
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancement of the mechanical properties of lysine-containing peptide-based supramolecular hydrogels by chemical cross-linking†
Libby J.
Marshall
 a,
Olga
Matsarskaia
b,
Ralf
Schweins
b and
Dave J.
Adams
*a
a,
Olga
Matsarskaia
b,
Ralf
Schweins
b and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bInstitut Laue-Langevin, 71 Avenue des Martyrs, CS 20156, 38042 Grenoble Cedex 9, France
First published on 30th August 2021
Abstract
Exposure of lysine-containing peptide-based gelators to the cross-linking agent glutaraldehyde allows tuning of gel mechanical properties. The effect of cross-linking depends on the position of the lysine residue in the peptide chain, the concentration of gelator and the conditions under which cross-linking takes place. Through control of these factors, cross-linking leads to increased gel strength.
Supramolecular hydrogels are soft, viscoelastic materials prepared by the self-assembly of small molecules (so-called gelators) via the formation of many non-covalent interactions.1 The structures formed during self-assembly trap large amounts of water, thus forming a material composed mostly of liquid but with solid-like properties.2 Peptide-based supramolecular hydrogels have many advantages including easy synthesis, potential for chemical modification, responsiveness to external stimuli, bioactivity and biocompatibility.2,3 However, such hydrogels often lack the mechanical strength required for certain applications owing to the non-covalent nature of the interactions holding the gel network together.3–5 Examples of such applications include scaffolds for cell and tissue engineering, where the mechanical properties of the scaffold need to mimic the cells’ native enviroment and are essential in determining the outcome of cellular differentiation;6 3D printing, where the gel must have sufficient stength to be extruded through a needle;7 and as drug delivery vehicles where sufficient stiffness ensures sustainable release of drug molecules.8
It is also very difficult to design gelators with a particular application in mind as the ability of a given molecule to form a hydrogel and the properties of any resulting gels are hard to predict.9 It is therefore important to design new methods for tuning the mechanical properties of well-studied gelators.
Many methods have been employed to improve the mechanical stability of peptide-based supramolecular hydrogels including control of gelation to ensure formation of a homogenous gel network,10 physical cross-linking using ions,11,12 exploitation of the chiral nature of peptides13 and mixing with polymer additives.14–16 However, these techniques only have a limited effect on gel properties. Recently, methods utilising chemical cross-linking to enhance the mechanical properties of supramolecular hydrogels have been explored. Such methods include native chemical ligation,17 disulfide formation,12,18 tyrosine dimerisation5 as well as the use of enzymes19 and cross-linking agents such as glutaraldehyde4 and Genipin.20
While chemical cross-linking in supramolecular hydrogels regularly increases gel stiffness, very few approaches also increase gel strength and some even decrease the gels’ resistance to strain.5 We therefore hope to expand the available methods for chemical cross-linking in order to further improve mechanical properties without compromising gel strength.
Previous work has successfully used glutaraldehyde (Fig. 1a) as a cross-linking agent via in situ formation of imine bonds and highlighted the importance of using dynamic chemistry to avoid interfering with self-assembly.4 We have used a similar approach here by incorporating a lysine (K) residue into the well-studied, peptide-based gelator 2NapFF (Fig. 1b). We expected that exposure to glutaraldehyde would result in cross-linking between primary amines on the side-chains of K residues on neighbouring fibres or within individual fibres (Fig. 2a) and thereby alter the strength and stiffness of the gels. However, it is impossible to predict how cross-linking will take place and its effect on gel properties. The gelators discussed here are 2NapFFK and 2NapKFF (Fig. 1c and d).
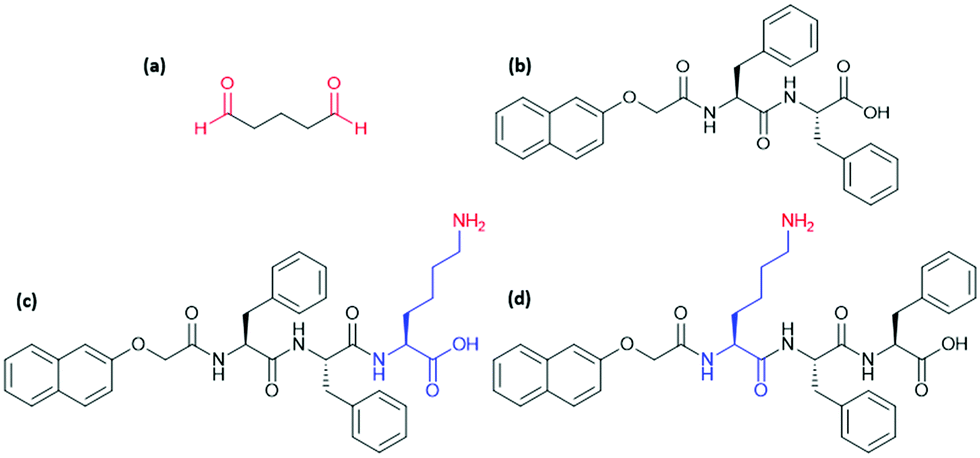 | ||
| Fig. 1 Chemical structures of (a) glutaraldehyde, (b) 2NapFF, (c) 2NapFFK and (d) 2NapKFF. The lysine residues are highlighted in blue and the groups involved in imine formation are highlighted in red. | ||
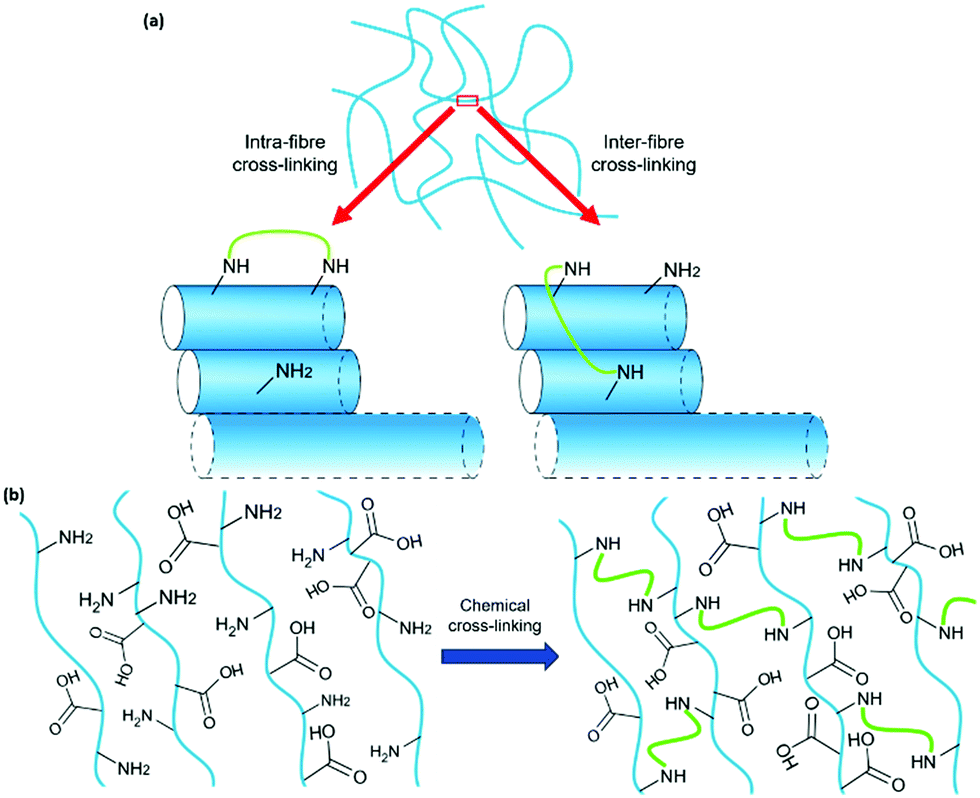 | ||
| Fig. 2 (a) Close up of a small section of the gel network. The gel network is formed by lateral association of individual 1D structures. These 1D structures were fit using SANS to a cylinder model combined with a power law. Cross-linking (green) between amines on K residues may occur within individual fibres (blue) or between neighbouring fibres. The arrangement of the fibres is purely hypothetical. (b) Schematic showing a hypothetical anti-parallel arrangement of fibres within a 2D section of the 3D gel network, highlighting how such an arrangement provides facile cross-linking (green) of amine groups on adjacent fibres. | ||
Previous work has shown the power of the FF motif in driving self-assembly alongside the aromatic N-terminal capping group through π–π stacking and the hydrophobic effect.2 The reported anti-parallel nature of peptide-based hydrogels containing FF may be beneficial in setting up the self-assembled structures for easy cross-linking between adjacent fibres, allowing self-assembly to facilitate cross-linking (Fig. 2b).21,22 We expected that the position of the K residue in the gelator would play a role in the final properties of the gels formed. While altering the position of the amine will affect its accessibility for cross-linking, it may also disrupt the π–π stacking interactions between aromatic rings and thereby alter self-assembly and the properties of the gels formed. It should be noted that the increased stability and possible changes in orientation of the gelator molecules caused by introduction of chemical bonds may drive self-assembly towards crystallisation rather than gel formation.23,24 Both gelators formed gels under all the conditions discussed here. We can expect that the gelator molecules in the gel state are kinetically trapped and lack the energy required to overcome the energy barrier to a more thermodynamically stable, crystalline state.25
Gels were prepared by first suspending the gelators in aqueous solution at high pH (∼11.6) by addition of 2 molar equivalents of NaOH (0.1 M). Such high pH ensures deprotonation of the C-terminal carboxylic acid group, rendering the molecules sufficiently polar to be dispersed in water.26 However, viscosity measurements of both gelators at high pH show shear-thinning (Fig. S2a, ESI†), indicative of the presence of worm-like micelles in solution.27 While stirring with glutaraldehyde at this pH reduces the viscosity of the gelator solutions, shear-thinning behaviour persists following the reaction with glutaraldehyde (Fig. S2b, ESI†), suggesting that worm-like micelles are still present.
Reducing the solution pH results in protonation of the C-terminal carboxylic acid, decreasing the polarity of the molecules and driving further association of self-assembled structures to exclude water.26 The self-assembled structures interact and entangle through the formation of non-covalent interactions as the pH continues to decrease until a 3D network is formed.26,28 Water is trapped within the network during self-assembly, giving the gel its viscoelastic properties.
The gelators were exposed to glutaraldehyde (1 molar equivalent) in two different conditions (Fig. S1, ESI†): stirring in solution at high pH (pH ∼ 11.6) for 24 hours before gelation and as gels at low pH (pH ∼ 3.5). Gel formation was triggered using glucono-δ-lactone (GdL) to reduce the pH, as described previously.29 All samples were left overnight (16 hours) after addition of GdL to allow gel formation. Time sweep data (Fig. S10, ESI†) shows that 16 hours are sufficient for compete gel formation. Glutaraldehyde was then added to the post-gelation samples, which were left undisturbed for a further 24 or 72 hours before rheology measurements. Since the glutaraldehyde had to be added to the top of the sample, a gradient of cross-linking may have been present in the post-gelation samples. This will have affected the results obtained from rheology measurements and is a limitation to this experimental design.
The control experiments were designed to account for gel age as well as the presence of glutaraldehyde.30 Pre-gelation samples were left to sit for 24 hours before rheology to ensure they were the same age as the 24 hour post-gelation samples and the 24 hour controls. Fig. 3, 4 and 5 show that, in most cases, the 24 hour and 72 hour control samples are not statistically different from a rheological perspective. Therefore, the differences between gels exposed to glutaraldehyde post-gelation for 24 and 72 hours are due to the interaction of the gel with glutaraldehyde and not the age of the gels.
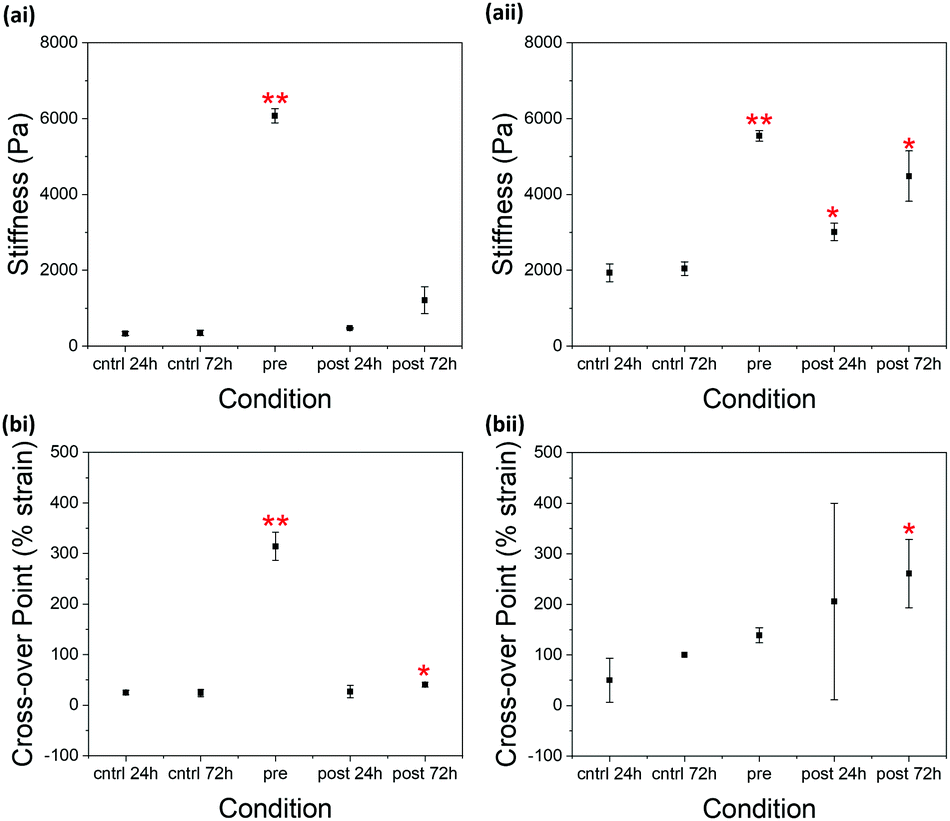 | ||
| Fig. 3 Scatter plots showing the (a) stiffness and (b) cross-over point of gels formed from 2NapFFK at concentrations of (i) 5 mg mL−1 and (ii) 10 mg mL−1 in different conditions. The stiffness was determined as the average G′ value in a selected section of linear viscoelastic region of the strain sweep (0.01–1% strain). This section was selected as is applicable to all samples. The cross-over point was taken as strain value within the strain sweep where G′′ crosses over G′. The points in the plot are the average from three samples. The error bars show the standard deviation between the three samples in each condition. Two-sample T-tests assuming unequal variances were performed between each triplicate of samples and the 24 hour control triplicate of samples. * = p > 0.05, ** p > 0.01. | ||
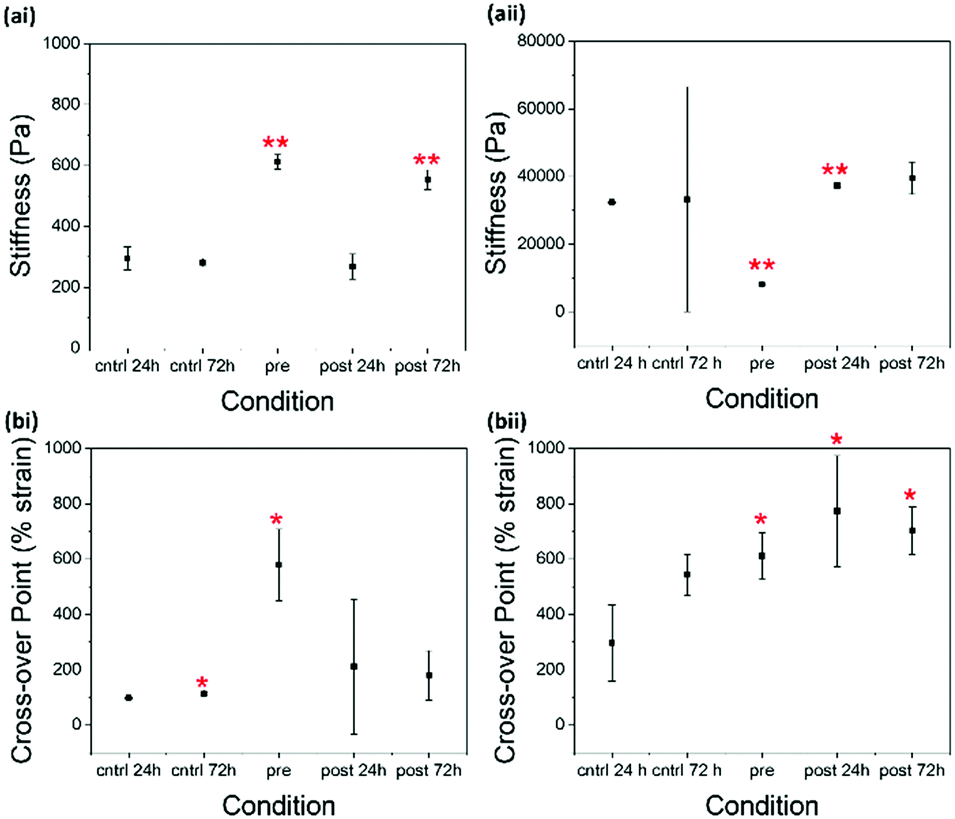 | ||
| Fig. 4 Scatter plots showing the (a) stiffness and (b) cross-over point of gels formed from 2NapKFF at concentrations of (i) 5 mg mL−1 and (ii) 10 mg mL−1 in different conditions. The stiffness was determined as the average G′ value in the linear viscoelastic region of the strain sweep (0.01–1% strain). This range was selected as is applicable to all samples. The cross-over point shows the strain value within the strain sweep where G′′ crosses over G′. The points in the plot are the average from three samples. The error bars show the standard deviation between the three samples in each condition. Two-sample T-tests assuming unequal variances were performed between each triplicate of samples and the 24 hour control triplicate of samples. * = p > 0.05, ** p > 0.01. In the case where the 72 hour control samples were statistically different from 24 hour control, the 72 hour post samples were compared to the 72 hour controls in the statistical test. | ||
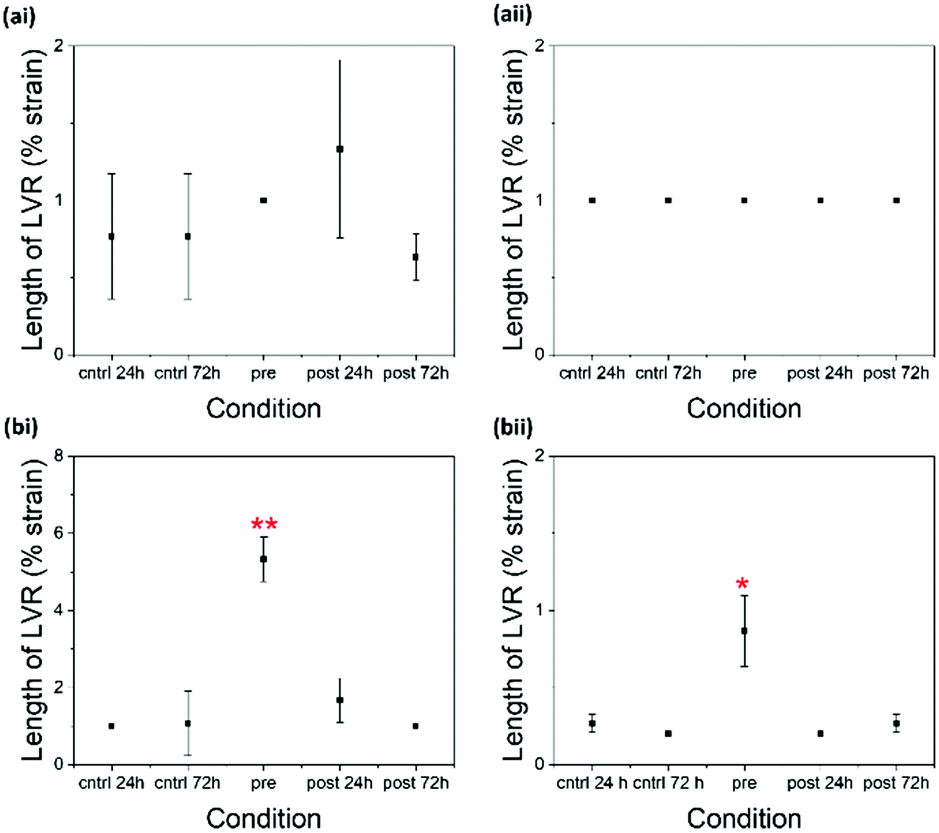 | ||
| Fig. 5 Scatter plots showing the length of the LVR recorded from gels composed of (a) 2NapFFK and (b) 2NapKFF at concentrations of (i) 5 mg mL−1 and (ii) 10 mg mL−1 in different conditions. The length of the LVR was taken as the strain value at which the G′ begins to decrease. The points in the plot are the average from three samples. The error bars show the standard deviation between the three samples in each condition. Two-sample T-tests assuming unequal variances were performed between each triplicate of samples and the 24 hour control triplicate of samples. * = p > 0.05, ** p > 0.01. | ||
Only slight increases in stiffness (G′) were observed in 2NapFFK 5 mg mL−1 (Fig. 3) when comparing the control samples and the samples exposed to glutaraldehyde for 24 hours post-gelation. This increase became more significant at a 2NapFFK concentration of 10 mg mL−1. The samples where glutaraldehyde was added post-gelation and left to react for 72 hours showed even greater increases in stiffness compared to the controls. Glutaraldehyde is therefore able to interact with the gelators within the gel network at low pH, resulting in increased gel stiffness. The degree of this increase depends on how much time glutaraldehyde is given to interact with the gels.
Unusually, stirring 2NapFFK (5 mg mL−1) with glutaraldehyde pre-gelation increased both the stiffness of the resulting gels and the strain required to completely break down the gel network (the strain at which G′ crosses over G′′). This can be explained by the pH at which glutaraldehyde is interacting with the gelator molecules. At high pH, more deprotonated amines are available for imine formation, resulting in more cross-linking and thereby a greater increase in G′ and G′′.31 It is also expected that the presence of the gel network at low pH makes it harder for glutaraldehyde to diffuse into the fibrous structures to interact with the amine groups. At a concentration of 10 mg mL−1, there is no obvious pattern in the change in the cross-over point of 2NapFFK. There could be a change in morphology at a concentration between 5 and 10 mg mL−1, making cross-linking more difficult.
Pre-gelation cross-linking 2NapFFK at a concentration of 5 mg mL−1 leads to an increase in G′ greater than that achieved by increasing the gelator concentration to 10 mg mL−1 (Fig. 3). A considerable increase in stiffness and resistance to strain can therefore be achieved without having to increase gelator concentration. By cross-linking prior to gelation, it is possible to use lower gelator concentrations while maintaining the desired gel strength.
For 2NapKFF (Fig. 4 and Fig. S3, ESI†), the control and the 24 hour post-gelation samples are virtually identical from a rheological perspective. The 72 hour post-gelation samples showed increased stiffness. 2NapKFF therefore needs more than 24 hours to sufficiently interact with glutaraldehyde at 5 mg mL−1. There was also an increase in the cross-over point of the 72 hour control and 24 hour control, suggesting the strength of these gels develops with age which may also contribute to the increased cross-over point of the 72 hour post-gelation samples. At 10 mg mL−1, there was a significant increase in stiffness after 24 hours of exposure to glutaraldehyde. The increased standard deviation in the 72 hour post-gelation samples prevents the difference in stiffness between these samples and the controls from appearing significant in the statistical tests used.
There is little difference between G′ and G′′ values of the pre- and 72 hour post-gelation 2NapKFF samples at 5 mg mL−1 (Fig. 4). However, there is a significant increase in the cross-over point of the pre-gelation samples compared to that of the control samples. The cross-over point of the 2NapKFF samples at a concentration of 10 mg mL−1 increases in all conditions compared to the 24 hour control samples. In addition, at both 5 mg mL−1 and 10 mg mL−1, the linear viscoelastic region (LVR) is considerably longer in the pre-gelation samples compared to both the controls and the post-gelation samples (Fig. 5 and Fig. S3, ESI†). This shows that the pre-gelation gels can withstand higher strain before the gel network begins to break down.18,32 This could be due to the increased proportion of amines available for cross-linking at high pH, as previously discussed. However, it is interesting that this increase in LVR was observed in 2NapKFF and not 2NapFFK, showing that the position of the K residue within the peptide chain plays a role in the gelators response to cross-linking. These observations are particularly exciting as most techniques previously used to modulate the mechanical properties of peptide-based supramolecular hydrogels result in increased stiffness (G′) but not increased mechanical strength. We highlight for all of these systems rheology has been performed on samples a maximum of 14 days after gel formation. No evidence of crystallisation was observed in these samples with only slight changes in rheological properties. Samples left undisturbed at room temperature for over a month showed no visible evidence of crystal formation or precipitation.
Another interesting observation is the significant decrease in the stiffness of pre-gelation samples formed from 2NapKFF at a concentration 10 mg mL−1. This reponse is in opposition to those observed in all other conditions shown here. The reduced stiffness could suggest inter-fibre cross-linking rather than intra-fibre cross-linking, which could disrupt lateral association of the fibres, resulting in reduced rigidity if the gel network.
The greater increase in stiffness in 2NapFFK compared to 2NapKFF could be explained by the position of the K residue. The K residue at the C-terminus is less sterically hindered and therefore more readily available for cross-linking.18 This will increase the efficiency of cross-linking resulting in the observed changes in mechanical properties. The drastic differences between the pre-gelation samples of 2NapFFK and 2NapKFF at 10 mg mL−1 confirms that the position of the K residue has a great effect on gel behaviour and provides a further level of control for tweaking gel properties.
Small angle neutron scattering (SANS, performed at instrument D11, Institut Laue – Langevin, Grenoble, France) was used to determine the effect of cross-linking pre- and post-gelation on the structures that constitute the gel network. The data from samples of 2NapFFK (5 mg mL−1, Fig. 6) suggests that cross-linking does not significantly change the nature of the fibres within the gel network as all three data sets could be fit to a standard cylinder model combined with a power law. However, cross-linking pre-gelation resulted in a considerable reduction in the length of the fibres, from 112 nm in the control sample to 40 nm in the pre-gelation sample. The control and post-gelation samples had lengths within experimental error of each other. The radii of the fibres were increased in both the pre- and post-gelation samples compared to the control sample, with the radii of the fibres in the post-gelation sample doubling in size. This indicates cross-linking between two adjacent fibres, consistent with the increased stiffness of the gels, as observed in rheology data.
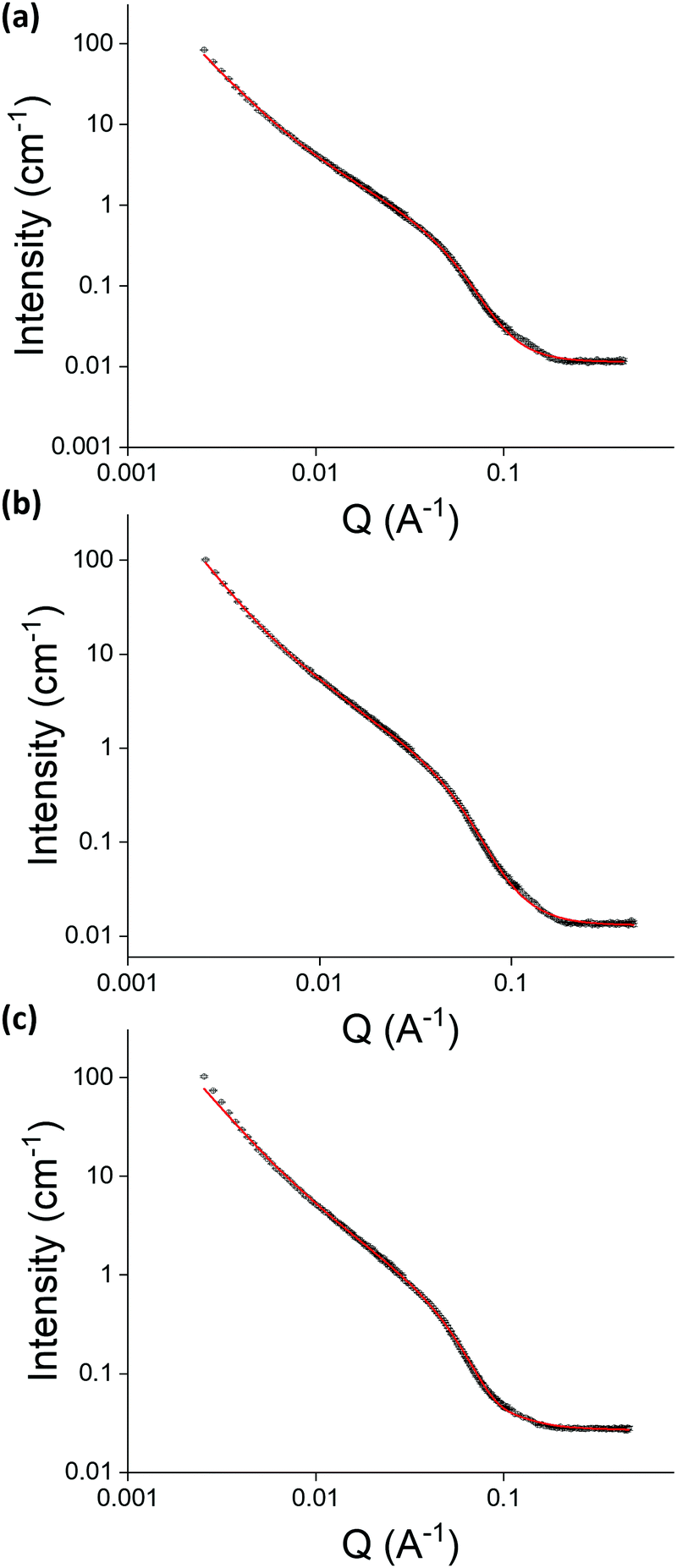 | ||
| Fig. 6 The SANS data collected (black) and fits (red) for (a) control, (b) cross-linking pre-gelation and (c) cross-linking post-gelation samples of 2NapFFK (5 mg mL−1). The data were fit to the range of a standard cylinder model combined with a power law. A full discussion of the fitting and parameters are available in the ESI† (Table S1). | ||
Observation of different changes in fibre structure depending on whether cross-linking was performed before or after gelation agrees with the observation of different changes in rheological behaviour when comparing pre- and post-gelation samples to control samples. Cross-linking before gelation results in a large reduction in length of the fibres and a slight increase in radii, while cross-linking post-gelation does not alter the length of the fibres but does greatly increase their radii. This fits with the theory that cross-linking post-gelation fortifies the already-formed structures in the gel network while cross-linking pre-gelation has a much greater effect on structure as cross-linking occurs before the gel network has formed. The length values from SANS fit may correspond to the Kuhn length rather than the entire length of the structures. This would suggest that the structures in the pre-gelation samples are much more flexible than those in the control and post-gelation samples. This would indicate that the presence of glutaraldehyde in the pre-gelation samples is interfering with lateral association during gel-formation.
To determine whether the observed changes in mechanical properties were the result of chemical cross-linking between gelator molecules we tried reacting a 5 mg mL−1 solution of 2NapFF with 1 molar equivalent of glutaraldehyde at pH 11.6. No colour change was observed (Fig. S22, ESI†). It was therefore concluded that glutaraldehyde is reacting with the amine group in the K-containing gelators and this reaction causes the colour change from white to pink in the pre-gelation samples and white to yellow in post-gelation samples (Fig. S21, ESI†). Previous work has shown similar colour changes on imine bond formation.14,18 It is expected that the pink colour in the pre-gelation samples is due to increased concentration of imine bonds compared to in the post-gelation samples. This hypothesis matches the greater changes observed in the rheological properties of the pre-gelation samples.
We also reacted 2NapFFK solution at pH 11.6 previously stirred with glutaraldehyde for 24 hours and gels exposed to glutaraldehyde pre- and post-gelation with the reducing agent NaBH4. If cross-linking had occurred via imine bond formation, reaction with NaBH4 would result in reduction of the imine bonds and thereby loss of colour. Loss of colour was observed in both samples (Fig. S23 and S24, ESI†), showing reduction of imine bonds and further confirming that cross-linking via imine bond formation had occurred.
To further prove that the observed changes in rheological properties were due to reaction between the primary amine on the K residue and glutaraldehyde, we performed a control experiment using the gelator 2NapFF (Fig. 1b) which does not contain a K residue. The control was performed at a concentration of 5 mg mL−1 as this is the concentration at which the greatest changes in rheology were observed for the other systems.
Figure S11 (ESI†) shows that the 2NapFF control and post-gelation samples were almost identical in terms of rheology. Therefore, glutaraldehyde does not alter the properties of gels formed from 2NapFF when added post-gelation. The pre-gelation samples showed a slight increase in stiffness (G′). However, the G′′ of these samples significantly increased, suggesting lower elasticity than the gels formed without glutaraldehyde. There is also not an order of magnitude between G′ and G′′, showing these samples were not “true gels”.33 Stirring with glutaraldehyde pre-gelation therefore does impact gel properties, perhaps by interfering with self-assembly after addition of the trigger.34 Importantly, no colour change was observed in any of the samples (Fig. S25, ESI†), showing no imine bond formation.
Finally, we performed 1H NMR spectroscopy on samples of each K-containing gelator with and without stirring with glutaraldehyde for 24 hours. The glutaraldehyde samples all showed a distinct imine peak between 8.0 and 9.5 ppm (Fig. S26, ESI†), further confirming that cross-linking is indeed taking place via imine formation.
We also synthesised and performed cross-linking on the final combination of the 2Nap capping group with two F residues and one K residue: 2NapFKF (Fig. S18, ESI†). Our results further confirm the importance of the position of the K residue in the peptide-chain in determining the behaviour of the gelators on cross-linking as 2NapFKF behaved differently to both 2NapFFK and 2NapKFF following cross-linking. While 2NapFFK showed increased stiffness in all cross-linking conditions and 2NapKFF showed increased stiffness following cross-linking in all conditions except pre-gelation at a 2NapKFF concentration of 10 mg mL−1, the stiffness of 2NapFKF decreased significantly following cross-linking pre-gelation at 5 mg mL−1 and decreased slightly following cross-linking pre-gelation at 10 mg mL−1 (Fig. S19, ESI†). The cross-over point of gels formed from 2NapFKF increased following cross-linking pre-gelation and post-gelation at both 2NapFKF concentrations, similar to 2NapFFK. The length of the LVR increased slightly following cross-linking pre-gelation at a 2NapFKF concentration of 10 mg mL−1. However, this increase was not as significant as observed in 2NapKFF.
Conclusions
Glutaraldehyde clearly has a significant effect on the mechanical properties of the gels studied so far, providing a simple strategy for tuning both strength and stiffness of peptide-based gelators. The position of the K residue in the gelator not only affects gel properties, but also how the gelator interacts with glutaraldehyde. The ability to achieve different degrees of change in mechanical properties depending on gelator concentration and whether the gelators are exposed to glutaraldehyde before or after gelation provides a further level of control. Such control is essential for fine-tuning gels for specific applications and increasing the potential uses of a single gelator. Expansion of known gelators is an attractive strategy due to the challenges associated with designing new gelators.2 We expect to see similar results with other peptide-based gelators by simply incorporating a K residue into the peptide chain and performing cross-linking with glutaraldehyde or a similar cross-linking agent. This work greatly expands the choice of tools available for optimising the properties of peptide-based hydrogels for specific applications and circumvents the need to design entirely new gelators from scratch.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. J. M. thanks the Leverhulme Trust for funding (RPG-2019-165). The experiment at the Institut Laue-Langevin was allocated beam time under experiment number 9-11-1970 (DOI: 10.5291/ILL-DATA.9-10-1670). This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement no. 654000.Notes and references
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- Y. Li, M. Qin, Y. Cao and W. Wang, Sci. China: Phys., Mech. Astron., 2014, 57, 849–858 CrossRef CAS.
- M. A. Khalily, M. Goktas and M. O. Guler, Org. Biomol. Chem., 2015, 13, 1983–1987 RSC.
- Y. Ding, Y. Li, M. Qin, Y. Cao and W. Wang, Langmuir, 2013, 29, 13299–13306 CrossRef CAS PubMed.
- L. Saunders and P. X. Ma, Macromol. Biosci., 2019, 19, 1800313 CrossRef PubMed.
- H. Jian, M. Wang, Q. Dong, J. Li, A. Wang, X. Li, P. Ren and S. Bai, ACS Appl. Mater. Interfaces, 2019, 11, 46419–46426 CrossRef CAS PubMed.
- K. Basu, A. Baral, S. Basak, A. Dehsorkhi, J. Nanda, D. Bhunia, S. Ghosh, V. Castelletto, I. W. Hamley and A. Banerjee, Chem. Commun., 2016, 52, 5045–5048 RSC.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- M. A. Greenfield, J. R. Hoffman, M. O. de la Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS PubMed.
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS PubMed.
- M. B. Taraban, Y. Feng, B. Hammouda, L. L. Hyland and Y. B. Yu, Chem. Mater., 2012, 24, 2299–2310 CrossRef CAS PubMed.
- P. R. A. Chivers and D. K. Smith, Chem. Sci., 2017, 8, 7218–7227 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC.
- J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC.
- J. P. Jung, J. L. Jones, S. A. Cronier and J. H. Collier, Biomaterials, 2008, 29, 2143–2151 CrossRef CAS PubMed.
- W. Y. Seow and C. A. E. Hauser, Adv. Healthcare Mater., 2013, 2, 1219–1223 CrossRef CAS PubMed.
- Y. Li, Y. Ding, M. Qin, Y. Cao and W. Wang, Chem. Commun., 2013, 49, 8653–8655 RSC.
- L. Chronopoulou, Y. Toumia, B. Cerroni, D. Pandolfi, G. Paradossi and C. Palocci, New Biotechnol., 2017, 37, 138–143 CrossRef CAS PubMed.
- A. N. Rissanou, E. Georgilis, E. Kasotakis, A. Mitraki and V. Harmandaris, J. Phys. Chem. B, 2013, 117, 3962–3975 CrossRef CAS PubMed.
- S. Fleming, P. W. J. M. Frederix, I. R. Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- J. Li, R. Xing, S. Bai and X. Yan, Soft Matter, 2019, 15, 1704–1715 RSC.
- K. Nagy-Smith, E. Moore, J. Schneider and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9816–9821 CrossRef CAS PubMed.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- C. Lin, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- N. Reddy, Y. Tan, Y. Li and Y. Yang, Macromol. Mater. Eng., 2008, 293, 614–620 CrossRef CAS.
- M. Bertasa, A. Dodero, M. Alloisio, S. Vicini, C. Riedo, A. Sansonetti, D. Scalarone and M. Castellano, Eur. Polym. J., 2020, 123, 1219–1223 CrossRef.
- B. P. Nowak and B. J. Ravoo, Soft Matter, 2020, 16, 7299–7304 RSC.
- L. Chen, S. Revel, K. Morris, D. G. Spiller, L. C. Serpell and D. J. Adams, Chem. Commun., 2010, 46, 6738–6740 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01136g |
| This journal is © The Royal Society of Chemistry 2021 |