
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of adsorption of ionic liquid constituents on the stability of layered double hydroxide colloids
Dóra
Takács
a,
Bojana
Katana
a,
Adél
Szerlauth
a,
Dániel
Sebők
b,
Matija
Tomšič
 c and
István
Szilágyi
*a
c and
István
Szilágyi
*a
aMTA-SZTE Lendület Biocolloids Research Group, Department of Physical Chemistry and Materials Science, University of Szeged, 1 Rerrich Béla tér, H-6720 Szeged, Hungary. E-mail: szistvan@chem.u-szeged.hu
bDepartment of Applied and Environmental Chemistry, University of Szeged, 1 Rerrich Béla tér, H-6720 Szeged, Hungary
cFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia
First published on 7th September 2021
Abstract
The influence of ionic liquid (IL) anions and cations on the charging and aggregation properties of layered double hydroxide (LDH) nanoparticles was systematically studied. Surface charge characteristics were explored using zeta potential measurements, while aggregation processes were followed in dynamic light scattering experiments in aqueous IL solutions. The results revealed that the aggregation rates of LDHs were sensitive to the composition of ILs leading to IL-dependent critical coagulation concentration (CCC) values being obtained. The origin of the interparticle forces was found to be electrostatic, in line with the classical Derjaguin–Landau–Verwey–Overbeek (DLVO) theory, as the experimental aggregation kinetics were in good agreement with the predicted data. The ion specific adsorption of IL anions led to different surface charge densities for LDHs, which decreased in the order Cl− > Br− > DCA− > SCN− > NO3− for counterions and BMIM+ > BMPYR+ > BMPY+ > BMPIP+ in the case of coions resulting in weaker electrical double layer repulsion in these sequences. Since van der Waals forces are always present and their strength does not depend significantly on the ionic strength, the CCC values decreased in the above order. The present results shed light on the importance of the interfacial arrangement of the IL constituent ions on the colloidal stability of particle dispersions and provide important information on the design of stable or unstable particle-ionic liquid systems.
1. Introduction
Layered double hydroxides (LDHs) represent a class of inorganic layered materials consisting of positively charged hydroxide sheets of divalent (e.g., Mg2+, Zn2+ or Ca2+) and trivalent (e.g., Al3+, Fe3+ or Cr3+) metal ions coordinated by hydroxide groups, as well as interlayer water and anions compensating the charge on the layers.1 The most common group of LDHs can be described by the general formula of [M(II)1−xM(III)x(OH)2][An−·mH2O], where M(II) and M(III) are the divalent and trivalent metal ions and An−·mH2O is the interlamellar charge-neutralizing anion in the hydrated state.2 The exploitation of the anion exchange capacity and lamellar structure – which are important properties of LDHs – is still a fascinating research topic, as these properties allow the direct intercalation of inorganic and organic anions.3–5Numerous LDH applications based on the intercalation or exchange of specific guest molecules have been comprehensively studied in the past decades. For example, polyoxometalate intercalated LDHs proved to be efficient catalysts in dehydrogenation, esterification and oxidation reactions.6 Anti-inflammatory, anti-cardiovascular and anti-cancer drugs were successfully immobilized in LDH supports and the obtained hybrids were used for biomedical therapies.7,8 Besides, the intercalation phenomenon was utilized in the removal of dye contaminants from the aqueous environment.9
However, strong attraction between the highly charged hydroxide layers and the interlayer anions hinders the access to the interlayer space in many cases. Delamination of LDHs into single layers is a possible way to obtain LDH systems for immobilization of guest molecules regardless of their size,10 but the delamination process is not straightforward in conventional media due to the high charge density, small interlayer distance and strong hydrophilicity.
One of the most promising candidates for the liquid phase delamination of LDHs is the ionic liquids (ILs), since they can be obtained using the desired molecular structures and physicochemical properties.11–13 ILs can be considered as molten salts and are generally composed of a bulky organic cation with an inorganic or organic anion. Their high chemical and thermal stability and extremely low vapor pressure generally make ILs less hazardous than organic solvents used in various applications.14,15 It is known that ILs are able to reduce the strength of attractive forces between charged surfaces due to the possible combined effect of their interfacial assembly or partial dissociation.16–18 Furthermore, the shielding of attractive interactions between the layers should also lead to the collapse of stacked structures and for instance, to the delamination of LDHs into unilamellar nanosheets. Although direct delamination of LDHs in ILs has not been reported yet, single layer nanosheets have been prepared in water–IL reversed emulsion19 as well as by intercalating IL constituents and performing subsequent in situ polymerization within the layers.20 The stabilization of the resulting delaminated LDH materials is a key point, which has not been explored in detail.
There is growing contemporary interest in application of LDH-IL systems. Accordingly, LDH supported palladium particles showed remarkable catalytic activity in nonaqueous ILs.21 IL impregnated LDH was synthesized and used as a solid electrolyte in batteries for advanced energy storage applications.22 IL assisted LDH synthesis led to the formation of particles of high specific surface area used for phosphate removal in water treatment processes.19 Finally, it was pointed out that LDH-IL systems can be applied in the preparation of ionogels23 or composite materials,24 since the combination of the features of the inorganic matrix and specific ILs may lead to synergistic effects in property selection and tuning. The above examples indicate a need for a comprehensive investigation of LDH-IL interactions and subsequently, on the interparticle forces across ILs. Nevertheless, only a handful of studies can be found in the literature that are focusing on particle aggregation in ILs25–29 and, to the best of our knowledge, no data are yet available for LDH-IL systems. Therefore, a systematic investigation on dispersion stability in such systems is required for a better understanding of the behavior of these complicated systems.
In this way, the first step is to gain knowledge of the interfacial assembly of IL constituents, since such a process is likely to play a crucial role in the stability of LDH particles in ionic media. Previous studies30–34 on other particle-IL systems pointed out that ion specific effects, similar to inorganic electrolytes,35–37 are important in the presence of IL constituents since they govern the adsorption of ions from solutions. Moreover, IL cations and anions can be arranged in sequences based on their adsorption affinity to a given surface.31,32,34
For standard inorganic salts, the extent of ion specific adsorption and its effect on the stability can be ordered in the Hofmeister series of anions and cations. This theory was originally developed to describe the influence of salt constituents on the stability of protein solutions as follows.38
| CO32− < SO42− < H2PO4− < F− < Cl− < Br− < NO3− < I− < ClO4− < SCN− |
| N(CH3)4+ < NH4+ < Cs+ < Rb+ < K+ < Na+ < Li+ < Mg2+ < Ca2+ |
In the past decades, a large number of papers reported on the applicability of such order in other phenomena. For instance, ion specific effects on the assembly of amphiphilic molecules were comprehensively studied.45 In addition, surface adsorption of cations followed the above order and the results of calculations revealed that the hydration of both the surface and the ions played a major role in the adsorption mechanism.46 Particle aggregation studies31,34–37,47,48 shed light on the fact that the critical coagulation concentration (CCC) data determined using the same particles in different electrolyte solutions can be ordered in the Hofmeister series too. However, the DLVO (Derjaguin, Landau, Verwey and Overbeek) theory,49 which describes the surface forces in the presence of salt constituents, failed to predict the dependence of such forces on the type of salt present and consequently, the trends in CCC values measured in various salt solutions.31,47,50–52
Besides, the CCC of particles in IL solutions were also found to be sensitive to the type of ion.31,32,34 Therefore, the traditional Hofmeister series of ions developed for the destabilization effect of ionic species in particle dispersions can be extended by using IL constituents. This issue can be adequately addressed in aqueous IL solutions by investigating the charging characteristics and aggregation properties of the colloidal particles.31,32,34 Nevertheless, no similar investigations have been performed with LDH particles yet.
The aim of the present study was to provide insight into the effect of the interfacial properties of IL constituents on the interparticle forces in LDH dispersions. Therefore, the basic charging and aggregation processes for positively charged LDH particles in diluted aqueous IL solutions were investigated. The type of IL anion (Cl−, Br−, DCA−, SCN−, and NO3−) and cations (BMIM+, BMPIP+, BMPYR+, and BMPY+) was systematically varied and thus, the specific effect of counter- and coions was studied. The surface charge properties were investigated using zeta (electrokinetic) potential measurements, while the aggregation features were assessed in time-resolved light scattering experiments. The aim was to discuss the generic dependence of the aggregation rates on the IL concentration and to address system specificities in the presence of different IL compounds.
2. Experimental
2.1. Materials
The ILs such as 1-butyl-3-methylimidazolium chloride (BMIMCl), 1-butyl-2-methylpyridinium chloride (BMPYCl), 1-butyl-1-methylpiperidinium chloride (BMPIPCl) and 1-butyl-1-methylpyrrolidinium chloride (BMPYRCl) were bought from IoLiTec GmbH, while 1-butyl-3-methylimidazolium bromide (BMIMBr), 1-butyl-3-methylimidazolium nitrate (BMIMNO3) and 1-butyl-3-methylimidazolium thiocyanate (BMIMSCN) were purchased from Sigma-Aldrich and 1-butyl-3-methylimidazolium dicyanamide (BMIMDCA) was obtained from Merck. Ultrapure water was used during the sample preparation from a VWR Purity TU+ device. All stock solutions and the water were adjusted to pH 9.0 with NaOH (AnalR NORMAPUR) and they were filtered prior to sample preparation using 0.1 μm syringe filters (Millex) to avoid dust contamination. The experiments were carried out at 25 °C. The particle concentration was kept at 10 mg L−1.The LDH particles used in the present work were prepared using the flash co-precipitation method followed by hydrothermal treatment.10,53 Briefly, a mixed metal ion solution was prepared by dissolving the salts in water (0.2 M Mg(NO3)2 and 0.1 M Al(NO3)3). The pH was set to 10 with 4.0 M NaOH solution. The alkali and salt solutions were mixed under an N2 atmosphere. After stirring vigorously for 30 minutes, the sample was centrifuged and washed with water. The slurry was redispersed and the resulting dispersion was transferred to an autoclave and treated in an oven at 120 °C for 24 hours. After cooling to ambient temperature, the sample was separated and dried at 50 °C overnight. To prove the successful synthesis of the LDH, powder X-ray diffraction (XRD) measurements were performed on a Bruker D8 Advanced diffractometer with Cu Kα (0.1542 nm) as a radiation source in the 2θ range from 5° to 80° with the 0.02° step. The distinctive XRD pattern of the pristine LDH is shown in Fig. 1. The diffraction patterns show the reflections reported earlier for LDH-based materials.53 The obtained LDH particles were then re-suspended in pH 9 water resulting in a stock dispersion of 5 g L−1 concentration, which was later diluted to reach the suitable particle concentration for the experiments.
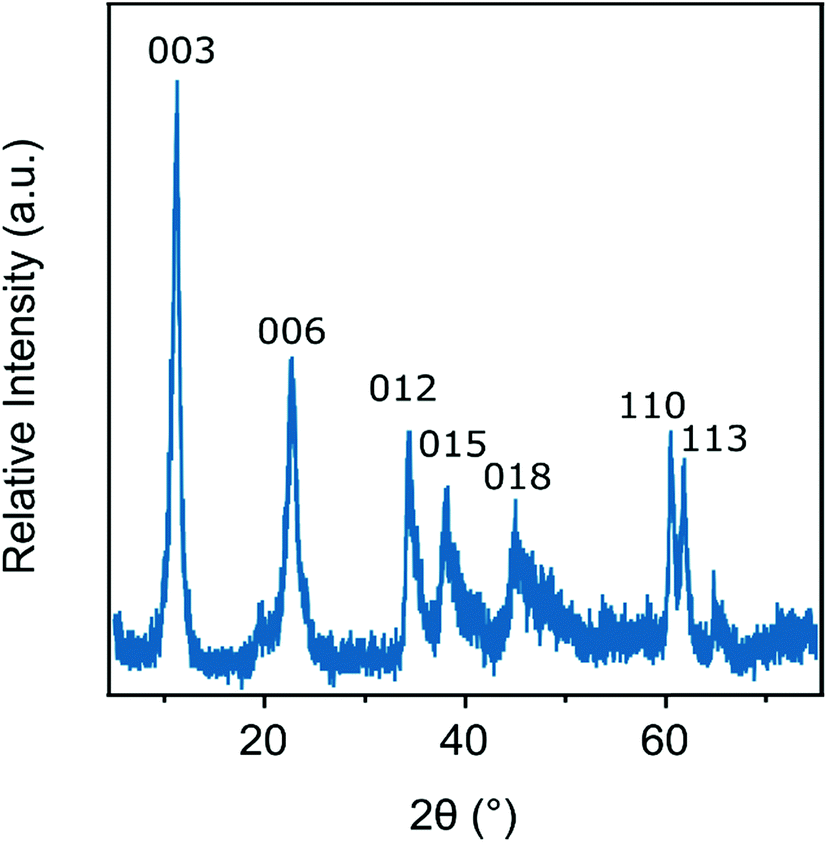 | ||
| Fig. 1 Powder XRD pattern of the LDH particles synthesized by flash coprecipitation and subjected to hydrothermal treatment. The Miller indices are indicated. | ||
2.2. Electrophoretic light scattering
A Litesizer 500 instrument (Anton Paar) equipped with a 40 mW semiconductor laser (658 nm wavelength) was used to measure the electrophoretic mobility of the particles in aqueous solutions of ILs. The samples were prepared by mixing the appropriate amount of ILs and water to obtain the desired concentration. The LDH particles were then added by the appropriate addition of the stock dispersion to obtain the final particle concentration. Prior to measurements, the samples were left to rest for 2 hours at room temperature, and then they were equilibrated for 1 additional minute in the instrument. The electrophoretic mobility (u) of each sample was measured five times, averaged and converted to zeta potential (ζ) value using the Smoluchowski equation54 as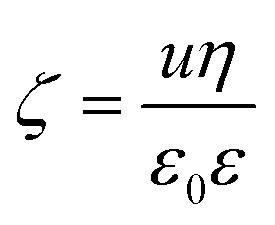 | (1) |
2.3. Dynamic light scattering
To explore particle aggregation, time-resolved dynamic light scattering (DLS) measurements were performed using Litesizer 500 instrument (Anton Paar) at a scattering angle of 175°. The same sample preparation procedure as in the electrophoretic mobility studies was followed, with the exception that the DLS measurements started immediately after addition of the desired volume of LDH stock dispersion. The total volume of each sample was 2.0 mL and the experiments were carried out in disposable polystyrene cuvettes.The hydrodynamic radius (Rh) of the particles was calculated by the Stokes–Einstein equation from the diffusion coefficient (D), which was extracted from the correlation function using the cumulant method.55 Time-resolved measurements were carried out to determine the apparent aggregation rate coefficients (kapp) as follows:56
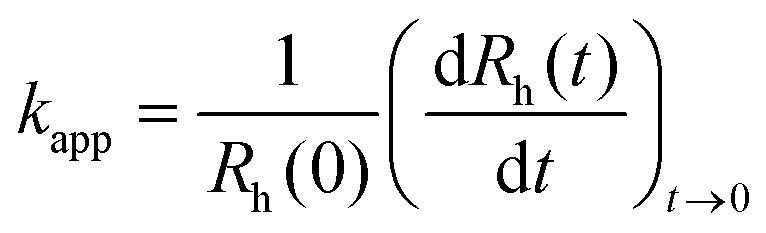 | (2) |
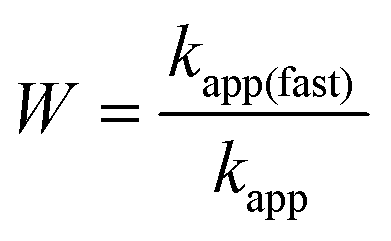 | (3) |
2.4. Refractometry
The refractive index (n) measurements were carried out on an Abbemat 3200 automatic one-wavelength refractometer (Anton Paar) at a wavelength of 589 nm. For the evaluation of the light scattering data, the n values were interpolated using a linear fit according to the following relation:| n = a·cIL + b | (4) |
| IL | Refractive index parameters | Viscosity parameters | |||
|---|---|---|---|---|---|
| a (M−1) | b | A (M−1/2) | B (M−1) | D (M−2) | |
| BMIMCl | 2.92 × 10−2 | 1.3325 | 3.77 × 10−1 | 2.87 × 10−4 | 3.89 × 10−1 |
| BMIMBr | 3.46 × 10−5 | 1.3325 | 2.93 × 10−3 | −3.30 × 10−10 | 4.17 × 10−7 |
| BMIMNO3 | 2.26 × 10−5 | 1.3325 | 2.94 × 10−3 | −3.40 × 10−10 | 3.66 × 10−7 |
| BMIMSCN | 3.61 × 10−2 | 1.3325 | −1.08 × 10−1 | 5.29 × 10−1 | 5.52 × 10−2 |
| BMIMDCA | 3.37 × 10−2 | 1.3326 | −1.63 × 10−1 | 6.86 × 10−1 | 2.06 × 10−2 |
| BMPYRCl | 2.89 × 10−2 | 1.3323 | 5.52 × 10−2 | 4.44 × 10−1 | 1.54 |
| BMPIPCl | 3.21 × 10−2 | 1.3320 | 1.43 × 10−1 | 3.08 × 10−1 | 3.95 × 10−1 |
| BMPYCl | 3.72 × 10−2 | 1.3321 | −2.88 × 10−2 | 4.38 × 10−2 | 3.92 |
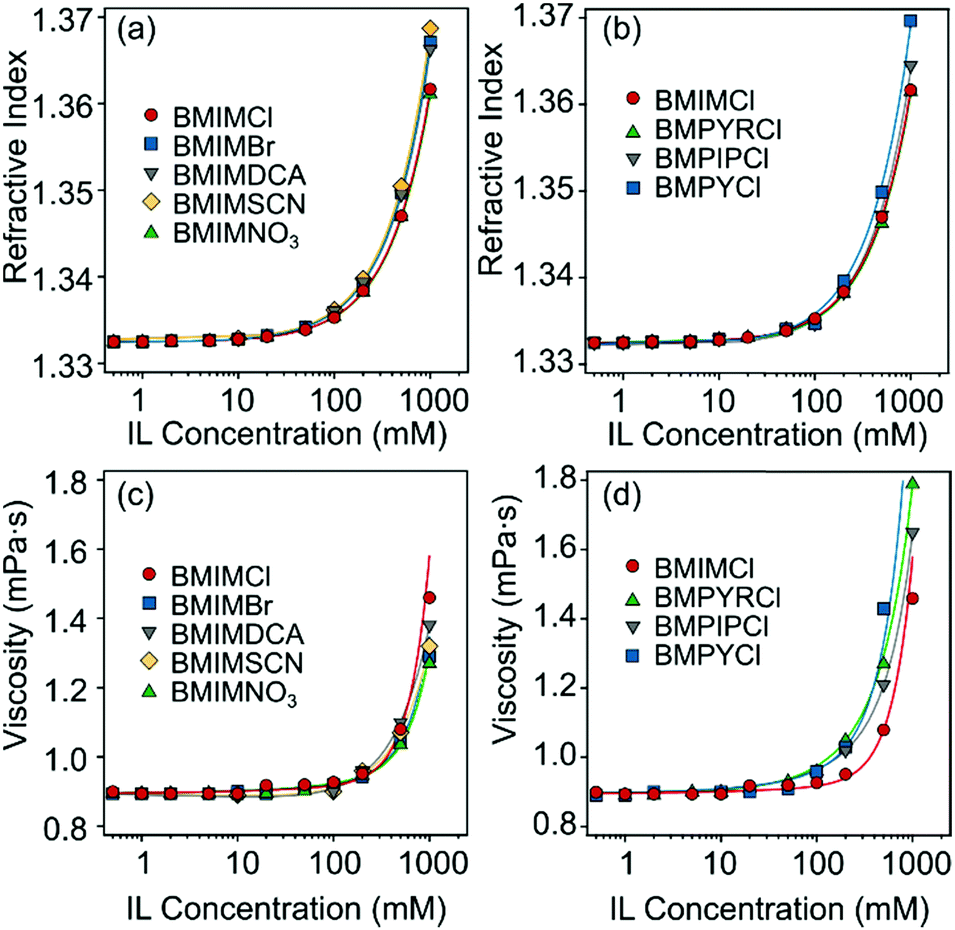 | ||
| Fig. 2 (a and b) Refractive indices and (c and d) viscosities of aqueous IL (BMIMCl, BMIMBr, BMIMNO3, BMIMSCN, BMIMDCA, BMPYCl, BMPIPCl, and BMPYRCl) solutions at different concentrations. The solid lines represent the fits obtained with eqn (4) (a and b) and (5) (c and d). | ||
2.5. Viscosimetry
A LVDV-II+ ProC/P viscometer (Brookfield) was used to measure the dynamic viscosities of the IL solutions in a cone-plate geometry (CPE-40 cone). The η of the different IL-water mixtures was determined by fitting shear stress versus shear rate data with the Casson model.57 The experimental η values were fitted as follows:58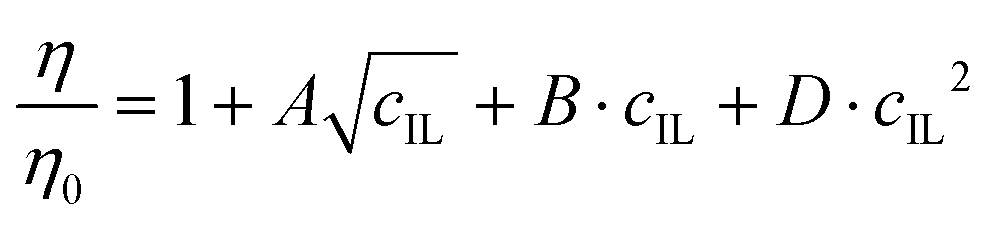 | (5) |
3. Results and discussion
The surface charge properties and colloidal stability of positively charged LDH particles were investigated in the presence of aqueous IL solutions by electrophoresis and DLS, respectively. The IL composition, i.e., the type of anions and cations, were systematically varied in the samples. Accordingly, BMIMCl, BMIMBr, BMIMNO3, BMIMSCN and BMIMDCA ILs were used to vary anions, while the influence of cations was investigated in the presence of BMIMCl, BMPYCl, BMPIPCl and BMPYRCl. Note that the anions are the counterions, while cations act as coions. Therefore, the effect of both counter and coions on the charging and aggregation features was assessed.Ion pair formation in ILs is an important phenomenon in aqueous solutions, which influence the ionic strength significantly. Accordingly, in dilute aqueous solutions of ILs, the ion association is generally weak and ILs tend to fully dissociate. However, as the concentration of ILs increases, ions may start to associate and form ion pairs.27,59 However, as it was pointed out earlier for BMIMCl,60 such ion pair formation is negligible in the concentration range used in our study. It was assumed that the situation is the same (i.e., no ion pairing) with the other ILs studied and the constituents are fully dissociated in solution. However, this can be different at the interface and the oppositely charged ions may form ion pairs with the IL components adsorbed on the particle surface. A similar effect was observed in particle aggregation studies earlier;31 however, no consensus view exists in this topic in the scientific community yet.
3.1. Effect of IL anions
First, the effect of IL anions on the surface charge properties and on the colloidal stability of LDHs was investigated in aqueous solutions of ILs at pH 9. Hence, the composition of ILs was systematically varied so that the ILs contained different anions (Cl−, Br−, DCA−, SCN−, and NO3−) and the BMIM+ cation. Note that under these experimental conditions the anions were the counterions. Therefore, they had the opposite sign of charge as the particle surface. The results on the zeta potentials are shown in Fig. 3a, which indicate that although the zeta potential values are system specific, the trend in their concentration dependence is very similar in all cases. Accordingly, they decrease by increasing the IL concentration (i.e., the ionic strength) due to the surface charge screening by the IL constituents and remained very close to zero at higher ionic strength. One observes that the potential values remained positive in the entire regime investigated due to the structural charge of the particles. The maximum in the trend of the measured zeta potential in the range of low concentrations is due to the electrokinetic effect, which is a typical observation for charged particles moving in an electric field.61 The magnitude of the zeta potential values at the same IL concentration decreases in the order Cl− > Br− > DCA− > SCN− > NO3− (Fig. 3a).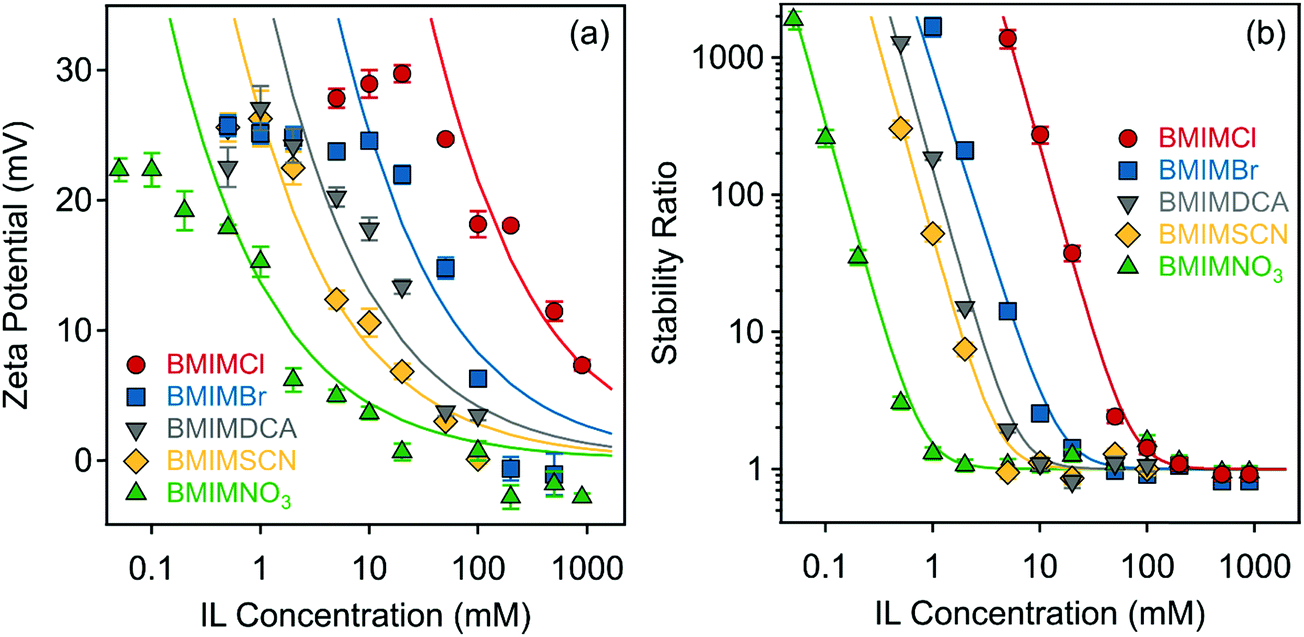 | ||
| Fig. 3 (a) Zeta potentials and (b) stability ratios of LDH particles in the presence of ILs composed of different anions (Cl−, Br−, DCA−, SCN− and NO3−) and the BMIM+ cation. The solid lines are the fits obtained with eqn (6) (a) and (7) (b). | ||
This tendency is also illustrated by the sequence of the surface charge densities (σ) shown in Table 2, which can be estimated from the ionic strength dependence of the potentials by the Debye–Hückel model:49,62
| σ = εε0κζ | (6) |
| IL cations | BMIM+ | BMIM+ | BMIM+ | BMIM+ | BMIM+ | BMPYR+ | BMPY+ | BMPIP+ |
|---|---|---|---|---|---|---|---|---|
| IL anions | Cl− | Br− | DCA− | SCN− | NO3− | Cl− | Cl− | Cl− |
| a Surface charge density determined with eqn (6). b Critical coagulation concentration calculated by eqn (7). c Apparent aggregation rate coefficient in the fast aggregation regime obtained by eqn (2). | ||||||||
| σ (mC m−2) | 16 | 6 | 3 | 2 | 1 | 14 | 8 | 4 |
| CCCb (mM) | 70 | 15 | 6 | 4 | 0.8 | 50 | 30 | 10 |
| k app(fast) (× 10−3 s−1) | 1.07 | 0.93 | 0.79 | 1.04 | 0.76 | 0.97 | 0.98 | 1.06 |
The values of the stability ratio were determined in the dispersions by time-resolved DLS measurements under identical experimental conditions (e.g., particle concentration, pH and IL concentration range) as the ones used in the electrophoretic studies, in order to allow a direct comparison of the tendencies in the measured zeta potential and stability ratio data. As shown in Fig. 3b, the general tendency was the same regardless of the type of the IL anions. Accordingly, slow aggregation and stable samples were observed at low IL concentrations, then the dispersions became unstable at high ionic strength as indicated by stability ratio values close to one. This behavior is typical for systems, in which the main interparticle forces originate from DLVO-type interactions, i.e., the superposition of the attractive van der Waals and the repulsive electrical double layer forces determines the colloidal stability of the dispersions. The observed slow and fast aggregation regimes are separated by the well-defined CCC values, which can be calculated from the stability ratio versus IL concentration plots as63
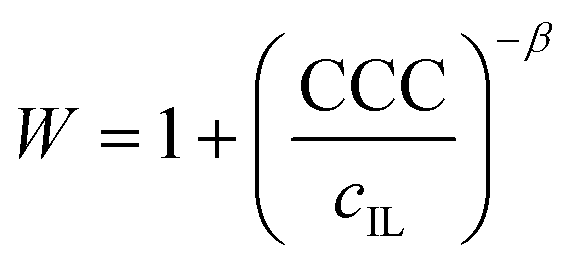 | (7) |
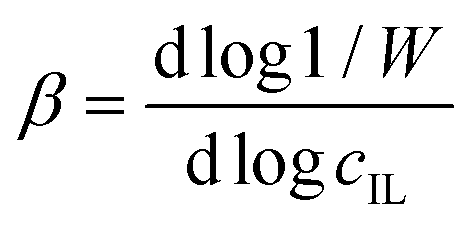 | (8) |
However, in the case of NO3− ions, the situation is more complicated because the surface charge and the CCC values indicated atypical behavior. If one considers the indirect Hofmeister series for positively charged hydrophobic LDH surfaces, these values should be higher than for the SCN− anion, as it was observed in the presence of standard inorganic electrolytes indicating the effect of the IL cation.37,48 Nevertheless, the lowest CCC was measured for this ion. Such a discrepancy from the traditional series is probably due to the different counterion affinity to the oppositely charged particles and the different extent of IL ion pair formation on the surface. It is known that the ions may associate in the bulk in various ILs.64,65 However, the IL constituents may also form ion pairs with the oppositely charged ions, while they are adsorbed on the particle surface.31 Accordingly, BMIM+ coions may adsorb on the LDH surface, which can induce a stronger co-adsorption of NO3− ions owing to associated IL molecules on the surface. Such a phenomenon then influences the surface charge and thus, the CCC as well. The adsorption of BMIM+ on the like-charged LDH surfaces will be further discussed later.
Looking at the zeta potential data before the CCC in the above system, one observes that they change only slightly (within 5 mV) before the CCC (0.8 mM); however, the decrease in the stability ratios is large in this regime. This is due to the low potential (around 20 mV) at limited IL concentrations.66 Such a low zeta potential is at the limit, where colloid particles are usually stable and thus, very small changes in the zeta potentials may give rise to sudden increase in the apparent aggregation rates and consequently, to a decrease in the stability ratios.
3.2. Effect of IL cations
Subsequently, the effect of the IL cations on the charging characteristics and aggregation features of LDH particles was also investigated. Zeta potentials and aggregation rates of positively charged particles were strongly influenced by the type of IL cations (BMIM+, BMPIP+, BMPYR+, and BMPY+) when the same (Cl−) counterion was used. Such a systematic experimental setup allows addressing the specific effect of different IL cations unequivocally. Fig. 4a indicates that the generic trend for the zeta potential values was similar to the one discussed in the previous section, i.e., the zeta potential values decreased as the concentration of the ILs increased due to charge screening.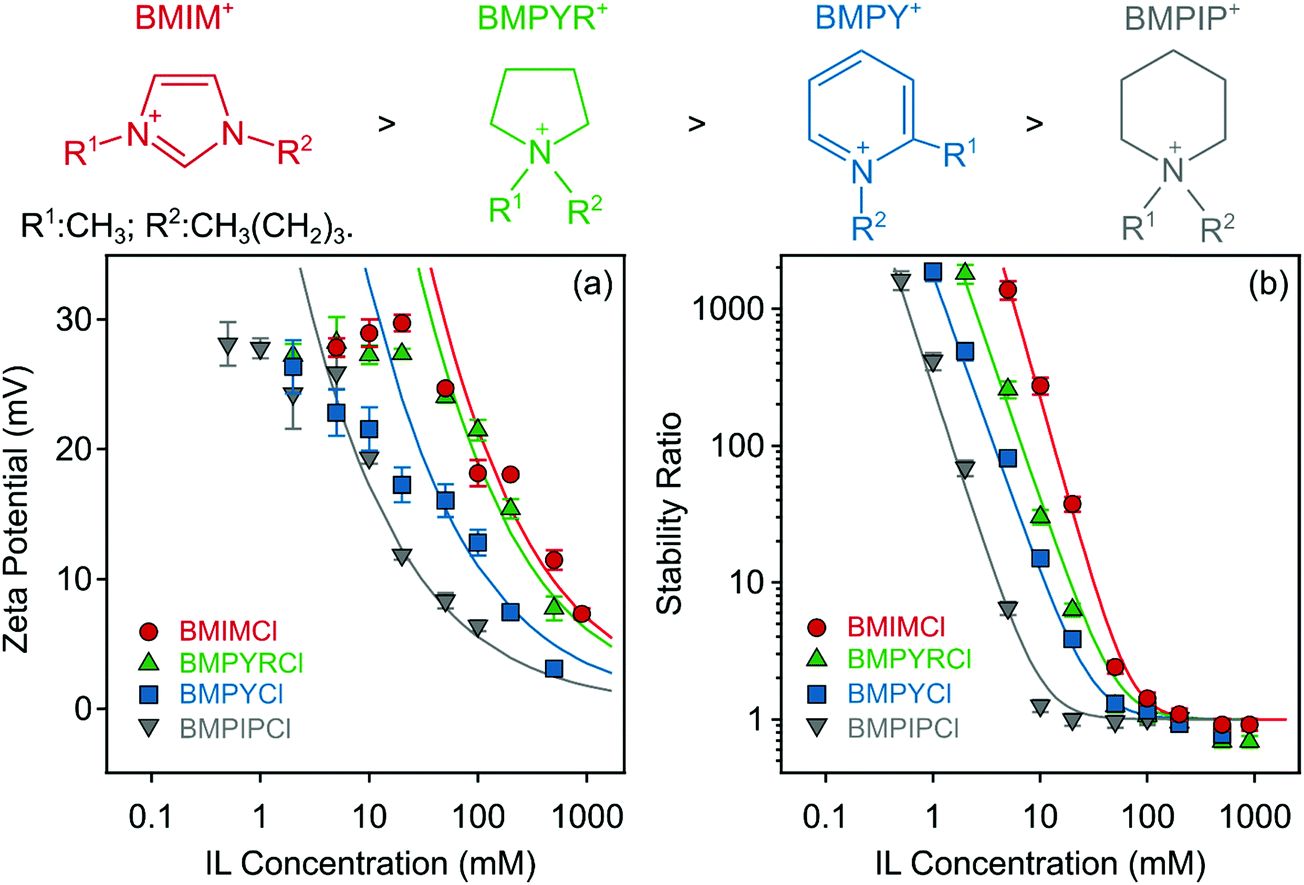 | ||
| Fig. 4 Zeta potentials (a) and stability ratios (b) of LDH particles in the presence of ILs composed of different cations (BMIM+, BMPIP+, BMPYR+, and BMPY+) and the Cl− anion. The solid lines are the fits obtained with eqn (6) (a) and (7) (b). The structures of the IL cations are presented on the top in the order of decreasing CCC from left to right. | ||
Also, the shape of the stability plots (Fig. 4b) was identical to those presented for varying anions (Fig. 3b). Accordingly, slow aggregation occurs at low IL concentrations and with increasing concentration, the stability ratio decreases until it reaches unity and subsequently settles at that value within the experimental error. Nevertheless, these observations are generic for colloidal systems containing charged particles in electrolyte solutions, but the actual values of the potentials and stability ratios vary by changing the composition of the ILs. An increase in these values can be observed in the BMPIP+ < BMPY+ < BMPYR+ < BMIM+ order at the same IL concentration indicating ion specific interactions with the like-charged surface of LDH particles. CCC values reported earlier for colloidal particles in the presence of various monovalent inorganic coions showed no clear dependence on the type of coion.47,67 However, when ILs were involved in the systematic study, the aggregation features were sensitive to the type of coion,31 due to short-range attraction between the IL constituents, as revealed in direct force measurements.30
3.3. Specificities in the fast aggregation regime
Ion specific effects on the aggregation rates in the fast aggregation regime, i.e., above the CCCs, were studied at high IL concentrations, where the viscosity of concentrated IL solutions can differ significantly from that of pure water.27 However, the viscosities of the studied IL solutions were the same within the experimental error in the investigated concentration range (Fig. 2c and d) and thus, the apparent aggregation rates (eqn (2)) were used for comparison with the experimental values to clarify system specificities. Fig. 5 shows that although the CCC depends sensitively on the type of the counter- and coion present, the fast aggregation rate coefficients obtained were almost identical regardless of the nature of the IL constituents.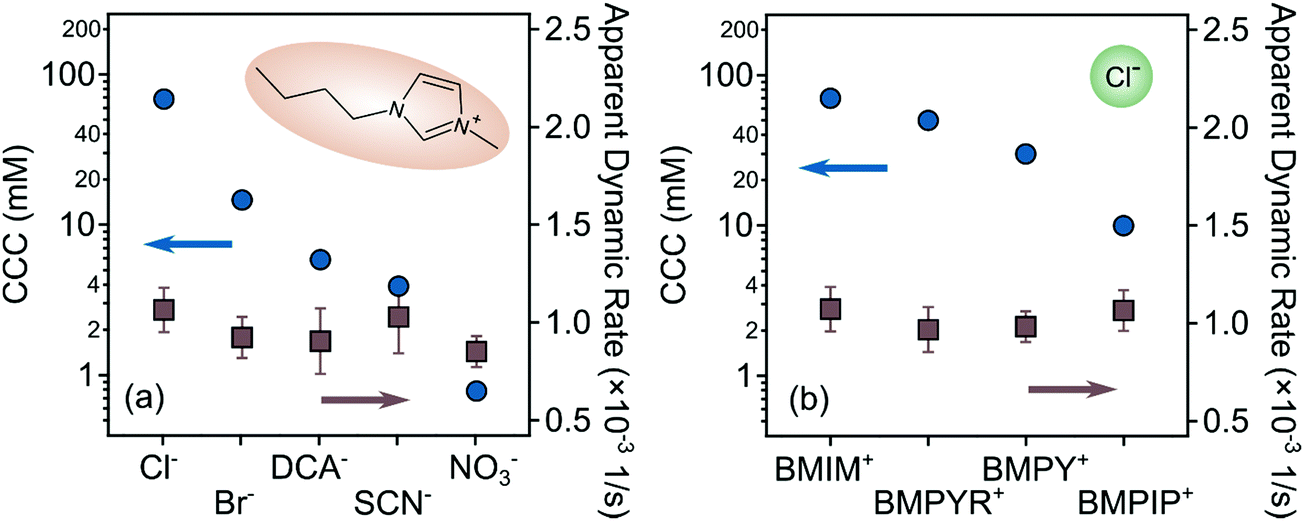 | ||
| Fig. 5 CCCs (left axis) and apparent fast aggregation rate coefficients (right axis) for LDH particles in the presence of (a) different IL anions and (b) cations. The rates were determined by averaging the aggregation rates obtained above the CCC in each system. | ||
These observations indicate that the origin of the attractive forces (i.e., van der Waals and possible hydrophobic interactions) is similar regardless of the type of ions. Similar findings were reported earlier for the latex particles in the presence of various monovalent electrolytes and ILs.31 Moreover, the highest value of the CCC in the case of BMIM+ clearly indicates the adsorption of these coions on the LDH surface, as was assumed already in the study of the IL anion effects in these systems. Such adsorption leads to somewhat higher surface charge density and subsequently, to stronger electrical double layer forces and to higher CCC.
The difference between the effect of anions and cations on the CCC values also deserves a discussion. As shown in Fig. 5, the CCCs vary within two orders of magnitudes for the anions being the highest for Cl− and the lowest for NO3−. Nevertheless, the changes in the cation series were moderate, and decreased by about an order of magnitude. This difference can be explained as follows. The counterions adsorb usually stronger to surfaces and ion specificity is more pronounced.68 Hence, modification in the surface charge upon counterion adsorption is larger, leading to a wider range of CCCs due to the strong relation between surface charge and aggregation features.47 Similar differences were observed in the surface charge density and CCC data once IL constituents were present as counter- or coions in latex31 and titania32 particle dispersions.
3.4. Prevailing interparticle forces
To further elucidate the origin of the interparticle forces between the LDH particles dispersed in aqueous IL solutions, the DLVO theory was used because it can quantitatively predict the CCC for given colloidal particles from the determined surface charge density values as49,66 | (9) |
The calculated and experimental data are shown in Fig. 6 and they indicate that the experimental CCCs could be well-predicted by DLVO theory. These findings further confirmed that the predominant interparticle forces are of a similar origin regardless of the type of IL ions. However, the CCC values are governed by the modification of the surface charge by adsorbing IL constituents, whose affinity is different leading to variation in the strength of the stabilizing electrical double layer forces.
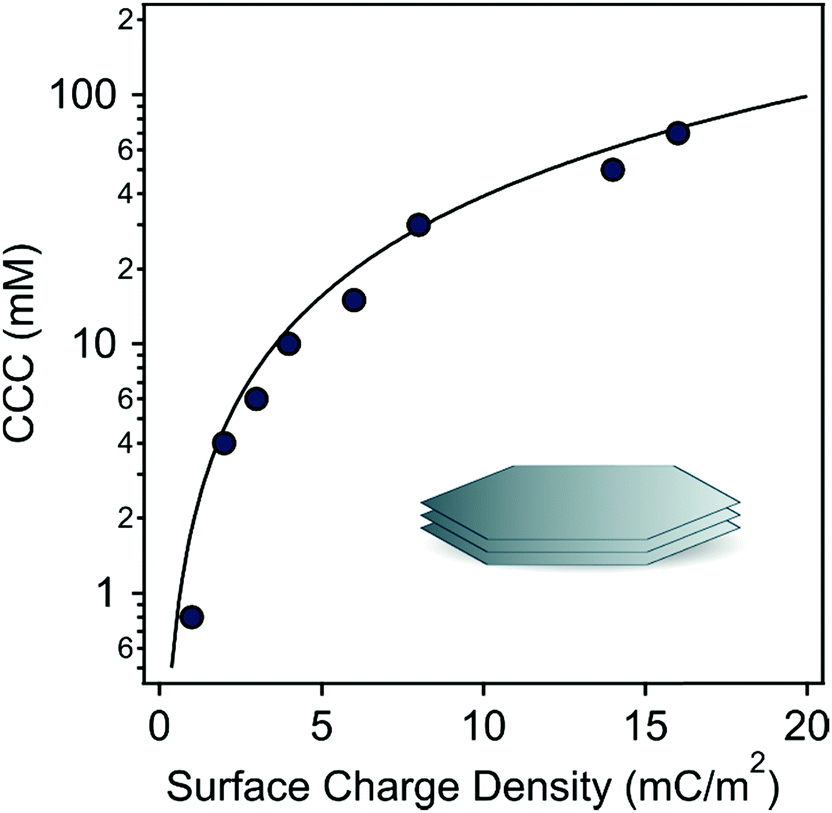 | ||
| Fig. 6 The experimentally obtained CCCs of LDH particles (symbols) versus surface charge densities. The solid line shows the DLVO prediction, which was calculated by eqn (9). | ||
4. Conclusions
The surface charge characteristics and aggregation kinetics of LDH particles were assessed in the presence of various ILs by light scattering techniques. ILs were composed of Cl−, Br−, DCA−, SCN− and NO3− anions as well as BMIM+ cations, while ILs containing Cl− anions with BMIM+, BMPYR+, BMPY+ and BMPIP+ cations were used to probe the effect of anions and cations, respectively. The deviation between the zeta potential and stability ratio data by varying the IL composition indicated specific ion adsorption on the particle surface in both situations. The trend in the obtained surface charge density values corresponds to the order of the CCCs, which partly follows the indirect Hofmeister series. It was also demonstrated that the aggregation rate in the fast aggregation regime does not depend on the type of IL constituent present in the system. This observation suggests that attractive forces acting in the fast aggregation regime are not influenced by the nature of the ions present. Moreover, it was found that the dependence of CCCs on the surface charge densities agrees well with DLVO theory. Although the origin of the interparticle forces was very similar in all systems regardless of the ILs present, ion specific effects were found to be important for the adsorption processes of the ILs on the surface. This observation shed light on the fact that IL ions interact strongly and specifically with the particle surface. Accordingly, the charging and aggregation properties were predominantly determined by the composition of the ILs, as they can modify the surface charge to different extents due to the different adsorption affinity of the IL constituents. The presented information provides useful insights for the scientific community working with LDH-IL dispersions developed for various purposes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Hungarian National Research, Development and Innovation Office (SNN131558) and the Slovenian Research Agency (research core funding No. P1-0201 and the project No. N1-0139 ‘Delamination of Layered Materials and Structure-Dynamics Relationship in Green Solvents’). T. D. is sponsored by the ELTE Márton Áron Special College. The support from the University of Szeged Open Access Fund (5463) is gratefully acknowledged.References
- P. J. Sideris, U. G. Nielsen, Z. H. Gan and C. P. Grey, Science, 2008, 321, 113–117 CrossRef CAS PubMed.
- Z. B. Cao, B. Li, L. Y. Sun, L. Li, Z. P. Xu and Z. Gu, Small Methods, 2019, 4, 1900343 CrossRef.
- J. Li, H. Z. Cui, X. J. Song, G. S. Zhang, X. Z. Wang, Q. Song, N. Wei and J. Tian, RSC Adv., 2016, 6, 92402–92410 RSC.
- F. L. Theiss, S. J. Couperthwaite, G. A. Ayoko and R. L. Frost, J. Colloid Interface Sci., 2014, 417, 356–368 CrossRef CAS PubMed.
- G. Mishra, B. Dash and S. Pandey, Appl. Clay Sci., 2018, 153, 172–186 CrossRef CAS.
- S. Omwoma, W. Chen, R. Tsunashima and Y. F. Song, Coord. Chem. Rev., 2014, 258, 58–71 CrossRef.
- Z. Gu, J. J. Atherton and Z. P. Xu, Chem. Commun., 2015, 51, 3024–3036 RSC.
- V. Rives, M. del Arco and C. Martin, Appl. Clay Sci., 2014, 88–89, 239–269 CrossRef CAS.
- M. N. Pahalagedara, M. Samaraweera, S. Dharmarathna, C. H. Kuo, L. R. Pahalagedara, J. A. Gascon and S. L. Suib, J. Phys. Chem. C, 2014, 118, 17801–17809 CrossRef CAS.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- Z. P. Zheng, W. H. Fan, S. Roy, K. Mazur, A. Nazet, R. Buchner, M. Bonn and J. Hunger, Angew. Chem., Int. Ed., 2015, 54, 687–690 CAS.
- R. Zirbs, K. Strassl, P. Gaertner, C. Schroder and K. Bica, RSC Adv., 2013, 3, 26010–26016 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- M. A. Gebbie, A. M. Smith, H. A. Dobbs, A. A. Lee, G. G. Warr, X. Banquy, M. Valtiner, M. W. Rutland, J. N. Israelachvili, S. Perkin and R. Atkin, Chem. Commun., 2017, 53, 1214–1224 RSC.
- D. A. Beattie, R. M. Espinosa-Marzal, T. T. M. Ho, M. N. Popescu, J. Ralston, C. J. E. Richard, P. M. F. Sellapperumage and M. Krasowska, J. Phys. Chem. C, 2013, 117, 23676–23684 CrossRef CAS.
- A. Sheehan, L. A. Jurado, S. N. Ramakrishna, A. Arcifa, A. Rossi, N. D. Spencer and R. M. Espinosa-Marzal, Nanoscale, 2016, 8, 4094–4106 RSC.
- T. R. Zhan, Y. M. Zhang, Q. Yang, H. H. Deng, J. Xu and W. G. Hou, Chem. Eng. J., 2016, 302, 459–465 CrossRef CAS.
- H. Benes, J. Kredatusova, J. Peter, S. Livi, S. Bujok, E. Pavlova, J. Hodan, S. Abbrent, M. Konefal and P. Ecorchard, Nanomaterials, 2019, 9, 618 CrossRef CAS PubMed.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127–14136 CrossRef CAS PubMed.
- Z. J. Wu, Z. K. Xie, J. Wang, T. Yu, Z. D. Wang, X. G. Hao, A. Abudula and G. Q. Guan, ACS Sustainable Chem. Eng., 2020, 8, 12378–12387 CrossRef CAS.
- E. Delahaye, Z. L. Xie, A. Schaefer, L. Douce, G. Rogez, P. Rabu, C. Gunter, J. S. Gutmann and A. Taubert, Dalton Trans., 2011, 40, 9977–9988 RSC.
- F. Seidi, M. Jouyandeh, S. M. R. Paran, A. Esmaeili, Z. Karami, S. Livi, S. Habibzadeh, H. Vahabi, M. R. Ganjali and M. R. Saeb, Colloids Surf., A, 2021, 611, 125826 CrossRef CAS.
- E. Vanecht, K. Binnemans, S. Patskovsky, M. Meunier, J. W. Seo, L. Stappers and J. Fransaer, Phys. Chem. Chem. Phys., 2012, 14, 5662–5671 RSC.
- M. Mamusa, J. Siriex-Plenet, F. Cousin, E. Dubois and V. Peyrea, Soft Matter, 2014, 10, 1097–1101 RSC.
- I. Szilagyi, T. Szabo, A. Desert, G. Trefalt, T. Oncsik and M. Borkovec, Phys. Chem. Chem. Phys., 2014, 16, 9515–9524 RSC.
- C. Guibert, V. Dupuis, J. Fresnais and V. Peyre, J. Colloid Interface Sci., 2015, 454, 105–111 CrossRef CAS PubMed.
- Z. Q. He and P. Alexandridis, Phys. Chem. Chem. Phys., 2015, 17, 18238–18261 RSC.
- V. Valmacco, G. Trefalt, P. Maroni and M. Borkovec, Phys. Chem. Chem. Phys., 2015, 17, 16553–16559 RSC.
- T. Oncsik, A. Desert, G. Trefalt, M. Borkovec and I. Szilagyi, Phys. Chem. Chem. Phys., 2016, 18, 7511–7520 RSC.
- P. Rouster, M. Pavlovic, T. Cao, B. Katana and I. Szilagyi, J. Phys. Chem. C, 2019, 123, 12966–12974 CrossRef CAS.
- B. Katana, D. Takács, F. D. Bobbink, P. J. Dyson, N. B. Alsharif, M. Tomšič and I. Szilagyi, Phys. Chem. Chem. Phys., 2020, 22, 24764–24770 RSC.
- B. Katana, D. Takacs, E. Csapo, T. Szabo, A. Jamnik and I. Szilagyi, J. Phys. Chem. B, 2020, 124, 9757–9765 CrossRef CAS PubMed.
- K. Higashitani, K. Nakamura, T. Fukasawa, K. Tsuchiya and Y. Mori, Langmuir, 2018, 34, 2505–2510 CrossRef CAS PubMed.
- J. L. Trompette and J. F. Lahitte, J. Phys. Chem. B, 2019, 123, 3859–3865 CrossRef CAS PubMed.
- W. Y. Yu, N. Du, Y. T. Gu, J. G. Yan and W. G. Hou, Langmuir, 2020, 36, 6557–6568 CrossRef CAS PubMed.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS.
- L. Medda, C. Carucci, D. F. Parsons, B. W. Ninham, M. Monduzzi and A. Salis, Langmuir, 2013, 29, 15350–15358 CrossRef CAS PubMed.
- D. Constantinescu, H. Weingartner and C. Herrmann, Angew. Chem., Int. Ed., 2007, 46, 8887–8889 CrossRef CAS PubMed.
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- H. I. Okur, J. Hladilkova, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed.
- J. Rubin, A. San Miguel, A. S. Bommarius and S. H. Behrens, J. Phys. Chem. B, 2010, 114, 4383–4387 CrossRef CAS PubMed.
- P. Lo Nostro and B. W. Ninham, Chem. Rev., 2012, 112, 2286–2322 CrossRef CAS PubMed.
- B. B. Kang, H. C. Tang, Z. D. Zhao and S. S. Song, ACS Omega, 2020, 5, 6229–6239 CrossRef CAS PubMed.
- D. F. Parsons and B. W. Ninham, Langmuir, 2010, 26, 6430–6436 CrossRef CAS PubMed.
- T. Oncsik, G. Trefalt, M. Borkovec and I. Szilagyi, Langmuir, 2015, 31, 3799–3807 CrossRef CAS PubMed.
- M. Pavlovic, R. Huber, M. Adok-Sipiczki, C. Nardin and I. Szilagyi, Soft Matter, 2016, 12, 4024–4033 RSC.
- D. F. Evans and H. Wennerstrom, The colloidal domain, John Wiley, New York, 1999 Search PubMed.
- D. F. Parsons, M. Bostrom, P. Lo Nostro and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- J. M. Peula-Garcia, J. L. Ortega-Vinuesa and D. Bastos-Gonzalez, J. Phys. Chem. C, 2010, 114, 11133–11139 CrossRef CAS.
- T. Lopez-Leon, J. L. Ortega-Vinuesa and D. Bastos-Gonzalez, ChemPhysChem, 2012, 13, 2382–2391 CrossRef CAS PubMed.
- D. G. Evans and R. C. T. Slade, in Layered Double Hydroxides, ed. X. Duan and D. G. Evans, 2006, vol. 119, pp. 1–87 Search PubMed.
- A. V. Delgado, F. Gonzalez-Caballero, R. J. Hunter, L. K. Koopal and J. Lyklema, J. Colloid Interface Sci., 2007, 309, 194–224 CrossRef CAS PubMed.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS PubMed.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- D. D. Joye, J. Colloid Interface Sci., 2003, 267, 204–210 CrossRef CAS PubMed.
- H. D. B. Jenkins and Y. Marcus, Chem. Rev., 1995, 95, 2695–2724 CrossRef CAS.
- M. Bester-Rogac, J. Hunger, A. Stoppa and R. Buchner, J. Chem. Eng. Data, 2011, 56, 1261–1267 CrossRef CAS.
- M. Bester-Rogac, A. Stoppa, J. Hunger, G. Hefter and R. Buchner, Phys. Chem. Chem. Phys., 2011, 13, 17588–17598 RSC.
- M. Borkovec, S. H. Behrens and M. Semmler, Langmuir, 2000, 16, 5209–5212 CrossRef CAS.
- G. Trefalt, I. Szilagyi and M. Borkovec, J. Colloid Interface Sci., 2013, 406, 111–120 CrossRef CAS PubMed.
- D. Grolimund, M. Elimelech and M. Borkovec, Colloids Surf., A, 2001, 191, 179–188 CrossRef CAS.
- A. Stoppa, J. Hunger, G. Hefter and R. Buchner, J. Phys. Chem. B, 2012, 116, 7509–7521 CrossRef CAS PubMed.
- H. K. Stassen, R. Ludwig, A. Wulf and J. Dupont, Chem. – Eur. J., 2015, 21, 8324–8335 CrossRef CAS PubMed.
- M. Galli, S. Saringer, I. Szilagyi and G. Trefalt, Colloids Interfaces, 2020, 4, 20 CrossRef CAS.
- T. Hegedus, D. Takacs, L. Vasarhelyi, I. Szilagyi and Z. Konya, Langmuir, 2021, 37, 2466–2475 CrossRef CAS PubMed.
- T. Sugimoto, T. C. Cao, I. Szilagyi, M. Borkovec and G. Trefalt, J. Colloid Interface Sci., 2018, 524, 456–464 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |