
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic syngas production using conjugated organic polymers†
Zhiwei
Fu
 a,
Anastasia
Vogel
a,
Martijn A.
Zwijnenburg
b,
Andrew I.
Cooper
*a and
Reiner Sebastian
Sprick
*ac
a,
Anastasia
Vogel
a,
Martijn A.
Zwijnenburg
b,
Andrew I.
Cooper
*a and
Reiner Sebastian
Sprick
*ac
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: sebastian.sprick@strath.ac.uk
First published on 18th January 2021
Abstract
A range of linear conjugated polymers is reported that promote photocatalytic CO2 reduction in water with a sacrificial hole-scavenger. Two photocatalysts containing dibenzo[b,d]thiophene sulfone were found to be the most active materials. A dibenzo[b,d]thiophene sulfone co-polymer with phenylene (P7) had the highest rate for producing CO, but also for the co-evolution of H2. The homopolymer of dibenzo[b,d]thiophene sulfone was found to be less active for CO production, but had a higher H2 production rate, which is explained by changes in the driving-force favouring proton reduction. The co-evolution of hydrogen is facilitated by residual palladium from the material synthesis. By varying the amount of palladium in the photocatalyst, syngas can be obtained with varying ratios of H2 to CO.
Introduction
The combustion of fossil fuels has resulted in increased concentrations of CO2 in the atmosphere, which in turn has caused more extreme weather events due to climate change.1 Much research is focused on replacing fossil fuels with ‘green’ solar derived alternative fuels, but it may ultimately be necessary to remove CO2 from the atmosphere to limit global warming.2 Carbon-capture and sequestration (CCS) is widely considered for this, but an alternative is to convert CO2, an unwanted side-product, into valuable chemicals through intermediates such as CO.3 For example, mixtures of hydrogen and CO (syngas) are used in large-scale industrial processes such as the Fischer–Tropsch process.4The electroreduction of CO2 into syngas has been studied widely, but this requires an additional bias to drive the reduction.4,5 Alternatively, photocatalytic CO2 reduction using sunlight and semiconductors as catalysts has been studied for the direct conversion of CO2 into value-added products.6 Most photocatalysts so far are either inorganic materials or molecular metal–organic compounds. Recently, there has been growing interest in using conjugated materials as photocatalysts due to their tunability and large synthetic diversity, initially for photocatalytic hydrogen production,7–13 but more recently also for CO2 reduction. Organic materials such as conjugated microporous polymers (CMPs),14,15 covalent triazine-based frameworks (CTFs),16,17 covalent organic frameworks (COFs)18–21 and unbranched conjugated polymers22 have been investigated for their potential as photocatalysts in CO2 reduction.
One of the challenges when using photocatalysts for syngas production is that different syngas compositions are required depending on its further processing, which could require either CO-rich or H2-rich mixtures4,23–25 This possess a challenge because the H2/CO ratios are difficult to control; hence, much research is focused on obtaining photocatalysts with high selectivity for CO over H2, with the H2 originating from a second, different process that is optimised for proton reduction.
Results and discussion
We started to explore if the co-evolution of both H2 and CO would be possible from a single material and whether the H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio could be controlled. For this, we prepared conjugated polymers via Suzuki–Miyaura polycondensation using Pd(0), which results in the formation of palladium particles within the material. This residual palladium has been shown previously to act as a co-catalyst for hydrogen production from water.26–28 Here, we study a range of photocatalysts: p-sexiphenylene,29 poly(p-phenylene) (P1),30 a carbazole-phenylene co-polymer (P4),30 a dibenzo[b,d]thiophene sulfone co-polymer (P7),30,31 the homopolymer of dibenzo[b,d]thiophene sulfone (P10),31 a pyrimidine–phenylene co-polymer (P29),32 a bipyridine–phenylene co-polymer (P30),32 poly(pyridine) (P31),32 and poly(benzothiadiazole) (P74).
:CO ratio could be controlled. For this, we prepared conjugated polymers via Suzuki–Miyaura polycondensation using Pd(0), which results in the formation of palladium particles within the material. This residual palladium has been shown previously to act as a co-catalyst for hydrogen production from water.26–28 Here, we study a range of photocatalysts: p-sexiphenylene,29 poly(p-phenylene) (P1),30 a carbazole-phenylene co-polymer (P4),30 a dibenzo[b,d]thiophene sulfone co-polymer (P7),30,31 the homopolymer of dibenzo[b,d]thiophene sulfone (P10),31 a pyrimidine–phenylene co-polymer (P29),32 a bipyridine–phenylene co-polymer (P30),32 poly(pyridine) (P31),32 and poly(benzothiadiazole) (P74).
All polymers were tested as photocatalysts for photocatalytic CO2 reduction under solar irradiation using a previously reported high-throughput work-flow.33,34 For this, vials were charged with photocatalysts (5 mg), CoCl2 (1 μmol), which acts as the co-catalyst, and 2,2′-bipyridine (BPy, 2 mg), which is required to form catalytically active Co centres.22,35–38
These vials were then transferred to a liquid handling robot, and inertised under CO2. The liquid handler then added water (1 mL), acetonitrile (MeCN, 3 mL), acting as an inert co-solvent, and triethanolamine (TEOA, 1 mL), which acts as the hole scavenger before capping the vials. After this, the samples were sonicated to disperse the photocatalysts and transferred to a solar simulator (AM1.5G, 1600 W xenon light source, air mass 1.5G filter, 350–1000 nm). The samples were irradiated with constant agitation for a specific time before measurement of the gaseous products using an automated gas chromatograph.
It was found that p-sexiphenylene, P31 and P74 showed little or no activity under the conditions used for either CO or H2 production (Fig. 1c). Photocatalysts P1, P4, P29, and P30 all produced CO with rates between 68.8 μmol g−1 h−1 for P30 and 291.9 μmol g−1 h−1 for P4 and selectivities between 37% for P30 and 59% for P4 because significant amounts of H2 were produced. Both P7 and P10 showed significantly higher rates of CO production: 959.1 μmol g−1 h−1 and 839.7 μmol g−1 h−1. The high CO production rates of P7 and P10 are accompanied by a high H2 evolution rate of 1523.7 μmol g−1 h−1 for P7 and an even higher H2 evolution rate of 2676.3 μmol g−1 h−1 for P10, which is in line with the observation that P10 is a better hydrogen evolution photocatalyst than P7.31 The high hydrogen evolution rates seem not to originate from oxidative dehydrogenation of TEOA as a side reaction,36 but may be explained by the presence of residual palladium, which acts as an efficient co-catalyst for proton reduction27,28 competing with cobalt sites for electrons. Together with the sluggish kinetics of CO2 reduction this results in poor CO production selectivity compared with other materials. No other products, such as methane, methanol, acetaldehyde or formate, were observed by gas chromatography and ion chromatography.
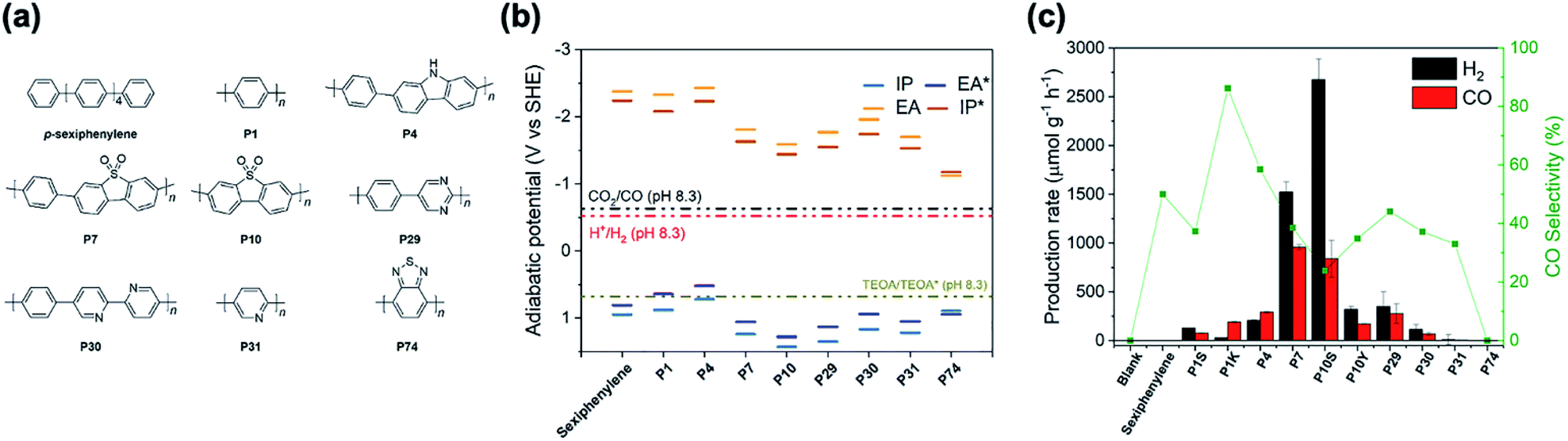 | ||
| Fig. 1 (a) Structures of the photocatalysts; (b) predicted redox potentials of the polymer photocatalysts taken from the literature (P1–P31)30–32,39 or calculated in this study (P74, see ESI†) and solution potentials (CO2 and proton reduction, TEOA oxidation, see ESI†) at pH 8.3 (the pH of the reaction mixture saturated with CO2) (c) evolution rates and selectivity of gaseous products produced by all photocatalysts in a high-throughput screening. Conditions: polymers (5 mg), CoCl2 (1 μmol), 2,2′-biyridine (2 mg) solvent (4 mL, MeCN/H2O = 3:1), TEOA (1 mL), solar simulator (5 hours), CO2 atmosphere. | ||
The role of palladium in the proton reduction reaction is further supported when looking at poly(p-phenylene) made via nickel catalysed Kumada-polycondensation (P1K) that contain residual nickel, which is a poorer hydrogen evolution co-catalyst. Hence, P1K has a higher CO evolution rate compared to P1 made via Suzuki–Miyaura polycondensation (189.7 vs. 77.7 μmol g−1 h−1), and a higher selectivity for CO production (86% vs. 37%). Similarly, P10 made via Yamamoto coupling (P10Y) has a higher selectivity for CO over H2 compared to the Suzuki–Miyaura polycondensation product (35% vs. 24%).
These observations show that palladium is, in general, a negative influence on CO/H2 selectivity. In keeping with this, literature reports of materials with good selectivities are usually made using metal-free conditions, hence, resulting in materials that have no residual metal present that facilitate hydrogen production.20 However, we note that some systems that were produced using Pd-catalysed cross-coupling reactions have been reported by other researchers to produce CO with good selectivity over H2 despite their Pd-content.16
The particularly poor CO selectivity for P10 compared to the structurally similar P7 can be explained by the fact that the EA (electron affinity; that is, the LUMO) of P10 is less negative than for P7 (ref. 31) (Fig. 1b) and that hence P10 thus has a reduced thermodynamic driving force for reduction relative to P7. As CO2 reduction is thermodynamically slightly less favoured than reduction of protons E(CO2,H+/CO) −0.63 V (ref. 40) E(H+/H2) −0.52 V vs. the standard hydrogen electrode, SHE, at pH 8.3, which is the experimentally measured pH of the reaction mixture saturated with CO2, this reduction in driving force has a more significant impact on CO2 reduction.
Zooming out from the question of what drives CO/H2 selectivity to what controls the overall activity of the different materials, we know from previous work on hydrogen evolution photocatalysts that the hydrogen evolution activity is often a trade-off between the polymers' optical properties (optical gap, the onset of light absorption), electronic properties (besides EA also IP, the ionisation potential, which controls the driving force for the oxidation of TEOA), and the dispersibility of the catalyst in the reaction medium. The oligomers and polymers with low activity are typically limited by one or more of these properties. p-Sexiphenylene and P1 adsorb limited visible light because they have the most blue-shifted optical gaps (see Fig. S5†). p-Sexiphenylene, P1 and P4 have the least positive IP values and thus the smallest driving force for the one-hole oxidation of TEOA (see Fig. 1B). P74 has the least negative EA value and thus the smallest driving force for proton or CO2 reduction. P29, P31 and especially P74, disperse very poorly in the reaction mixture. The most active materials, P7 and P10, in contrast are some of the most dispersible materials and are among the materials with the most positive IP values and, hence, the largest driving force for TEOA oxidation, while also having a significant driving force for proton and CO2 reduction. Also, especially for P10, these two polymers absorb a significant part of the visible spectrum.
We next went on to explore the performance of P7 (5 mg) with different amounts of CoCl2 and BPy in a high-throughput screening experiment. It was found that an increased amount of CoCl2, while keeping its ratio with BPy constant, decreases the amounts of both H2 and CO produced (Fig. S-2 and Table S-2†), particularly when 5 μmol or 10 μmol CoCl2 were used and the selectivity for CO was highest with 1 μmol CoCl2 and 2 mg Bpy. Similar results were found when increasing the amount of CoCl2 while keeping the amount of BPy constant (Fig. S-2 and Table S-3†), while control experiments showed that CoCl2 and Bpy on their own in the absence of photocatalyst produce only negligible amounts of H2 and CO, showing that the process is indeed photocatalytic and driven by the polymer photocatalyst (Table S-4†). Without BPy or CoCl2, we find that CO production rates are very low (Fig. S-17†), which is consistent with other reports that show that both are required to form catalytically active Co centres.22,35–38 We used 1 μmol CoCl2 and 2 mg Bpy hereafter as the optimised conditions for photocatalytic CO2 reduction with P7. Residual palladium from the synthesis seems to facilitate competing hydrogen production. We therefore explored if controlling the concentration of residual palladium offers a pathway to producing syngas, hence mixtures of hydrogen and CO, in different ratios. For this, we simply varied the amount of [Pd(PPh3)4] used in the synthesis of P7 from 0.1 mol% to 5 mol%, which gave materials with varying amounts of residual palladium, ranging from 0.043 wt% up to 1.444 wt% as determined by inductively coupled plasma optical emission spectrometry (ICP-OES) measurements. While the amounts of residual palladium do not match the amounts used in the polycondensation reaction, we found that materials that were made using more palladium also contained more residual palladium, with an almost linear increase of residual palladium content of the polymer with increased amount of palladium catalyst used in the polymerisation (Fig. S-3†). These samples, denoted P7-0.1% to P7-5% indicating the amount of [Pd(PPh3)4] used in the synthesis of each P7 sample, were then characterised to rule out that any other differences in properties beyond the palladium content that could affect the performance of these materials. UV-Vis absorption spectra measured in solid-state showed that the optical properties were not affected by the amount of palladium used in the polycondensation reaction with very little difference in their absorption on-sets (Fig. S-4†). Similarly, photoluminescence spectra show no difference in the maximum emission wavelength position (Fig. S-7†) and FT-IR spectra show no noticeable differences (Fig. S-8†). Secondary electron microscopy shows that all materials consist of irregular shaped micrometre sized particles in all cases (Fig. S-10†) and transmission electron microscopy (Fig. S-11†) show the presence of large Pd clusters in the case of P7-1% and P7-5% with the latter clearly showing a larger number of Pd clusters. A previous X-ray absorption spectroscopy study of the residual Pd in P10 has shown though that only the largest Pd clusters could be resolved and most Pd resides as very small clusters within the photocatalyst, which might also be the case here.41
Taken together, all characterisation for the materials made using different amounts of catalyst in the synthesis (in the range of 0.1–5.0 mol%) indicates little difference in their physical properties, except for their palladium content. We therefore went on to test P7-0.1% to P7-5% as photocatalysts for CO2 reduction under solar irradiation (Fig. 2a). The effect of the amount of residual Pd on the CO production rates for P7-0.1% and P7-0.5% was found to be insignificant with rates of 680.1 μmol g−1 h−1 and 660.4 μmol g−1 h−1. P7-1% and P7-2% show a slight reduction in CO evolution rates (567.4 μmol g−1 h−1 and 539.8 μmol g−1 h−1), and P7-3% and P7-5% show a significant reduction in their CO evolution rates (465.8 μmol g−1 h−1 and 340.1 μmol g−1 h−1). By contrast, it was found that the hydrogen evolution rates are much more affected by the palladium content with an increase of the H2 evolution rate increasing from 275.0 μmol g−1 h−1 for P7-0.1% to 711.9 μmol g−1 h−1 for P7-1% and 845.9 μmol g−1 h−1 for P7-2%. Going to P7-3% and P7-5% a significant increase in the rate was again observed (1130.2 μmol g−1 h−1 and 1677.3 μmol g−1 h−1). This shows that the palladium concentration profoundly effects the activity of these photocatalysts for hydrogen production and can also be observed in a reduction of the photoluminescence intensity with increased palladium content (Fig. S-9†), possibly due to enhanced trapping and quenching of excitions.41 The residual Pd also effects the ability to reduce carbon dioxide, in particular at higher palladium concentrations. The observation that the hydrogen evolution rate in the limit of low Pd concentrations (<0.2 wt%, Fig. 2b) significantly increases with the amount of Pd is a common observation27,28 and shows that excitons or polarons after dissociation of excitons do not reach Pd to facilitate proton reduction,41 while abundant Co is present allowing for efficient carbon dioxide reduction. The initial steep increase in activity is followed by a region that is less effected by the increase in palladium, which has also been observed for other conjugated materials27,28 before significantly increased H2 evolution rate, as more palladium relative to the amount of cobalt loaded on the materials is available for proton reduction. The ratios of H2/CO for P7-0.1% to P7-1% range from 0.4:1 to 1.25:1. H2 rich syngas ranging from 1.6:1 to 4.9:1 (H2/CO) were produced by P7-2% to P7-5%. This makes it possible to produce syngas for the generation of aldehydes via hydroformylation of alkene with a ratio of 1:1H2/CO as well as the methanol synthesis and F–T synthesis of alkanes requiring a ratio of 2:1H2/CO.4 The lifetime of the excited states of P1 and P7 were studied by time-correlated single photon counting (TCSPC) experiments. The average weighted photoluminescence emission lifetime (Table S-6†) of P7 (τavg = 1.13 ns, λem = 475 nm) was similar with that (τavg = 1.05 ns, λem = 475 nm) in MeCN purged with CO2. When measured in MeCN/H2O/TEOA suspension, the average emission lifetime of P7 (τavg = 0.80 ns, λem = 475 nm) was longer than that of P1 (τavg = 0.5 ns, λem = 453 nm), in line with the observed activity.
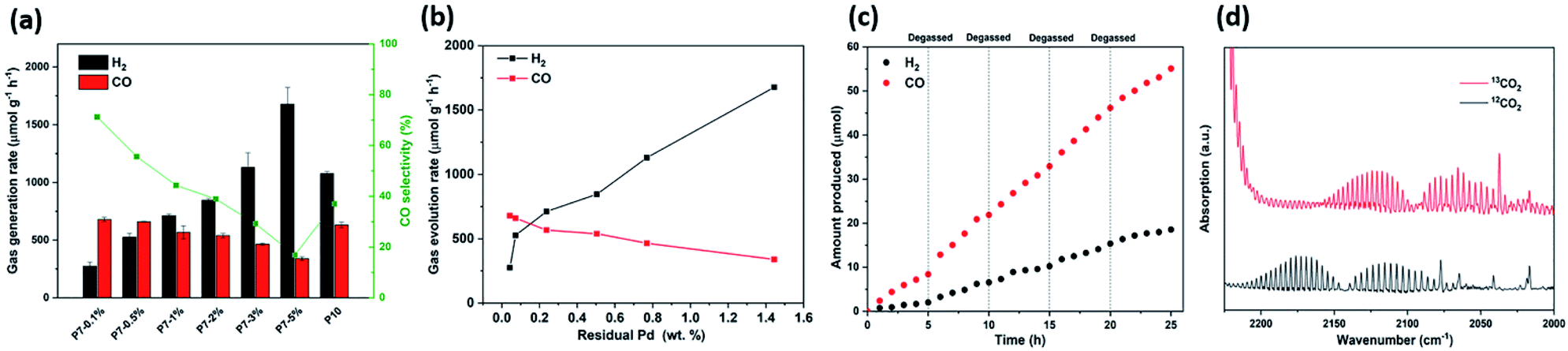 | ||
| Fig. 2 (a) Photocatalytic CO and H2 production of P7-0.1% to P7-5% (5 mg) from MeCN/water/TEOA mixtures (25 mL, 3/1/1) under simulated solar irradiation (1 sun, AM 1.5G); (b) correlation between the amount of palladium and the CO and hydrogen evolution rates of P7-0.1% to P7-5%; (c) cycling experiments of P7-0.1% (5 mg) under simulated solar irradiation (1 sun, AM 1.5G) over 25 hours irradiation with intermitted degassing every 5 hours; (d) FT-IR spectra of the gaseous products after 1 hour of P7 in MeCN/H2O/TEOA solution using either 12CO2 and 13CO2 under solar irradiation (1 sun, AM 1.5G). | ||
Longer-term experiments with intermittent degassing every 5 hours showed good stability of the sample (P7-0.1%) over 25 hours producing CO rich syngas (Fig. 2c). Using isotopically labelled 13CO2 as the carbon source resulted in the formation of 13CO strongly suggesting that CO2 was the source of the produced CO (Fig. 2d). Furthermore, the action spectrum of P7-0.1% follows broadly speaking the light absorption profile also supporting the hypothesis that the CO production is indeed photocatalytic.
Conclusions
In conclusion, we show that linear conjugated polymers can be used as photocatalysts for CO2 reduction to CO in the presence of a sacrificial hole scavenger. Polymer P7 was found to be the most active photocatalyst for CO2 reduction, while the structurally related P10 performs far worse in terms of CO selectivity, producing a similar amount of CO but much more hydrogen. This difference in selectivity can be rationalised by differences in the predicted thermodynamic driving force for CO2 and proton reduction for P7 and P10. Residual palladium was found to result in significant hydrogen production, which was found to compete with CO production at high loadings. Within a certain concentration range, Pd acts as a co-catalyst for H2 production without reducing the CO evolution rates significantly, potentially allowing for syngas production, if the production efficiencies can be increased, within a range of H2/CO ratios that are adjustable by varying the Pd concentration.Experimental section
General methods
All reagents were obtained from Sigma-Aldrich, TCI, ABCR, Fisher Scientific or Fluorochem and used as received. Cobalt(II) chloride hexahydrate (BioReagent), 2,2′-bipyridyl (Reagent Plus, ≥99%) and triethanolamine (≥99.0%) were purchased from Sigma-Aldrich. Acetonitrile (HPLC gradient grade), N,N′-dimethylformamide (GC Headspace Grade) were obtained from Fisher Scientific. Water for the CO2 reduction experiments was purified using an ELGA LabWater system with a Purelab Option S filtration and ion exchange column (ρ = 15 MΩ cm−1) without pH level adjustment. Reactions were carried out under nitrogen atmosphere using standard Schlenk techniques. Photocatalysts P1K,30 P1S,30 P4,30 P7,30 P10,31 P29,32 P30,32 and P31 (ref. 32) were prepared according to literature procedures.Synthesis of P74
4,7-Dibromo-2,1,3-benzothiadiazole (412 mg, 1.4 mmol), 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester) (543 mg, 1.4 mL), N,N-dimethylformamide (40 mL) and K2O3 (aqueous, 2 M, 8 mL) were combined and degassed with nitrogen for 30 minutes. Then [Pd(PPh3)4] (40 mg) was added and heated to 150 °C for 2 days. After cooling to room temperature, the reaction mixture was poured into water, the solids were filtered off and washed with methanol. The polymer was purified using a Soxhlet extraction with chloroform to give P74 as a dark powder (387 mg, quant.). Anal. calcd for (C6H2N2S)n: C, 53.72; H, 1.50; N, 20.88%; found C, 49.13; H, 0.95; N, 18.26%High-throughput CO2 reduction experiments
Polymer (5 mg), CoCl2 (0.5–10 μmol) and 2,2′-bipyridine (1.0–20 mg) were added into sample vials (V = 12.5 mL) and purged with carbon dioxide in a Sweigher Chemspeed Technologies robotic platform for 3 hours. A liquid handling system transferred water/solvent/hole-scavenger mixture (water/acetonitrile/triethanolamine, 1:3:1, 5 mL) from stock jars inside the system into the sample vials. A capper/crimper tool was then used to automatically seal the vials under CO2 atmosphere. All sample vials were ultrasonicated in an ultrasonic bath for 5 minutes before illumination using a solar simulator (AM1.5G, Class AAA, IEC/JIS/ASTM, 1440 W xenon, 12 × 12 in, MODEL:94123A) for the time specified while constantly being redispersed with a rocker/roller device. Samples of gaseous products were analysed using a Shimadzu 2014 HS-GC gas chromatograph equipped with a ShinCarbon ST micropacked column (Restek 80–100 mesh, 2 m length, 0.53 mm inner diameter) and a thermal conductivity detector. All samples were duplicated to ensure reproducibility within a run.
CO2 Reduction experiments
A quartz flask was charged with polymer powder (5 mg), 2,2′-bipyridine (10 mg), cobalt(II) chloride (5 μmol), a mixture of acetonitrile, water and triethanolamine (3:1:1 vol, 25 mL), and sealed with a septum. The resulting suspension was ultrasonicated for 20 minutes and then purged with CO2 for 30 minutes. The mixture was illuminated an Oriel Instruments LSH-7320 Solar Simulator (IEC ABA certified) with 1 Sun output. Laser adjustment of the instrument was used to adjust the distance of the reaction flask to the light source. Samples of gaseous products were taken with a gas-tight syringe and run on a Shimadzu GC-2014 gas chromatograph equipped with a ShinCarbon ST micropacked column (Restek 80–100 mesh, 2 m length, 0.53 mm inner diameter) and a thermal conductivity detector. Gases dissolved in the reaction mixture and the pressure increase generated by the evolved gases was neglected in the calculations.
Determination of external quantum efficiency for CO production
The external quantum efficiencies of CO production were determined using monochromatic LED light (λ = 395, 405, 420, 490 and 515 nm). The reactions were conducted on the same photochemical experimental setup under the optimised reaction conditions. For the experiments, P7-0.1% (1 mg) 2,2′-bipyridine (2 mg) and cobalt(II) chloride (1 μmol) were suspended in acetonitrile, water and triethanolamine (3:1:1 vol. mixture, 5 mL). The illuminated area was 8 cm2 and the light intensity was measured by a ThorLabs PM100D Power and Energy Meter Console with a ThorLabs S120VC photodiode power sensor. The EQE was calculated using the following equation:| EQE% = 2 × [(nCO) × NA × h × c] × 100%/(I × S × t × λ) |
Isotopic labelling experiments
13CO2 Labelling experiments were carried on a Bruker Vertex 70V Fourier-transform infrared spectrometer with an argon-purged custom-made gas IR cell. A vial containing the photocatalyst, 2,2′-bipyridine and cobalt(II) chloride in a mixture of acetonitrile/water/triethanolamine (3:1:1) was purged with 13CO2 (Sigma-Aldrich, 99 atom% 13C, <3 atom% 18O) for 3 minutes. Then the resulting suspension was illuminated for 1 hour using an Oriel Instruments LSH-7320 Solar Simulator (IEC ABA certified) with 1 Sun output. A sample of the gas headspace (1000 μL) was injected into the gas IR cell and the IR spectrum was measured (32 scans with a resolution of 0.5 cm−1). Background was measured using Ar purged cell and subtracted from the measurement.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) for financial support under Grant EP/N004884/1. Z. Fu thanks the China Scholarship Council for a PhD studentship. A. V. thanks the European Union's Horizon 2020 research and innovation programme (Marie-Skłodowska-Curie Individual Fellowship) for financial support under grant agreement No. 796322. R. S. S. thanks the University of Strathclyde for financial support through The Strathclyde Chancellor's Fellowship Scheme. Dr G. Neri and Prof. A. Cowan are thanked for help with the FT-IR measurements. I. Heath-Apostolopoulos is acknowledged for help with the calculations. R. Clowes is thanked for help with high-throughput measurements. L. Liu is acknowledged for SEM measurements and Dr C. M. Aitchison TEM measurements.References
- T. M. L. Wigley, Science, 2005, 307, 1766–1769 CrossRef CAS.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS.
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS.
- S. R. Foit, I. C. Vinke, L. G. J. de Haart and R. A. Eichel, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CrossRef CAS.
- W. Zhu, J. Fu, J. Liu, Y. Chen, X. Li, K. Huang, Y. Cai, Y. He, Y. Zhou, D. Su, J. J. Zhu and Y. Lin, Appl. Catal., B, 2020, 264, 118502 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- J. Jayakumar and H. H. Chou, ChemCatChem, 2020, 12, 689–704 CrossRef CAS.
- Y. Miseki and K. Sayama, Adv. Energy Mater., 2019, 9, 1801294 CrossRef.
- L. Y. Ting, J. Jayakumar, C. L. Chang, W. C. Lin, M. H. Elsayed and H. H. Chou, J. Mater. Chem. A, 2019, 7, 22924–22929 RSC.
- C.-L. Chang, W.-C. Lin, C.-Y. Jia, L.-Y. Ting, J. Jayakumar, M. H. Elsayed, Y.-Q. Yang, Y.-H. Chan, W.-S. Wang, C.-Y. Lu, P.-Y. Chen and H.-H. Chou, Appl. Catal., B, 2020, 268, 118436 CrossRef CAS.
- A. F. M. EL-Mahdy, A. M. Elewa, S. W. Huang, H. H. Chou and S. W. Kuo, Adv. Opt. Mater., 2020, 8, 2000641 CrossRef CAS.
- W. H. Wang, L. Y. Ting, J. Jayakumar, C. L. Chang, W. C. Lin, C. C. Chung, M. H. Elsayed, C. Y. Lu, A. M. Elewa and H. H. Chou, Sustainable Energy Fuels, 2020, 4, 5264–5270 RSC.
- Y. Chen, G. Ji, S. Guo, B. Yu, Y. Zhao, Y. Wu, H. Zhang, Z. Liu, B. Han and Z. Liu, Green Chem., 2017, 19, 5777–5781 RSC.
- H. P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T. X. Wang, A. Thomas and B. H. Han, ACS Catal., 2019, 10, 3959–3968 CrossRef.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem.–Eur. J., 2018, 24, 17454–17458 CrossRef CAS.
- S. Zhang, S. Wang, L. Guo, H. Chen, B. Tan and S. Jin, J. Mater. Chem. C, 2019, 8, 192–200 RSC.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton, L. Liu, X. Wu, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, S. Jin, Y. Xiang, P. Wang, B. Jiang, F. Ichihara, M. Oshikiri, X. Meng, Y. Li, W. Matsuda, J. Ma, S. Seki, X. Wang, H. Huang, Y. Wada, H. Chen and J. Ye, Nat. Commun., 2020, 11, 1149 CrossRef.
- B. Acharya, P. Roy and A. Dutta, Biofuels, 2014, 5, 551–564 CrossRef CAS.
- S. Hernández, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- M. B. Ross, Y. Li, P. De Luna, D. Kim, E. H. Sargent and P. Yang, Joule, 2019, 3, 257–264 CrossRef CAS.
- L. Li, Z. Cai, Q. Wu, W. Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- S. Yanagida and S. Matsuoka, in Optical Materials Technology for Energy Efficiency and Solar Energy Conversion XI: Photovoltaics, Photochemistry, Photoelectrochemistry, es. A. Hugot-Le Goff, C.-G. Granqvist and C. M. Lampert, 1992, vol. 1729, p. 243 Search PubMed.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- C. B. Meier, R. Clowes, E. Berardo, K. E. Jelfs, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Chem. Mater., 2019, 31, 8830–8838 CrossRef CAS.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS.
- M. Zhou, S. Wang, P. Yang, C. Huang and X. Wang, ACS Catal., 2018, 8, 4928–4936 CrossRef CAS.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2014, 2, 353–358 CrossRef CAS.
- S. Wang, B. Y. Guan, Y. Lu and X. W. Lou, J. Am. Chem. Soc., 2017, 139, 17305–17308 CrossRef CAS.
- J. J. Walsh, C. Jiang, J. Tang and A. J. Cowan, Phys. Chem. Chem. Phys., 2016, 18, 24825–24829 RSC.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, gas production data, UV/Vis, FT-IR and PL spectra, SEM, TEM images, TCSPC data and DFT results for P74 and TEA oxidation potentials. See DOI: 10.1039/d0ta09613j |
| This journal is © The Royal Society of Chemistry 2021 |