in the charge states are intrinsic features of single phase HEF7.
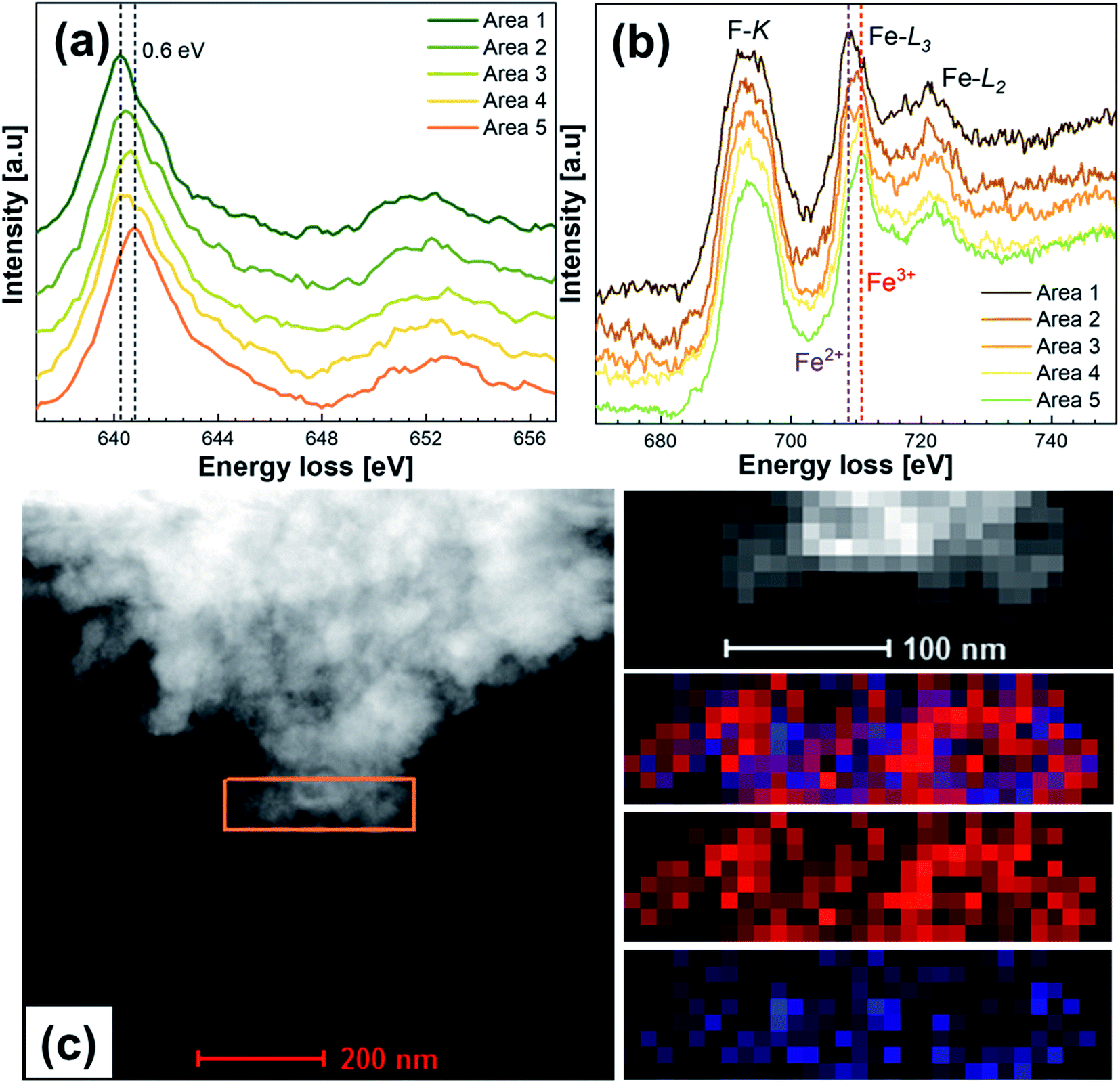 | ||
| Fig. 5 Valence state characterization of HEF7. EELS spectra of the (a) Mn L2,3 edge and (b) Fe L2,3 edge; (c) EELS mapping results on the marked area denoting the presence of Fe2+ (red) and Fe3+ (blue). | ||
Further, the surface chemical state of the HEF7 sample was probed with X-ray photoelectron spectroscopy (XPS). The relevant spectra of the constituent elements are shown in Fig. 6 and the corresponding reference spectra in Fig. S5–S13.† The survey spectrum showing the overview of constituents in HEF7 (binding energy range of 0–1350 eV) is given in Fig. S14.†
 | ||
| Fig. 6 XPS spectra of (a) Zn 2p, (b) Cu 2p, (c) Mg 1s (d) Zn Auger LMM kinetic energy spectra, (e) Co 2p, (f) Mn 2p, (g) F 1s, (h) Ni 2p and (i) Fe 2p of HEF7 nanoparticles. | ||
In the Zn 2p spectrum (Fig. 6a), a single doublet with the 2p3/2 component at 1023.1 eV binding energy and a full width at half maximum (FWHM) intensity of 2.3 eV is observed. As determining the Zn oxidation state purely from the Zn 2p peak is difficult, the Zn LMM Auger line is also considered (see Fig. 6d). The Zn LMM Auger line is observed at a kinetic energy of 985.4 eV and thus Zn can be assigned to the Zn2+ state.41 This agrees with our measurements on pure ZnF2 (see Fig. S5†) as a reference that shows similar values as Zn 2p3/2 is identified at 1022.9 eV with a FWHM intensity of 1.9 eV and a Zn LMM line at 985.2 eV. In the Cu 2p region (Fig. 6b), Cu ions in HEF7 show very interesting spectra with 2 main peaks at 933.4 and 937.3 eV. The peak at 937.3 eV with a FWHM intensity of 3.5 eV is analogous to Cu 2p3/2 of pure CuF2 measurements shown in Fig. S6.† Additionally, satellites of the Cu2+ state appear at 942.0–945 eV in the spectrum. However, the additional peak at 933.4 eV (FWHM: 2.3 eV) in the Cu 2p3/2 spectrum of HEF7 could be attributed to a reduced state of Cu ions or possibly the CuO or Cu(OH)2 state.42 According to this peak assignment, the majority of Cu ions at the surface of HEF7 (64% of total Cu ions) are attributed to Cu2+.42 As shown in Fig. 6c, the Mg 1s peak in the HEF7 sample is successfully assigned to Mg2+ ions in Mg–F bonding at 1305.4 eV with a FWHM intensity of 2.5 eV (ref. 43 and 44) (pure MgF2 XPS spectrum is given in Fig. S7†). The Co 2p spectrum (Fig. 6e) of HEF7 shows one doublet with a multiplet structure and a characteristic satellite. The main peak position at 783 eV as well as the satellites at 6.3 eV higher than the main peak indicates that the Co ions in HEF7 appear as Co2+ in a Co–F bonding situation. Additionally, the fitting parameters of HEF7 match with those of pure CoF2 as presented in Fig. S8.† To interpret the Ni 2p3/2 spectrum (Fig. 6h) in HEF7, it is compared to the Ni 2p3/2 spectrum of the pure NiF2 powder sample. The overlap of the Ni LMM and Fe LMM line in the Co 2p spectra is ignored, because of the low intensity of the Auger peaks. The peak overlay in Fig. S9† shows a similar binding energy and FWHM intensity of the main peak. Therefore, the Ni 2p3/2 spectrum of HEF7 was fitted with the multiplet parameters of Ni 2p3/2 in NiF2 and can be attributed to the Ni2+ state. However with this method the existence of a minor contribution of Ni3+ ions cannot be fully excluded, since the binding energies of Ni2+ and Ni3+ ions are very close to each other, at least in oxide samples.45–49 In the peak fitting of the Ni spectrum, overlapping F KLL, Mn LMM, and Fe 2s peaks are taken into consideration. The majority of F ions (Fig. 6g) at 685.4 eV with a FWHM of 2 eV are attributed to the F ions in the crystal structure and have a binding energy similar to that of binary fluorides (shown in Fig. S5–S12†).The low-intensity peak at 686.7 eV might probably occur through the ball milling process by forming C–F contaminations on the surface.50 In the Mn 2p spectra (Fig. 6f), a broad doublet can be observed. The Mn 2p3/2 peak can be fitted with two sets of multiplets and including the overlapping Ni LMM Auger peaks that indicates approximately 60% Mn3+ and 40% Mn2+ on the surface of HEF7. The multiplet sets of Mn2+ and Mn3+ are taken from the MnF2 and MnF3 reference measurements shown in Fig. S10 and S11,† respectively. Identification of the chemical state of Fe ions in HEF7 using XPS is challenging. In HEF7, the Fe 2p (Fig. 6i) overlaps with low intensity F plasmons and the Co LMM Auger line. Both aspects increase the background intensity and hamper reliable peak and background positioning. Furthermore, the overlapping Co Auger line hinders direct comparison to the FeF2 reference. Therefore, oxidation state analysis of the Fe ions in HEF7 from XPS was not possible. In summary, XPS confirms that Mg, Zn, and Co ions are present in the 2+ state, even at the outermost surface of the HEF particles. For Cu and Mn, mixed valence states were found at the surface, including around 64% of Cu ions and 40% of Mn ions in the 2+ state. Also, the majority of Ni ions can be considered as Ni2+. The Fe oxidation state could not be analyzed from XPS because of intense peak overlap. We assume that the discrepancy between XPS and EELS results regarding the oxidation state of Cu and Mn is surface related, since EELS pertains to the bulk material while XPS only to the surface. This indicates that a different surface state compared to the bulk material is apparent, was as well indicated by MS measurements and most probably an effect of the ball milling procedure.
Finally, to analyze the anionic site and to identify the presence of F− in the complex crystal structure, solid-state nuclear magnetic resonance (NMR) spectroscopy was carried out on pristine HEF7 and MEF4 nanoparticles. Exact probing of fluorine, which is a direct neighbor of the transition metal elements, is quite challenging due to often unpaired electronic spins. Among the starting binary fluorides (precursors described in detail in the Experimental section) MgF2, ZnF2, and NiF2 illustrated clear resonance peaks in the NMR spectra (refer Fig. S15†). Other binary fluorides, exhibited no signals as the unpaired electronic spin density at the F− sites leads to strong line broadening and very fast nuclear spin relaxation as F− sites are bound directly to paramagnetic ions.51–54Fig. 7 shows the F-NMR spectra of 4- and 7-element containing fluoride powders (MEF4 and HEF7). It should be noted that the overall intensity for both samples is extremely small, compared to the spectra of the binary samples (Fig. S15†), since all contributions are strongly broadened. MEF4, (CuNiFeZn)F2, still shows some residual peaks in the region between 0 and −400 ppm, which might hint at some residual clustering of some of the metals (comparable data of the NMR data of binary fluorides can be found in Fig. S15†). For HEF7, (CuNiFeCoZnMnMg)F2, no signals appear anymore, indicating that the electronic situation present in pure MgF2, ZnF2, NiF2 and MEF4 has changed in HEF7 materials and the relaxation time is much lower. This is noteworthy, since cations of binary fluorides showing a signal in 19F-MAS-NMR and of those not showing a signal are mixed with similar ratios in MEF4 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and HEF7 (3:4), respectively. Whether this is a direct result of the high entropy, cannot be confirmed with certainty from the data available here.
:2) and HEF7 (3:4), respectively. Whether this is a direct result of the high entropy, cannot be confirmed with certainty from the data available here.
 | ||
| Fig. 7 19F-MAS-NMR spectra of MEF4 and HEF7. Due to the high degree of cation mixing, peak-absence is noted in HEF7. | ||
To probe the HEF7 activity for the OER, linear sweep voltammetry (LSV) experiments were performed. For comparison, IrO2, a benchmark material for the OER, was also measured. Fig. 8a exhibits the LSV curves of HEF7 and IrO2 recorded at a scan rate of 5 mV s−1 in 1 M KOH. HEF7 shows superior activity, reaching 10 mA cm−2 at an overpotential of 292 mV, to IrO2, which required a higher overpotential (340 mV) for the same current density. The lower Tafel slope, 39 mV dec−1, was obtained for HEF compared to 45.2 mV dec−1 for IrO2, suggesting a faster kinetic charge transfer (Fig. 8b). Electrochemical impedance spectroscopy (EIS) was employed to further analyze the interfacial charge transfer processes at increasing potential from 1.25 V to 1.70 V with an interval of 0.05 V (Fig. 8c) and (Fig. S16†). In order to gain further insight into EIS, the data were fitted using the equivalent circuit shown in the inset of Fig. 8c, using the Zview software (Scribner Associates, Southern Pines, NC). The fitting data are added as solid lines and data as dots. In this potential range, mass transfer is not a limiting factor, as no Warburg behavior was observed. According to the Nyquist plots at 1.55 V (Fig. 8c), while HEF7 had a charge transfer resistance (Rct) of 6.305 Ω, the IrO2 had an approximately 2-fold higher Rct value, 12.143 Ω. The EIS data indicate that the superior OER activity of HEF7 to IrO2 is correlated with the faster charge transfer, which is well matched with the Tafel measurements. To avoid the artifacts from gas evolution during the electrolysis reaction, a voltage range of 1.3 V to 1.55 V for the EIS measurements was adopted. The deviation in the lower frequency region might be caused due to bubble formation during electrolysis as reported.55,56Fig. 8d and S17† show the double-layer capacitances (Cdl) values of the two samples. The Cdl value of HEF7 turned out to be approximately 4 times smaller than that of IrO2, implying that the higher activity of HEF7 is not arising from the electrochemical surface areas (ECSA) of the sample. Thus, if averaged by the ECSA, the resulting difference in activity, i.e., current density, between the catalysts would be even larger than that based on the geometric area of the electrodes (Fig. 8a). Given that such metrics have been commonly employed for benchmarking, Fig. 8e indicates that HEF7 is highly competitive with other high entropy derivative catalysts (such as HEO, HEPF, and HE-MOFs) recently reported for the OER, involving the lowest Tafel slope and second lowest overpotential to deliver current density.18,57,58 In comparison with reported catalytic activities of the binary fluorides such as CoF2,59 FeF2 (ref. 60) and NiF2,61 HEF7 was found to have improved performance for the OER, which is a strong indication of the benefits arising from the high entropy concept. With the implementation of the high entropy concept, it is possible to incorporate many different elements into a single-phase structure, thereby enabling various catalytic centers in a single compound. The unique catalytic activities of HEM emerge from the complex interaction between different elements in a single-phase solid solution. Compared with single metal catalysts, HEF provides a platform of synergetic effects of highly dispersed active sites, abundant fluorine sites and diffusion channels.58,62,63 Hence improved OER kinetics may arise due to low charge transfer resistance, the low polarization resistance values and lower Tafel slopes. In addition, fluorine possessing the highest electronegativity is ideal for achieving enhanced electrocatalytic activity.
 | ||
| Fig. 8 (a) Linear sweep voltammetry (LSV) curves, (b) Tafel plots of HEF7 (red solid line) and IrO2 (grey dashed line) in 1 M KOH, (c) Nyquist plots of HEF7 (red circle) and IrO2 (grey square) recorded at potentials of 1.45 V, 1.50 V and 1.55 V, (d) double layer capacitances (Cdl) of HEF and IrO2, (e) the equivalent circuit used for EIS data fitting and (f) Comparison of the Tafel slopes and overpotentials required at a current density of 10 mA cm−2 among the catalysts in this work and other high entropy derivatives. (HEPF: high entropy perovskite fluoride, HEO: high entropy oxide, HE-MOF: high entropy metal organic framework). | ||
In order to evaluate the stability of the catalysts, accelerated degradation tests (ADT) were conducted by scanning the potential between 1.4 V and 1.65 V for 3000 cycles at a scan rate of 50 mV s−1 in 1 M KOH. Fig. 9a exhibits the polarization curves of HEF and IrO2 before and after the ADT. The catalytic activity of HEF after ADT remained nearly identical to the initial activity before ADT, indicating good catalytic stability comparable to IrO2. Furthermore, chronopotentiometry measurements were performed to assess the long-term performance at 10 and 50 mA cm−2 for 6 h, respectively (Fig. 9b). During 12 h of electrolysis in total, for HEF, no significant increase in the corresponding potentials was observed, staying at lower potentials than IrO2 (Fig. 9b). The XRD patterns of HEF7 were recorded prior to the chronopotentiometry experiment which lasted for 12 h and then compared with the ones measured again afterwards. As shown in Fig. S18,† the two diffractograms had no detectable difference despite the long-time test, which is in good agreement with the robust catalytic performance. According to the XRD patterns acquired after the long term cycling process, prominent peaks of the rutile phase indicate that the structure is well maintained after the long electrolysis process.
 | ||
| Fig. 9 Electrochemical stability tests (a) LSV curves of HEF and IrO2 before and after accelerated degradation tests (ADT) (b) chronopotentiometry measurements of the OER at 10 and 50 mA cm−2 for 6 h respectively using HEF (red) and IrO2 (grey). | ||
Due to the small crystallite size and the preparation of the electrode, it would be very difficult to detect changes using XRD. Further investigation after cycling might be out of the scope of this study.
Conclusions
In summary, this report shows the preparation, characterization and promising application of novel rutile-type high entropy fluorides. A successful low-cost synthesis route could be shown, which details the preparation of multi-cationic high entropy fluorides by a simple mechanochemical process. This method evades the need for a high-temperature preparation and quenching process. Based on the combined investigations from XRD, ICP-OES, TEM, NMR and EDX-EELS studies, it is shown that all pristine HEF samples crystallized in a phase-pure rutile structure with the presence of agglomerated fine nanoparticles. The identification of the local structure of the HEF compounds was probed by F-NMR and XPS. The local structural disorder of the HEF compounds arose presumably due to the synthesis process and the resulting variation in the oxidation states of Fe was detected utilizing Mössbauer spectroscopy. Finally, the OER activity of tailored HEF was validated for electrocatalytic applications and HEF exhibited a lower overpotential and Tafel slope in comparison to reference commercial IrO2. As reported for high entropy oxides, multi-cation substitution in HEF materials may as well provide an interesting path for tailoring the electrochemical properties of conversion electrodes for energy storage applications.Experimental section
Synthesis methodology
Commercially available analytical grade transition metal difluorides with purity >99% (such as MnF2, FeF2, CoF2, NiF2, CuF2, ZnF2, and MgF2; Alfa Aesar) were used for this work. All the chemicals were used for the synthesis without any further purification steps. These HEF materials were prepared by a dry long-term high energy milling process carried out at 500 rpm for 48 h in a WC jar (WC ball to powder weight ratio as 20:1). Using this route, HEF samples containing equimolar ratios of 4, 5, 6 and 7 elements were produced. All samples were prepared in an Ar atmosphere (H2O < 0.5 ppm, O2 < 0.5 ppm).
Characterization methodology
All the pristine HEF samples were characterized with powder X-ray diffraction (XRD) patterns collected in the range from 10° to 90° (2θ) on a Bruker D8 diffractometer with Cu Kα radiation (λ = 1.54 Å) equipped with a LYNEXE detector. A step size of 0.02° and 4 s time per step at 30 kV and 40 mA was used. Rietveld refinements were performed by using the TOPAS software (TOPAS 5, Bruker) to identify the phase composition and lattice parameters. LaB6 (NIST 660a) was used as the reference material to determine the instrumental intensity distribution for the refinement of XRD data. The structural model for pristine HEF7 was created using 3D visualization software, VESTA (visualization for electronic and structural analysis). The elemental analysis of all HEF powders was performed by inductively coupled plasma optical emission spectroscopy (ICP-OES) (iCAP 7600DUO from Thermo Fisher Scientific). About 5–10 mg of the samples (weighing accuracy ±0.05 mg) were dissolved in 6 mL hydrochloric acid and 2 mL nitric acid at 353 K for four hours in a graphite oven. The analysis of the elements was accomplished with four different calibration solutions and an internal standard (Sc). The range of the calibration solutions did not exceed a decade. Two or three wavelengths of each element have been used for calculations. The amount of fluorine was estimated using an ion-selective electrode (perfectION, Mettler Toledo). The obtained residue was diluted and pH was leveled between 5 and 8 using both 5 M NaOH solution and TISAB (total ionic strength adjustment buffer) solution. The concentration of fluorine was found by the method of standard addition. TEM studies (SAED, HR-TEM, EDX and EELS) were conducted using an FEI Titan 80–300 microscope, with a CEOS image spherical aberration corrector, a HAADF-STEM detector (Fischione model 3000), an EDX detector and a Tridiem Gatan image filter. An accelerating voltage of 300 kV was used for imaging. Elemental maps were constructed using EDS. Micrograph measurements were conducted on the as-synthesized HEF nanoparticles (without air exposure) dispersed on a holey carbon-coated gold grid inside an Ar filled glovebox. Since the fluoride-based cathode materials are prone to being beam sensitive, low voltage imaging was necessary under certain imaging conditions. Room temperature 19F-MAS-NMR (magic-angle spinning) data were acquired with a Bruker 200 MHz spectrometer at a field of 4.7 T, corresponding to a Larmor frequency of 188.3 MHz. Spinning was performed at 25 kHz in 2.5 mm rotors; NMR spectra were acquired with a Hahn-Echo experiment (π/2 pulse of 0.5 μs duration). Liquid CFCl3 was used as the reference for the chemical shift calibration. X-ray photoelectron spectroscopy (XPS) measurements were performed on a K-Alpha+ instrument (Thermo Fisher Scientific) with a monochromatic Al-Kα X-ray source (1486.6 eV) and 400 μm spot size. A K-Alpha+ charge compensation system was applied to prevent localized charge buildup during analysis using 8 eV electrons and low-energy Ar ions. Data acquisition and processing were carried out using the Thermo Avantage software.64,65 The spectra were fitted with one or more Voigt profiles. The binding energies are reported with respect to the C 1s peak of hydrocarbons at 285.0 eV. The analyzer transmission function, Scofield sensitivity factors65 and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2M formalism.6657Fe Mössbauer spectroscopy data were gathered in transmission geometry using a spectrometer with a moving source of 57Co in a Rh matrix and a triangular velocity variation. All reported isomer shifts of samples are reported relative to the bcc phase-Fe measured at room temperature. Catalyst ink, mixed with 1 mL deionized water, 0.25 mL 2-propanol, 10 μL Nafion solution (5 wt%) and 2 mg of catalysts, was used for electrochemical measurements. Standard IrO2 (99.9%, ABCR GmbH, as-received) was used to make the ink. The resulting ink was sonicated for at least 1 h and then loaded via drop-casting (20 μL) on plasma-treated carbon cloth, 0.25 cm2 (ELAT-hydrophilic plain cloth, Fuel Cell Store). Electrochemical tests were carried out on a multi-channel potentiostat/galvanostat (VSP300, Biologic) electrochemical workstation. A lab-made double junction Ag/AgCl electrode with saturated KCl filling solution served as a reference electrode and a Pt wire was used as a counter electrode. The linear sweep voltammetry (LSV) curves were obtained at a scan rate of 5 mV s−1 in 1 M KOH (1 N, Merck KGaA). The LSV experiment was conducted at 1 mV s−1 for Tafel slope analysis. The ohmic drop was corrected at a rate 90% with the current interrupt (CI) method. All electrochemical measurements in this work were carried out in the reversible hydrogen electrode (RHE) scale calculated as in eqn (1):| E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.197 V + 0.0592 × pH | (1) |
Overpotentials (η) for the oxygen evolution reaction (OER) were obtained by subtracting 1.23 V from the experimental potentials.
The Tafel equation given in eqn (2) was used for calculating the Tafel slope:
 | (2) |
Electrochemical impedance spectroscopy (EIS) measurements were performed at applied constant potentials from 1.3 V to 1.55 V with an interval of 0.05 V in a frequency range from 100 KHz to 10 mHz. An AC sinusoidal potential of 5 mV was applied as the perturbation signal. Chronopotentiometry measurements were conducted to test the long-term stability at 10 and 50 mA cm−2 for each 6 h. The double-layer capacitances (Cdl) of the catalysts were obtained from CVs in the potential range from 1.18 V to 1.32 V (V vs. RHE) in 1 M KOH. The scan rates were from 5 mV s−1 to 100 mV s−1 (5, 10, 20, 40, 60, 80 and 100 mV s−1). ΔJ = |ja − jc| at 1.27 V were plotted versus the scan rate and subsequently the slope of the plot was fitted and calculated, which is equal to the value twice the corresponding Cdl.
X-ray diffraction (XRD) analysis for the catalytic samples were performed using an X'Pert Philips diffractometer in Bragg–Brentano geometry with monochromatic CuKα radiation (0.1541 nm) and a fast Si-PIN multi-strip detector. The XRD spectra were collected at a scan rate of 0.02 degree per s. The HEF7 particles were detached and collected by sonicating the HEF-deposited carbon cloth electrode in ethanol for 30 min. The obtained suspension was drop-cast on a zero-diffraction plate and then dried in a 75 °C oven for 30 min prior to XRD measurement.
Author contributions
P. A. S. synthesized the materials, performed the experiments and wrote the manuscript. Y. C. prepared the materials and characterized the materials and wrote the manuscript. S. L. performed the catalytic measurements and helped writing the manuscript. K. W. performed TEM and R. A. performed XPS measurements. A. S. and R. K. conducted Mössbauer spectroscopy and S. I. NMR spectroscopy. S. S. B. and H. H. provided fruitful ideas and corrected the manuscript. Q. W., M. B. and B. B. supervised the synthesis and experiments, directed the project and wrote the manuscript. All the authors contributed to the data analysis, discussion and manuscript preparation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
P. A. S. acknowledges the MERAGEM graduate school and the Ministry of Science, Research and Arts of the State of Baden Württemberg for funding the research. Y. C. acknowledges the financial support from the China Scholarship Council (CSC). P. A. S. thanks Prof. Wolfgang Bessler (HS Offenburg) for the fruitful discussions. Q. S., B. B. and H. H. acknowledge the support of EnABLES, a project funded by the European Union's Horizon 2020 research and innovation program under grant agreement no. 730957. M. B. acknowledges the support of the German Research Foundation (DFG) project no. SE 1407/4-2. The authors acknowledge the Centre for Electrochemical Energy Storage Ulm-Karlsruhe (CELEST). The authors acknowledge the support from the Karlsruhe Nano Micro Facility (KNMF), Helmholtz research infrastructure at Karlsruhe Institute of Technology (KIT). The authors thank Prof. Xile Hu for providing experimental support in carrying out the electrochemical measurements at EPFL. S. L. gratefully acknowledges funding from European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 838367. The authors acknowledge Dr Thomas Bergfeldt (IAM-AWP, KIT) and David Stenzel for the ICP-OES measurements. We acknowledge support by the KIT-Publication Fund of the Karlsruhe Institute of Technology.References
- J. W. Yeh and S. J. Lin, J. Mater. Res., 2018, 33, 3129–3137 CrossRef CAS.
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 7936, 295–309 CrossRef.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 1–9 CrossRef.
- J. W. Yeh, S. K. Chen, S. J. Lin, J. Y. Gan, T. S. Chin, T. T. Shun, C. H. Tsau and S. Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- B. Gwalani, R. M. Pohan, J. Lee, B. Lee, R. Banerjee, H. J. Ryu and S. H. Hong, Sci. Rep., 2018, 8, 1–9 CrossRef CAS.
- Q. Wang, A. Sarkar, D. Wang, L. Velasco, R. Azmi, S. S. Bhattacharya, T. Bergfeldt, A. Düvel, P. Heitjans, T. Brezesinski, H. Hahn and B. Breitung, Energy Environ. Sci., 2019, 12, 2433–2442 RSC.
- A. Sarkar, B. Breitung and H. Hahn, Scr. Mater., 2020, 187, 43–48 CrossRef CAS.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J. P. Maria, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- D. Bérardan, S. Franger, D. Dragoe, A. K. Meena and N. Dragoe, Phys. Status Solidi RRL, 2016, 10, 328–333 CrossRef.
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC.
- Y. Qin, J. X. Liu, F. Li, X. Wei, H. Wu and G. J. Zhang, J. Adv. Ceram., 2019, 8, 148–152 CrossRef CAS.
- J. Gild, Y. Zhang, T. Harrington, S. Jiang, T. Hu, M. C. Quinn, W. M. Mellor, N. Zhou, K. Vecchio and J. Luo, Sci. Rep., 2016, 6, 2–11 CrossRef.
- T. Jin, X. Sang, R. R. Unocic, R. T. Kinch, X. Liu, J. Hu, H. Liu and S. Dai, Adv. Mater., 2018, 30, 1707512 CrossRef.
- A. Sedegov, S. Vorotilo, V. Tsybulin, K. Kuskov and D. Moscovskikh, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 558, 1–7 Search PubMed.
- A. Sarkar, R. Djenadic, N. J. Usharani, K. P. Sanghvi, V. S. K. Chakravadhanula, A. S. Gandhi, H. Hahn and S. S. Bhattacharya, J. Eur. Ceram. Soc., 2017, 37, 747–754 CrossRef CAS.
- X. Chen and Y. Wu, J. Am. Ceram. Soc., 2019, 1–7 Search PubMed.
- K. Zhang, W. Li, J. Zeng, T. Deng, B. Luo, H. Zhang and X. Huang, J. Alloys Compd., 2020, 817, 153328 CrossRef CAS.
- T. Wang, H. Chen, Z. Yang, J. Liang and S. Dai, J. Am. Chem. Soc., 2020, 142, 4550–4554 CrossRef CAS.
- X. Hua, R. Robert, L. S. Du, K. M. Wiaderek, M. Leskes, K. W. Chapman, P. J. Chupas and C. P. Grey, J. Phys. Chem. C, 2014, 118, 15169–15184 CrossRef CAS.
- M. G. Cottam and D. J. Lockwood, Low Temp. Phys., 2019, 45(1), 78–91 CrossRef CAS.
- F. Waltz, M. A. Swider, P. Hoyer, T. Hassel, M. Erne, K. Möhwald, M. Adlung, A. Feldhoff, C. Wickleder, F. W. Bach and P. Behrens, J. Mater. Sci., 2012, 47, 176–183 CrossRef CAS.
- A. Rehmer, K. Scheurell, G. Scholz and E. Kemnitz, Nanomaterials, 2017, 7, 1–13 CrossRef.
- W. Tong and G. G. Amatucci, J. Power Sources, 2017, 362, 86–91 CrossRef CAS.
- F. Wang, S. W. Kim, D. H. Seo, K. Kang, L. Wang, D. Su, J. J. Vajo, J. Wang and J. Graetz, Nat. Commun., 2015, 6, 1–9 CAS.
- J. Kohl, D. Wiedemann, S. Nakhal, P. Bottke, N. Ferro, T. Bredow, E. Kemnitz, M. Wilkening, P. Heitjans and M. Lerch, J. Mater. Chem., 2012, 22, 15819–15827 RSC.
- J. Li, L. Xu, K. Wei, S. Ma, X. Liu, Y. Zhao and Y. Cui, Ionics, 2020, 26, 3367–3375 CrossRef CAS.
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. S. Hu, Angew. Chem., Int. Ed., 2020, 59, 264–269 CrossRef CAS.
- C. Li, K. Chen, X. Zhou and J. Maier, npj Comput. Mater., 2018, 4, 1–15 CrossRef.
- Y. Han, J. Hu, C. Yin, Y. Zhang, J. Xie, D. Yin and C. Li, J. Mater. Chem. A, 2016, 4, 7382–7389 RSC.
- D. Cao, C. Yin, D. Shi, Z. Fu, J. Zhang and C. Li, Adv. Funct. Mater., 2017, 27, 1701130–1701139 CrossRef.
- H. Chen, J. Fu, P. Zhang, H. Peng, C. W. Abney, K. Jie, X. Liu, M. Chi and S. Dai, J. Mater. Chem. A, 2018, 6, 11129–11133 RSC.
- F. Wang, R. Robert, N. A. Chernova, N. Pereira, F. Omenya, F. Badway, X. Hua, M. Ruotolo, R. Zhang, L. Wu, V. Volkov, D. Su, B. Key, M. Stanley Whittingham, C. P. Grey, G. G. Amatucci, Y. Zhu and J. Graetz, J. Am. Chem. Soc., 2011, 133, 18828–18836 CrossRef CAS.
- A. P. O'Mullane, M. Escudero-Escribano, I. E. L. Stephens and K. Krischer, ChemPhysChem, 2019, 20, 2900–2903 CrossRef.
- X. Cui, B. Zhang, C. Zeng and S. Guo, MRS Commun., 2018, 8, 1230–1235 CrossRef CAS.
- S. Ramasamy, J. Jiang, H. Gleiter, R. Birringer and U. Gonser, Solid State Commun., 1990, 74, 851–855 CrossRef CAS.
- M. C. Troparevsky, J. R. Morris, M. Daene, Y. Wang, A. R. Lupini and G. M. Stocks, Jom, 2015, 67, 2350–2363 CrossRef CAS.
- J. W. Stout and S. A. Reed, J. Am. Chem. Soc., 1954, 76, 5279–5281 CrossRef CAS.
- M. J. Armstrong, A. Panneerselvam, C. O'Regan, M. A. Morris and J. D. Holmes, J. Mater. Chem. A, 2013, 1, 10667–10676 RSC.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925–946 CrossRef CAS.
- V. Šepelák, A. Feldhoff, P. Heitjans, F. Krumeich, D. Menzel, F. J. Litterst, I. Bergmann and K. D. Becker, Chem. Mater., 2006, 18, 3057–3067 CrossRef.
- C. D. Wagner, Anal. Chem., 1975, 47, 1201–1203 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- S. Da-Ming, S. Zhao-Qi, L. Ai-Xia and X. Zhi-Yuan, Vacuum, 1999, 52, 383–386 CrossRef.
- E. Quesnel, L. Dumas, D. Jacob and F. Peiró, J. Vac. Sci. Technol., A, 2000, 18, 2869–2876 CrossRef CAS.
- R. Azmi, M. Masoumi, H. Ehrenberg, V. Trouillet and M. Bruns, Surf. Interface Anal., 2018, 50, 1132–1137 CrossRef CAS.
- C. R. Mariappan, V. Kumar, R. Azmi, L. Esmezjan, S. Indris, M. Bruns and H. Ehrenberg, CrystEngComm, 2018, 20, 2159–2168 RSC.
- J. Wang, D. Stenzel, R. Azmi, S. Najib, K. Wang, J. Jeong, A. Sarkar, Q. Wang, P. A. Sukkurji, T. Bergfeldt, M. Botros, J. Maibach, H. Hahn, T. Brezesinski and B. Breitung, Electrochem, 2020, 1, 60–74 CrossRef.
- R. Azmi, V. Trouillet, M. Strafela, S. Ulrich, H. Ehrenberg and M. Bruns, Surf. Interface Anal., 2018, 50, 43–51 CrossRef CAS.
- V. Kumar, C. R. Mariappan, R. Azmi, D. Moock, S. Indris, M. Bruns, H. Ehrenberg and G. Vijaya Prakash, ACS Omega, 2017, 2, 6003–6013 CrossRef CAS.
- S. Mori, P. Cong, Y. Shinden and H. Nanao, Tribol. Lett., 2004, 17, 83–89 CrossRef CAS.
- G. Scholz, S. Breitfeld, T. Krahl, A. Düvel, P. Heitjans and E. Kemnitz, Solid State Sci., 2015, 50, 32–41 CrossRef CAS.
- A. Sadoc, M. Body, C. Legein, M. Biswal, F. Fayon, X. Rocquefelte and F. Boucher, Phys. Chem. Chem. Phys., 2011, 13, 18539–18550 RSC.
- Y. Guo, S. Wuttke, A. Vimont, M. Daturi, J. C. Lavalley, K. Teinz and E. Kemnitz, J. Mater. Chem., 2012, 22, 14587–14593 RSC.
- N. A. Matwiyoff, L. B. Asprey, W. E. Wageman, M. J. Reisfeld and E. Fukushima, Inorg. Chem., 1969, 8, 750–753 CrossRef CAS.
- K. Elsøe, M. R. Kraglund, L. Grahl-Madsen, G. G. Scherer, J. Hjelm, S. H. Jensen, T. Jacobsen and M. B. Mogensen, Fuel Cells, 2018, 18, 640–648 CrossRef.
- S. Siracusano, S. Trocino, N. Briguglio, V. Baglio and A. S. Aricò, MDPI Mater., 2018, 11, 1–15 Search PubMed.
- X. Zhao, Z. Xue, W. Chen, X. Bai, R. Shi and T. Mu, J. Mater. Chem. A, 2019, 7, 26238–26242 RSC.
- D. Wang, Z. Liu, S. Du, Y. Zhang, H. Li, Z. Xiao, W. Chen, R. Chen, Y. Wang, Y. Zou and S. Wang, J. Mater. Chem. A, 2019, 7, 24211–24216 RSC.
- Z. Liu, H. Liu, X. Gu and L. Feng, Chem. Eng. J., 2020, 397, 125500 CrossRef CAS.
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, ACS Catal., 2020, 10, 4019–4047 CrossRef CAS.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W. G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef CAS.
- H. Li, H. Zhu, S. Zhang, N. Zhang, M. Du and Y. Chai, Small Struct., 2020, 1, 2000033 CrossRef.
- G. M. Tomboc, T. Kwon, J. Joo and K. Lee, J. Mater. Chem. A, 2020, 8, 14844–14862 RSC.
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- H. Shinotsuka, S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 2015, 47, 871–888 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10209a |
| ‡ Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2021 |