
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding gel-to-crystal transitions in supramolecular gels†
Demetra
Giuri
a,
Libby J.
Marshall
 b,
Claire
Wilson
b,
Annela
Seddon
*cd and
Dave J.
Adams
*b
b,
Claire
Wilson
b,
Annela
Seddon
*cd and
Dave J.
Adams
*b
aDipartimento di Chimica Giacomo Ciamician, Alma Mater Studiorum, Università di Bologna, Via Selmi, 2, 40126, Bologna, Italy
bSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
cSchool of Physics, HH Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK. E-mail: annela.seddon@bristol.ac.uk
dBristol Centre for Functional Nanomaterials, HH Wills Physics Laboratory, University of Bristol, Tyndall Avenue, Bristol, BS8 1TL, UK
First published on 12th July 2021
Abstract
Most supramolecular gels are stable or assumed to be stable over time, and aging effects are often not studied. However, some gels do show clear changes on aging, and a small number of systems exhibit gel-to-crystal transitions. In these cases, crystals form over time, typically at the expense of the network underpinning the gel; this leads to the gel falling apart. These systems are rare, and little is known about how these gel-to-crystal transitions occur. Here, we use a range of techniques to understand in detail a gel-to-crystal transition for a specific functionalised dipeptide based gelator. We show that the gel-to-crystal transition depends on the final pH of the medium which we control by varying the amount of glucon-δ-lactone (GdL) added. In the gel phase, at low concentrations of GdL, and at early time points with high concentrations of GdL, we are able to show the nanometre scale dimensions of the self-assembled fibre using SAXS; however there is no evidence of molecular ordering of the gel fibres in the WAXS. At low concentrations of GdL, these self-assembled fibres stiffen with time but do not crystallise over the timescale of the SAXS experiment. At high concentrations of GdL, the fibres are already stiffened, and then, as the pH drops further, give way to the presence of crystals which appear to grow preferentially along the direction of the fibre axis. We definitively show therefore that the gel and crystal phase are not the same. Our work shows that many assumptions in the literature are incorrect. Finally, we also show that the sample holder geometry is an important parameter for these experiments, with the rate of crystallisation depending on the holder in which the experiment is carried out.
Introduction
Low molecular weight gels are formed by the self-assembly of small molecules into one-dimensional structures1–3 which entangle and cross-link to form a network.4,5 There are many classes of molecule that have been shown to be effective gelators, and many solvents that can be gelled depending on the exact gelator used.1,6,7 Designing low molecular weight gelators is difficult.1 A number of ways have been used to find and design effective gelators. Commonly, a gelator is found and then close structural analogues prepared; it is not uncommon here for many not to form gels. Computational approaches are becoming more important.8–12 Solvent parameters can be used to provide a means of identifying which solvents a particular molecule will gel.13 One approach that is widely discussed is the use of information accessed from crystal structures. There is an argument that there is a link between the gel and crystal phase or at least there is sufficient structural similarity to infer packing in the gel from a crystal structure.14–22 Important motifs that lead to specific types of one-dimensional interaction in certain crystal structures have been used to guide design of further analogues. It is not uncommon with this approach to grow crystals in one solvent mixture and infer information as to packing to design gelators for a completely different solvent system. In some cases, it has been shown that the packing in the gel and in a crystal phase are not the same, although these were accessed at different concentrations.23Functionalized dipeptides are one class of effective hydrogelator.2,24–29 Typically, such dipeptides form gels in a number of ways, but one successful method is a pH switch.24,25 Here, a solution of the dipeptide is prepared at high pH (typically >9) which forms a micellar dispersion of the gelator; gels are formed when the pH is decreased.24 Many of the dipeptides that can be used to prepare gels do so by forming long anisotropic structures such as cylindrical or elliptical fibers, which entangle and laterally associate to form the network.24,25 Generally, the assumption is that the observed properties of the gels do not change over time. Of relevance here, a small number of dipeptides and functionalized dipeptides form gels from which crystals form over time.20,30–35 This gel-to-crystal transition results in the gel becoming weaker and eventually falling apart leading to phase separation into crystals and a solution.
The packing in dried stalks prepared from the gel phase can be determined using fiber X-ray diffraction.30,31 Single crystal X-ray diffraction can be used to determine the packing in the crystal phase. This implies that the packing is different in the gel and crystal phase,30,31 although there may be an effect of drying the gel. However, how the transition from gel-to-crystal occurs is not understood. Others have also shown that crystals can be grown from the gel phase in a range of systems.14,36–44 This can be spontaneous.45 For example, Andrews et al. have shown a case where a metastable gel transforms to a crystal phase, leading them to suggest that for their case the gelation is the first stage of the crystallisation process.46 Alternatively, the transition to the crystal phase can be induced by freezing.47
Here, we follow the entire process from solution-to-gel-to-crystal. We show conclusively that packing within the gel and crystal phase are not the same, showing that many of the assumptions in this field are likely incorrect. We also show that there is a significant effect on the outcome of the self-assembly in moving from H2O to D2O, providing another parameter for optimizing and using these systems, and also that there is a significant effect on the rate of the transition from gel-to-crystal depending on the geometry of the container in which the gelation occurs.
We focus on the gels formed from (2S)-2-(2S)-2-{2-naphthalen-2-yloxyacetamido}-3-phenylpropanamido-3-phenylpropanoic acid (2NapAA; Fig. 1). Whilst we have a large library of these materials, 2NapAA is a rare example that forms a gel from which crystals form over time.31 Most other examples either form stable gels or directly crystallize or precipitate.
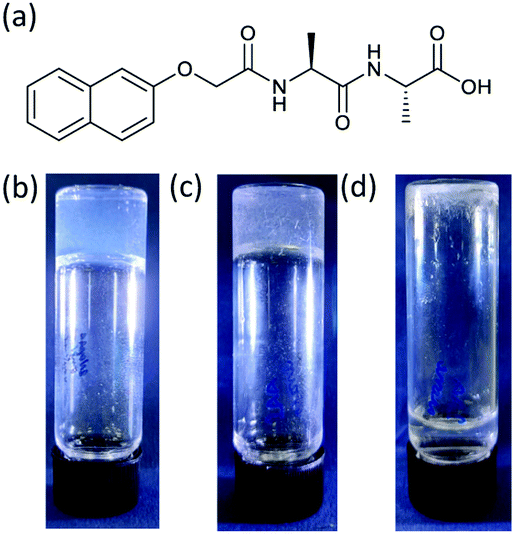 | ||
| Fig. 1 (a) The chemical structure of 2NapAA; photographs of (b) a gel formed from 2NapAA; (c) crystals forming within the gel phase (d) crystals sedimented to the bottom of the sample tube with no remaining gel phase. | ||
Results and discussion
Free-flowing stock solutions of 2NapAA were prepared at pH 10.5 at a concentration of 5 mg mL−1. Gelation was then triggered by a slow reduction in pH from approximately 10.5 to around 3.5 induced by the hydrolysis of GdL.48 As described previously,31 initially a metastable gel is formed, followed by a slow crystallization from the gel phase. Here, we show that the rate of crystallization increases when larger quantities of GdL are used, as this results in a faster decrease in the pH of the system. By careful control over the amount of GdL used, crystals suitable for X-ray diffraction can be grown directly from the gel phase (see below).31 Crystal formation required a minimum concentration of GdL of 6 mg mL−1 at this concentration of 2NapAA, correlating with when the pH decreases below 4. This correlation with a specific pH suggests that the crystallization is driven by a specific degree of protonation.To exemplify the different behavior on the basis of relative rates of gelation and crystallization, three concentrations of GdL were chosen for further study, 4 mg mL−1 (denoted AA4), 20 mg mL−1 (AA20) and 36 mg mL−1 (AA36). In all cases, a fixed concentration of 2NapAA was used (5 mg mL−1). The samples were followed with time by rheology (Fig. 2). It should be noted that direct suspension of 2NapAA at low pH does not lead to gel formation (Fig. S14, ESI†). The rate of hydrolysis of GdL is temperature dependent.49 Hence, there is also an effect of temperature on the rate of transition from solution-to-gel-to-crystal (Fig. S15 and S16, ESI†).
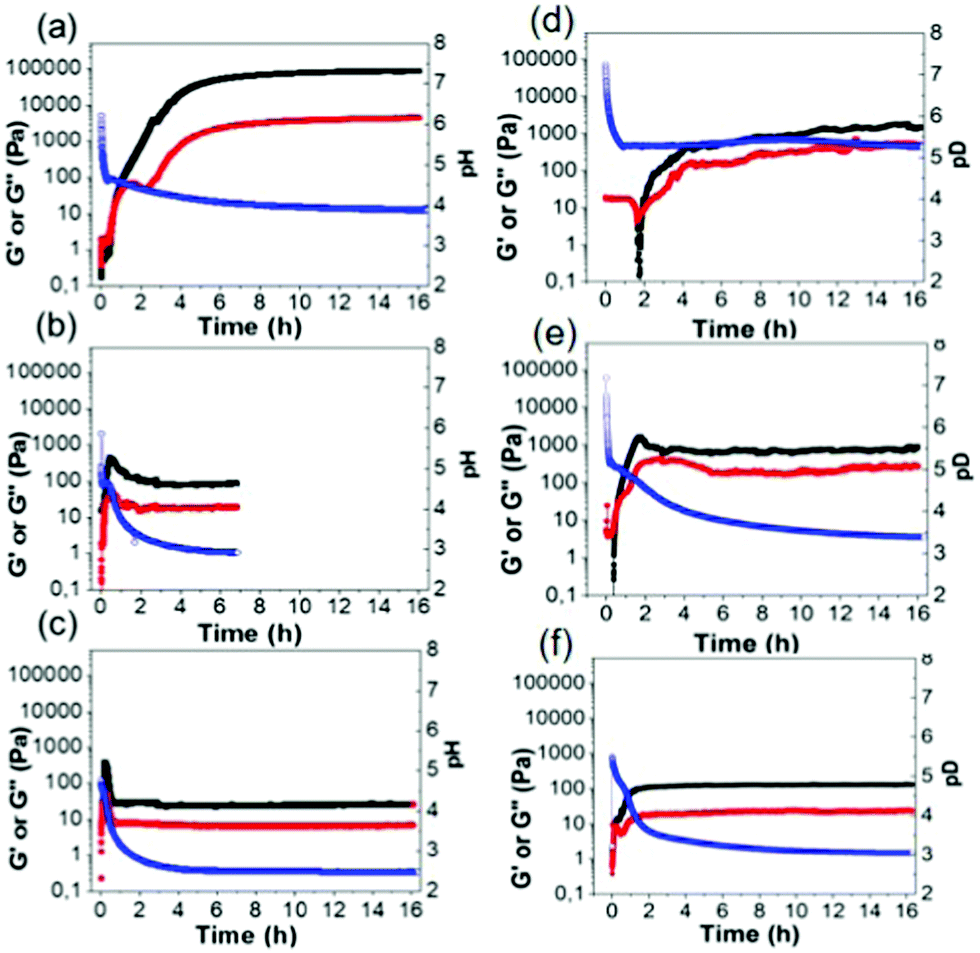 | ||
| Fig. 2 Time sweep experiments for (a) AA4, (b) AA20 and (c) AA36 in H2O and (d) AA4(D), (e) AA20(D) and (f) AA36(D) in D2O. In all cases, the black data represent G′, the red data represent G′′ and the blue data show the pH or pD. The relatively noisy data in some cases is due to crystallization occurring during the experiment. This is also why not all experiments were carried out for the same length of time. | ||
The slowest pH change occurred with the lowest amount of GdL (AA4). Here, the pH drops initially at a relatively quick rate before reaching the apparent pKa of the system (5.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31). At this point, the rate of decrease in the pH changes due to buffering of the system by the 2NapAA. From the time-sweep rheology, the storage modulus (G′) and loss modulus (G′′) begin to increase at the point where the apparent pKa is reached. Initially, both G′ and G′′ are very similar before G′ starts to dominate significantly over G′′. There is a decrease in G′′, before it starts to increase again. We have reported similar behavior previously for another system;50 the decrease in G′′ implies there is a structural transition occurring in the solutions as the sample reaches the gel point where G′ becomes greater than G′′. The final gels are stiff (G′ of around 100 kPa) and these gels are stable to further change for at least two weeks. For AA20, G′ and G′′ both increase as the pH decreases, but at a pH of below 4.1, G′ and G′′ decrease before becoming constant. This correlates with the onset of crystallization. For AA36, this behavior happens faster than for AA20 as expected from the faster decrease in pH.
31). At this point, the rate of decrease in the pH changes due to buffering of the system by the 2NapAA. From the time-sweep rheology, the storage modulus (G′) and loss modulus (G′′) begin to increase at the point where the apparent pKa is reached. Initially, both G′ and G′′ are very similar before G′ starts to dominate significantly over G′′. There is a decrease in G′′, before it starts to increase again. We have reported similar behavior previously for another system;50 the decrease in G′′ implies there is a structural transition occurring in the solutions as the sample reaches the gel point where G′ becomes greater than G′′. The final gels are stiff (G′ of around 100 kPa) and these gels are stable to further change for at least two weeks. For AA20, G′ and G′′ both increase as the pH decreases, but at a pH of below 4.1, G′ and G′′ decrease before becoming constant. This correlates with the onset of crystallization. For AA36, this behavior happens faster than for AA20 as expected from the faster decrease in pH.
There can be differences for this kind of system in some cases when carrying out the self-assembly and gelation in D2O compared to in H2O.51,52 Primarily here, this is because the rate of hydrolysis of GdL is different in H2O compared to D2O,49 leading to changes in the rate of gelation, although in some cases the micellar structures formed at high pH can also differ.51 2NapAA is more prone to crystallization in D2O (Fig. 2; note the samples in D2O have a “(D)” after the name). Even with 4 mg mL−1 of GdL, crystallization occurs and the rheological data with time show a very different profile in H2O and D2O (Fig. 2a and d).
The evolution of structure in these systems was followed using optical microscopy. For AA4, large spherulitic domains (∼0.7 mm) can be seen in sample AA4 after 50 minutes (Fig. S1 and S2, ESI†). No sign of crystal formation was observed over several hours. For AA20, both spherulitic domains and crystals appear after 10 minutes after the addition of GdL (Fig. S4, ESI†). The spherulitic domains are smaller compared to those in sample AA4 while the crystals reach a radius of about 1.4 mm after 1 hour. The formation of crystals over this time period correlates with the time sweep rheology. Finally, the kinetics of crystal formation for AA36 is even faster: many small and thin crystals appear 2 minutes after the addition of the GdL, which grow with time (Fig. S6, ESI†). Similar structures were formed in the systems in D2O, although the growth of crystals is slower with smaller crystals at early time; the crystals for AA20(D) are thinner and longer as compared to those of AA20, which are larger and more regular in shape (see Table S1, ESI†). Powder X-ray diffraction (pXRD) was performed on the crystals after the growth inside the gels, with all crystals collected directly from their solution. All patterns matched the calculated pattern determined from the crystal structure (Fig. S9 and S10, ESI†).
To understand how the gel phase converts to the crystal phase, we used confocal microscopy and small angle scattering. Both of these techniques allow the sample to be monitored without drying artefacts, which are known to be an issue in this class of gels.53 Incorporating Nile Blue and using confocal microscopy for AA20, spherulitic domains with dimensions of about 50 μm are clearly visible in the gel phase 5 minutes after the addition of the GdL (Fig. 3a). At this point, no crystals are formed and the spherulites are formed from what appear to be fibrous structures. The spherulitic domains seem to become smaller with time (Fig. 3), which is presumably because the gel network is converting to a crystal phase. Indeed, after 1 hour, it is possible to image larger, straight crystals in addition to small spherulitic domains (Fig. 3c). This again shows that the gel state and the crystal state are different.
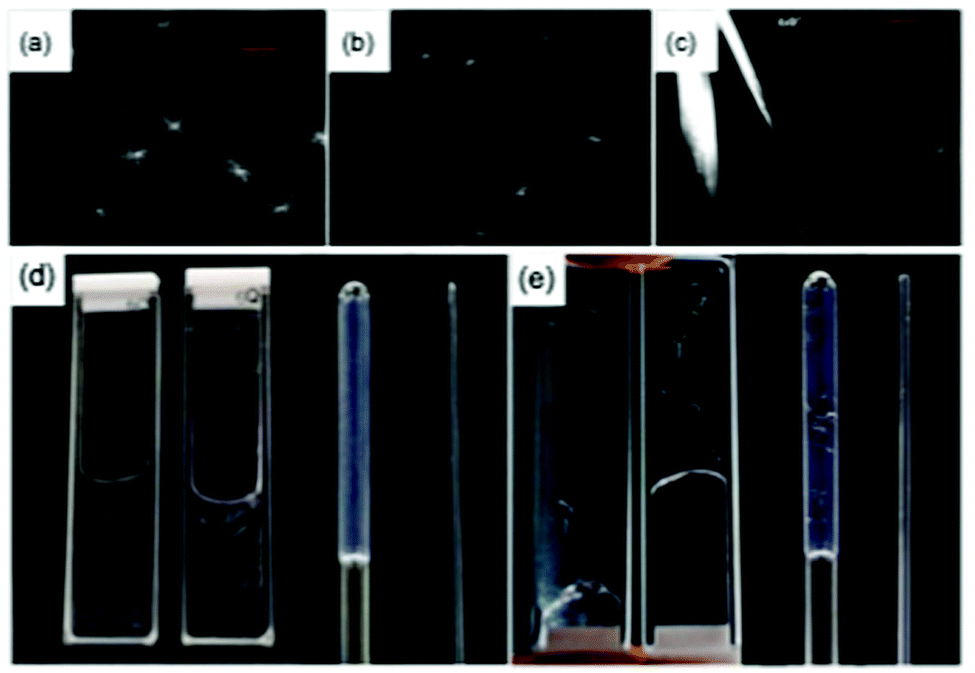 | ||
| Fig. 3 Confocal microscopy images of AA20 after (a) 5 minutes; (b) 15 minutes; (c) 60 minutes. In all cases, the scale bar shows 50 μm. (d) and (e) show photographs of samples prepared from (d) AA4 19 hours after adding GdL and (e) AA20 1 hour after addition of GdL. In both (d) and (e), the same stock solutions were used to prepare all samples. The photographs show from left to right samples in a 1 mm thickness cuvette, a 2 mm thickness cuvette, a 4 mm diameter NMR tube and a 1.5 mm diameter capillary. | ||
In carrying out the above experiments, it became clear that the kinetics of gel and crystal formation are highly dependent on the geometry of the sample holders used. As examples, images of AA4 and AA20 in different geometry sample holders are shown in Fig. 3d and e respectively. Clear differences were observed which were dependent only on the geometry of the holder. The formation of crystals in sample AA4 started after 30 minutes in the 1 mm path quartz cuvette and after 2 hours in the 2 mm path cuvette, while in the NMR tube and the capillary gels were formed without crystallization even after several days. For sample AA20, crystals formed in all the holders, starting after 10 minutes from the addition of the GdL. The crystals are bigger in the NMR tube and in the 2 mm cuvette, smaller and fewer in the 1 mm cuvette and very small and few in the capillary. These observations complicate comparison across techniques which typically use different geometries of sample environment. The rheology and pH data in Fig. 2 were collected in the same geometry, but the microscopy for example has to be collected in a different shape holder. Hence, direct comparison of timescales is potentially difficult. Elsewhere, it has been noted that the surface chemistry on which the gel forms can lead to differences in the networks.54 Here, the two cuvettes and capillary are formed from quartz, whilst the NMR tube is borosilicate glass. As such, there may be some effect, but the differences observed in the degree of crystallization even within the quartz holders imply that this is not the dominating difference. We note that confinement is known to affect the crystallisation process.55 This is reflected in our experiments, where crystallisation did not occur in 1.5 mm SAXS capillaries but was easily observed in 3.5 mm capillaries, and in NMR tubes.
Hence, when following the gel-to-crystal transition using small angle X-ray scattering (SAXS) and wide angle X-ray scattering (WAXS), Fig. 4, we found we needed to adapt exact quantities of GdL due to a need to choose thin capillaries.
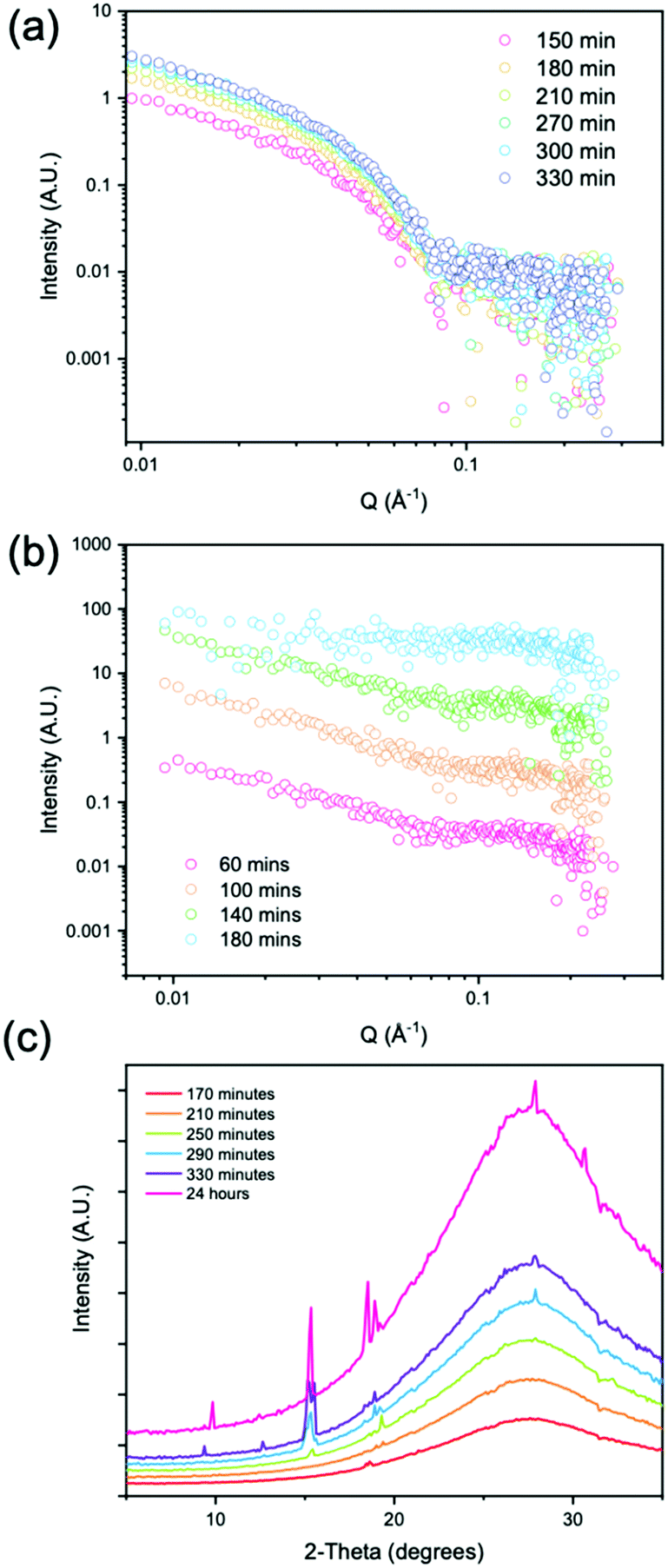 | ||
| Fig. 4 (a) SAXS data for low concentration of GdL showing gelation and increase in Kuhn length with time; (b) SAXS data for high concentration of GdL showing breakdown of gel prior to crystallization over time. The data are offset on the y-axis for clarity; (c) WAXS pattern showing the growth of peaks during the crystallization of 2NapAA over time. Data are offset for clarity. No background subtraction has been applied due the difficulty of exact subtraction of the capillary and solvent in the high Q region. | ||
We followed the evolution of the scattering as the pH decreased using either 4 mg mL−1 of GdL (denoted low concentration) and 8 mg mL−1 of GdL (denoted high concentration). The low concentration led to gelation only with no evidence of crystallization whereas the high concentration led to rapid gelation followed by a gel-to-crystal transition. At the low concentration of GdL, stable gelation occurred, and as expected from data from related systems,56 the SAXS scattering intensity increased in intensity over time. The data could be fitted to a flexible cylinder model with a radius which at early time points has a value of 2.9 nm, and which reaches a value of 4 nm after 210 minutes. Over further time points, as the pH decreases, there is little change in the radius, with the overall length being outside that which can be accessed readily with this method. Importantly however, the Kuhn length increases over time, starting at a value of approximately 5 nm, and reaching a value of approximately 20 nm by 270 minutes. This shows that the fibres are becoming more rigid with time (Fig. 4a and Table S3, ESI†). From WAXS data taken of the gels at low concentrations of GdL, there are no peaks visible, which suggests that there is no ordered molecular packing in the gel fibres.
At the high concentrations of GdL, the data at early times can be best fit to a cylinder model with at the first two time points, the radius being approximately 3.7 nm. The radius at the final time point is 3.0 nm, which can be explained by the fact that during the measurement, the gel to crystal transition is occurring. As a Kuhn length can no longer be fitted, this implies that the persistence length also lies outside the accessible range, i.e., that the persistence length continues to increase as the pH is decreased further (Table S4, ESI†). This differs from our previous data for gel phases where the system tends to a certain persistence length. This suggests that the 2NapAA crystallization is driven by this ability to increase the persistence length as the pH is decreased and hence charge is removed. At longer times, the signal intensity in the SAXS is lost, implying that the structures underpinning the gel phase are no longer present.
Prior to 170 minutes, there are no peaks present in the WAXS. After 170 minutes, the first peak appears in the WAXS which coincides with the loss of the structure in the SAXS. Initially, the WAXS shows the presence of a single peak at Q = 1.32 Å−1 which has a real space value of 4.6 Å. This distance corresponds closely to the molecular separation along the fiber axis as found by fiber diffraction of the dried gel phase.31 When compared to the simulated pXRD pattern, this distance matches well with the 111 plane (Fig. 5a). This implies that growth of the crystal begins preferentially along the fibre axis, which is also indicated by the increase in the persistence length.
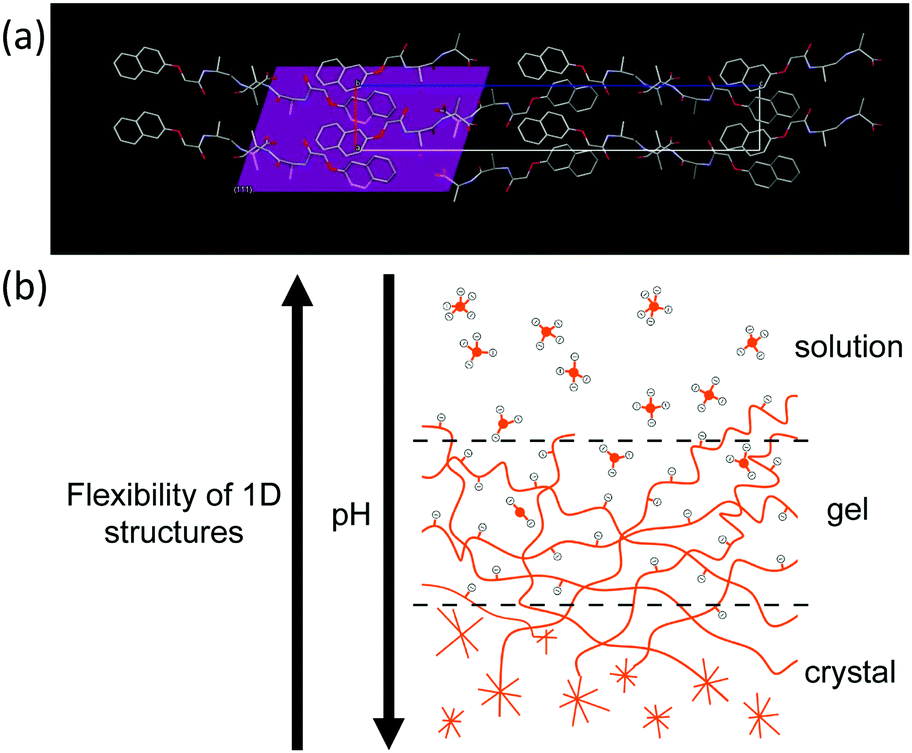 | ||
| Fig. 5 (a) Single crystal structure of 2NapAA showing the 111 plane, with initial plane of growth; (b) cartoon showing the solution-to-gel-to-crystal transition for 2NapAA. As the pH decreases, charge is removed and the flexibility of the structures decreases, resulting in crystal formation. | ||
At 250 minutes, a second peak appears which has a Q value of 1.09 Å−1 (5.76 Å) and comparison with the simulated pXRD indicates that this is the 1 0 1 plane. At 290 minutes, a peak at 1.96 Å−1 appears which has a real space value of 3.2 Å. This value is close to that for the 124 plane found by pXRD. This remains constant throughout the time series. Over the course of the experiment, further peaks in the WAXS continue to appear, the positions of which are given in Table S5 (ESI†) and which all have a corresponding peak in the simulated pXRD pattern.
These results show that the gel phase and crystal phase of 2NapAA are not the same. In the gel phase, at low concentrations of GdL, and at early time points with high concentrations of GdL, we are able to show the nanometre scale dimensions of the self-assembled fibre using SAXS; however, there is no evidence of molecular ordering of the gel fibres in the WAXS. At low concentrations of GdL, these self-assembled fibres stiffen with time but do not crystallise over the timescale of the SAXS experiment. At high concentrations of GdL, the fibres are already stiffened, and then, as the pH drops further, give way to the presence of crystals which appear to grow preferentially along the direction of the fibre axis and with the same molecular spacing as we see in our previous work on dried gels.
Overall, these data show that the initially formed structures are flexible fibres with a radius of around 4 nm. Over time, the persistence length increases as the charge is removed driven by a decrease in the pH. When sufficient charge has been removed, the rigidity is such that transformation to the crystal phase is possible. The unusual gel-to-crystal transformation seen for 2NapAA as compared to most other examples of dipeptide-based gelators can be explained by the tendency to increase persistence length as charge is removed (Fig. 5b). This does not seem to occur for other examples. The exact reason for this is unclear but could be due to a blockier removal of charge as opposed to a gradual removal of charge uniformly along the fibre as we have suggested for other examples.50
Conclusions
We have shown here for 2NapAA that the gel-to-crystal transition depends on the pH of the medium. Above pH 4.1, there is little evidence of crystallisation, but crystals appear once the pH decreases below this value. The rate of crystallisation and the dimensions of the crystals depend on the rate of pH change. We also highlight that there is a significant effect of the shape and size of the sample holder in which the experiment is carried out.We have followed the gelation and gel-to-crystal transition in its entirety using SAXS and WAXS. In the gel phase, at low concentrations of GdL, and at early time points with high concentrations of GdL, we are able to show the nanometre scale dimensions of the self-assembled fibre using SAXS. The fibres gradually stiffen with time, but at no stage is there evidence of molecular ordering within these gel fibres from the WAXS. At high concentrations of GdL, the fibres are already stiffened and crystals form as the pH drops further. The spherical domains of fibres that can be imaged by microscopy gradually disappear and are replaced by crystals, which appear to grow preferentially along the direction of the fibre axis.
We highlight that these data clearly show that the gel phase and the crystal phase are not the same. As such, we would urge caution in interpreting or understanding the gel phase in such systems by extrapolating from crystal structures. This work therefore provides new insight into this type of gel.
Author contributions
Conceptualization – DG, AS, DA; data curation – all; formal analysis – all; funding acquisition – DG, AS, DA; investigation – all; methodology – all; project administration – DA; resources – AS, DA; supervision – DA; writing – all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DG thanks the University of Bologna, Marco Polo funding. LJM thanks the Leverhulme Trust for funding (RPG-2019-165). DA and BD thank the EPSRC for funding (EP/S019472/1 and EP/L021978/1). This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547. The Ganesha X-ray scattering apparatus was purchased under EPSRC Grant ‘Atoms to Applications’ (EP/K035746/1).Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- R. Eelkema and A. Pich, Adv. Mater., 2020, 32, 1906012 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- S. Sathaye, A. Mbi, C. Sonmez, Y. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 34–68 CAS.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- P. W. J. M. Frederix, I. Patmanidis and S. J. Marrink, Chem. Soc. Rev., 2018, 47, 3470–3489 RSC.
- M. G. Corradini and M. A. Rogers, Curr. Top. Food Sci. Technol., 2016, 9, 84–92 Search PubMed.
- R. Van Lommel, J. Zhao, W. M. De Borggraeve, F. De Proft and M. Alonso, Chem. Sci., 2020, 11, 4226–4238 RSC.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- A. Rajbhandary, W. W. Brennessel and B. L. Nilsson, Cryst. Growth Des., 2018, 18, 623–632 CrossRef CAS.
- W. Liyanage, W. W. Brennessel and B. L. Nilsson, Langmuir, 2015, 31, 9933–9942 CrossRef CAS PubMed.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS PubMed.
- W. Liyanage, N. M. B. Cogan and B. L. Nilsson, ChemNanoMat, 2016, 2, 800–804 CrossRef CAS.
- F. M. Menger, Y. Yamasaki, K. K. Catlin and T. Nishimi, Angew. Chem., Int. Ed. Engl., 1995, 34, 585–586 CrossRef CAS.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- S. Kralj, O. Bellotto, E. Parisi, A. M. Garcia, D. Iglesias, S. Semeraro, C. Deganutti, P. D’Andrea, A. V. Vargiu, S. Geremia, R. De Zorzi and S. Marchesan, ACS Nano, 2020, 14, 16951–16961 CrossRef CAS PubMed.
- I. Kapoor, E.-M. Schön, J. Bachl, D. Kühbeck, C. Cativiela, S. Saha, R. Banerjee, S. Roelens, J. J. Marrero-Tellado and D. D. Díaz, Soft Matter, 2012, 8, 3446–3456 RSC.
- P. Terech, N. M. Sangeetha and U. Maitra, J. Phys. Chem. B, 2006, 110, 15224–15233 CrossRef CAS PubMed.
- A. Vidyasagar and K. M. Sureshan, Angew. Chem., Int. Ed., 2015, 54, 12078–12082 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506–6521 CAS.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- A. D. Martin and P. Thordarson, J. Mater. Chem. B, 2020, 8, 863–877 RSC.
- R. Das, B. Gayakvad, S. D. Shinde, J. Rani, A. Jain and B. Sahu, ACS Appl. Bio Mater., 2020, 3, 5474–5499 CrossRef CAS.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144–4156 RSC.
- K. A. Houton, K. L. Morris, L. Chen, M. Schmidtmann, J. T. A. Jones, L. C. Serpell, G. O. Lloyd and D. J. Adams, Langmuir, 2012, 28, 9797–9806 CrossRef CAS PubMed.
- A. D. Martin, J. P. Wojciechowski, M. M. Bhadbhade and P. Thordarson, Langmuir, 2016, 32, 2245–2250 CrossRef CAS PubMed.
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Zamith Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927–935 RSC.
- T. Yuan, Y. Xu, J. Fei, H. Xue, X. Li, C. Wang, G. Fytas and J. Li, Angew. Chem., Int. Ed., 2019, 58, 11072–11077 CrossRef CAS PubMed.
- O. Bellotto, S. Kralj, R. De Zorzi, S. Geremia and S. Marchesan, Soft Matter, 2020, 16, 10151–10157 RSC.
- M. Guo, Q. Yin, Y. Li, Y. Huang, Z. Zhang and L. Zhou, Chem. Lett., 2017, 46, 1292–1295 CrossRef CAS.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2008, 2248–2250, 10.1039/B801913D.
- R. Mohanrao, K. Hema and K. M. Sureshan, Nat. Commun., 2020, 11, 865 CrossRef CAS PubMed.
- J. Liu, F. Xu, Z. Sun, Y. Pan, J. Tian, H.-C. Lin and X. Li, Soft Matter, 2016, 12, 141–148 RSC.
- Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884–889 CrossRef CAS.
- Y. Xu, C. Kang, Y. Chen, Z. Bian, X. Qiu, L. Gao and Q. Meng, Chem. – Eur. J., 2012, 18, 16955–16961 CrossRef CAS PubMed.
- G. Fichman, T. Guterman, J. Damron, L. Adler-Abramovich, J. Schmidt, E. Kesselman, L. J. W. Shimon, A. Ramamoorthy, Y. Talmon and E. Gazit, Sci. Adv., 2016, 2, e1500827 CrossRef PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- E. C. Barker, A. D. Martin, C. J. Garvey, C. Y. Goh, F. Jones, M. Mocerino, B. W. Skelton, M. I. Ogden and T. Becker, Soft Matter, 2017, 13, 1006–1011 RSC.
- T. Guterman, M. Levin, S. Kolusheva, D. Levy, N. Noor, Y. Roichman and E. Gazit, Angew. Chem., Int. Ed., 2019, 58, 15869–15875 CrossRef CAS PubMed.
- J. L. Andrews, E. Pearson, D. S. Yufit, J. W. Steed and K. Edkins, Cryst. Growth Des., 2018, 18, 7690–7700 CrossRef CAS.
- X. Liu, J. Fei, A. Wang, W. Cui, P. Zhu and J. Li, Angew. Chem., Int. Ed., 2017, 56, 2660–2663 CrossRef CAS PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS PubMed.
- A. Z. Cardoso, A. E. Alvarez Alvarez, B. N. Cattoz, P. C. Griffiths, S. M. King, W. J. Frith and D. J. Adams, Faraday Discuss., 2013, 166, 101–116 RSC.
- K. McAulay, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, N. Khunti, N. Cowieson, H. Cui, A. Seddon and D. J. Adams, Langmuir, 2020, 36, 8626–8631 CrossRef CAS PubMed.
- T. R. Canrinus, F. J. R. Cerpentier, B. L. Feringa and W. R. Browne, Chem. Commun., 2017, 53, 1719–1722 RSC.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- M. G. F. Angelerou, A. Sabri, R. Creasey, P. Angelerou, M. Marlow and M. Zelzer, Chem. Commun., 2016, 52, 4298–4300 RSC.
- F. C. Meldrum and C. O'Shaughnessy, Adv. Mater., 2020, 32, 2001068 CrossRef CAS PubMed.
- K. McAulay, L. Thomson, L. Porcar, R. Schweins, N. Mahmoudi, D. J. Adams and E. R. Draper, Org. Mater., 2020, 02, 108–115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2053987. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sm00770j |
| This journal is © The Royal Society of Chemistry 2021 |