
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Safely probing the chemistry of Chernobyl nuclear fuel using micro-focus X-ray analysis†
Hao
Ding
a,
Malin C.
Dixon Wilkins
 a,
Clémence
Gausse
a,
Lucy M.
Mottram
a,
Shikuan
Sun
a,
Martin C.
Stennett
a,
Daniel
Grolimund
b,
Ryan
Tappero
c,
Sarah
Nicholas
c,
Neil C.
Hyatt
*a and
Claire L.
Corkhill
*a
a,
Clémence
Gausse
a,
Lucy M.
Mottram
a,
Shikuan
Sun
a,
Martin C.
Stennett
a,
Daniel
Grolimund
b,
Ryan
Tappero
c,
Sarah
Nicholas
c,
Neil C.
Hyatt
*a and
Claire L.
Corkhill
*a
aNucleUS Immobilisation Science Laboratory, Department of Materials Science and Engineering, The University of Sheffield, S1 3JD, UK. E-mail: c.corkhill@sheffield.ac.uk; n.c.hyatt@sheffield.ac.uk
bSwiss Light Source, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland
cBrookhaven National Laboratory, NSLS-II, Upton, NY 11973, USA
First published on 10th February 2021
Abstract
Detailed chemical analysis of the solidified molten fuel still residing in the stricken Chernobyl reactor unit 4 are inferred using multi-modal micro-focus X-ray analysis of a low-radioactivity proxy. A fascinating mixture of molten UO2, nuclear fuel cladding, concrete, stainless steel and other nuclear reactor components, these materials behaved like lava, solidifying to form a complex, highly radioactive glass-ceramic. Using element-specific chemical probes (micro-X-ray fluorescence and X-ray absorption spectroscopy), coupled with micro-diffraction analysis, the crystalline phase assemblage of simulants of these heterogeneous materials was established, which included “chernobylite” and a range of compositions in the (U1−xZrx)O2 solid solution. Novel insight to nuclear accident fuel chemistry was obtained by establishing the oxidation state and local coordination of uranium not only in these crystalline phases, but uniquely in the amorphous fraction of the material, which varied depending on the history of the nuclear lava as it flowed through the reactor. This study demonstrates that micro-focus X-ray analysis of very small fractions of material can yield rich chemical information, which can be applied to nuclear-melt down materials to aid decommissioning and nuclear fuel management at nuclear accident sites.
Introduction
The accident that occurred at the 4th reactor of the Chernobyl Nuclear Power Plant on April 28th 1986 resulted in complete melting of the reactor core with severe consequences. It generated large amounts of highly radioactive nuclear fuel-containing materials, including so-called lava-like fuel-containing materials (LFCMs),1–4 which have also been identified at the Fukushima Daiichi nuclear power plant. LFCM, a mixture of nuclear fuel and melted reactor components (stainless steel, concrete etc.), collected from different locations within the Chernobyl reactor, are classified as either black or brown based on their colour,3,5 influenced by the U and Fe content.3 Investigation of a limited number of samples of these materials has been conducted but analysis was restricted by their high radioactivity, complicating the handling and characterisation process.6–11To gain deeper insight into the properties of materials arising from nuclear reactor accidents, in a safe and efficient manner, a suite of simulant brown and black LFCM samples with much lower radioactivity, i.e. excluding fission products, were synthesised and characterised.12–14 Detailed bulk analysis of these simulant materials was reported and the morphology and mineralogy were found to closely approximate those of real LFCMs. In the present study, micro-focus synchrotron X-ray analysis was used to investigate several representative regions of these materials. By combining the analyses from micron-resolved chemical probes (X-ray fluorescence, μ-XRF and X-ray Absorption Spectroscopy, μ-XAS) and diffraction analysis (μ-XRD), it was possible to accurately identify crystalline phases, determine the local U chemistry in crystalline and amorphous phases, and reconstruct the paragenetic sequence, i.e., the chronology of crystallisation of phases from the lava melt. Further analysis of Extended X-ray Absorption Fine Structure (EXAFS) spectra were used to quantitatively identify the local environment of U, providing novel insights to nuclear accident material chemistry. Understanding these interactions and U behaviours are key to building a complete understanding of nuclear fuels in accident scenarios. We show that rich chemical information can be obtained by these methods on extremely small, μm-sized, samples; such size reduction on real nuclear fuel materials would drastically lower the hazard associated with their analysis.
Experimental
Materials and synthesis
The detailed preparation of simulant Chernobyl brown and black LFCMs was reported in full in our previous study.13 Both brown and black LFCMs were synthesised using the same method. Their compositions, derived from those of real LFCM, are somewhat different from one another, with brown LFCM containing more U and black LFCM containing no Fe (see ESI Table 1†). For convenience, the materials and preparation of these samples, developed within the HADES facility,15 and a short description of their similarity to real LFCM, are briefly described here. The batched compositions of brown and black LFCMs were based on an average of all analysed samples in the literature, but with additional ZrO2 added to encourage the crystallisation of zircon (ZrSiO4). The corresponding precursors including SiO2 (Lochaline Quartz Sand 99.6%), CaCO3 (Fisher 98%), ZrO2 (Aldrich 99%), Na2CO3 (Alfa Aesar 98%), BaCO3 (Alfa Aesar 99%), Al(OH)3 (Acros 95%), Mn2O3 (Aldrich 99%), stainless steel 316 (Fe/Cr18/Ni10/Mo3, Goodfellow), Mg(OH)2 (Sigma-Aldrich 99.9%), and UO2 (BDH) were mixed, and then calcined in alumina crucibles under a reducing atmosphere (5% H2 in 95% N2) at 1500 °C for 4 h, followed by a second dwell at 720 °C for 72 h. The second heating step was added to facilitate crystallite growth. Analysis by X-Ray Diffraction (XRD) and Scanning Electron Microscopy, coupled with Energy Dispersive X-ray Spectroscopy (SEM/EDS), demonstrated that the simulant LFCMs accurately reflected the phase assemblage, microstructure and mechanical properties of real brown and black LFCM.12–14 Thermal characterisation of the simulants confirmed that the crystallisation temperatures were consistent with those of naturally-occurring versions of the LFCM minerals.13 Furthermore, the corrosion rates of U from the simulant LFCMs were found to be similar to those determined from limited studies of real LFCM.13Micro-focus X-ray analysis
Samples were prepared for μ-focus measurements (μ-XRF, μ-XANES and μ-XRD) by sectioning mounting on a 250 μm thick Spectrosil fused quartz slide. The sample was then thinned and polished to a thickness of 50 μm by standard metallographic procedures. The micro-focus measurements (excluding μ-EXAFS) were conducted at the Swiss Light Source, Paul Scherrer Institute, Switzerland, on the microXAS-X05LA beamline.16 A fixed-exit double crystal monochromator (Si (111) and Si (311) crystals) was used, maintaining consistent and stable beam angles and offsets for all experiments. The energy was calibrated by measuring XANES of Zr and Y foil standards. The spot size of the monochromatic beam as focused on the samples was tuned to 1 μm × 1 μm by a Kirkpatrick–Baez (KB) mirror system with a fixed incident X-ray energy of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 eV. Samples were mounted on a manipulator 80 mm behind the KB mirror (total focus distances of 150 mm horizontal × 270 mm vertical). For the micro-focus XRD measurements, the angles and distances between the area detector (DECTRIS Eiger 4M) and samples were calibrated by measuring a silicon standard. The accessible 2θ range was 3.5 to 42.6° 2θ with a resolution of 0.02°. All samples were scanned in transmission mode by the beam in a raster pattern. X-ray fluorescence (XRF) spectra were simultaneously measured, using a Si drift-detector (KETEK) with 4% energy resolution.
100 eV. Samples were mounted on a manipulator 80 mm behind the KB mirror (total focus distances of 150 mm horizontal × 270 mm vertical). For the micro-focus XRD measurements, the angles and distances between the area detector (DECTRIS Eiger 4M) and samples were calibrated by measuring a silicon standard. The accessible 2θ range was 3.5 to 42.6° 2θ with a resolution of 0.02°. All samples were scanned in transmission mode by the beam in a raster pattern. X-ray fluorescence (XRF) spectra were simultaneously measured, using a Si drift-detector (KETEK) with 4% energy resolution.
Uranium LIII edge X-ray Absorption Near Edge Spectroscopy (XANES) data were collected over the energy range 17060–17325 eV, with a resolution of 0.2 eV. Energy calibration was conducted based on the K edge (17038 eV) of an Y foil standard. Four measurements of each point of interest were collected in fluorescence mode and averaged. The threshold energy (E0) and edge point were determined as the first inflection point and white line position of the spectra respectively. The oxidation state of U has a linear influence on the minimum excitement energy, where it is shifted due to a change in effective nuclei charge as the U valence changes. The linear function was established based on data collected from a range of standard U-containing compounds, which have similar chemical environments and electron configurations to the unknown samples. Standards measured included: UO2 (average U oxidation state (OS) 4 and coordination number (CN) 8); CaUO4 (average OS 6 and CN 8); UMoO5 (average OS 5 and CN 7); LaUO4 (average OS 5 and CN 8); UTiO5 (average OS 6 and CN 7); UTi2O6 (average OS 4 and CN 6); and Ca3UO6 (average OS 6 and CN 6).
The average oxidation states of standards and samples were determined by examining the energy position of the XANES spectra, compared to the linear relationship between oxidation state and energy position of a range of U-containing standards. The U LIII XANES of compounds incorporating the uranyl species are characterised by a post-edge resonance or shoulder at ca. 15 eV above the white line, which arises from multiple scattering associated with the UO22+ oxocation.17 As no such uranyl shoulder was observed in the spectra of any point of interest, Ca3UO6 and CaUO4 were selected as the reference compounds for U(VI) in linear combination fitting, to minimise the influence of the shoulder caused by uranyl species seen at the U LIII edge (CaUO4 has a particularly long linear O–U–O moiety, closer to a bond length indicative of U–O double-bonding, and Ca3UO6 consists of simple regular (UO6) octahedra with 6 equivalent oxygen positions).18 XANES spectra of pure synthetic USiO4 were also collected for use as a silicate U(IV) standard for description of the U coordination in the zircon and glass phases.19,20
Micro-focus Extended X-ray Absorption Fine Structure (EXAFS) data were collected at the National Synchrotron Light Source II (NSLS-II), Brookhaven National Laboratory, USA, on beamline 4BM (XFM). The measurements were taken under similar conditions as the measurements at the Swiss Light Source, but with a wider energy range of 17015–17730 eV. The spot size was 2 μm × 1 μm. The raw data were normalized and a Fourier transform applied over the k-range 3.0–10.0 Å−1 using Athena and Artemis, parts of the Demeter software package.21 Scattering paths including amplitude, phase shift, mean free path and the initial path lengths were calculated by employing the FEFF code program in Artemis. The fitting was conducted by fixing the amplitude at 0.95, using the same Debye–Waller factors for all paths comprising the first oxygen shell, and refining the interatomic distance and coordination numbers simultaneously.
Reconstruction of the crystalline phase assemblage
Azimuthal integration of individual 2D μ-diffraction patterns and summation of the whole area were applied using the XRDUA software package to construct 1D diffraction patterns.22 The phase assemblage was determined by matching the diffraction peaks with materials previously reported in the ICSD PDF database. The location and relative abundances of the elements of interest were obtained from the related μ-XRF maps. With the use of both μ-XRD and μ-XRF data, the phase assemblage was reconstructed using an in-house MATLAB model that automated diffraction pattern indexing and plotted an image showing the relative distribution of phases. The model was composed of three main functions that were performed consecutively: (1) location of peaks: by searching through individual XRD patterns, the peaks were determined by matching within the range of standard PDF reflection and over a proper threshold of intensity; (2) identification of phases: all peaks corresponding to the same phase were assigned with a specific value which were shown as the same colour in the final map. If the spectrum had more than one phase, the phase with relative higher intensity was assumed to be the major phase (even though it is possible to show an overlap pixel in a different colour, the distinction of phases was blurred to illustrate in a single map); (3) build map: collecting the assigned value of all pixels, the topography was constructed, with different colours representing the corresponding crystalline phases.Results and discussion
LFCM mineral phase identification and stoichiometry analysis
The phases identified in simulant brown LFCM were: zircon [ZrSiO4; PDF 00-033-1485], which manifest as large (approx. 40–60 μm) prismatic particles, often, but not always containing U; cubic Zr-containing UO2 (c-(U1−xZrx)O2; PDF 04-019-4898) and tetragonal U-containing ZrO2 (t-(U1−xZrx)O2; PDF 04-020-6305) crystallites, 5–15 μm in size; which were clustered together into fused aggregates by monoclinic ZrO2 with U (m-(Zr1−xUx)O2). These crystallites were embedded within a calcium aluminosilicate glass matrix, which also contained U and Zr (Fig. 1). | ||
| Fig. 1 Micro-focus analysis of simulant brown LFCM. Showing distribution of (a) U-Lα and (b) Zr-Kα fluorescence signals; diffraction patterns of each identified phase present including (c) m-ZrO2, (d) t-(U1−xZrx)O2, (e) m-(Zr1−xUx)O2, (f) (Zr1−xUx)SiO4, (g) c-(U1−xZrx)O2, (h) calcium aluminosilicate glass; and (i) 2D μ-diffraction map reconstructed from XRD patterns taken at each individual pixel in the μ-XRF maps shown in (a) and (b). The black line in (i) indicates the region that was examined using a line analysis, detailed in Fig. 8. | ||
Black LFCM exhibited a similar phase assemblage to brown LFCM, albeit with distinctly different morphology. Both “fused” (Fig. 2a–d and ESI Fig. 1†) and dendritic crystalline morphologies were observed (Fig. 2f–h and ESI Fig. 2†). Fe–Ni alloy particles [Fe–Ni; PDF 00-047-1405] (Fig. 2e), arising from the incorporation of stainless steel were observed. Their distribution, on the edge of fused particles (Fig. 2d, bottom left), results from the capture of molten metal during crystal growth, in agreement with previous studies of fuel-cladding interactions at high temperature.23
 | ||
| Fig. 2 μ-focus fluorescence analysis of black LFCM. Showing distribution of (a) Fe-Kα, (b) Zr-Kα and (c) U-Lα fluorescence signals; and (d) the 2D μ-diffraction map reconstructed from XRD patterns, all for the aggregated morphology; (e) the μ-diffraction pattern of FeNi phase with m-ZrO2; the distribution of (f) U-Lα and (g) Zr-Kα fluorescence signals, and (h) 2D μ-diffraction map reconstructed from XRD patterns, all for the dendritic morphology. | ||
Using the U-Lα and Zr-Kα counts obtained by μ-XRF, the ratio, r = U/(Zr + U), in each of the three (U1−xZrx)O2 phases was determined (Fig. 3a). The phase identified as c-(U1−xZrx)O2 had U ratios (r) in the range of 0.2 < r < 1.0 while m-(Zr1−xUx)O2 had r in the range 0.0 < r < 0.1. The tetragonal phase was intermediate, with 0.1 < r < 0.5. Quantification of the deviation in (111) reflection position, which shifts to a higher position 2θ as the Zr content increases due to a contraction of the unit cell associated with the substitution of Zr4+ (ionic radius = 0.84 Å) for U4+ (ionic radius = 1.00 Å) in the UO2 structure,24 was also used to approximate the stoichiometry. Assuming Vegard's Law, the U:Zr ratio of c-(U1−xZrx)O2 should follow an approximately linear relationship with shift in reflection position, within the range of 0.2 to 1 mole fraction U (Fig. 3a and b). The range of stoichiometry approximated for this phase from μ-XRD was x = 0 to 0.8, in good agreement with the qualitative μ-XRF analysis. The same principle was applied to t-(U1−xZrx)O2, giving x = 0.2 to 0.9. It was not possible to apply this methodology to obtain the stoichiometry of m-(Zr1−xUx)O2 for several reasons: (i) the mole fraction of U in was less than 0.1, therefore, any shift in the XRD reflection resulting from a change in stoichiometry of this phase would be below the resolution of μ-XRD analysis (0.02°); (ii) since no monoclinic phase exists for UO2, Vegard's law is not applicable; and (iii) for the monoclinic crystal system, the unit cell parameters have more than one independent variable (a ≠ b ≠ c; β ≠ 90), therefore, any peak shift against the lattice parameter is not expected to be a linear, or an approximately linear relationship.
 | ||
| Fig. 3 Colour gradient maps of brown LFCM derived from μ-XRF and μ-XRD analysis. Showing (a) the normalised μ-XRF U/U + Zr ratio, where a value of 1 is equal to UO2 and 0 corresponds to regions where no U-Lα was observed; (b) the U fraction in c-(U1−xZrx)O2 estimated by the normalised peak shift of the cubic (111) peak in the μ-XRD patterns acquired for each pixel, where a value of 1 indicates no shift of this reflection; (c) the full width at half maximum (FWHM) of the (111) reflection in the μ-XRD patterns acquired for each pixel, where a value of 0.02 indicates a peak with 0.02° (2θ) width; and (d) the phase distribution of the regions where (U,Zr)O2 phases are present. | ||
The full width at half maximum reflection intensity (Fig. 3c) is influenced by both the U/Zr ratio and the size of the crystallite. The FWHM was observed to change where crystals were clustered, indicating that different compositions of crystallite were agglomerated. Indeed, Fig. 3d shows that most individual pixels contained more than one phase. The FWHM tended to broaden from the centre to the edge of c-(U1−xZrx)O2, indicating an increase in the Zr concentration and a decrease in crystallite size. Accordingly, more Zr-rich compositions (t-(U1−xZrx)O2 and m-(Zr1−xUx)O2) were found to cluster around c-(U1−xZrx)O2. Differentiation between m-ZrO2 and m-(Zr1−xUx)O2 was more challenging from examination in the shift of reflection positions, since both crystallise in the same structure. These two phases were thus identified using a combination of μ-XRD patterns and U Lα emission in the μ-XRF, by comparing the relative abundance of U at the corresponding pixels to the average U abundance in the glassy matrix.
The reflections observed in the summed whole area diffraction pattern were broadened when compared with the reflections observed at individual pixels. Due to the extended dwell times during the heat treatment of the simulant LFCMs, it is unlikely that this is caused by internal stresses. This reflection broadening therefore suggests that the simulant material contains compositions from across the whole solid solution of (U1−xZrx)O2.
Zircon (ZrSiO4) phases were also abundant (Fig. 1) and, like their “chernobylite” counterparts in real LFCM,25 the crystallites contained U. In the μ-XRF maps, regions of zircon were observed with a U-rich edge and U-depleted centre (Fig. 4). The major diffraction peak in the U-rich edge region (marked A, Fig. 4b) was shifted to a lower angle than the centre U-depleted region due to the substitution of U for Zr in the zircon/coffinite (USiO4) solid solution, Zr1−xUxSiO4. Assuming only zircon was present within the area sampled, the stoichiometry was approximated from μ-XRF analysis to range from x = 0–0.2. The upper bound of x reported here is slightly higher than that reported for chernobylite in real LFCM (x = 0.05–0.10).25,26 The abundance of U in Zr1−xUxSiO4 has also been reported in simulant Fukushima molten core-concrete interaction (MCCI) products, at a higher value of x than chernobylite (x = 0.19–0.27).27 This discrepancy is likely a result of the slower cooling time for real LFCM, which allows for thermodynamic equilibration of U during Zr1−xUxSiO4 formation.19
 | ||
| Fig. 4 Analysis of Zr1−xUxSiO4 formed in simulant brown LFCM. Showing (a) μ-XRF map of the U distribution showing locations of μ-XRD patterns; and (b) μ-XRD patterns taken in the U-rich zircon outer-edge and the U-poor zircon centre. | ||
Average uranium oxidation state and local coordination in mineral phases and glass matrix
Points representative of the different U environments were selected (ESI Fig. 3 and 4†) and analysed by U LIII-edge μ-X-ray Absorption Near Edge Spectroscopy (μ-XANES). The average oxidation states of all spectra collected from the different regions (Fig. 5a), as-estimated from linear combination fits using the range of standards listed in the Method section, was between 4.0 and 4.5 (Fig. 5b); the crystallites in the brown and black LFCMs had mean oxidation states of 4.2 ± 0.1 and 4.3 ± 0.1, respectively. This is in good agreement with the phases identified by μ-XRD and μ-XRF, the majority of which are U4+-containing minerals. The estimated average oxidation states of these selected points were classified into three groups based on their distribution (Fig. 5c): (i) (U1−xZrx)O2 spectra were observed to concentrate around the energy position of UO2; (ii) Zr1−xUxSiO4 spectra were clustered slightly above the energy position of USiO4, and below that of UO2; and (iii) spectra acquired from the glassy matrix clustered at slightly higher energies. This shows the clear difference in the local environment around U between materials containing silicate anions (USiO4, as well as U-containing zircon), and those containing independent oxygen anions (UO2, UTi2O6). | ||
| Fig. 5 μ-focus X-ray Absorption Near Edge Spectroscopy (XANES) of simulant LFCM. (a) XANES spectra of U standards and representative locations in LFCM; (b) average U oxidation states of points acquired in the sample (open symbols) compared with a range of U standards of known valence (closed symbols, labelled) as a function of X-ray energy; and (c) detailed view of the mean oxidation state of points sampled within brown (open diamonds) and black (closed diamonds) LFCM, and their classification according to the phase of their origin (note the different scale compared with b). | ||
Analysis of the points representing areas of (U1−xZrx)O2 of all types were observed to concentrate around the energy position of UO2, with estimated oxidation states slightly above 4.0+. U LIII XANES spectra of these spots closely resemble that of the UO2 standard with regard to the position, intensity and width of the whiteline (Fig. 5a). The spectra in the glass matrix had the highest average oxidation state (4.4 ± 0.1) due to the trend of increasing solubility of U as the valence increases from tetravalent to hexavalent. However, the estimated oxidation state of the point in the glass region was below 4.5, indicating that the concentration of dissolved U in the glass matrix remained low.
Representative crystallites of c-(U1−xZrx)O2 and t-(U1−xZrx)O2 were further analysed by fitting of the Extended X-ray Absorption Fine Structure (EXAFS). The k3-weighted EXAFS spectra and corresponding Fourier transform (FT) were fit as shown in Fig. 6 and ESI Fig. 5.† The fitting results (see ESI Table 2†) show that the O-shell distance of c-(U1−xZrx)O2 (2.32 ± 0.01 Å) and t-(U1−xZrx)O2 (2.27 ± 0.01 Å) were shorter than that of the expected sum of ionic radii for 8-fold co-ordination (2.37 Å).24 The calculated bond valence sums for c-(U1−xZrx)O2 (4.3 v.u.) and t-(U1−xZrx)O2 (4.5 v.u.) based on the fitted model were consistent with the estimated oxidation state result. The bond length changes agree well with the observation made from analysis of the μ-XRD pattern that the Zr4+ dopants led to a unit cell contraction. The number of second neighbour U (and Zr) atoms was estimated by application of a linear constraint, with the sum of the weighted paths set equal to 12, consistent with a fluorite related structure. The number of second-neighbour U atoms in c-(U1−xZrx)O2 (8.7 ± 1.0) was higher than that of t-(U1−xZrx)O2 (6.0 ± 1.0), consistent with the higher U/Zr ratio determined from μ-XRF and expected from the phase diagram.28 The c-(U1−xZrx)O2 phase also had a longer mean distance of the first U–U and U–Zr shell (3.83 ± 0.02 Å) compared to the t-(U1−xZrx)O2 (3.73 ± 0.03 Å) phase, consistent with an expansion of the unit cell with increased U/Zr ratio.
 | ||
| Fig. 6 Local coordination analysis of U in simulant LFCM by μ-EXAFS. Showing the spectra and model fit of (a) c-(U1−xZrx)O2 phase in brown LFCM μ-EXAFS spectrum in radial space and (b) the corresponding k3-weighted μ-EXAFS spectrum; (c) t-(U1−xZrx)O2 phase in brown LFCM μ-EXAFS spectrum in radial space and (d) the corresponding k3-weighted μ-EXAFS spectrum; and (e) a Zr1−xUxSiO4 phase in black LFCM μ-EXAFS spectrum in radial space and (f) the corresponding k3-weighted μ-EXAFS spectrum. | ||
The Zr1−xUxSiO4 (Fig. 6e and f) was fitted with two shells of O: 4 shorter bonds (2.26 ± 0.11 Å) and 4 longer bonds (2.36 ± 0.13 Å). The calculated bond valence contributions for these two bonds were 0.64 v.u. and 0.49 v.u. with a sum of 4.5 v.u. based on the fitted model. The distance of these two bonds is slightly shorter compared with the previous reports of the U coordination environment in USiO4,29 which is attributable to the presence of the smaller Zr4+ (ionic radius = 0.84 Å) species.
Regions located within the glassy matrix of brown and black LFCMs were selected (ESI Fig. 6 and 7†) and analysed by fitting of the μ-EXAFS data. The k3-weighted EXAFS spectra and corresponding Fourier transform (FT) were fitted shown as Fig. 7. For the brown LFCM (Fig. 7a and b), an initial model of one O-shell reported in previous U(IV)-containing silicate glasses,22 did not adequately fit the structural environment of U(IV). Thus, a two O-shell model was applied, and fitted satisfactorily, and results of structural parameter refinement are summarized in ESI Table 3.† It was fitted with 2.4 ± 0.6 U–O paths at a short distance of 2.14 ± 0.03 Å and 3.8 ± 1.1 U–O paths at longer distance 2.32 ± 0.02 Å. This model, implying a distorted 2 + 4 co-ordination of U(IV)O6 polyhedra in the brown LFCM glass phase, has a mean contact distance (2.25 Å) close to that reported for undistorted U(IV)O6 polyhedra (2.28 Å) reported in silicate glasses.30 There was no second shell contribution detected during analysis, indicating the U present in this region was incorporated within the glass structure rather than a crystalline structure, in good agreement with a previous study on U(IV) glasses.30 The calculated bond valence sum (4.1 v.u.) based on the fitted model agrees well with the shift of white line position (0.2 eV to higher energy position compared with UO2 standard). The distorted 2 + 4 co-ordination of the U(IV)O6 environment in the brown glass phase is unusual, but is similar to that observed in the crystal structure of brannerite, UTi2O6.31 The shorter and longer U–O paths have associated bond valence contributions of 0.92 v.u. and 0.56 v.u., which suggest that the U(IV) is co-ordinated to non-bridging or non-framework oxygens (since co-ordination to bridging oxygens would clearly result in unreasonable (Si,Al)–O bond lengths and polyhedral distortion).30
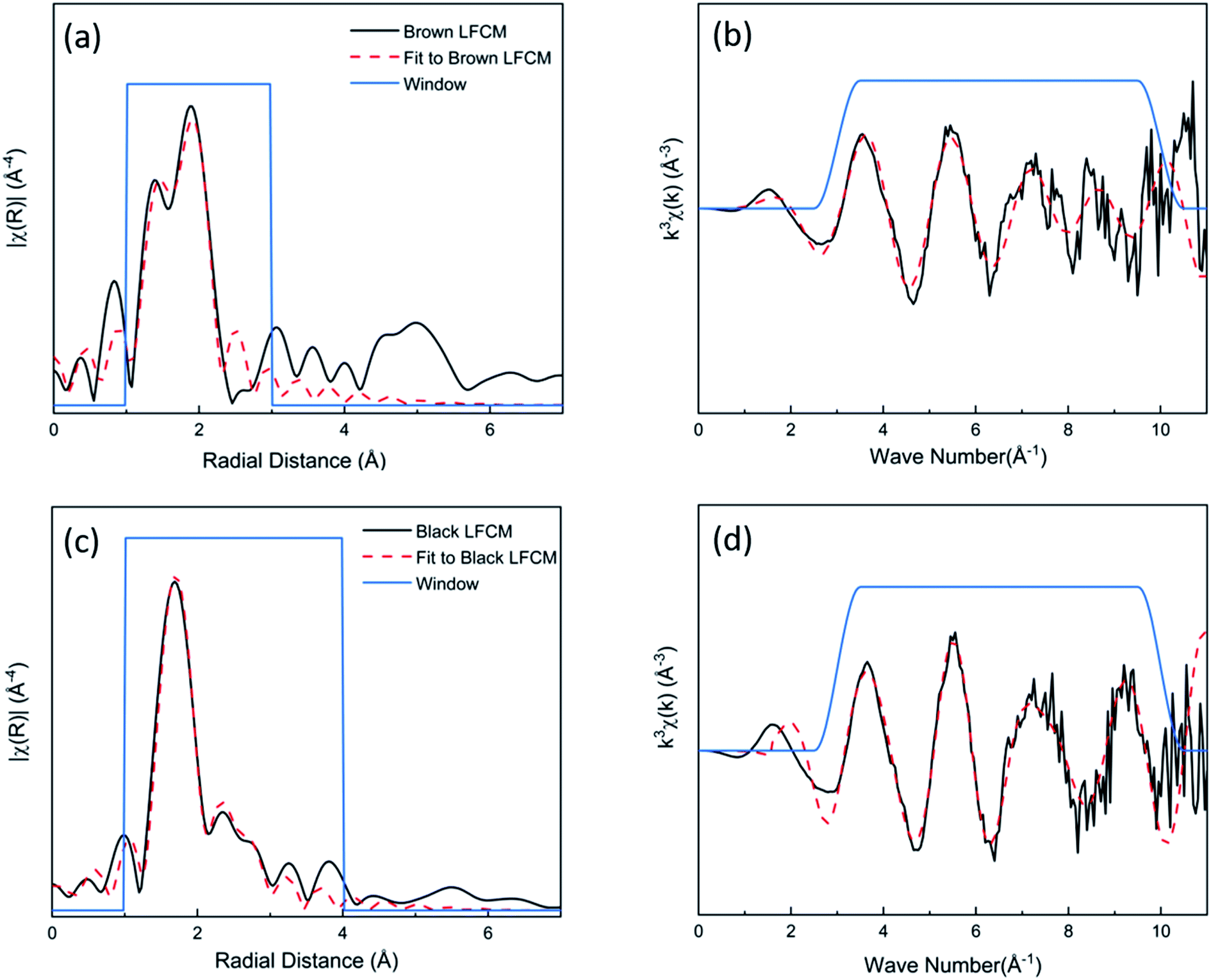 | ||
| Fig. 7 Local coordination analysis of U in the glass matrix simulant LFCM by μ-EXAFS analysis. Showing the spectra and model fit of (a) brown LFCM glass μ-EXAFS in radial space and (b) the corresponding k3-weighted EXAFS spectrum; (c) black LFCM glass μ-EXAFS in radial space and; (d) the corresponding k3-weighted EXAFS spectrum. | ||
In contrast to the brown LFCM glass matrix, the main peak for the black LFCM glass (Fig. 7c and d) μ-EXAFS data was fitted by two O-shells with 4.5 ± 0.8 oxygens at a short distance of 2.25 ± 0.02 Å and 3.3 ± 0.6 oxygens at a longer distance of 2.44 ± 0.03 Å. This indicates that U(IV) is present in 8 coordinated sites, which is similar to the UO8 polyhedra of USiO4. The calculated bond valence sum (4.2 v.u.) based on the fitted O-shell agrees well with the whiteline position shift (0.4 eV to higher energy position). The shorter and longer U–O paths had associated bond valence contributions of 0.64 v.u. and 0.38 v.u respectively, which suggest that the U(IV) is likely co-ordinated to (Si,Al)–O bond with lower bond strengths. The second neighbour Si-shell was observed and added into the model at the distance 3.19 ± 0.03 Å, improving the fit. These data are in agreement with studies of Zr–silicate glasses;32 Zr(IV) shows the same type of higher coordinated sites (ZrO8) in highly polymerised melts, which are considered to be a precursor to the crystallization of zircon.
These results provide evidence that differences in the local structure of U may exist between the crystalline phases and the glass matrices of brown and black LFCM, highlighting the heterogeneous properties of nuclear meltdown materials, on the atomic scale. However, no significant change was detected in the U oxidation states of these phases and materials, which all exhibited mean oxidation states of below 5.0. Such information may be useful to the decommissioning and secure management of LFCM, for example, in designing storage and disposal options where knowledge of U oxidation state can inform the disposal containment system, with reducing conditions preferred to retain U in its insoluble U(IV) oxidation state.33,34 Prior to extraction from the reactor unit, where humid conditions have persisted, U(IV) in the LFCM will readily oxidise to the highly soluble U(IV). This has resulted in the formation of secondary uranium phases at the surface of LFCM35 that can be released into the air as aerosol particles, which are a respirable hazard during decommissioning processes.36 Given that the local coordination of U in the glass matrix of brown and black LFCM was different, and that in our previous work we showed that LFCM corrosion by water proceeded in a similar mechanism to glass corrosion,13 it is possible that the U released during glass corrosion, and hence the formation of secondary uranium phases may be different for the brown and black LFCM. Further work is required to determine the corrosion mechanisms and verify this hypothesis.
Insights to the paragenesis of simulant Chernobyl LFCM
The heat treatment regime used in the synthesis of the simulant LFCM, including a period of rapid cooling from 1500 °C, and an extended dwell at 720 °C, was sufficient to promote crystallisation of the same phases that are observed in real LFCM.3 However, it was found that the diffusion of species was limited when compared to real LFCM, which was allowed to cool over a long period of time. For example, one consequence of the shorter cooling time in simulant samples is the presence of a relatively high proportion of t-(U1−xZrx)O2 at temperatures below the cubic to tetragonal phase transition in the UO2–ZrO2 phase diagram.28 This indicates that diffusion remained limited, leading to some regions being unable to completely transition to the cubic equilibrium structure.Where clusters of phases formed, a major fraction of the c-(U1−xZrx)O2 and t-(U1−xZrx)O2 crystallites were surrounded by either m-ZrO2 or m-(Zr1−xUx)O2 phases. Given that the ratio of U in the (U,Zr)O2 phases was shown by μ-XRF and μ-XRD to decrease, from cubic > tetragonal > monoclinic, this suggests that c-(U1−xZrx)O2 is the first to nucleate from the melt. As U is depleted from the local region of the melt, t-(U1−xZrx)O2 will crystallise, followed by m-(Zr1−xUx)O2. When no further U remains in that region, m-ZrO2 can form. A relatively high proportion of m-ZrO2 was formed when compared with real LFCM, which we attribute to the addition of excess Zr to the batch. A linear μ-XRD analysis with patterns selected from a cross section from the centre of an agglomerated mass of phases towards the glass matrix (depicted by the line in Fig. 1i) confirm this sequence (Fig. 8, ESI Fig. 8†). The centre of the agglomeration (pixel 1, Fig. 8) was indexed as c-(U1−xZrx)O2; moving towards the outside of the agglomerated mass of crystallites, the reflections relating to c-(U1−xZrx)O2 decreased at the same time as those indexed as t-(U1−xZrx)O2 increased in intensity. From pixel 15 onwards, and on the outer edge of the agglomeration, μ-XRD reflections of m-(Zr1−xUx)O2 and m-ZrO2 phases were observed, which then disappeared as the line scan passed into the glass matrix, characterised by a lack of diffraction peaks. The final phase observed was zircon, which was located in an adjacent agglomeration of crystallites (Fig. 1i).
 | ||
| Fig. 8 μ-XRD patterns showing the phase assemblage across the linear section of simulant brown LFCM shown in Fig. 1i as a black arrow. The section begins at pixel 1, in a region containing c-(U1−xZrx)O2 and ends at pixel 40, in a region containing ZrSiO4. The corresponding diffraction patterns are taken from each individual pixel across the linear section. Note that the large area of high intensity between ∼4 and 8° 2θ in the diffraction patterns corresponds to the diffuse scattering arising from the underlying amorphous matrix. | ||
U-containing zircon was distributed in a regions of fused morphology, associated with c-(U1−xZrx)O2 and m-ZrO2 on the outer edges, and t-(U1−xZrx)O2, which were present on top of the zircon. The presence of m-ZrO2, not widely observed in real LFCM,3 is thought to accompany zircon formation where there is an excess of Zr. It is thought that zircon is formed from molten Zr–U–O reaction with a silicate (SiO2) melt.3,25 The proximity of t-(U1−xZrx)O2 to zircon suggests that this process, at least in the simulant LFCM, is also accompanied by the formation of a tetragonal (U1−xZrx)O2, according to:
| Zr–U–O–Si (melt) → (Zr1−xUx)SiO4 + t-(U1−xZrx)O2 + m-ZrO2 | (1) |
Brown LFCM contained more zircon than black LFCM,12 and contained larger crystallites, indicating that zircon formation was more favourable in the former composition. Analysis of the local coordination of U in the glass matrix found that in simulant brown LFCM, U was present in a U(IV)O6-type environment, while in the simulant black LFCM it was in a UO8-type environment, similar to the coordination in coffinite, the U-end member of the U–zircon solid solution (Zr1−xUxSiO4). In the black LFCM, therefore, U was preferentially retained in a coffinite-like environment within the glass instead of nucleating as (Zr1−xUx)SiO4.
Oscillatory zonation of U, similar to that observed in chernobylite in real brown LFCM,37 was observed in U-rich zircon in the present study. Such zoning is attributed to differential growth rates of crystal faces during cooling.38 Previous analysis of the simulant brown LFCM showed very faint zoning throughout the crystallite,12 however, only two zones were apparent in the μ-XAS analysis. Since the temperature of U-rich zircon formation in this simulant material was found to be similar to chernobylite in real LFCM (crystallites were formed at ∼1245 ± 5 °C and estimates made from analysis of real LFCM gave a range of between 1000–1250 °C),13,37 the differences observed between the simulant and real samples are most likely due to differences in the cooling regime. A faster cooling regime will retard crystal growth, and this may prevent formation of more alternating zones. An alternative hypothesis is that oscillatory zones form when contaminant elements are incorporated into the crystal structure during growth under conditions of cooling from a melt.39 Since the simulant brown LFCM contained a limited number of elements when compared with the real Chernobyl LFCM which, for example, also contained all of the fission products expected to arise in nuclear fuel, in addition to other impurities sourced from materials within the reactor, it is also possible that the lack of complex oscillatory zoning is due to the simplified chemical composition used.
Conclusion
This study of simulant Chernobyl LFCM using a μ-focus XAS approach, integrating μ-XRF, μ-XAS and μ-XRD, demonstrates that it is possible to develop a highly detailed and complementary data set from which to accurately identify crystalline phases, determine local U chemistry in crystalline and amorphous phases, and to reconstruct the paragenetic sequence of phase formation in nuclear fuel formed in accident scenarios. These techniques have built significantly on previous studies of nuclear melt-down materials, by determining the U chemistry in the individual μm-scale components, including the amorphous phase, highlighting that chemical heterogeneity extends to the atomic scale. Importantly, these data can be acquired from μm-sized fractions of material. Size reduction of highly radioactive materials, such as fuels resulting from a nuclear accident, significantly lowers the risk associated with performing detailed investigations. Therefore, it could be possible to acquire the same detailed chemical insights as developed here, for real nuclear accident fuels.Data availability
Data will be made available upon reasonable request to the corresponding author.Code availability
Matlab codes that were used to analyze the μ-XRF and μ-XRD are written for Matlab2018a with Signal Processing toolbox. These codes are freely available in Github (https://github.com/Hding8).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded by the UK Engineering and Physical Science Research Council (EPSRC) under grant agreement numbers EP/N017374/1, EP/R01924X/1, EP/S020659/1 and EP/S01019X/1. MCDW is grateful to the Nuclear Decommissioning Authority for PhD sponsorship. This research utilised the HADES/MIDAS facility at the University of Sheffield established with financial support from EPSRC and BEIS, under grant EP/T011424/1. We acknowledge the Paul Scherrer Institut, Villigen, Switzerland for provision of synchrotron radiation beamtime at beamline X05 of the SLS and would like to thank Dario Ferreira Sanchez for assistance. This research used beamline 4-BM of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We also wish to thank Dr Stephanie Szenknect for kind provision of synthetic coffinite standard, and Dr Bruce Ravel for beamline bananagrams.References
- A. N. Kiselev and K. P. Checherov, At. Energy, 2001, 91, 967–975 CrossRef CAS.
- S. V. Ushakov, B. E. Burakov, S. I. Shabalev and E. B. Anderson, MRS Online Proc. Libr., 1996, 465, 1313 CrossRef.
- B. E. Burakov, E. B. Anderson, S. I. Shabalev, E. E. Strykanova, S. V. Ushakov, M. Trotabas, J. Y. Blanc, P. Winter and J. Duco, Mater. Res. Soc. Symp. Proc., 1997, 465, 1297–1308 CrossRef CAS.
- A. A. Borovoi, A. S. Lagunenko and E. M. Pazukhin, At. Energy, 1998, 84, 295–299 CrossRef CAS.
- S. A. Bogatov, A. A. Borovoi, A. S. Lagunenko, E. M. Pazukhin, V. F. Strizhov and V. A. Khvoshchinskii, Radiochemistry, 2008, 50, 650–654 CrossRef CAS.
- A. A. Shiryaev, B. E. Burakov, I. E. Vlasova, M. S. Nickolsky, A. A. Averin and A. V. Pakhnevich, Mineral. Petrol., 2020, 114, 489–499 CrossRef CAS.
- D. W. Akers, S. M. Jensen and B. K. Schuetz, US Nuclear Regulatory Commission Report no. NUREG/CR-6195, 1994, DOI:10.2172/1014080.
- A. A. Shiryaev, I. E. Vlasova, B. E. Burakov, B. I. Ogorodnikov, V. O. Yapaskurt, A. A. Averin, A. V. Pakhnevich and Y. V. Zubavichus, Prog. Nucl. Energy, 2016, 92, 104–118 CrossRef CAS.
- Y. V. Dubasov, E. a. Smirnova and S. I. Malimonova, Radiochemistry, 2012, 54, 298–307 CrossRef CAS.
- Y. A. Teterin, A. S. Baev and S. A. Bogatov, J. Electron Spectros. Relat. Phenomena, 1994, 68, 685–694 CrossRef CAS.
- V. A. Zheltonozhskii, M. V. Zheltonozhskaya, N. V. Kulich, M. D. Bondar’kov, A. M. Maksimenko and E. M. Pazukhin, Radiochemistry, 2011, 53, 550–554 CrossRef CAS.
- S. T. Barlow, D. J. Bailey, A. J. Fisher, M. C. Stennett, C. L. Corkhill and N. C. Hyatt, MRS Adv., 2017, 2, 609–614 CrossRef CAS.
- S. T. Barlow, D. J. Bailey, A. J. Fisher, M. C. Stennett, C. Gausse, H. Ding, V. A. Krasnov, S. Yu. Sayenko, N. C. Hyatt and C. L. Corkhill, npj Mater. Degrad., 2020, 4, 3 CrossRef CAS.
- C. Paraskevoulakos, J. P. Forna-Kreutzer, K. R. Hallam, C. P. Jones, T. B. Scott, C. Gausse, D. J. Bailey, C. A. Simpson, D. Liu, C. Reinard, C. L. Corkhill and M. Mostafavi, Mater. Design., 2021, 201, 109502 CrossRef CAS.
- N. C. Hyatt, C. L. Corkhill, M. C. Stennett, R. J. Hand, L. J. Gardner and C. L. Thorpe, IOP Conf. Ser. Mater. Sci. Eng., 2020, 818, 012022 CrossRef CAS.
- C. Borca, D. Grolimund, M. Willimann, B. Meyer, K. Jefimos, J. Vila-Comamala and C. David, J. Phys. Conf. Series., 2009, 186, 012003 CrossRef.
- C. Den Auwer, E. Simoni, S. Conradson and C. Madic, Eur. J. Inorg. Chem., 2003, 21, 3843–3859 CrossRef.
- R. B. King, Chem. Mater., 2002, 14, 3628–3635 CrossRef CAS.
- X. Guo, S. Szenknect, A. Mesbah, S. Labs, N. Clavier, C. Poinssot, S. V. Ushakov, H. Curtius, D. Bosbach, R. C. Ewing, P. C. Burns, N. Dacheux and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6551–6555 CrossRef CAS.
- S. Szenknect, D. Alby, M. López García, C. Wang, R. Podor, F. Miserque, A. Mesbah, L. Duro, L. Zetterström Evins, N. Dacheux, J. Bruno and R. C. Ewing, Sci. Rep., 2020, 10, 1–10 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- W. De Nolf, F. Vanmeert and K. Janssens, J. Appl. Cryst., 2014, 47, 1107–1117 CrossRef CAS.
- C. Journeau, F. Sudreau, S. Magne and G. Cognet, Mat. Sci. Eng. A., 2001, 299, 249–266 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- E. B. Anderson, B. E. Burakov and E. M. Pazukhin, Radiochim. Acta, 1992, 60, 149–152 Search PubMed.
- E. M. Pazukhin, Radiochemistry, 1994, 36, 109–154 Search PubMed.
- T. Kitagaki, K. Yano, H. Ogino and T. Washiya, J. Nucl. Mater., 2017, 486, 206–215 CrossRef CAS.
- I. Cohen and B. E. Schaner, J. Nucl. Mater., 1963, 9, 18–52 CrossRef CAS.
- S. Labs, C. Hennig, S. Weiss, H. Curtius, H. Zänker and D. Bosbach, Environ. Sci. Technol., 2014, 48, 854–860 CrossRef CAS.
- F. Farges, C. W. Ponader, G. Calas and G. E. Brown, Geochim. Cosmochim. Acta, 1992, 56, 4205–4220 CrossRef CAS.
- J. T. Szymanski and J. D. Scott, Can. Mineral., 1982, 20, 271–279 CAS.
- F. Farges, C. W. Ponader and G. E. Brown, Geochim. Cosmochim. Acta, 1991, 55, 1563–1574 CrossRef CAS.
- G. R. Choppin and B. E. Stout, Sci. Total Environ., 1989, 83, 203–216 CrossRef CAS.
- I. Grenthe, J. Drożdżynński, T. Fujino, E. C. Buck, T. E. Albrecht-Schmitt and S. F. Wolf, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer Netherlands, Dordrecht, 2006, pp. 253–698 Search PubMed.
- B. E. Burakov, E. E. Strykanova and E. B. Anderson, Mat Res. Soc. Symp. Proc., 1997, 465, 1309–1311 CrossRef CAS.
- V. P. Badovskii, A. E. Melenevskii, Yu. V. Morozov, I. A. Ushakov and V. N. Shcherbin, Radiochemistry, 2014, 56, 311–318 CrossRef CAS.
- P. Pöml, B. Burakov, T. Geisler, C. T. Walker, M. L. Grange, A. A. Nemchin, J. Berndt, R. O. C. Fonseca, P. D. W. Bottomley and R. Hasnaoui, J. Nucl. Mater., 2013, 439, 51–56 CrossRef.
- R. T. Pidgeon, Contrib. Mineral. Petrol., 1992, 110, 463–472 CrossRef CAS.
- G. Vavra, Contrib. Mineral. Petrol., 1990, 106, 90–99 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta09131f |
| This journal is © The Royal Society of Chemistry 2021 |