
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A-site deficient chromite with in situ Ni exsolution as a fuel electrode for solid oxide cells (SOCs)†
Diana-María
Amaya-Dueñas
 *a,
Guoxing
Chen
b,
Anke
Weidenkaff
bc,
Noriko
Sata
a,
Feng
Han
a,
Indro
Biswas
a,
Rémi
Costa
*a and
Kaspar Andreas
Friedrich
ad
*a,
Guoxing
Chen
b,
Anke
Weidenkaff
bc,
Noriko
Sata
a,
Feng
Han
a,
Indro
Biswas
a,
Rémi
Costa
*a and
Kaspar Andreas
Friedrich
ad
aGerman Aerospace Center (DLR), Institute of Engineering Thermodynamics, Pfaffenwaldring 38-40, D-70569 Stuttgart, Germany. E-mail: diana.amayaduenas@dlr.de; remi.costa@dlr.de
bInstitute of Materials and Earth Sciences, Technische Universität Darmstadt, Alarich-Weiss-Str. 2, D-64287 Darmstadt, Germany
cFraunhofer IWKS, Materials Recycling and Resource Strategies, Brentanostraße 2a, D-63755 Alzenau, Germany
dInstitute of Building Energetics, Thermal Engineering and Energy Storage (IGTE), University of Stuttgart, Pfaffenwaldring 31, D-70569 Stuttgart, Germany
First published on 1st February 2021
Abstract
A-site deficient lanthanum strontium chromite perovskite La0.65Sr0.3Cr0.85Ni0.15O3−δ (L65SCrN) decorated by in situ exsolution of Ni nanoparticles was synthesized and implemented as a fuel electrode on a 5 cm × 5 cm electrolyte-supported cell (ESC) for solid oxide cells (SOCs) with an active surface of 16 cm2. The stoichiometric formulation La0.70Sr0.3Cr0.85Ni0.15O3−δ (L70SCrN) was also prepared in order to evaluate the reducibility and behavior towards Ni exsolution with respect to L65SCrN. This comparison was assessed by means of X-ray diffraction (XRD) and thermogravimetric analysis (TGA) in a reducing atmosphere. Metallic Ni was successfully detected using XRD on the A-site deficient formulation after TGA treatment. Surface analysis by means of X-ray photoemission spectroscopy (XPS) revealed a relative enrichment in Cr3+. Ni exsolution was investigated on the L65SCrN formulation by annealing in a reducing atmosphere at 500 °C and 900 °C for 3 hours. The Ni nanoparticle size (from ∼8 up to 100 nm) and morphology were characterized by means of scanning electron microscopy (SEM). Furthermore, L65SCrN was screen printed onto a 90 µm thick CGO20-3YSZ-CGO20 electrolyte on which the oxygen electrode La0.58Sr0.4Fe0.8Co0.2O3−δ (LSCF) was printed on the other side. With ideal contacting, the electrochemical cell performance of the L65SCrN fuel electrode was demonstrated to be comparable to those of the state-of-the-art Ni-based cermets: ASRDC_Total at −0.3 A cm−2 was calculated to be 0.676 Ω cm2 in co-electrolysis operation. Reversible operation (rSOC) at 860 °C with a H2O/H2 ratio of 1 could be shown and co-electrolysis operation (H2O/CO2 = 2) at −0.45 A cm−2 and 860 °C with a voltage degradation of less than 3.5 mV/1000 hours could be demonstrated for 950 hours. Even though L65SCrN showed promising results for SOC operation, further investigations of Ni exsolution in doped chromites by varying temperature, time and pO2 are proposed for a detailed understanding and optimization of the Ni nanoparticle size.
Introduction
Since the industrial revolution, carbon-rich fossil feedstocks have played an important role in our daily life in order to fulfil our needs for energy demand and for a broad range of household and commercial products. Nowadays, the chemical industry relies on crude oil, coal and natural gas to produce the key building blocks such as olefins and aromatics. Nevertheless, the improvement of the corresponding synthesis processes in terms of selectivity and energy consumption or the development of alternative routes has become a major priority for the modern industry due to limited recoverable natural oil reserves and growing environmental considerations regarding greenhouse gas emissions.1CO2 is emitted in increasing amounts due to the growing need for power generation (coal-based plants) and industrial products, such as steel and chemicals, e.g. ethylene production by oxidative coupling of methane (OCM).1 This greenhouse gas is also an essential feedstock for numerous chemical synthesis processes in combination with hydrogen. In some processes, CO2 is pre-reduced at high temperature with hydrogen through the reverse water gas shift (RWGS) reaction yielding CO – a more reactive molecule – as an essential building block for downstream chemical synthesis. Methanol, which is an important multipurpose intermediate commonly used for the production of various chemicals, is currently produced from syngas (H2 + CO) which can also be generated via catalytic steam or autothermal reforming of methane.1
In the light of syngas production and methanol synthesis, there are significant economic and environmental interest in valorizing renewable carbon sources. For this reason, since the last 10 years, the conversion of plant-derived materials (biomass) and CO2 has attracted attention from industry and academia with the aim of producing fuels and bulk chemicals using direct electrosynthesis routes with a reduced CO2 footprint.2
Power-to-X concepts intend to convert excess renewable power into diverse fuels and chemicals that can be used for large capacity energy storage.3,4 Among the various concepts, the technologies based on Solid Oxide Cells (SOCs) operating at temperatures typically around 750–850 °C enable conversion of electricity at a high efficiency into valuable fuels (hydrogen or hydrocarbons) by means of high temperature electrolysis (HTE) without the need of precious catalysts. Interestingly, due to fast kinetics SOCs enable the simultaneous electrolysis of H2O–CO2 at high temperature into syngas, which can be further used for large-scale production of methanol and other green fuels and chemicals through the Fischer–Tropsch (F–T) synthesis.1,5,6 Moreover, SOCs offer the unique advantage of enabling reversible operation. i.e. either energy storage or electricity production. In the energy storage mode, electrical energy from renewable sources is converted to valuable fuels (hydrogen or hydrocarbons) by means of HTE, while in discharge mode these fuels could be used for power production through fuel cell operation.4 A reversible Solid Oxide Cell (rSOC) system could effectively ensure large storage capacity and grid balancing.
State-of-the-art SOCs rely on Ni-based cermet components, owing to the excellent electrical conductivity and high catalytic activity of Ni towards H2O–CO2 splitting and hydrogen dissociation reactions at high temperatures. Ni–Zr0.85Y0.15O2−δ (Ni–YSZ) cermet fuel electrodes – typically used in the so-called Anode-Supported Cells (ASCs) – have been largely investigated in either operating modes. When operated in fuel cell mode, the electrodes are susceptible to poisoning with different fuel gas impurities such as sulfur species that have deleterious effects on performance, especially in reformate gases, and long-term stability.7,8 Moreover, they suffer from irreversible degradation when exposed to re-oxidation reactions.9 These cermet electrodes are prone to Ni agglomeration leading to loss of electrical percolation and diminution of the triple-phase-boundary (TPB) length.10 When operated in electrolysis, they suffer from irreversible microstructural alterations, especially at high temperatures, high current densities and high pH2O.11 In co-electrolysis operation, carbon formation has been observed at the electrode–electrolyte interface with a reactant conversion of ∼67% at 875 °C, causing microstructural alterations accompanied by a deactivation of the active sites.12 By contrast, Ni–Ce1−xGdxO2−δ (Ni–CGO) based fuel electrodes – typically used in the Electrolyte-Supported Cells (ESCs) – have also been investigated as fuel electrode materials because of their catalytic properties and CGO phase enhanced tolerance against carbon formation.13 Nevertheless, due to the large content of metallic Ni, such Ni–CGO cermet electrodes are also vulnerable to dimensional alterations caused by grain coarsening upon redox cycling i.e. repeated alternation of oxidizing and reducing atmospheres, which adversely affects the apparent electronic conductivity leading to an increase of the ohmic resistance (Rohm) and the gas transport properties of the electrode.14
The wide operating range of SOC-based electrochemical reactors requires robust and durable fuel electrodes with high performance in either operating mode: fuel cell or electrolysis operation. This implies performance and durability in a broad range of partial pressures (pH2, pH2O, pO2, pCO and pCO2) and a given dimensional stability nearly independent of the atmosphere.
Perovskite-based oxides (ABO3) have been proposed as alternative materials to the Ni cermets as fuel electrodes for SOCs because of their outstanding stability in both reducing and oxidizing atmospheres and their flexibility in terms of composition, that enables a wide variety of doping elements on their A- and B-sites to tune their electrocatalytic properties. As a fuel electrode, high catalytic activity can be achieved when the A-site is a lanthanide and/or alkaline-earth cation and the B-site a transition metal cation such as Mn, Co, Fe, Ni, Cr and Ti.15
Strontium titanates have been widely studied and have shown remarkable performance as fuel electrodes in steam electrolysis on the laboratory scale, where the perovskite's surface has been decorated with catalytically active Ni and Fe nanoparticles.16 It has been reported that surface decoration with catalytically active nanoparticles can be achieved using redox exsolution methods, where a catalytically active metal (i.e. Ni or Fe) is incorporated into the crystal lattice of the perovskite backbone under oxidizing conditions and is released (exsolved) on the surface as metal nanoparticles, either by exposure to a reducing atmosphere or by applying a large cathodic overpotential.17,18 It is generally admitted that exsolution is favoured upon A-site deficiency: when the oxygen vacancy concentration is high enough to partially destabilize the perovskite lattice due to the high deficiency on A- and O-sites, metal particles from the B-site exsolve while charge balance of the lattice is maintained.19 A recent study by Neagu et al. about Ni exsolution on lanthanum–calcium doped titanates and lanthanum–cerium doped titanates by in situ observation with environmental transmission microscopy (ETEM) showed that the exsolution phenomena and thus the shape of the resulting nanoparticles are significantly affected by the temperature and the oxygen partial pressure (pO2),20 being important operating parameters for the rSOC reactors.
Lanthanum chromites present an alternative towards strontium titanates as another perovskite family that can also host B cations to be exsolved in situ on their surface to enhance the electrocatalytic activity. (La,Sr)(Cr,M)O3 perovskites (M = Mn, Fe, Co and Ni) have been recently investigated for H2O electrolysis, CO2 electrolysis and H2O–CO2 co-electrolysis: mostly in stoichiometric formulations21 and a few with A-site deficiency.17,22 However, the Ni exsolution phenomena on lanthanum chromites upon temperature and atmosphere variation remain unclear, and the performance of such perovskite electrodes still needs to be improved in order to achieve comparable results with the typical Ni-cermet fuel electrodes.
Given the operating conditions of rSOC reactors with a focus on Solid Oxide Electrolysis Cell (SOEC) applications, the lack of Ni exsolution research on chromites arouses the interest to investigate the performance of Ni-decorated chromites as fuel electrodes for SOCs, raising as well the importance to evaluate their durability and performance in either mode on rSOC reactors.
In this paper, we focus on the exploration of the A-site deficient chromite La0.65Sr0.3Cr0.85Ni0.15O3−δ (L65SCrN) fuel electrode decorated with Ni nanoparticles for SOC applications with the aim: (i) of evaluating the Ni exsolution as a function of temperature in La0.65Sr0.3Cr0.85Ni0.15O3−δ and (ii) of characterizing the electrochemical performance of the screen printed La0.65Sr0.3Cr0.85Ni0.15O3−δ fuel electrode on a 5 cm × 5 cm ESC in fuel cell operation (SOFC), in H2O electrolysis and H2O–CO2 co-electrolysis operation (SOEC) at high temperature.
Experimental procedures
LSCrN synthesis
La0.65Sr0.3Cr0.85Ni0.15O3−δ (L65SCrN) and La0.70Sr0.3Cr0.85Ni0.15O3−δ (L70SCrN) ceramic powders were prepared using the glycine nitrate combustion method described in Sun et al.23 According to these two formulations, stoichiometric amounts of La(NO3)2·6H2O (99.9% REO Alfa Aesar), Sr(NO3)2 (98% Alfa Aesar), Ni(NO3)2·6H2O (98% Alfa Aesar) and Cr(NO3)2·9H2O (98.5% Alfa Aesar) were dissolved in deionized water and mixed with glycine (J.T.Baker™). The glycine molar ratio for the total content of metal cations was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Next, these solutions were stirred and heated on a hot plate until a dark green-colored gel was formed. Previous thermogravimetric measurements in synthetic air performed on these gels indicated that the solvent evaporation takes place at ∼91 °C followed by an exothermic self-combustion reaction at ∼220 °C.24 Therefore, in this study, the gels were heated up to ∼220 °C where self-combustion occurred. Finally, the resulting ceramic precursors were calcined in air at a rate of 3 °C min−1 up to 1400 °C for one hour since it was the minimal firing temperature at which a perovskite phase could be achieved,24 which is consistent with previous studies on lanthanum chromites.25
:1. Next, these solutions were stirred and heated on a hot plate until a dark green-colored gel was formed. Previous thermogravimetric measurements in synthetic air performed on these gels indicated that the solvent evaporation takes place at ∼91 °C followed by an exothermic self-combustion reaction at ∼220 °C.24 Therefore, in this study, the gels were heated up to ∼220 °C where self-combustion occurred. Finally, the resulting ceramic precursors were calcined in air at a rate of 3 °C min−1 up to 1400 °C for one hour since it was the minimal firing temperature at which a perovskite phase could be achieved,24 which is consistent with previous studies on lanthanum chromites.25
Characterization of LSCrN powders
Crystalline structure was investigated using X-ray diffraction (XRD) with a RIGAKU diffractometer operating at 40 kV and 30 mA with a Cu-Kα1,2 radiation source and a Bragg–Brentano configuration in the range of 2θ from 20–80° with a scanning rate of 0.4° min−1. Crystalline phases were identified from the ICDD database. A different scanning rate of 0.1° min−1 was used for the as-prepared and reduced L65SCrN samples, where phases were identified and quantified by Rietveld analyses using the FullProf.2k program suite.Morphology and microstructure were observed with a scanning electron microscope Zeiss ULTRA PLUS SEM (Carl Zeiss AG, Germany) in combination with energy-dispersive X-ray spectroscopy (EDX) for elemental analysis, where a Bruker XFlash 5010 detector was operated at 125 eV with the Quantax 400 Software. The spatial resolution was ∼100 nm and elements with atomic numbers higher than 4 (Boron) could be detected.
Surface chemistry investigations with X-ray photoemission spectroscopy (XPS) were carried out using a system with a base pressure of 2 × 10−10 mbar, with a hemispherical analyzer (ESCALAB250, ThermoFisher Scientific) and a monochromated Al Kα source with an X-ray energy of 1486.74 eV (XM1000, ScientaOmicron). The peak shape analysis was carried out with Unifit 2013, applying convoluted Gaussian/Lorentzian profiles and a Shirley background function.26–28 The surface stoichiometry of the occurring atoms/signals was calculated using the numerically fitted peak areas, photoionization cross sections reported by Yeh and Lindau29 and instrumental transmission functions given by the manufacturer.
The reducibility of the as-prepared L65SCrN and L70SCrN powders was characterized by means of thermogravimetric analysis (TGA) in a reducing atmosphere (5% H2–Ar) with the analyzer Netzsch Jupiter 449C at a heating rate of 3 °C min−1 from 25 °C to 1200 °C. Such reducibility analysis by TGA was accompanied by a temperature-programmed reduction (TPR) performed on the flow-through quartz reactor TPDRO 1100 (Thermo Scientific, Italy), in which L65SCrN and L70SCrN powder specimens were introduced using quartz glass wool as a support. A thermocouple (type K) was placed in a thin quartz glass tube next to the specimen to monitor the temperature. The oven temperature was monitored and controlled by another thermocouple. The specimens were pretreated in the reactor tube with flowing Ar gas at a flow rate of 20 mL min−1 and by heating at a rate of 10 °C min−1 from 30 °C to 150 °C with a holding time of 60 min. After cooling the specimen back to 30 °C, the TPR process was initiated with a reducing gas mixture of 5% H2–Ar at a constant flow rate of 20 mL min−1 and a heating rate of 5 °C min−1 from 30 °C to 1000 °C with a holding time of 60 min at 1000 °C. Under these conditions, the sample temperature was ∼1000 °C while the oven temperature was 1100 °C. The exhaust gas from the reactor was analyzed using a Thermal Conductivity Detector (TCD).
Cell manufacturing
The L65SCrN electrocatalyst was implemented as fuel electrode into an electrolyte-supported cell (ESC) by screen printing, using a commercial square substrate (5 cm × 5 cm and 90 µm of thickness) of 3 mol% Y2O3-doped ZrO2 electrolytes double-side coated with ca. 5 µm of Ce0.8Gd0.2O2−δ (CGO20-3YSZ-CGO20) from Kerafol GmbH, Germany. The fuel electrode ink was prepared by dispersing the L65SCrN powder in a solution (94 wt% α-Terpineol and 6 wt% ethyl cellulose) with a powder to solution ratio of 2:1, followed by mixing with the 3-roll milling machine EXAKT 80E EL. The prepared ink was printed on the electrolyte using the screen printer Aurel model 900 (Aurel automation s.p.a, Italy). The half-cell was fired at 1200 °C for 1 hour in air with a heating rate of 3 °C min−1. Afterwards, the oxygen electrode was printed on the other half of the cell with a commercial ink of La0.58Sr0.4Fe0.8Co0.2O3−δ (LSCF). The printed area for both electrodes was 16 cm2 (4 cm × 4 cm). Platinum paste was brushed on the sintered fuel electrode surface for current collection. Finally, the cell was fired at a rate of 3 °C min−1 to 1050 °C in air and held for one hour.
Electrochemical characterization
The electrochemical performance of the L65SCrN fuel electrode in the ESC architecture was studied on the test bench described elsewhere.24 The fuel electrode was contacted with a platinum mesh and the oxygen electrode with a gold mesh. A gold frame was used as a sealant between the fuel and the air side. For commissioning, the cells were heated (3 °C min−1) to 900 °C with N2 (1 SLPM) and air (1 SLPM) for sealing purposes and subsequently reduced with H2 (1 SLPM) for 1 hour on the fuel side. Afterwards, the operating temperature was adjusted to 860 °C. Electrochemical experiments were carried out in different fuel gas mixtures shown in Table 1. The equilibrium gas phase compositions and the theoretical OCV according to the Nernst voltage were calculated with the software CANTERA.30| Operation mode | % H2 | % H2O | % CO2 |
|---|---|---|---|
| FC | 90 | 10 | — |
| FC-EC | 50 | 50 | — |
| EC | 20 | 80 | — |
| Co-EC | 5 | 63.7 | 31.3 |
Electrochemical Impedance Spectroscopy (EIS) was performed in galvanostatic mode with the workstation Zahner PP-240 in a frequency range from 50 mHz to 100 kHz. The amplitude of the current stimulus was 500 mA. Distribution of relaxation times (DRT) calculations were carried out with the impedance analysis and modelling software ec-idea31 and the equivalent circuit model-fit of the impedance data with the commercially available program ZView®.32
In fuel cell (FC) mode, the polarization curves (i–V) were measured from OCV to 0.8 A cm−2 at a rate of 0.012 A s−1 and in electrolysis (EC) and co-electrolysis (co-EC) modes from OCV to −1.0 A cm−2 at a rate of −0.012 A s−1. For all operating modes the total fuel gas flow was maintained at 1 SLPM, except for co-EC, which was kept at 0.8 SLPM.
Results and discussion
A comparative assessment towards LSCrN reducibility and Ni exsolution was performed between stoichiometric L70SCrN and A-site deficient L65SCrN perovskite powder samples. Furthermore, the temperature effect on the Ni exsolution was investigated on the L65SCrN and the electrochemical performance was evaluated in a full cell assembly with an ESC architecture, in which the L65SCrN perovskite was implemented as fuel electrode.Nickel exsolution assessment on LSCrN powders
Phase identifications of both the as-prepared ceramic powders L70SCrN and L65SCrN, as well as in reduced conditions (with 5% H2–Ar at 3 °C min−1 from 25 °C to 1200 °C) were performed by XRD in order to verify that the perovskite phase was stable after the reduction treatment. X-ray diffractograms of the L70SCrN before and after reduction are shown in Fig. 1. No NiO secondary phase or other impurities could be identified. Interestingly, no metallic Ni could be detected on the reduced L70SCrN sample as one may have expected upon reduction. However, a few nanoparticles could be observed in the SEM image of the reduced L70SCrN powder in Fig. S1† in the ESI. This suggests that the total amount of metallic Ni in the reduced L70SCrN (originating from the reduction of a possible NiO secondary phase as well as from the likely to occur exsolution of metallic Ni upon reduction of the host perovskite) remained below the detection level of the XRD analysis, i.e. a phase content of less than 1 wt%.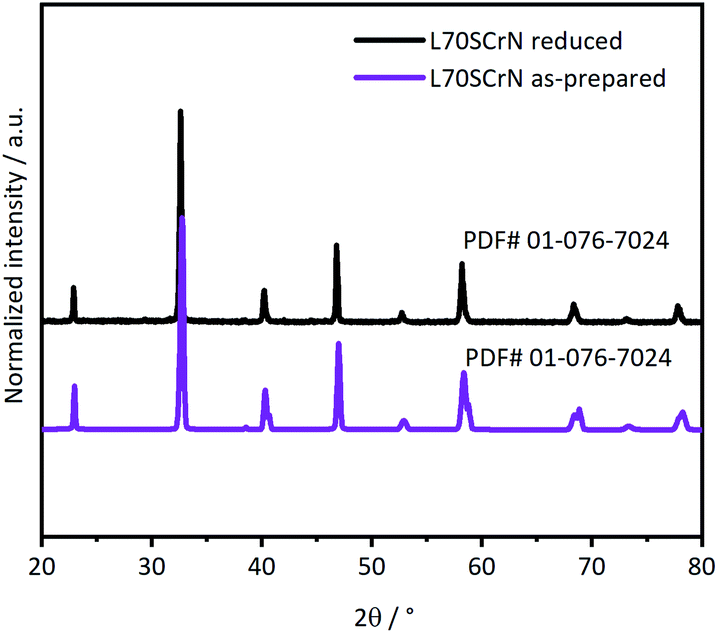 | ||
| Fig. 1 XRD patterns of the as-prepared L70SCrN powder and reduced in 5% H2–Ar up to 1200 °C at 3 °C min−1. Crystal systems were identified with ICCD as the perovskite chromite phase: PDF# 01-076-7024. | ||
In contrast, secondary phases could be identified for L65SCrN in both the as-prepared and reduced samples (Fig. 2a and b). Therefore, Rietveld analyses were performed with the aim of quantifying those secondary phases. An orthorhombic lattice (space group 62, Laue class mmm) was calculated for the as-prepared L65SCrN sample with parameters to be a = 5.496 Å, b = 5.450 Å, c = 7.737 Å and V = 231.762 Å3. A secondary phase was identified as nickel oxide (NiO) with cubic lattice (space group 225, Laue class m![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and lattice parameters a = 4.176 Å and V = 72.851 Å3. Although the NiO content was 4.22 mol%, it was not considered to be a detrimental impurity since it would be reduced operando into metallic Ni, being also catalytically active and electronically conductive.
m) and lattice parameters a = 4.176 Å and V = 72.851 Å3. Although the NiO content was 4.22 mol%, it was not considered to be a detrimental impurity since it would be reduced operando into metallic Ni, being also catalytically active and electronically conductive.
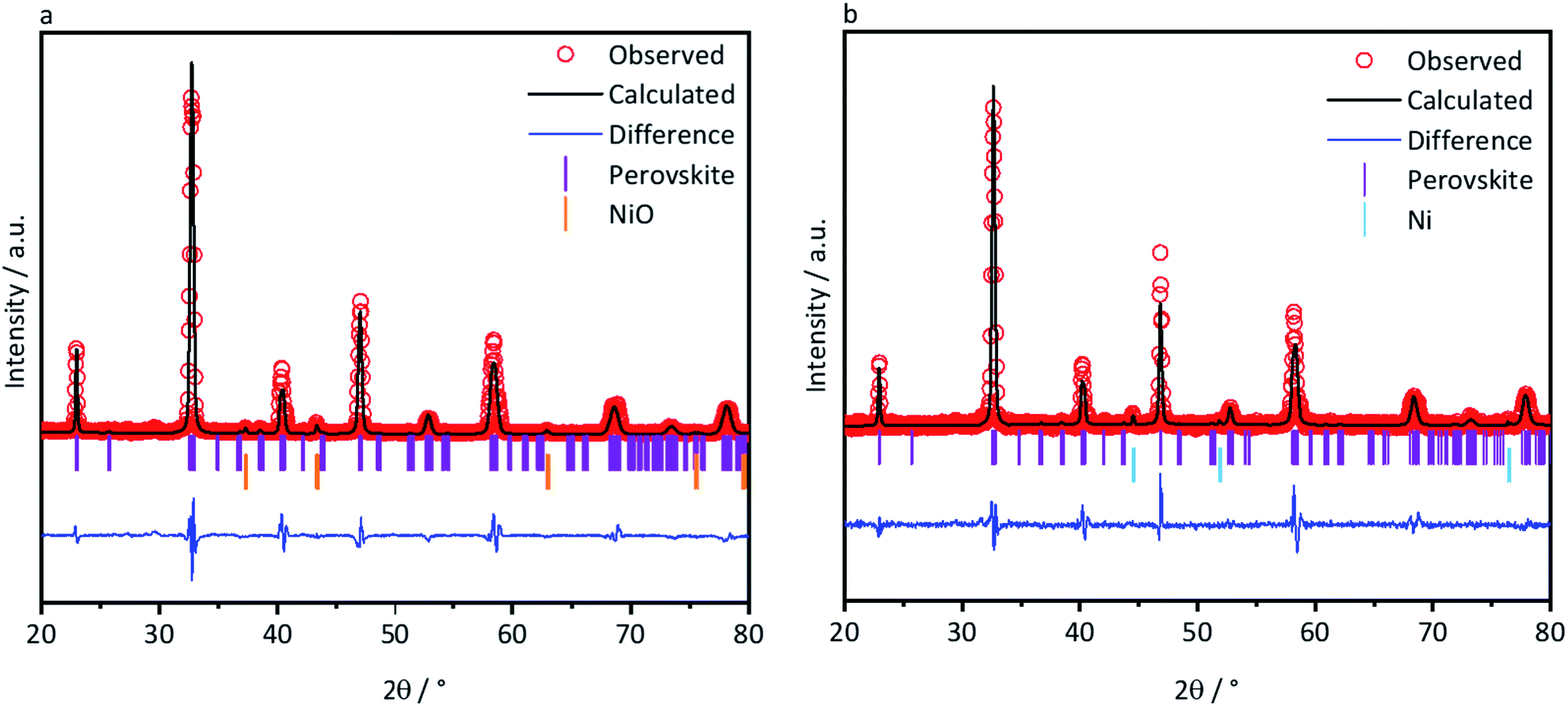 | ||
| Fig. 2 XRD patterns of the as-prepared L65SCrN powder (a) and reduced (b) in 5% H2–Ar up to 1200 °C at 3 °C min−1. Crystallographic parameters and the concentration of secondary phases were calculated from Rietveld refinement. | ||
For the reduced L65SCrN sample, an expanded orthorhombic lattice (also space group 62, Laue class mmm) was calculated (a = 5.465 Å, b = 7.758 Å, c = 5.506 Å and V = 233.409 Å3). A secondary phase of metallic Ni was calculated to be 4.83 mol% with a cubic lattice (space group 225, Laue class mm) with a = 3.524 Å and V = 43.761 Å3, from which the metallic Ni characteristic peak (111) was identified at 44.4°. This corroborates that metallic Ni can be achieved upon exposure of L65SCrN to a reducing atmosphere at high temperatures. It is pertinent to note that the amount of metallic Ni after reduction (4.83 mol%) is greater than the content of NiO (4.22 mol%) in the as-prepared sample, indicating that the metallic Ni has been effectively exsolved from the L65SCrN matrix under these reducing conditions. The characteristic peak of the perovskite at 47.2° shifted slightly to a lower diffraction angle (46.9°), which corresponds to an expansion of the perovskite orthorhombic lattice (from 231.762 Å3 to 233.409 Å3). This expansion could be due to the loss of Ni2+ cations from the lattice that are reduced to Ni0 and exsolved on the surface in correlation with the consumption of vacancies on the A-site, but also due to the oxygen loss.33,34 A change in the Cr4+/Cr3+ ratio in the host perovskite matrix could also contribute to this expansion, taking into account that the ionic radius in an octahedral environment for Cr4+ is 0.55 Å and for Cr3+ is 0.615 Å.35 To better assess this, the as-prepared and reduced L65SCrN powders were also investigated by means of XPS.
At first, the XPS studies could not conclusively confirm the presence of a metallic nickel phase in the reduced L65SCrN sample. As the typically used signal of the Ni2p electrons overlaps with the very distinct 3d signals of Lanthanum of an unusual quadruplet shape, the deconvolution of the traces of metallic nickel was not possible this way. Similar to Nenning and Fleig,25 the Ni3p region was used (Fig. 3a), which also partially overlaps with the Cr3s signal, but the chemical structure of the surface chromium could be evaluated by means of the Cr2p signal (Fig. 3b) and added as boundary to the numerical model for the nickel 3p region, with its low cross section. While it was possible to identify two occurring Ni species in both the as-prepared and reduced L65SCrN samples, which are attributed to oxidic Ni2+ (∼67.2 eV) and surface Ni(OH)2 (∼69.6 eV),36 the clear evidence for a Ni0 species is lost in the signal noise. However, the signal deconvolution, which was performed using the aforementioned chromium signature and a fixed Lorentzian peak width for the as-prepared sample, converged to a result with Gaussian line widths of 2.2 eV (Ni2+) and 2.6 eV (Ni(OH)2), which correspond to the overall findings of broader signals for the latter. For the reduced sample, it converged to Gaussian linewidths of 3.0 eV (Ni2+) and 2.2 eV (Ni(OH)2) – which has to be considered a false solution, due to the resulting linewidths. The deconvolution with three components, Ni2+, Ni(OH)2 and Ni0, however, could not be calculated successfully. The accuracy of the peak fit, which relies on the data quality, may be argued, but the indirect indicator supports the findings from the XRD evaluation.
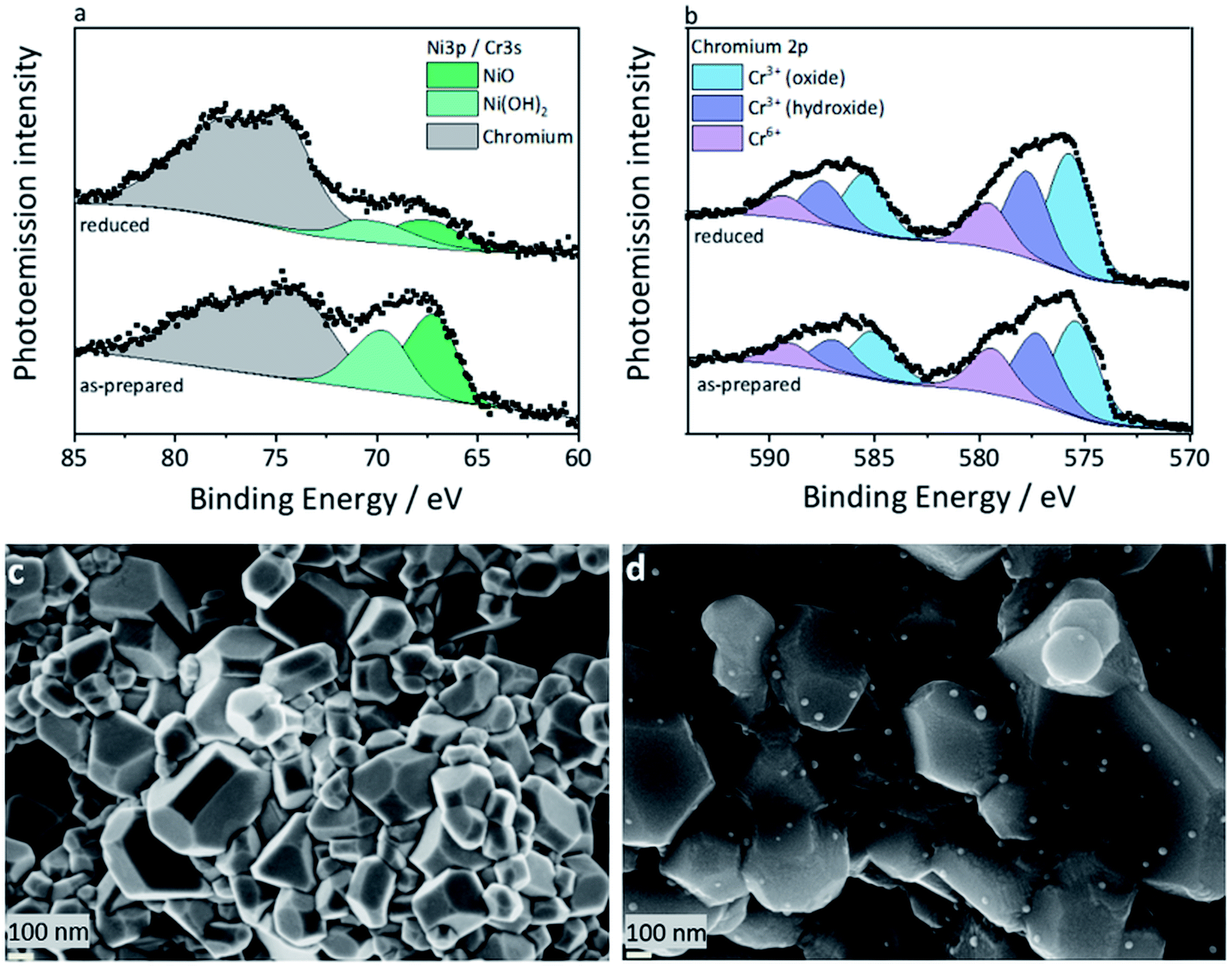 | ||
| Fig. 3 (a) Photoemission (XPS) spectra of the overlapping nickel 3p and chromium 3s region. The chromium signature was fed into the model based on the data obtained from more distinct chromium 2p signals shown in (b). The linewidth of the NiO peak fit of the reduced sample reveals that an additional (metallic) nickel component at ∼65 eV on the surface of the reduced sample may be hidden, but could not be resolved. (b) Photoemission spectra of the chromium 2p region. Possible Cr4+ cannot be distinguished from the predominant oxidic Cr3+. (c) SEM image of the as-prepared L65SCrN powder sample and (d) reduced L65SCrN powder sample (subjected to a thermal treatment with 5% H2–Ar at a ramp of 3 °C⋅min−1 from 25 °C to 1200 °C). Both (c) and (d) powder samples were analyzed by XRD and XPS. | ||
It has to be noted that the high surface sensitivity of this method might misguide the interpretation if not considered. The information depth, i.e. 3λ, where λ is the escape depth of the relevant electrons, is below 10 nm,37 which means that only the topmost surface of the crystallites is visible to this method. This surface sensitivity may also explain the presence of Ni(OH)2 that can easily form on top of the nickel exsolved surface particles.38
The nickel ratio was calculated with the Ni3p signal for the as-prepared and reduced samples. The concentration of nickel on the surface was 11.2 at% for the as-prepared sample and 3.8 at% for the reduced sample (Table 2). These results, which may seem surprising at first, could be explained by an agglomeration of surface nickel in nanoparticles upon reduction. Since their typical particle size (Fig. 3d) is larger than the information depth, that is less than 10 nm, they may not be fully probed by XPS. This is an important finding because this suggests that nickel on the surface of the L65SCrN sample changes its distribution, one can expect homogeneous on the surface of the as-prepared perovskite, to a more heterogeneous distribution where nickel is agglomerated and likely concentrated into nanoparticles during reduction. This indicates that the top surface of the perovskite after reduction tends to be depleted in nickel, yielding an overall reduction of the nickel concentration on the surface of the material analyzed by XPS.
| Surface species | As-prepared | Reduced |
|---|---|---|
| Cr3+ (oxide) | 10.7 at% | 14.2 at% |
| Cr3+ (hydroxide) | 7.8 at% | 10.3 at% |
| Cr6+ | 5.2 at% | 5.4 at% |
| Ni3p (total) | 11.2 at% | 3.8 at% |
The ratio between the surface states of chromium was determined by a peak fit of the Cr3p region. The separated components were identified, according to systematic studies by Biesinger et al.,39 to be Cr3+ (oxide) at ∼575.7 eV, Cr3+ (hydroxide) at ∼577.6 eV, and Cr6+ at 579.5 eV. A Cr4+ state, as a possible cause for the observed change in lattice parameters, has been discussed in earlier works on LSCr perovskites,40 but cannot be distinguished with this method due to the very close binding energies of Cr3+ and the anomalous Cr4+.41,42 Considering the inherent surface sensitivity of photoemission spectroscopy, the bulk properties of the investigated perovskite crystallites are not accessible anyway, and the surface states of chromium are not relevant to the lattice parameters. However, the surface stoichiometry (Table 2) of these chromium species can give indirect insight. The reduced L65SCrN sample shows an abundance of surface Cr3+ (both oxidic and hydroxide), which would be consistent with the mentioned lattice expansion observed by XRD (Fig. 2).
In order to better understand and highlight the role of the A-site deficiency in the Ni exsolution, TGA and TPR were performed in a reducing atmosphere (5% H2–Ar) on the as-prepared L70SCrN and L65SCrN powders. Since no other volatile species or compounds are expected to be formed during such thermal treatments, the net weight loss measurement by TGA is attributed to the net loss of oxygen, assuming that the oxygen from the perovskite lattice (and from the NiO secondary phase) reacted with hydrogen to form H2O.34 The TGA and TPR profiles are shown in Fig. 4a and b, respectively. In Fig. 4a, the maximum onset for weight loss, corresponding to the maximum value of the derivate (DTGMax), occurs at 442 °C for L65SCrN and at 472 °C for L70SCrN.
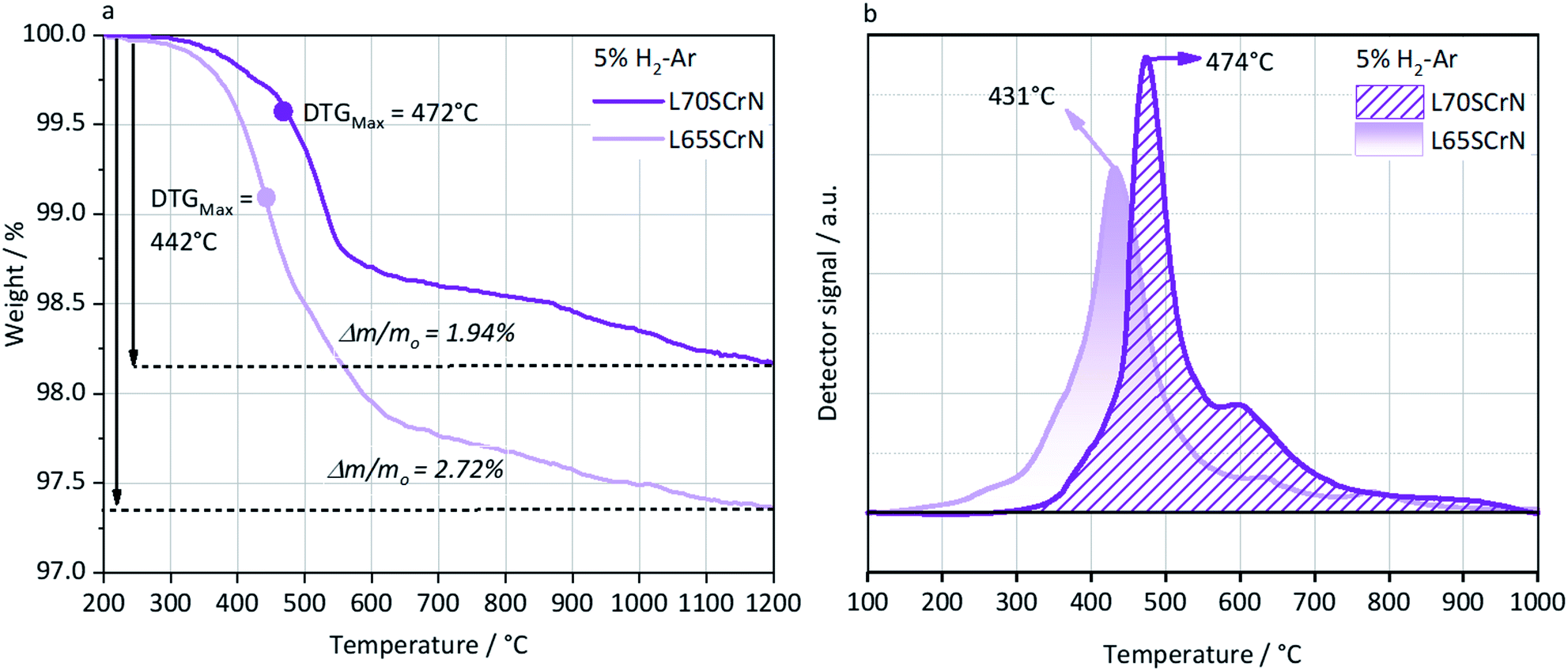 | ||
| Fig. 4 (a) TGA of L65SCrN and L70SCrN in 5% H2–Ar up to 1200 °C at 3 °C min−1. The maximum of the derivative function, i.e. where the mass loss kinetics is the fastest was calculated and is indicated by the DTGMax. (b) TPR of L65SCrN and L70SCrN in 5% H2–Ar up to 1000 °C at 5 °C min−1. | ||
The main peaks in the TPR profiles (Fig. 4b) occur at 431 °C and 474 °C for L65SCrN and L70SCrN, respectively. These values are in good agreement with the maxima of the TGA derivatives considering the difference of these measurements such as temperature increment. One should point out that these maxima are in the temperature range where phase transitions are expected to take place during the reduction of the perovskite.43 Along the temperature range of the TGA measurement (until 1200 °C), the total weight losses were 1.94 wt% for L70SCrN and 2.72 wt% for L65SCrN. Assuming that the NiO secondary phase (4.22 mol%) of the L65SCrN powder was fully reduced over the thermal treatment, it accounts for 0.04 wt% losses in the TGA experiment, yielding a net weight loss of 2.68 wt% for L65SCrN. Interestingly, both perovskites showed a continuous weight loss above 600 °C at a comparable rate, since the gap between the corresponding signals of the two phases remained at ∼0.78 wt%. For the specific case of L70SCrN, this evolution contrasts with the observation reported by Sun et al.44 This suggests a continuous evolution of the exsolution process upon temperature increase under reducing conditions. Considering SOC operation, this observation is of particular interest if temperature variations during operation are considered. From the TGA results, the oxygen deficiencies (δ), which are related to the oxygen vacancy concentration, were calculated using the method described by Myung et al.18 Upon reduction, an ABO3 perovskite loses oxygen turning to the ABO3−δ form with a corresponding change of mass from mABO3 to mABO3−δ. Since the quantity of perovskite moles is conserved, it is possible to assume the following relation:
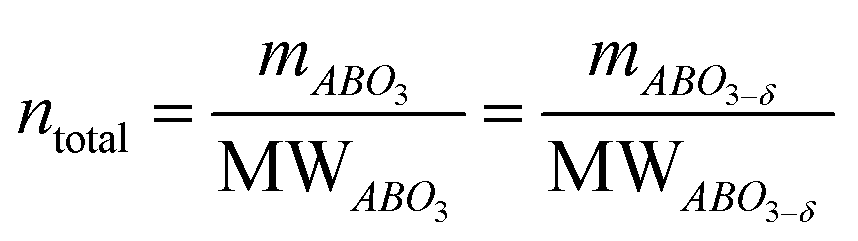 | (1) |
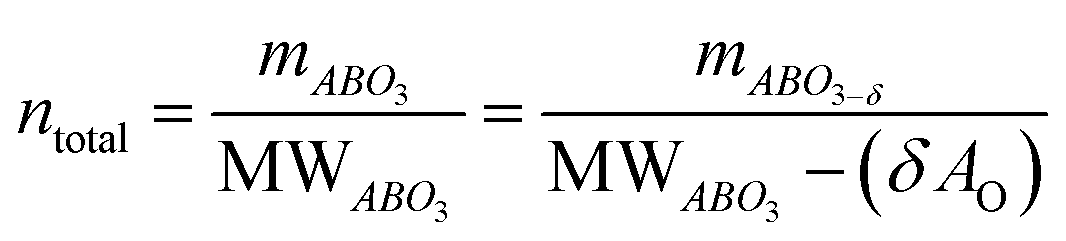 | (2) |
Knowing that the weight loss measured by TGA is given by:
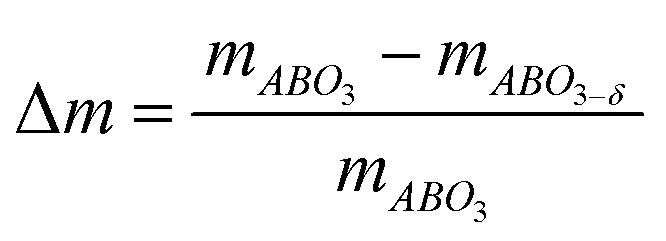 | (3) |
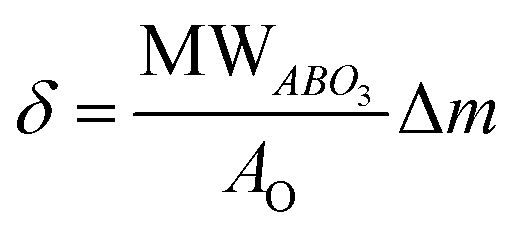 | (4) |
For the cases of L65SCrN and L70SCrN, i.e. with Ni cations on the B-site that can be reduced and forced out of the lattice, the reduction and exsolution processes of nickel will result in a consumption of the oxygen vacancies, so that it is not possible to relate directly net oxygen mass loss to oxygen deficiency. Nonetheless, though the initial oxygen stoichiometry was not determined in each of the compounds, a net specific oxygen consumption δO can be calculated in analogy with eqn (4). Therefore, by considering the net weight losses of 1.94 wt% and 2.68 wt%, the net oxygen consumption for each perovskite was calculated to be δO(L70SCrN) = 0.27 (mol O/mol L70SCrN) and δO(L65SCrN) = 0.36 (mol O/mol L65SCrN) for L70SCrN and L65SCrN, respectively. The significant difference in the net specific oxygen consumption upon reduction between L65SCrN and L70SCrN suggests that the A-site deficiency enhances the reducibility of the LSCrN, which seems to favor the exsolution of metallic Ni. To better understand the influence of the temperature and time on the formation of metallic nickel, the Ni nanoparticles' morphology was investigated in two isothermal reducing treatments: below and above 600 °C. These treatments were performed on L65SCrN since it showed superior reducibility and are explained and detailed in the following section.
Influence of the temperature on the Ni exsolution on L65SCrN
L65SCrN powder samples were exposed to pure hydrogen at different temperatures. For comparison, powder samples were annealed in hydrogen at either 500 °C or 900 °C for an annealing time of 3 hours. After reduction, the presence of the metallic Ni concomitant with the perovskite phase was confirmed in the XRD patterns (Fig. 5). However, the presence of impurity traces such as SrO2 and La2O3 could also be noted for the reduced samples and traces of Sr3(CrO4)2 for the as-prepared sample. SEM imaging of the two powder samples, i.e. L65SCrN reduced at 500 °C for 3 hours and L65SCrN reduced at 900 °C for 3 hours, revealed different morphologies (Fig. 6). For the L65SCrN sample reduced at 500 °C for 3 hours, one can observe spherical well-dispersed nanoparticles on the perovskite surface of a diameter of ∼8 nm (Fig. 6a, surface type 1) to ∼30 nm (Fig. 6a, surface type 2). The presence of the nanoparticles is correlated with a local enrichment in nickel which suggests that those nanoparticles are very likely made of metallic nickel. These observations are detailed in the ESI, in Fig. S2 and S3.† The corresponding EDS maps of Ni and Cr are shown in Fig. S4 and S5† respectively. Interestingly, it could be suggested that the exsolution of Ni nanoparticles on the surface of the perovskite grains may depend on the crystallographic orientation: surfaces denoted by type 1 appear to have qualitatively higher Ni nanoparticle density than type 2 surfaces (Fig. 6a). Though it was not possible to determine the specific crystallographic orientations of those surfaces, this strongly suggests that exsolution of nanoparticles is influenced by the surface characteristics of the perovskite grains. This is in agreement with the observations made on titanates by Neagu et al. as they found that during exsolution the particles remained socketed in the [110] crystallographic orientation with respect to the perovskite lattice, which is one of the key structural features that provides exsolved nanoparticles their stability.20 Such an orientation relationship is in accordance with previous reports, whereby the diffusion direction for B-site cations in perovskite lattices is along the [110] orientation.19,20,45 On the L65SCrN sample reduced at 900 °C for 3 hours coarser Ni nanoparticles of an irregular shape of ∼30 nm up to 100 nm could be observed (Fig. 6b). Such nanoparticle growth is consistent with the observations made in the corresponding XRD pattern (red pattern in Fig. 5) that reveals a more intense Ni characteristic peak. A closer inspection of the different surfaces could not reveal a variation in nanoparticle density, which appears qualitatively lower than the one on the sample reduced at 500 °C. This lower particle density on the surface of the perovskite and their coarser particle size suggest a growth mechanism of the Ni nanoparticles that takes place upon temperature increase. | ||
| Fig. 5 XRD patterns of the as-prepared L65SCrN powders (green pattern) reduced in H2 at 500 °C for 3 hours (blue pattern) and reduced in H2 at 900 °C for 3 hours (red pattern). The host chromite perovskite phase was identified with the PDF #01-076-7024 for all three patterns. | ||
 | ||
| Fig. 6 SEM images of Ni exsolution after 3 hour treatment under a H2 atmosphere at (a) 500 °C and (b) 900 °C. | ||
This is in good agreement with the observation made by Jo et al. on Co exsolved nanoparticles on strontium titanates, where the average grain size increased while the Co nanoparticle density decreased.46
Comparable observation of Ni coarsening on chromites was reported by Sauvet et al. after methane reforming experiments on A-site stoichiometric La0.70Sr0.3Cr0.95Ni0.05O3−δ between 750–850 °C.47 Kobsiriphat et al. attempted to explain Ni and ruthenium nucleation in the chromites La0.8Sr0.2Cr0.69Ni0.31O3−δ and La0.8Sr0.2Cr0.82Ru0.18O3−δ under reduction in dry hydrogen at 800 °C for different times.48 They observed that the Ni nanoparticles had coarsened significantly from a particle size of ∼10–15 nm (after 3 hours) to an average hemisphere diameter of 50–60 nm after 311 hours of reduction. In contrast, for the Ru-doped chromite after 311 hours of reduction at 800 °C, there was no significant change in the Ru nanoparticles since their size did not exceed 10 nm. From these observations and by analogy to thin-film nucleation, they concluded that particle coarsening may be due to a fast surface diffusion, which would allow nuclei to be fed by adatoms yielding larger and more widely spaced nuclei.48 Therefore, they suggested that the faster Ni particle coarsening was likely explained by larger Ni surface diffusivities in comparison to Ru on the chromite, although quantitative data on chromites are not available.48 This would be consistent with the above-mentioned observations made on the nickel concentration determined by XPS on the surface of the as-prepared and reduced L65SCrN samples. Such high surface diffusivity was highlighted by Sakai et al., who investigated the chromium diffusion in lanthanum chromites between ∼700–1400 °C by 50Cr tracer diffusion and secondary ion mass spectrometry (SIMS).49 They estimated that independently of the temperature, the grain boundary diffusion coefficient was 105 times larger than the bulk diffusion coefficient.49 Interestingly, this behavior has also been observed in other perovskite families, such as strontium titanates SrTi0.75Co0.25O3−δ, where exsolved Co particles diffuse onto the existing Co nanoparticles rather than nucleating in new locations at the grain boundaries, due to the increment of Co diffusivity at high temperatures (above 700 °C). In this case, the distances between the particles previously nucleated are assumed to be shortened.46 Another interesting observation was made by Kousi et al. on La0.7Ce0.1Co0.3Ni0.1Ti0.6 O3−δ (LCCNT) where they identified different sizes on the exsolved Ni–Co nanoparticles in the bulk as compared to the ones exsolved on the surface: the bulk particles were smaller (∼10 nm) than the ones exsolved on the surface ∼40 nm, noting as well that on the bulk the nanoparticle population was significantly higher. These conjectures were made based on a SEM cross-section evaluation, where it is possible to identify the surface and the bulk.50
Moreover, it is possible to correlate the differences in the observed Ni exsolution morphologies at 500 °C and 900 °C upon reduction (Fig. 6) with the formation of oxygen vacancies, since the exsolution is favored when these vacancies reach a high concentration.19 The formation of oxygen vacancies upon reduction may take place either in a surface site or in a bulk site.51 It has been claimed that there is a strong correlation between the bulk and the surface kinetics, which indicates that not only the oxygen vacancy concentration but also pO2 plays a significant role in the surface exchange processes in mixed ionic and electronic conductors (MIEC) such as chromites.52
Another approach was made by Gao et al. who investigated Ni exsolution phenomena on a Sc-based A-site deficient perovskite La0.4Sr0.4Sc0.9Ni0.1O3−δ (LSSN) at different temperatures and annealing times. They proposed that Ni exsolution could be considered as a chemically driven heterogeneous phase transformation, being a consequence of four physical processes: diffusion, reduction, nucleation and growth. They found that the nucleation is affected by: mechanical stresses, related strains on the perovskite lattice, metallic Ni wetting angles, and A-site and oxygen vacancies. These factors significantly determine where the nucleation would take place. Moreover, parameters such as the atmosphere (e.g. pO2 and pH2), annealing time and temperature may affect the particle growth.53
In the case of A-site dopant diffusion, a study carried out on manganite-based perovskite oxides, LnMnO3, demonstrated that the surface oxygen vacancy attracts the dopant (that partially substituted the host on the A-site) driving it to the surface on the host sublattice.33 It has been assessed by DFT (density functional theory) that the elastic and electrostatic interactions of the dopant with the surrounding lattice are driving forces for dopant segregation on perovskite compounds. The factors that may affect these driving forces are the dopant size, the lattice parameter, and the distribution of charged vacancies.33 However, the diffusion phenomenon from the segregating cations should be carefully studied since the surface composition depends both on thermodynamics and kinetics,33 which could be also the case for the B-site dopant diffusion in other perovskite systems.
Regarding L65SCrN in the present study, it is unclear how the Ni enrichment on the surface originating from the bulk may occur. If we consider the assumption by Gao et al. that the mass transport process during the Ni exsolution is critical for the particle growth, it is likely that such growth results from the Ni2+ ion diffusion followed by the reduction reaction to metallic Ni, which may be limited by two possible models: mechanical energy effects (as a function of the strain activation energy) or limited Ni supply.53 They found that the Ni particles preferably nucleate on the surface rather than in the bulk due to their tendency to decrease the strain activation energy.53 Using DFT calculations, they concluded that these models are likely to represent the actual Ni exsolution mechanism on LSSN. This contrasts with the observations reported on lanthanum titanate-based fuel electrodes for which the particle – substrate interaction prevails and thus stabilizes the Ni exsolved nanoparticles on the surface in the temperature range between ∼650–900 °C in reducing atmospheres.20 For instance, Neagu et al. observed directly the Ni exsolution process by ETEM on two different compositions with widely different content of exsolvable Ni: La0.43Ca0.37Ti0.94Ni0.06O3 and La0.8Ce0.1Ti0.6Ni0.4O3. They found that the location of the particle-socket did not change during the timescale of the experiment, indicating that the nanoparticle was locked/socketed in place once it was formed (exsolved).20 More precisely, they found during the experiment that additional particles formed within the nanoscale proximity of the first ones but neither of them moved nor drifted under the environmental transmission microscope (ETEM) electron beam. This revealed that particle–support interactions are strongly dominant over particle–particle interactions for titanates.20 Contrary to the case of lanthanum chromites, particle–particle interaction prevails at high temperatures.
Although lanthanum titanates do not seem to exhibit the same behavior as strontium titanates, lanthanum scandates or lanthanum chromites towards Ni particle growth, recent studies highlighted that for perovskite oxide-based electrocatalysts, there is a strong influence of the gas atmosphere, i.e. pO2 in correlation with the temperature, on the shape of the exsolved nanoparticles,20,25 which is susceptible to affect their electrocatalytic performance.
In this study, the reduction of L65SCrN at low temperatures (∼500 °C) yielded the finest and well-dispersed Ni exsolved nanoparticles. Such operating conditions are still far below from the typical operating temperatures (T > 800 °C) of ESC in SOC applications. Since it is shown that Ni particle–particle interactions prevail at high temperature in chromites, yielding particle coarsening, it is an important aspect to consider for the implementation of L65SCrN as a fuel electrode into a SOC stack because it may affect the morphological stability of the reactive surfaces. Possibly, the Ni exsolved particle size could be optimized with a rigorous investigation on the Ni exsolution phenomena in these A-site deficient chromites. Synthesis and processing parameters may also play a significant role: porous structures may be one key factor for Ni exsolution.53 For a better understanding, additional investigation of the Ni concentration profiles across the perovskite grains upon reduction as well as the Ni particle size evolution as a function of temperature, annealing time, gas atmosphere, grain size and porosity would be necessary, e.g. by TEM and TOF-SIMS, accompanied by DFT modeling.33,46,53
Therefore, it is questionable how stable a L65SCrN electrocatalyst may perform at the stack level since pO2 gradients are usually observed along the gas channels: for instance, high pH2 at the inlet and high pH2O at the outlet are characteristic of FC operation, while in EC mode high pH2O at the inlet and high pH2 at the outlet are typical (assuming high fuel and steam utilizations for both modes). Moreover, reversible operation (FC-EC) would expose the electrocatalyst alternatively to pO2 gradients.
In the following section, we focus on the electrochemical performance of cells with the L65SCrN fuel electrode upon variation of the operating conditions.
SOC electrochemical performance with the L65SCrN electrocatalyst as a fuel electrode
The electrochemical performance of a full cell with an ESC architecture and a L65SCrN fuel electrode has been evaluated in FC, FC-EC, EC and co-EC modes as described in Table 1.Performance evaluation in FC, FC-EC and EC operation
The measured open circuit voltage (OCV) was 1.25 V at 900 °C with pure H2/air, demonstrating appropriate gas tightness of the sealing. For the three different operating modes, the measured OCV was slightly above (∼20 mV) the theoretical Nernst potential E:54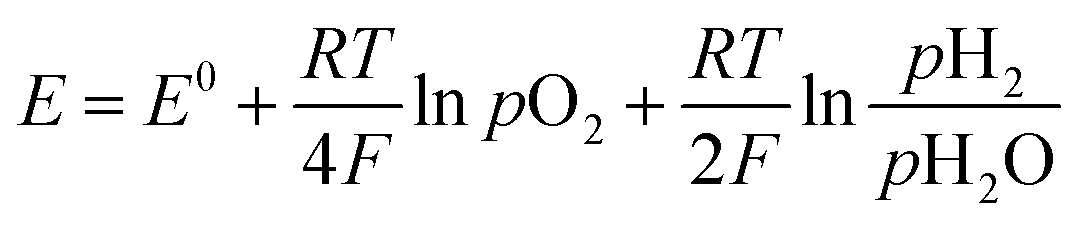 | (5) |
Such a difference was assigned to a deviation of the inlet gas composition due to inaccuracy of the steam supply mass flow control.
Polarization curves at 860 °C in fuel cell (FC), reversible (FC-EC) and electrolysis (EC) operation are shown in Fig. 7a. In FC mode, i.e. with a 90% H2–10% H2O fuel gas mixture, the oscillations observed find their origin in pH2O fluctuations from the steam supply due to marginal operation. In reversible mode FC-EC, i.e. with a 50% H2–50% H2O fuel gas mixture, the I–V characteristics evolve continuously from either side of the OCV which reflects the reversible functionality of the full cell and thus the L65SCrN electrocatalyst in either mode.
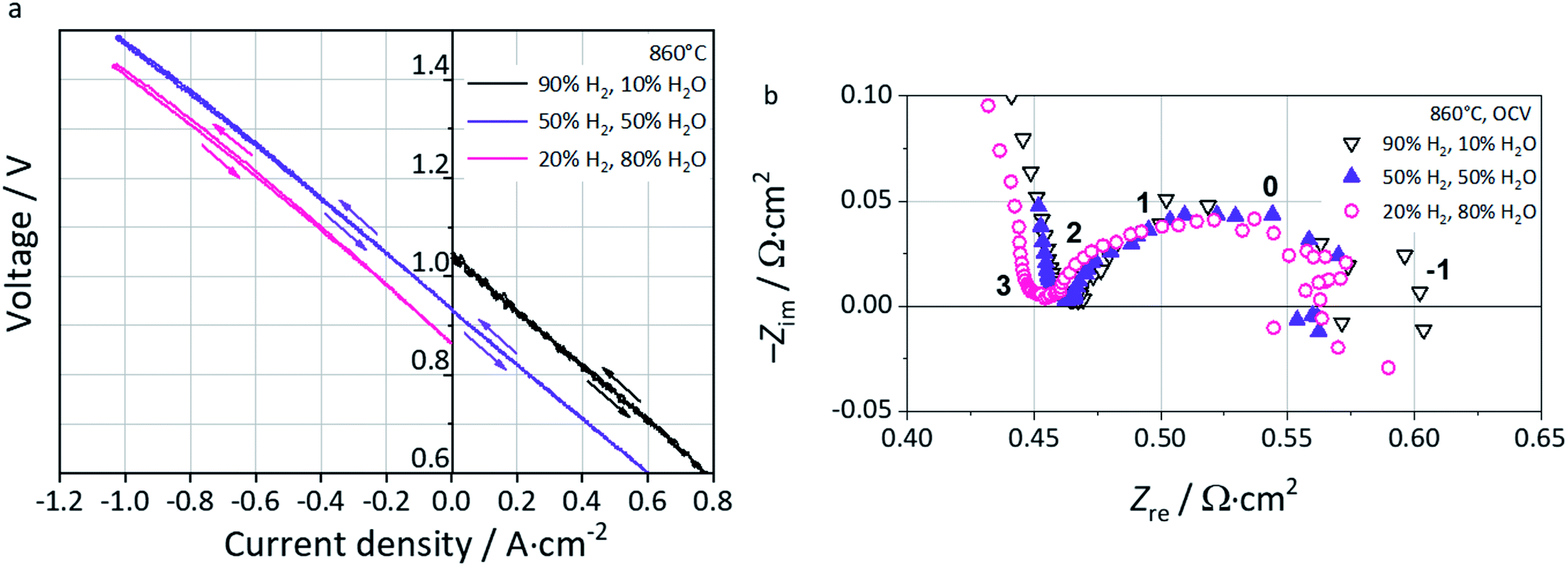 | ||
| Fig. 7 (a) Polarization curves at 860 °C in FC, FC-EC and EC modes. (b) Nyquist plot at OCV for fuel gas compositions 90% H2–10% H2O (black), 50% H2–50% H2O (purple) and 20% H2–80% H2O (pink) at 860 °C with indication of frequency decades. | ||
In EC mode, with a 20% H2–80% H2O fuel gas mixture, the I–V characteristics show a nearly linear evolution from OCV until an inflection point which corresponds to the thermoneutral voltage of steam electrolysis (∼1.29 V).3 Above this value, the slope of the polarization curve decreases yielding a curve flattening. This is explained by the exothermal nature of the steam electrolysis at higher cell voltages, causing a net heat production. Since the oven of the test bench was operated isothermally, this heating effect cannot be controlled at the cell level causing a net increment of the temperature and enhancing the electrode reaction kinetics.3
EIS data recorded near OCV conditions are shown in the Nyquist Plot of Fig. 7b. The ohmic resistance Rohm is estimated to be 0.47 Ω cm2 with 10% H2O, 0.46 Ω cm2 with 50% H2O, and 0.45 Ω cm2 with 80% H2O, suggesting a sensitivity of this parameter to pH2 and thus pO2 in the feed gas. As a p-type conductor, this increment of Rohm upon increase of pH2 could be explained by a decrease of the conductivity in L65SCrN, since the positively charged oxygen vacancies that are created upon reduction hinder the transportation of electrical holes,55 and decrease the effect of the alkaline earth doping on the electrical conductivity of lanthanum chromites.56 However, it is important to note that the ohmic resistance values are comparable with commercial ESC references, since they are slightly lower than that for a state-of-the-art Ni-CGO fuel electrode tested also at 860 °C with an estimated value of 0.55 Ω cm2.3
At low frequencies, i.e. below 1 Hz, the EIS data were scattered due to the small voltage variations induced by fluctuations in the steam supply, which made difficult to determine accurately the total area specific resistance from these spectra. Therefore, the total area specific resistance (ASRDC_Total) was calculated as the slope from the polarization curves at ±0.3 A cm−2 (linear range) where the influences of gas conversion and concentration polarization are expected to be minimal.
These values correspond to 0.58 Ω cm2 (FC-10% H2O), 0.54 Ω cm2 (FC-50% H2O), 0.56 Ω cm2 (EC-50% H2O) and 0.57 Ω cm2 (EC-80% H2O). Interestingly, the measured values are in the same order of magnitude than the ones reported by Schefold et al. on a commercial ESC: ASRDC_Total = 0.5 Ω cm2, which consisted of an electrolyte of 3YSZ, a LSCF air electrode, screen-printed CGO layers as the diffusion barrier between the electrolyte and electrodes and a Ni-CGO fuel electrode operated in steam electrolysis (EC-75% H2O) at 850 °C.57
Performance evaluation in co-EC operation
Electrochemical characterization of this cell with the L65SCrN fuel electrode was performed in co-electrolysis mode at 860 °C (co-EC) with the fuel gas composition 5% H2, 63.7% H2O and 31.3% CO2.ASRDC_Total at −0.3 A cm−2 was calculated from the polarization curve (Fig. 8a) to be 0.676 Ω cm2. This value lies in the same order of magnitude (being lower) as the one reported for an ESC reference with a Ni-CGO fuel electrode tested in co-electrolysis (25% H2, 25% H2O, 25% CO2, 25% CO) at 830 °C which showed an ASRDC_Total of 0.84 Ω cm2 at −0.3 A cm−2.58
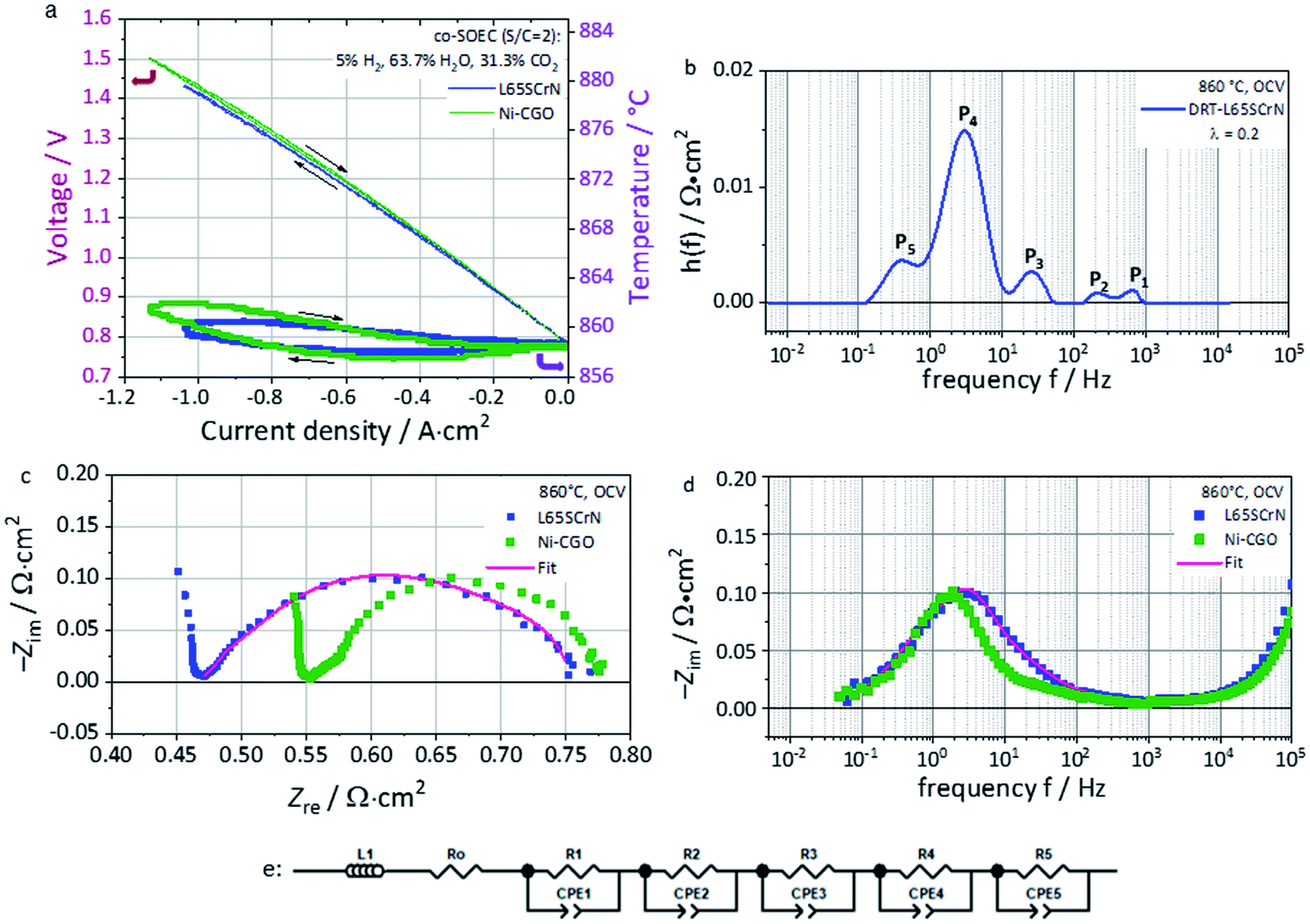 | ||
| Fig. 8 (a) Polarization curves in co-electrolysis mode (co-EC) with the fuel gas composition of 5% H2, 63.7% H2O and 31.3% CO2 at 860 °C of the L65SCrN fuel electrode ESC (blue pattern) and state-of-the-art Ni-CGO fuel electrode ESC (green pattern). (b) DRT calculation of the L65SCrN impedance spectra using (c) the Nyquist plot and (d) the Imaginary impedance plot at OCV at 860 °C with fuel gas composition 5% H2, 63.7% H2O and 31.3% CO2. (e) ECM proposed with the fitting on the L65SCrN impedance spectra. | ||
 | ||
| Fig. 9 Cross-sectional view of the ESC implementing the L65SCrN electrocatalyst as the fuel electrode after rSOC operation at 860 °C. | ||
Even though these conditions are not directly comparable, this first approach suggests promising performance of the L65SCrN electrocatalyst as a fuel electrode. The polarization curve shows a linear evolution from OCV until the thermoneutral voltage of co-electrolysis that is determined under the tested conditions to ∼1.32 V lying between the steam electrolysis value (1.29 V) and CO2 electrolysis (1.46 V).3,59 Above this point, the curve flattens due to the decrement in the ASRDC_Total value, probably enhanced by the temperature increment in this exothermal regime.3
From the EIS spectra recorded at OCV (Fig. 8c and d), a Rohm of 0.47 Ω cm2 was identified for L65SCrN (blue pattern). Furthermore, the polarization resistance ASRAC_Pol was estimated to be 0.29 Ω cm2, which can be directly compared with the estimation by Dueñas et al. on an ESC with the Ni-CGO fuel electrode under the same operating conditions (green pattern): 0.23 Ω cm2.3
Regarding the electrode polarization processes, they are dominated by one main contribution which suggests a convolution of the electrode losses with the gas losses (conversion and diffusion processes).
A calculation of the distribution of relaxation times (DRT) was performed with the aim of identifying relevant processes within the L65SCrN fuel electrode-cell. Five contributions or processes could be identified (Fig. 8b), which allowed proposing the equivalent circuit model (ECM) shown in Fig. 8e, with good fitting of the EIS spectra in Fig. 8c and d (χ2 = 1.55 × 10−4). The proposed ECM comprised an ohmic resistance Ro of 4.69 × 10−1 Ω cm2 to model the ohmic losses and a series connection of five RQ-elements, where Q represents a constant-phase element (CPE).
Five different physical and electrochemical processes were identified as shown in Table 3, where the resulting peak frequency (fDRT_peak) and the polarization resistance values (area under the peaks) from the DRT analysis were used to calculate the capacitance of an ideal RC element from the general eqn (6):60
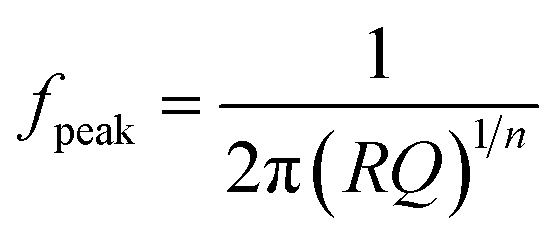 | (6) |
| Peak # | Cell component | Physical/electrochemical origin | ECM elements65,66 | Characteristic frequency |
|---|---|---|---|---|
| 1 | Oxygen electrode | LSCF/CGO interfacial double-layer capacitance61 | R 1 = 4.23 × 10−3 Ω cm2, Q1 = 6.09 × 10−2 s0.97/Ω cm2, n = 0.97 | ∼7.96 × 102 Hz |
| 2 | LSCF charge transfer61 | R 2 = 6.25 × 10−3 Ω cm2, Q2 = 1.28 × 10−1 F cm−2, n = 1 | ∼1.99 × 102 Hz | |
| 3 and 4 | Fuel electrode | L65SCrN fuel electrode electrochemical reaction | R 3 = 3.32 × 10−2 Ω cm2, Q3 = 2.06 × 10−1 s0.99/Ω cm2, n = 0.99 | ∼2.45 × 101 Hz |
| R 4 = 1.71 × 10−1 Ω cm2,Q4 = 3.00 × 10−1 F cm−2, n = 1 | ∼3.11 Hz | |||
| 5 | Fuel electrode (+oxygen electrode) | Gas diffusion (perpendicular to cell) and gas conversion (parallel to the cell in co-flow configuration) including contact mesh and flow field | R 5 = 6.92 × 10−2 Ω cm2, Q5 = 3.69 F cm−2, n = 1 | ∼3.70 × 10−1 to 6.23 × 10−1 Hz |
For the case of the LSCF charge transfer process (Peak#2) and the LSCF/CGO interfacial double-layer capacitance (Peak#1), characteristic frequencies were identified with comparable values as the ones reported by Yurkiv et al.61 For the electrochemical process on the L65SCrN fuel electrode, there is a lack of reported EIS data, which makes difficult the precise identification of the processes.
Nonetheless, since no additional high frequency processes are visible in the EIS spectra it is suspected that the L65SCrN charge transfer process is correlated with large chemical capacitance linked to its oxygen non-stoichiometry which overlaps with the gas conversion process. For this impedance feature, the DRT analysis suggests two processes (Peak#3 and Peak#4) at a frequency between ∼3–25 Hz that, by analogy with the behavior of MIEC materials reported by Adler et al.,62 are likely to be connected to each other and be characteristic of the response of the L65SCrN. A last process (Peak#5) at low frequencies between 0.1 Hz and 1 Hz was attributed to the gas losses where there is an overlapping of the RWGS reaction (catalytic conversion),63 the diffusion in the electrodes and the gas conversion processes (within the gas channels). At frequencies above 103 Hz, it was not possible to fit the EIS spectrum due to an artifact of the measurement.
Overall, this corresponds to a first approach to model and understand the electrochemical behavior of an ESC with a L65SCrN fuel electrode in co-electrolysis. However, this would need to be confirmed and further investigated to understand in detail the different electrochemical processes that take place within the cell and does not preclude other approach to better reflect the behavior of the electrode materials. Especially, with the proposed model, if two connected processes are reasonable to consider for the impedance feature of L65SCrN, a parametric study varying temperature, current density and gas compositions of the fuel electrode with DRT studies will be needed in order to understand the response of the L65SCrN fuel electrode and make a clear process assignment.64 After the above described electrochemical tests were performed, the tested ESC was observed by SEM (Fig. 9), where no delamination or mismatch at none of the interfaces platinum current collector-L65SCrN, L65SCrN-CGO20, CGO20-3YSZ and CGO20-LSCF could be observed on the polished cross section, which suggests good thermo-mechanical compatibility between the different cell components. Elemental analysis performed by EDS revealed the presence of silicon, likely in the form of SiO2 species, within the CGO20 barrier layer (Fig. S6 and Table S1 in the ESI†). Therefore, long-term evaluation in co-electrolysis with ultra-pure feed water was performed with a new cell in order to exclude any exogenous sources of degradation and clearly assess the stability of the L65SCrN fuel electrode in operation in view of SOC applications.
Evaluation of the long-term stability in co-EC operation
Long-term steam co-electrolysis was carried out for 950 hours under galvanostatic conditions at a fixed current density of −0.46 A cm−2 at 860 °C with an initial voltage of ∼1.3 V (Fig. 10a). EIS measurements at OCV (Fig. 10c and d) were performed in order to monitor the evolution upon operation. Compared to the previous cell (Fig. 8), a higher Rohm was measured likely due to a contact issue between the electrodes and the current collectors for this long-term test. However, the polarization resistance (Rpol), for testing times above 677 hours, but possible already from the first 100 hours, coincides with the one reported in Fig. 8c, being ca. ∼0.3 Ω cm2 for both cases. The same accounts with the imaginary part of the impedance ZIm from Fig. 8d and 10d, which are in the same order of magnitude, i.e. close to −0.1 Ω cm2 in both cases.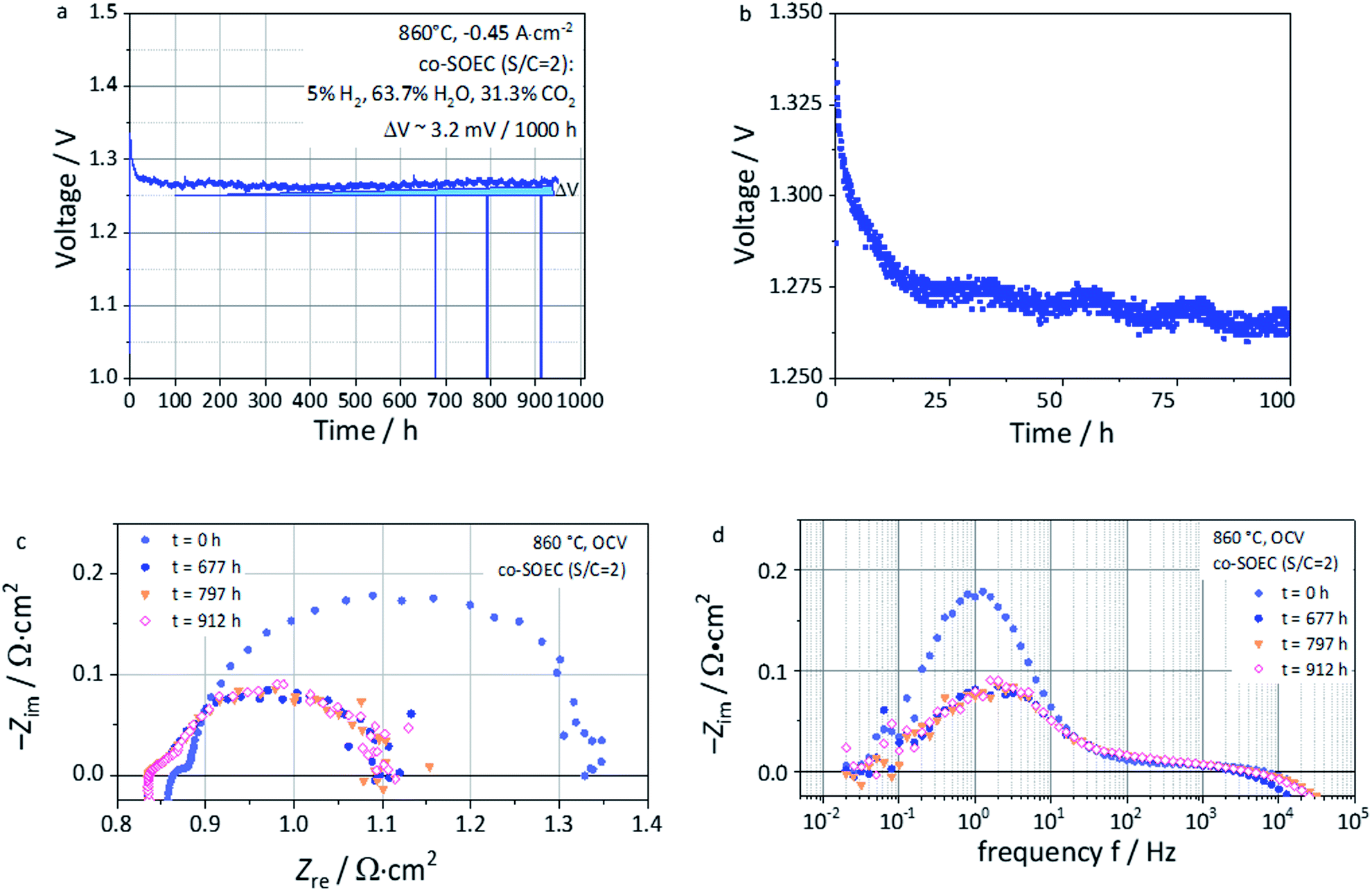 | ||
| Fig. 10 (a) Voltage vs. time during long-term co-electrolysis (co-EC) with the fuel gas composition of 5% H2, 63.7% H2O and 31.3% CO2 at 860 °C and −0.46 A cm−2 of the L65SCrN fuel electrode ESC (b) Zoom of (a) until 100 hours of operation. (c) Nyquist plot and (d) imaginary impedance plot at OCV and 860 °C during long-term study (951 hours). | ||
The first 100 hours of the test are marked by a decreasing voltage of the cell, meaning an improvement of the cell performance (Fig. 10a and b). This improvement is expressed by a decrease of both Rohm and Rpol over time, which could be due to the following factors:
(i) the oxygen electrode needed ca. 100 hours to be correctly contacted, due possibly to the gold mesh current collector stabilization.
(ii) A removal of impurities that are present in a trace amount in CO2 that may have been adsorbed on the surface of the perovskite and poisoned the electrode during the 24 hours of cell operation at OCV, before the start of the galvanostatic durability test.
(iii) An activation process of the L65SCrN fuel electrode to accommodate the defect chemistry under the testing conditions (∼100 hours) during which one can speculate further nickel exsolution to occur.
This last feature presents some analogy with the phenomenon reported by Neagu et al. on titanates,20 who reported such evolution in a few minutes time-frame and not in several hours as for this study. As this behavior of titanates was reported in pure hydrogen, it is reasonable to consider the gas composition to play a significant role in this observation. After the first 100 hours of operation, the voltage evolved nearly linearly. Cyclic fluctuations of the water supply resulted in voltage oscillation of about ∼1–2 mV. The corresponding voltage increment due to the degradation could be estimated as 3.2 mV/1000 h. This is about ∼1.6 times lower than the value reported by Schefold et al., on an ESC cell with a Ni-CGO cermet fuel electrode operated in steam electrolysis (EC-75% H2O) at 850 °C and −0.7 A cm−2 who measured 5.1 mV/1000 h.57 Despite the difference under operating conditions and the lower applied current density, such a low voltage degradation rate reported for an ESC with a L65SCrN fuel electrode is promising. After cooling, SEM investigation of the non-polished surface of the L65SCrN fuel electrode after 950 hours of co-electrolysis operation was performed (Fig. 11). As expected, the contamination of the electrode by silicon was negligible, suggesting a minimal impact on the transport properties of the L65SCrN fuel electrode (EDS analysis shown in the ESI in Fig. S7 and Table S2†). Investigation of the surface of the L65SCrN perovskite grains revealed the presence of Ni exsolved nanoparticles well distributed over the whole area of the analyzed sample. This is illustrated in a representative manner in Fig. 11 and S8.† The average size of those exsolved nanoparticles was estimated in the range of 10 to 25 nm on this sample, which is significantly lower than the one estimated on the powder sample at 900 °C in pure hydrogen (Fig. 6b). Considering the 950 hour operating time, this observation suggests that particle coarsening was limited and did not occur under the testing conditions. This is consistent with observations reported where the size of the exsolved nanoparticle increases until a critical stable radius, as a function of the temperature and time.46,53 Knowing that the oxygen partial pressure pO2 was about ∼10−24 bar in the case of powder sample reduction at 900 °C,67 and only ∼10−15 bar in this case of long-term co-electrolysis,30 it seems that pO2 plays the most important role in the morphology of the exsolved nanoparticles. The relative predominance of the parameters that affect the Ni exsolution morphology could be qualitatively ranked as follows: pO2 > temperature > time. Since electrolysis operation implies reducing conditions to the fuel electrode, it is nonetheless reasonable to expect that the applied current density and thus the overpotential at the L65SCrN fuel electrode is going to have an additional influence on the exsolved nickel nanoparticles. Since A-site deficiency and exsolution are intimately correlated, it is difficult to decorrelate the impact of the two phenomena on the cell performance. Working with an A-site deficient perovskite is of high interest with perspective of industrialization since it enables to control the impurities (as the B-site elements) in the produced materials. ESCs are intrinsically characterized by a high operating temperature to counter-balance the ohmic losses induced by the thickness of the electrolyte. This feature limits the impact of the electrocatalysis on the overall cell performance. However, given the contingencies of the ESC cell configuration, we believe that the observed behavior and performance is to a large extent due to the exsolution of nickel.
 | ||
| Fig. 11 (a) and (b) SEM images with Ni nanoparticles after 950 hour co-electrolysis operation (co-EC) with the fuel gas composition of 5% H2, 63.7% H2O and 31.3% CO2 at 860 °C and −0.46 A cm−2 of the L65SCrN fuel electrode ESC. | ||
Conclusions
Lanthanum strontium chromite perovskites with partial nickel substitution were investigated for the sake of developing an alternative electrocatalyst to traditional cermets as the fuel electrode for SOC applications. These materials have been synthesized and their propensity to exsolve Ni nanoparticles under exposure to a reducing atmosphere has been investigated ex situ. Introduction of a deficiency up to 5% on the A-site of the perovskite was shown to be effective to enhance the exsolution capability of the synthesized material (L65SCrN), compared to a full stoichiometric perovskite (L70SCrN).The density and particle shape of the exsolved nanoparticles on the surface of the perovskite were shown to be sensitive to the crystallographic orientations of the surfaces and pO2. This behavior is consistent with the observations made on other families of perovskites with exsolution of nanoparticles such as titanates. However, in contrast to the titanates, the evolution in the shape, size and coverage ratio of the Ni nanoparticles, upon temperature increase characterized by a particle coarsening, suggests that particle–particle interactions prevail over particle–substrate interactions on the surface of lanthanum chromites. This significant morphological change of the nanoparticles under operating conditions could affect their catalytic activities over time and thus impact the overall performance and durability of an electrode made of these materials.
Tested on ESCs and with optimal contacting solutions, L65SCrN electrodes demonstrated performance levels that are comparable with the ones of state-of-the-art cermet fuel electrodes in fuel cell, electrolysis and co-electrolysis operation: representative conditions for a reversible SOC system. Excellent voltage stability was reported in co-electrolysis operation over 950 hours with a voltage degradation of about 3.2 mV/1000 hours. Qualitatively, it is suggested that pO2 is the main factor governing the particle size followed by the temperature and then time. This suggests that the nanoparticles can be dimensionally stable when the system is operated isothermally, or when exsolution takes place at a temperature higher than the nominal operating temperature of the cell. Therefore, considering a SOC stack implementing cells with L65SCrN fuel electrodes, one can reasonably expect that the exsolution would take place during the commissioning of the stack, yielding coarsened Ni nanoparticles that are dimensionally stable during operation, fulfilling the durability requirements.
However, as a disadvantage the high temperature thermal treatment that is usually performed for stack commissioning would yield coarsened nanoparticles that may impede further electrode performance optimization. One aspect to consider and being advantageous would be to maintain the exsolved nanoparticles as fine as possible to optimally improve the performance of L65SCrN electrodes by tuning for instance pO2.
Additional investigation of the exsolution phenomena by varying parameters such as temperature, time and pO2 would be thus needed in order to better understand the mechanisms of exsolution. Another important aspect to evaluate is how the Ni nanoparticle size impacts the performance of the electrode and how to fine tune the exsolution parameters for maximizing electrocatalytic activity. This would enable improving the presented L65SCrN fuel electrode and developing a durable electrode morphology for rSOC applications.
Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 5th February 2021, in which there was an error in a value of oxygen partial pressure quoted in the text. This has now been corrected, and the Royal Society of Chemistry apologises for this error.Acknowledgements
We would like to thank the University of Bayreuth, Chair for Electrical Energy Systems, for providing the impedance analysis and modelling software ec-idea (https://www.ec-idea.uni-bayreuth.de). Also, the German Academic Exchange Service (DAAD) is acknowledged for the PhD scholarship of Diana-María Amaya-Dueñas with the award DLR/DAAD Research Fellowships – Doctoral Studies, 2017.References
- E. V. Kondratenko and U. Rodemerck, in Perovskites and Related Mixed Oxides: Concepts and Applications, ed. P. Granger, V. I. Parvulescu, S. Kaliaguine and W. Preiller, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 1st edn, 2016, pp. 517–537, Recent Progress in Oxidative Conversion of Methane to Value-Added Products Search PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 350 CrossRef.
- D. M. Amaya Dueñas, M. Riedel, M. Riegraf, R. Costa and K. A. Friedrich, Chem. Ing. Tech., 2020, 92, 45–52 CrossRef.
- S. Santhanam, M. P. Heddrich, M. Riedel and K. A. Friedrich, Energy, 2017, 141, 202–214 CrossRef.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- G. Kasiraman, B. Nagalingam and M. Balakrishnan, Energy, 2012, 47, 116–124 CrossRef CAS.
- M. Riegraf, M. P. Hoerlein, R. Costa, G. Schiller and K. A. Friedrich, ACS Catal., 2017, 7, 7760–7771 CrossRef CAS.
- M. Riegraf, A. Zekri, M. Knipper, R. Costa, G. Schiller and K. A. Friedrich, J. Power Sources, 2018, 380, 26–36 CrossRef CAS.
- R. Costa, F. Han, P. Szabo, V. Yurkiv, R. Semerad, S. K. Cheah and L. Dessemond, Fuel Cells, 2018, 18, 251–259 CrossRef CAS.
- T. L. Skafte, J. Hjelm, P. Blennow and C. R. Graves, in Proceedings of 12th European SOFC & SOE Forum 2016, ed. European Fuel Cell Forum, Lucerne, 2016, pp. 8–27, Quantitative review of degradation and lifetime of solid oxide cells and stacks Search PubMed.
- M. P. Hoerlein, M. Riegraf, R. Costa, G. Schiller and K. A. Friedrich, Electrochim. Acta, 2018, 276, 162–175 CrossRef CAS.
- Y. Tao, S. D. Ebbesen and M. B. Mogensen, J. Electrochem. Soc., 2014, 161, F337–F343 CrossRef CAS.
- V. Duboviks, R. C. Maher, M. Kishimoto, L. F. Cohen, N. P. Brandon and G. J. Offer, Phys. Chem. Chem. Phys., 2014, 16, 13063–13068 RSC.
- A. Faes, A. Hessler-Wyser, A. Zryd and J. V. Herle, Membr, 2012, 2, 585–664 CrossRef CAS.
- H. Zhu, P. Zhang and S. Dai, ACS Catal., 2015, 5, 6370–6385 CrossRef CAS.
- G. Tsekouras, D. Neagu and J. T. S. Irvine, Energy Environ. Sci., 2013, 6, 256–266 RSC.
- V. Kyriakou, D. Neagu, E. I. Papaioannou, I. S. Metcalfe, M. C. M. van de Sanden and M. N. Tsampas, Appl. Catal., B, 2019, 258, 117950 CrossRef CAS.
- J.-h. Myung, D. Neagu, D. N. Miller and J. T. S. Irvine, Nature, 2016, 537, 528–531 CrossRef CAS.
- D. Neagu, T.-S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef.
- D. Neagu, V. Kyriakou, I.-L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS.
- F. M. Sapountzi, S. Brosda, K. M. Papazisi, S. P. Balomenou and D. Tsiplakides, J. Appl. Electrochem., 2012, 42, 727–735 CrossRef CAS.
- X. Zhang, Y. Song, G. Wang and X. Bao, J. Energy Chem., 2017, 26, 839–853 CrossRef.
- Y. Sun, J.-H. Li, M.-N. Wang, B. Hua, J. Li and J.-L. Luo, J. Mater. Chem. A, 2015, 3, 14625–14630 RSC.
- D. M. Amaya Dueñas, G. Chen, A. Weidenkaff, N. Sata, F. Han, G. Schiller, R. Costa and K. A. Friedrich, ECS Trans., 2019, 91, 1751–1760 CrossRef.
- A. Nenning and J. Fleig, Surf. Sci., 2019, 680, 43–51 CrossRef CAS.
- R. Hesse, T. Chassé and R. Szargan, Fresenius. J. Anal. Chem., 1999, 365, 48 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 55, 4709 CrossRef.
- A. Proctor and P. M. A. Sherwood, Anal. Chem., 1982, 54, 13 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- D. G. Goodwin, H. K. Moffat and R. L. Speth, Cantera: An object-oriented software toolkit for chemical kinetics, thermodynamics, and transport processes, Version 2.2.0, 2015 Search PubMed.
- M. Hahn, S. Schindler, L.-C. Triebs and M. A. Danzer, Batteries, 2019, 5, 43 CrossRef CAS.
- D. Johnson, ZView Electrochemical Impedance Software, Version 2.3b, Scribner Associates, Inc., 2000 Search PubMed.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS.
- A. J. Carrillo, K. J. Kim, Z. D. Hood, A. H. Bork and J. L. M. Rupp, ACS Appl. Energy Mater., 2020, 3, 4569–4579 CrossRef CAS.
- Database of Ionic Radii, http://abulafia.mt.ic.ac.uk/shannon/ptable.php, accessed July 2020.
- A. K. Opitz, A. Nenning, C. Rameshan, M. Kubicek, T. Götsch, R. Blume, M. Hävecker, A. Knop-Gericke, G. Rupprechter, B. Klötzer and J. Fleig, ACS Appl. Mater. Interfaces, 2017, 9(41), 35847–35860 CrossRef CAS.
- M. P. Seah and W. A. Dench, Surf. Interface Anal., 1979, 1, 1–11 CrossRef.
- M. Lorenz and M. Schulze, Surf. Sci., 2000, 454–456, 234–239 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- N. Gunasekaran, N. Bakshi, C. B. Alcock and J. J. Carberry, Solid State Ionics, 1996, 83, 145–150 CrossRef CAS.
- I. Ikemoto, K. Ishii, S. Kinoshita, H. Kuroda, M. A. Alario Franco and J. M. Thomas, J. Solid State Chem., 1976, 17, 425–430 CrossRef CAS.
- F. Garbassi, E. Mello Ceresa, G. Basile and G. C. Boero, Appl. Surf. Sci., 1982-1983, 14, 330–350 Search PubMed.
- D. Papargyriou, D. N. Miller and J. T. S. Irvine, J. Mater. Chem. A, 2019, 7, 15812 RSC.
- Y. Sun, J. Li, Y. Zeng, B. S. Amirkhiz, M. Wang, Y. Behnamiana and J. Luo, J. Mater. Chem. A, 2015, 3, 11048–11056 RSC.
- R. A. De Souza, M. S. Islam and E. Ivers-Tiffée, J. Mater. Chem., 1999, 9, 1621–1627 RSC.
- Y.-R. Jo, B. Koo, M.-J. Seo, J. K. Kim, S. Lee, K. Kim, J. W. Han, W. Jung and B.-J. Kim, J. Am. Chem. Soc., 2019, 141, 6690–6697 CrossRef CAS.
- A. L. Sauvet and J. T. S. Irvine, Solid State Ionics, 2004, 167, 1–8 CrossRef CAS.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, M. Shah, L. D. Marks and S. A. Barnett, J. Electrochem. Soc., 2010, 157, B279–B284 CrossRef CAS.
- N. Sakai, K. Yamaji, T. Horita, H. Negishi and H. Yokokawa, Solid State Ionics, 2000, 135, 469–474 CrossRef CAS.
- K. Kousi, D. Neagu, L. Bekris, E. Cali, G. Kerherve, E. I. Papaioannou, D. J. Payne and I. S. Metcalfe, J. Mater. Chem. A, 2020, 8, 12406–12417 RSC.
- A. Ladavos and P. Pomonis, in Perovskites and Related Mixed Oxides: Concepts and Applications, ed. P. Granger, V. I. Parvulescu, S. Kaliaguine and W. Preiller, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 1st edn, 2016, pp. 369–387, Methane Combustion on Perovskites Search PubMed.
- C. Pirovano, A. Rolle and R.-N. Vannier, in Perovskites and Related Mixed Oxides: Concepts and Applications, ed. P. Granger, V. I. Parvulescu, S. Kaliaguine and W. Preiller, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 1st edn, 2016, pp. 169–184, Perovskite and Derivative Compounds as Mixed Ionic–Electronic Conductors Search PubMed.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- O. A. Baturina and A. E. Smirnova, in New and Future Developments in Catalysis: Batteries, Hydrogen Storage and Fuel Cells, ed. S. L. Suib, Elsevier, 2013, pp. 69–97, Catalytic Processes Using Fuel Cells, Catalytic Batteries, and Hydrogen Storage Materials Search PubMed.
- Y.-J. Yang, T.-L. Wen, H. nTu, D.-Q. Wang and J. Yang, Solid State Ionics, 2000, 135, 475–479 CrossRef CAS.
- S. P. Jiang, L. Liu, K. P. Ong, P. Wu, J. Li and J. Pu, J. Power Sources, 2008, 176, 82–89 CrossRef CAS.
- J. Schefold, A. Brisse, A. Surrey and C. Walter, Int. J. Hydrogen Energy, 2019, 45, 5143–5154 CrossRef.
- M. Preininger, B. Stoeckl, V. Subotić, F. Mittmann and C. Hochenauer, Appl. Energy, 2019, 254, 113695 CrossRef CAS.
- X. Sun, M. Chen, S. H. Jensen, S. D. Ebbesen, C. Graves and M. Mogensen, Int. J. Hydrog. Energy, 2012, 37, 17101–17110 CrossRef CAS.
- A. Leonide, PhD dissertation, Karlsruhe Institute of Technology (KIT), 2010.
- V. Yurkiv, R. Costa, Z. Ilhan, A. Ansar and W. G. Bessler, J. Electrochem. Soc., 2014, 161, F480–F492 CrossRef CAS.
- S. B. Adler, J. A. Lane and B. C. H. Steele, J. Electrochem. Soc., 1996, 143, 3554–3564 CrossRef CAS.
- J.-C. Njodzefon, C. R. Graves, M. B. Mogensen, A. Weber and J. Hjelm, J. Electrochem. Soc., 2016, 163, F1451–F1462 CrossRef CAS.
- S. Dierickx, A. Weber and E. Ivers-Tiffée, Electrochim. Acta, 2020, 355, 136764 CrossRef CAS.
- M. R. Shoar Abouzari, F. Berkemeier, G. Schmitz and D. Wilmer, Solid State Ionics, 2009, 180, 922–927 CrossRef CAS.
- C. H Hsu and F. Mansfeld, Corrosion, 2001, 57, 747–748 CrossRef.
- M. H. Pihlatie, A. Kaiser, M. Mogensen and M. Chen, Solid State Ionics, 2011, 189, 82–90 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SEM images of the exsolved Ni nanoparticles. See DOI: 10.1039/d0ta07090d |
| This journal is © The Royal Society of Chemistry 2021 |