
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoparticles endowed with low-temperature colloidal stability by cyclic polyethylene glycol in ethanol†
M. Ali
Aboudzadeh
 ab,
Joscha
Kruse
ab,
Maria
Sanromán Iglesias
ab,
Daniele
Cangialosi
ab,
Angel
Alegria
bc,
Marek
Grzelczak
*ab and
Fabienne
Barroso-Bujans
*abd
ab,
Joscha
Kruse
ab,
Maria
Sanromán Iglesias
ab,
Daniele
Cangialosi
ab,
Angel
Alegria
bc,
Marek
Grzelczak
*ab and
Fabienne
Barroso-Bujans
*abd
aDonostia International Physics Center (DIPC), Paseo Manuel Lardizábal 4, 20018 Donostia–San Sebastián, Spain. E-mail: fbarroso@dipc.org; Tel: +34 94301 8803
bMaterials Physics Center, CSIC-UPV/EHU, Paseo Manuel Lardizábal 5, 20018 Donostia–San Sebastián, Spain. E-mail: marek.g@csic.es; Tel: +34 94301 8971
cDepartamento de Polímeros y Materiales Avanzados: Física, Química y Tecnología, University of the Basque Country (UPV/EHU), Apartado 1072, Donostia–San Sebastián 20080, Spain
dIKERBASQUE – Basque Foundation for Science, María Díaz de Haro 3, E-48013 Bilbao, Spain
First published on 29th July 2021
Abstract
The colloidal stability of metal nanoparticles is tremendously dependent on the thermal behavior of polymer brushes. Neat polyethylene glycol (PEG) presents an unconventional upper critical solution temperature in ethanol, where phase segregation and crystallization coexist. This thermal behavior translated to a PEG brush has serious consequences on the colloidal stability in ethanol of gold nanoparticles (AuNPs) modified with PEG brushes upon cooling. We observed that AuNPs (13 nm diameter) stabilized with conventional linear PEG brushes (Mn = 6 and 11 kg mol−1) in ethanol suffer from reversible phase separation upon a temperature drop over the course of a few hours. However, the use of a polymer brush with cyclic topology as a stabilizer prevents sedimentation, ensuring the colloidal stability in ethanol at −25 °C for, at least, four months. We postulate that temperature-driven collapse of chain brushes promotes the interpenetration of linear chains, causing progressive AuNP sedimentation, a process that is unfavorable for cyclic polymer brushes whose topology prevents chain interpenetration. This study reinforces the notion about the importance of polymer topology on the colloidal stability of AuNPs.
Introduction
The colloidal stability of nanoparticles (NPs) is very important for their storage, transportation and use. In particular, metal NPs are highly in demand in applications related to biomedicine, sensing, and catalysis, among others.1–4 The applicability of nanoparticles is often conditioned by their colloidal stability in the liquid phase under extreme conditions, which is tackled by the proper design of the stabilizing organic shell.4,5Poly(ethylene glycol) (PEG) is one of the most representative polymer brushes used for stabilizing gold nanoparticles (AuNPs) in polar media. A PEG shell suppresses the formation of protein corona, controlling the cell uptake and in vivo clearance.6 PEG provides the AuNPs with a neutral surface charge, chemical stability, and hydrophilicity, and very importantly, the ability to modulate the colloidal stability by external stimuli (e.g. temperature, ionic strength, pH, pressure, solvent change or even solvent evaporation).7–9 In fact, PEGylation has been successfully employed to transfer AuNPs and nanorods from one phase to another.9–11 Another benefit of using PEG as a capping agent is the long-term colloidal stability of nanoparticles and therefore the prolonged shelf life of a sample.
The solvation of polymer brushes located on a particle's surface determines the overall diameter of the NP. In good solvent conditions, a polymer chain is extended due to favorable polymer-solvent interactions whereas in bad solvent conditions, polymer–polymer interactions dominate causing chain collapse.12 These changes in the physical behavior of polymer brushes can cause aggregation of NPs. PEG brushes reversibly shrink in water with increasing temperature as a consequence of the decreasing solubility of PEG, as observed in PEG brushes on iron oxide NPs13 and in oligo(ethylene glycol)–alkane brushes on AuNPs.14
In our previous study we observed that cyclic PEG (CPEG) brushes chemically grafted to AuNPs behave different than their linear analogues when subjected to external stimuli like temperature or salt.15 These differences can be attributed to the fact that CPEG brushes have to accommodate in less space near the surface than the linear chains, thus, causing increased concentration of polymer segments near the surface. As a result, the cyclic chains are forced to stretch more than the linear chains. A study on CPEG chains physisorbed on AuNPs also demonstrated different stability compared with physisorbed and chemisorbed linear PEG (LPEG) against heating, freezing and salt addition.16 In those cases, the cyclic topology provided higher colloidal stability, likely due to a stronger surface adsorption of cycles, as suggested by the authors,16 based on previous works on the adsorption of cyclic PEG on silica nanoparticles of 25 nm in diameter17 and other experimental and computational studies. Thiolated 18-crown-6 crown ethers (a cyclic oligomer of PEG) were also used as capping agents for AuNPs demonstrating endowing NPs with reversible aggregation–disaggregation upon temperature cycling.18 Other interesting thermoresponsive cyclic polymer brushes reported to date are based on polyoxazolines.19 This polymer exhibits a lower critical solution temperature (LCST) in water. Interestingly, the cyclic brush provided a reversible thermoresponsive behavior to the NPs, in contrast to the analogous linear brush.19
In a number of experiments, we have observed that the storage of an ethanol dispersion containing highly concentrated AuNPs stabilized with PEG at −25 °C loses colloidal stability, which is then restored at room temperature. Although literature data lacks the experimental evidence of such behavior, we found it important to address this issue since the stability of PEG in media other than water raises technological relevance. Ethanol is the second most important solvent after water for biological applications. In ethanol, neat PEG exhibits an upper critical solution temperature (UCST).20 That is, the polymer exhibits phase separation upon cooling. However, PEG crystallizes from solution before phase separation occurs in ethanol, rendering the UCST of this system thermodynamically inaccessible.20
In this study, we demonstrate that CPEG can be used as a AuNP ligand with longer-term stability in ethanol at low temperatures compared to LPEG, an important property for storage and immediate use. A plausible mechanism is given based on experimental data obtained by 1H NMR, differential scanning calorimetry (DSC), and UV-Vis spectroscopy of AuNP@PEG colloids in ethanol, as well as on the study of the phase behavior of neat CPEG and LPEG in ethanol at low temperatures.
Experimental section
Synthesis of citrate-capped AuNP
AuNP@citrate with a core diameter (d0) of 13.2 nm was prepared according to the inversed Turkevich method21 as described previously.15Synthesis of aqueous AuNP@PEG
AuNP@CPEG and AuNP@LPEG were synthesized by ligand exchange from AuNP@citrate using the protocol described in our previous report.15 CPEG and LPEG of Mn = 6 and 11 kg mol−1, both containing 2,4-dinitrobenzene as a protector group for thiol (Scheme 1),22 were used for grafting and as reference polymers in calorimetric and microscopy studies. | ||
| Scheme 1 Gold nanoparticles stabilized with polyethylene glycol (PEG) of different topologies: (a) cyclic and (b) linear. Structure of CPEG and LPEG used in this study. | ||
Transfer of AuNP@PEG from water to ethanol
12 mL of aqueous AuNP@PEG samples were centrifuged at 7000 rpm for 60 min to remove the water phase. Afterwards, 6 mL of ethanol were added into the centrifuge tubes, which were immersed in an ultrasonic bath for 5 min. After 24 h of storage at room temperature, the NPs were concentrated by centrifugation (6000 rpm, 90 min) and diluted by the addition of 6 mL of ethanol.Optical microscopy
A Leica Microsystems AF6000-DFC microscope was used to monitor the formation of crystal structures upon cooling in CPEG and LPEG 6k dissolved in ethanol at a nominal concentration of 5 wt% (some of the ethanol may have evaporated during sample preparation). Samples were deposited between two round glass coverslips separated by a ring of 0.5 mm thickness. An objective of 50×/0.5 was used. A Linkam temperature control unit (THMS 600), equipped with a temperature programmer, was employed for the temperature-dependent studies. Images were continuously recorded from 20 to −60 °C, and back to 20 °C, at heating and cooling rates of 20 °C min−1.Near infrared spectroscopy
The phase transitions of CPEG and LPEG in ethanol (5 wt%) were measured in a FT/IR 6300 spectrometer from Jasco. A 0.1 cm path length quartz cuvette was used for the sample solution. A Linkam temperature control unit (THMS 600), equipped with a temperature programmer, was employed for the temperature-dependent studies. Single beam (SB) spectra were continuously monitored during cooling and heating cycles at a rate of 1 and 20 °C min−1. The sample chamber was purged with nitrogen gas during the whole experiment to avoid water condensation. The change in the solution transmittance is reported from the SB values at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 according to T(%)cooling = 100 × (SBf − SB)/(SBf − SB0) and T(%)heating = 100 × (SB − SB0)/(SBf − SB0), where SB0 and SBf are the initial and final SB values.
500 cm−1 according to T(%)cooling = 100 × (SBf − SB)/(SBf − SB0) and T(%)heating = 100 × (SB − SB0)/(SBf − SB0), where SB0 and SBf are the initial and final SB values.
UV-Vis spectroscopy
UV-Vis spectra were recorded on an Agilent 8453A UV-visible spectrophotometer equipped with deuterium and tungsten lamps (190–1000 nm range, 0.5 nm resolution). Quartz Hellma cuvettes of 0.1 cm path length were used.Dynamic light scattering (DLS)
Hydrodynamic diameters (DH) of the nanoparticles in ethanol and water were measured using a DLS instrument (Zetasizer Nano Series, Malvern Instruments, Malvern, UK). This instrument determines the NP size from intensity–time fluctuations of a laser light (633 nm) scattered from a sample at an angle of 173°. The final value of DH is obtained by averaging the peak values from three different measurements that were analyzed in 13 runs of 30 s each. All measurements were conducted at 20 °C.Differential scanning calorimetry
DSC data were registered on a Q2000 TA Instrument equipped with a liquid nitrogen cooling system. The system was calibrated with indium melting. All samples were measured in sealed aluminum pans. A helium flow rate of 25 mL min−1 was used throughout. Ethanol PEG solutions (∼15 mg specimens) of 4, 5 and 6 wt% were cycled at least twice from 25 °C to −150 °C and heated back to 25 °C at a rate of 20 °C min−1 (both cooling and heating). All the runs were completely reproducible. Neat PEGs (∼5 mg specimens) were first cooled from room temperature to −150 °C and heated to 80 °C at 20 °C min−1. Then, the samples were cooled to −150 °C and heated back to 80 °C at 20 °C min−1 (both cooling and heating). The last cycle was used to evaluate the thermal transitions. The redispersion of AuNP@PEG in ethanol was measured by previously cooling ∼10 mg specimens to −25 °C in a freezer for 5 days, followed by a quick transfer to the DSC platform, which was previously cooled at −150 °C. Then, the sample was heated at a rate of 30 °C min−1 (1st heating), cooled to −150 °C and heated back to 35 °C at a rate of 30 °C min−1 (2nd heating).Nuclear magnetic resonance
1H NMR spectra of AuNP@PEG were recorded at 500.15 MHz on a Bruker Avance Neo in ethanol-d6. The variable-temperature data were recorded at 25 °C and from 20 to −30 °C in steps of 10 °C, and finally at 25 °C again. The sample was equilibrated for 5 min at each temperature before data acquisition.Results
AuNP@PEG in ethanol
Colloidal gold nanoparticles modified with cyclic and linear PEG (AuNP@CPEG and AuNP@LPEG) were stabilized in ethanol starting from their synthesis in water.15 Ligand exchange and transfer from water to ethanol were verified by UV-Vis spectroscopy, exhibiting a shift of the localized surface plasmon resonance (LSPR) from 517 nm (AuNP@citrate) to 522 nm (AuNP@CPEG 6k, water) upon ligand exchange and a shift to 523 nm (AuNP@CPEG 6k, ethanol) upon solvent transfer (Fig. 1a). The nanoparticles maintained good colloidal stability in ethanol (Fig. 1b).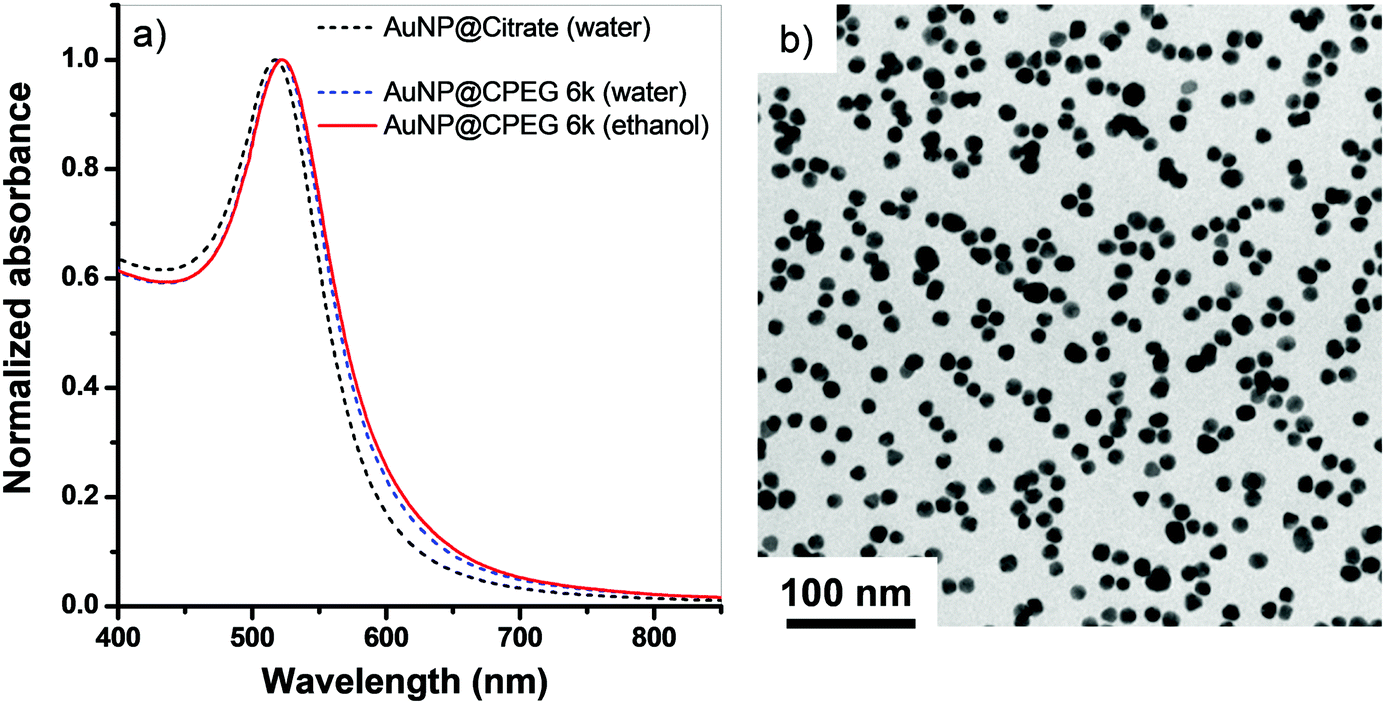 | ||
| Fig. 1 (a) Normalized absorption spectra of AuNP@CPEG 6k dispersed in ethanol after being phase transferred from water. Spectra of AuNP@citrate. (b) TEM micrograph of AuNP@CPEG 6k. | ||
The grafting densities (σ) of CPEG and LPEG brushes on AuNPs, estimated by thermogravimetry,15 were similar for each molecular weight pair (Table 1). Hydrodynamic diameters, DH, of the nanoparticles in ethanol were determined using DLS. The brush height, H, is calculated from H = (DH − d0)/2, where d0 is the metallic core diameter (13.2 ± 0.4 nm as determined by SAXS).15 Smaller H values for CPEG brushes than for LPEO brushes were confirmed (Table 1), in agreement with a previous study on AuNP@PEG in water.15 In ethanol, the H values were consistently higher than in water, indicating that the PEG brushes are slightly more extended in ethanol.
| AuNP@PEG | Molecular weight | Grafting density | Brush height in water at 20 °C | Brush height in ethanol at 20 °C |
|---|---|---|---|---|
| M n (kg mol−1) | σ (chains per nm2) ± 10% | H (nm) | H (nm) | |
| CPEG | 6.5 | 0.4 | 14.4 ± 0.3 | 16.7 ± 0.4 |
| LPEG | 5.8 | 0.4 | 16.4 ± 1.0 | 18.6 ± 0.6 |
| CPEG | 11.2 | 0.3 | 18.1 ± 0.4 | 20.0 ± 0.3 |
| LPEG | 10.7 | 0.3 | 22.2 ± 0.2 | 24.5 ± 0.3 |
Colloidal stability of AuNP@PEG in ethanol
The colloidal stability of AuNP@PEG 11k in ethanol ([Au°] = 1 mM) was verified by cooling the samples in a freezer to −25 °C (Fig. 2). After 12 h, the sedimentation of AuNP@LPEG was clearly observed while AuNP@CPEG remained dispersed. Redispersion of AuNP@LPEG occurred by warming the sample back to room temperature. Similar results were observed for AuNP@PEG 6k with [Au°] = 0.9 and 1.7 mM (Fig. S1, ESI†). The redispersion of nanoparticles, monitored by UV-Vis spectroscopy, showed that AuNP@LPEG 11k is able to recover the maximum absorption in about 90 min, while the AuNP@CPEG 11k remains practically unchanged (Fig. 3). The wavelength at the LSPRmax slightly blue-shifted upon the NP redispersion, from 523 to 521 nm for both samples. | ||
| Fig. 2 Pictures of AuNP@CPEG 11k and AuNP@LPEG 11k ([Au°] = 1 mM), dispersed in ethanol upon cooling to −25 °C in a freezer and warming back to room temperature. | ||
 | ||
| Fig. 3 Absorption spectra of AuNP@LPEG 11k and AuNP@CPEG 11k in ethanol immediately after being taken out from the freezer and introduced in the spectrometer at 25 °C. [Au°] = 1 mM. The absorbance in the peak maxima was normalized to 1 at the final time. Inset: Time-dependent change of LSPR maximum (522 nm) for AuNP@LPEG 11k and AuNP@CPEG 11k. | ||
In order to explain such phenomenology, the thermal behavior of neat CPEG and LPEG polymers in ethanol was studied.
Behavior of neat PEG in ethanol
To understand the reversible aggregation of nanoparticles we studied the temperature behavior of neat polymers in ethanol. CPEG and LPEG of Mn = 6 and 11 kg mol−1 dissolved well in ethanol after gentle heating. At room temperature, the solutions at a concentration of 5 wt% remain macroscopically transparent indicating total solubility, in contrast to previous results on LPEG of higher molecular weight (Mn = 83 kg mol−1) which were crystalline in solution at room temperature.20 Under the optical microscope, micrometer length scale spherical structures emerged at −10 °C gaining size and number with a further temperature drop (Fig. 4). Upon heating, the structures vanished indicating a reversible process. Naked-eye inspection showed that the cooled solution of polymer at −30 °C is a solid-like paste, corroborating previous observations on LPEG of 83 kg mol−1, where the formation of a new phase composed of stacked core/layer platelets in ethanol was suggested.20 The core containing mainly ethanol is constrained between two layers of crystalline PEG in a single platelet. The platelets further stack together and form a lamellar structure.20 | ||
| Fig. 4 Images taken on an optical microscope (and pictures on the right) of LPEG 6k in ethanol at a nominal concentration of 5 wt% during (a) cooling and (b) heating. | ||
CPEG forms a solid-like paste at −30 °C similar to LPEG in ethanol. Under the microscope, spherulite-like structures started to appear at −5 °C upon cooling and started to vanish at 0 °C upon heating (Fig. 5). A reduction in their size is observed at 0 °C before their total disappearance in liquid ethanol at 10 °C.
 | ||
| Fig. 5 Images taken on an optical microscope of CPEG 6k in ethanol at a nominal concentration of 5 wt% during heating. | ||
To gain further insight into the changes of the polymer solution with temperature and cooling rate, we measured the transmittance of CPEG and LPEG 6k at 2 wt% and 5 wt% in ethanol at cooling rates of 1 and 20 °C min−1 using near infrared (NIR) light at 10500 cm−1, a wavenumber where the samples do not exhibit absorption bands (Fig. S2, ESI†). The data of Fig. 6 show a decrease of the solution transmittance upon cooling in all samples, which is in line with the experiments in Fig. 4 and 5. The formation of spherulite-like structures is undoubtedly linked to an increase of the light dispersion and therefore, to a reduction of the light transmittance. This process is reversible upon heating (Fig. S3, ESI†).
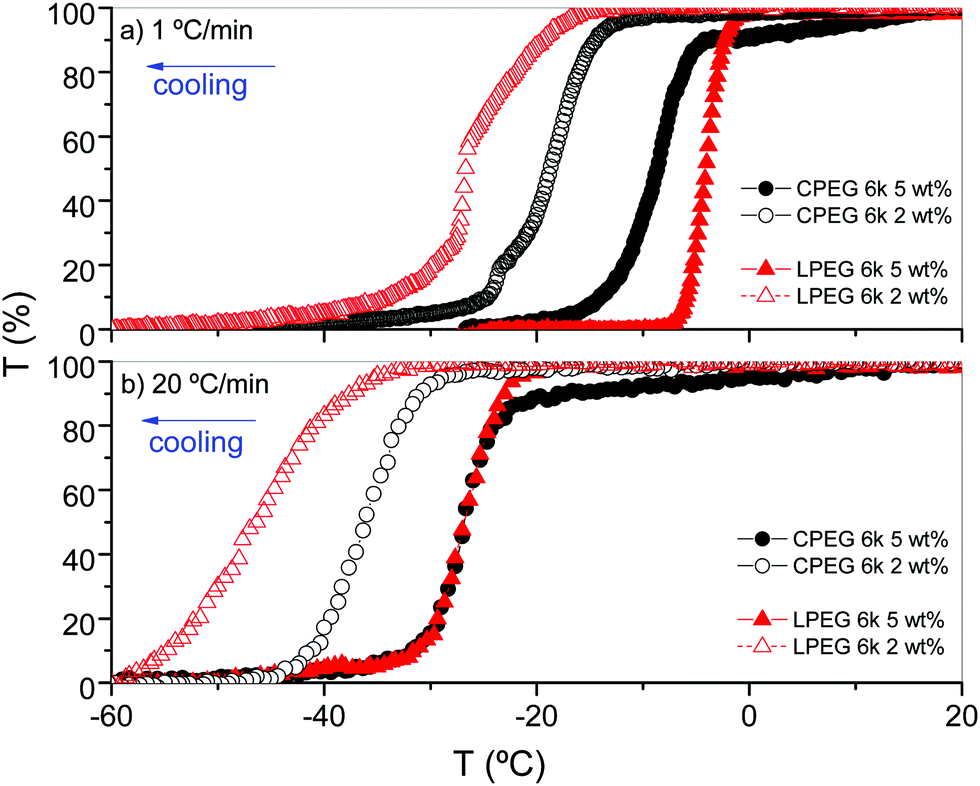 | ||
| Fig. 6 Solution transmittance, T(%)cooling, of CPEG 6k and LPEG 6k in ethanol (c = 2 and 5 wt%) at cooling rates of (a) 1 °C min−1 and (b) 20 °C min−1. | ||
Some differences can be observed in the thermal behavior between CPEG and LPEG as expected from their distinct topologies.23 At a 1 °C min−1 cooling rate (Fig. 6a), the light transmittance of LPEG of 5 wt% begins to decrease at −2 °C, and that of 2 wt% at −17 °C. Conversely, the decrease in light transmittance of CPEG is gradual at both concentrations up to a certain temperature from which it starts to drop; T(5 wt%) = −5 °C and T(2 wt%) = −13 °C. By increasing the cooling rate to 20 °C min−1 (Fig. 6b), the transition temperature decreases compared to that measured at 1 °C min−1. The dependence of the transition temperature with polymer concentration is notably higher for LPEO than CPEO at both cooling rates. These observations on the polymer concentration and cooling rate dependence underline the kinetic nature of a crystallization process, as proved in the next experiments of DSC. The distinct behavior of phase separation between CPEG and LPEG can be attributed to their distinct topologies, as previously observed in aqueous solutions of poly(N-isopropylacrylamide).23 Steric constraints will undoubtedly affect the chain collapse and the packing of chain segments in a distinct manner.
DSC data of CPEG and LPEG at concentrations of 5 wt% in ethanol (Fig. 7a, Mn = 6 kg mol−1) exhibit exothermic peaks under cooling at temperatures below those observed in the optical microscope and NIR experiments. The detection of different, although related, events such as the phase segregation and crystallization in this polymer system with the different experimental techniques could explain such differences. Upon heating, an endothermic peak is observed. Both the exothermic and endothermic processes occur at lower temperatures than that of crystallization (Tc) and melting (Tm) in the neat polymer. By comparing the enthalpies of the endothermic process in LPEG and CPEG in ethanol with the melting enthalpy (ΔHm) of the neat PEG (Table 2), lower but comparable melting values can be observed for the polymers in solution. These results suggest that the thermal events detected in the DSC correspond to the crystallization and melting of PEG in ethanol, in agreement with the observation of PEG crystals by small angle neutron scattering (SANS).20 By increasing the molecular weight, the crystallization and melting temperatures in the neat polymer and in solution slightly increase (Fig. 7b). Moreover, the crystallization temperature decreases with decreasing PEG concentration (Fig. S5, ESI†), and it is not detectable at concentrations below 4 wt%. However, we know from the transmittance experiments that at concentrations as low as 2 wt%, a clear phase segregation occurs with the concomitant formation of a solid-like paste. Finally, by reducing the cooling rate (Fig. S6, ESI†), Tc increases in agreement with expectations,24 and with the transmittance experiments.
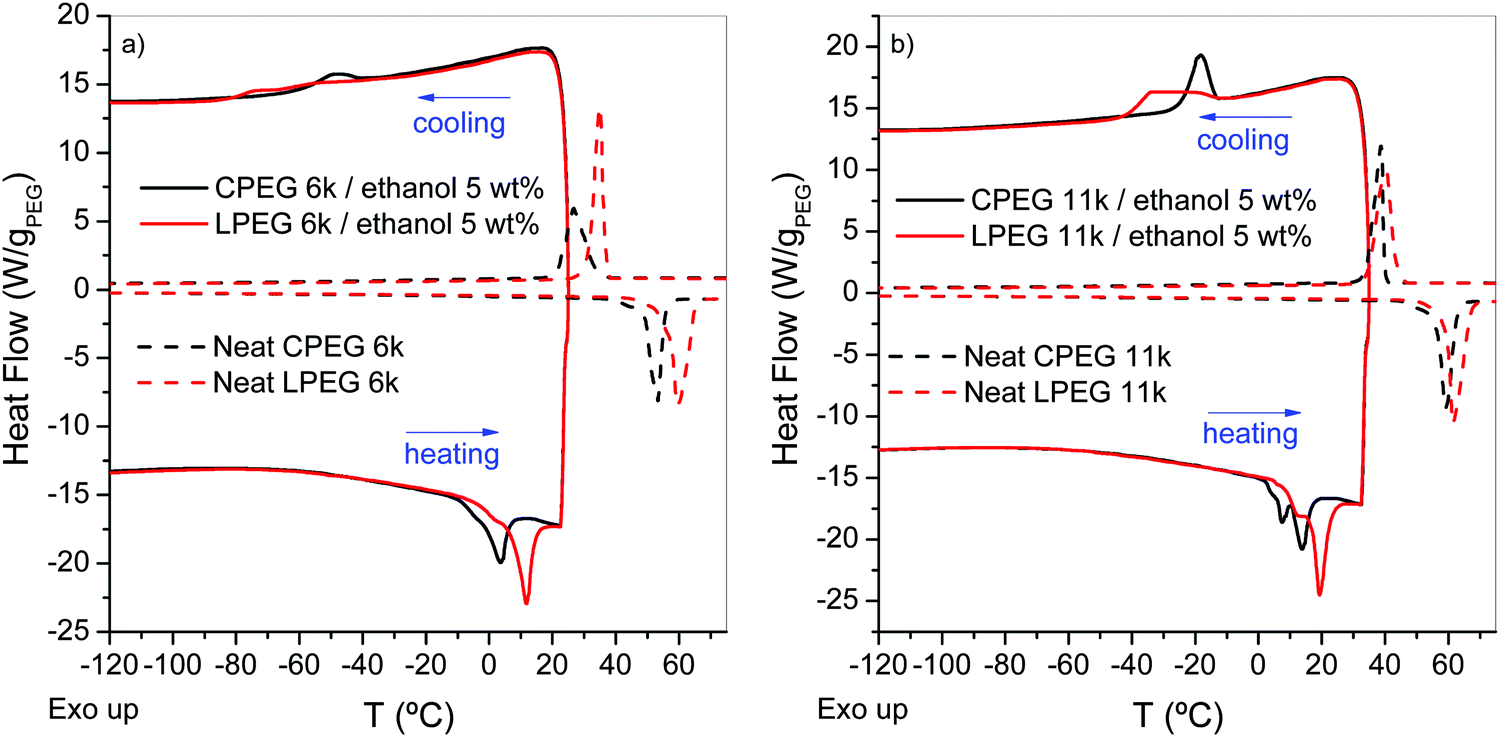 | ||
| Fig. 7 Cooling and heating DSC scans (20 °C min−1) of CPEG and LPEG at 5 wt% in ethanol. Heat flow has been normalized to the amount of PEG in the sample. Heat flow data of neat CPEG and LPEG reference samples. (a) Mn = 6 kg mol−1 and (b) = 11 kg mol−1. | ||
| Sample | Neat PEG | PEG/ethanol (5 wt%) | AuNP@PEG/ethanol ([Au°] = 10 mM, [PEO] = 0.03 wt%) | ||||
|---|---|---|---|---|---|---|---|
| T m (K) | ΔHm (kJ mol monomer−1) | T m (K) | ΔHma (J gtotal−1) | ΔHmb (kJ mol monomer−1) | T endo (K) | ΔHa (J gtotal−1) | |
| a Melting enthalpy is referred to as the total mass of sample including the ethanol. b Melting enthalpy is normalized to the amount of PEG monomers in the sample. | |||||||
| LPEG 6k | 332 | 7.0 | 276, 284 | 5.6 | 4.9 | 263 | 3.0 |
| LPEG 11k | 334 | 7.5 | 285, 292 | 6.6 | 5.8 | 269 | 3.3 |
| CPEG 6k | 326 | 4.9 | 271, 278 | 4.0 | 3.5 | ∼245 | 0.5 |
| CPEG 11k | 332 | 6.1 | 280, 287 | 5.0 | 4.4 | ∼250 | −0.5 |
The Tm values obtained from the melt and solution (Fig. 7 and Table 2) exhibit large differences between the cyclic and linear topology. The Tm of CPEG is in all cases lower than that of LPEG, in agreement with a previous study on the crystallization kinetics of CPEG from the melt.25 Our results suggest that the crystallization of CPEG in the solution might follow different kinetics than that of its linear analogue.
Discussion
The studies of the phase behavior of neat polymers revealed that both CPEG and LPEG exhibit an UCST in ethanol upon cooling. Spherulite-like structures grow together with the formation of long-range network structures as evidenced by the formation of a solid-like paste. The temperature dependence of this thermal transition is slightly different for each topology in agreement with a previous study.23 Since the thermal behavior of a polymer brush is not necessarily the same as in the neat polymer,26 a description of the brush phenomenology is rather difficult. Notwithstanding this, we can provide a plausible explanation for the cooling-induced sedimentation of AuNP@LPEG in ethanol based on previous and subsequent experiments.AuNP@LPEG sediments over 12 h whereas AuNP@CPEG remains stable for at least four months at −25 °C. The slow sedimentation kinetics of AuNP@LPEG inhibits its monitoring by dynamical or isothermal DSC. Thus, we focused on measuring the redispersion of nanoparticles in ethanol. We transferred the samples cooled for 5 days in a fridge at −25 °C to the DSC platform (previously cooled at −150 °C), and then heated to 35 °C. An endothermic peak followed by an exothermic process were clearly detected during the first heating run but were no longer detected in a second heating (Fig. 8a and b). In contrast to linear brushes, the endothermic process in AuNP@CPEG was barely detected (Fig. 8c and d). The whole enthalpy of the redispersion process was obtained by integration of the endothermic and exothermic process (Table 2). As noticed, ΔH is about 3 J g−1 of the total mass sample (AuNP@PEG + ethanol) for LPEG, whereas it is about 0 for CPEG. The normalization of ΔH values to the mass of PEG in the sample ([PEG] = 0.03 wt%) gives a value of the order of 400 kJ molmonomer−1 for linear brushes. These amounts largely exceed the ΔHm of PEG/ethanol suggesting that the thermal events found in AuNP@PEG are associated in greater part to other processes different from PEG melting, for example the polymer solvation (with an order of magnitude of 1 kJ mol−1)27 and other thermal phenomena as discussed below.
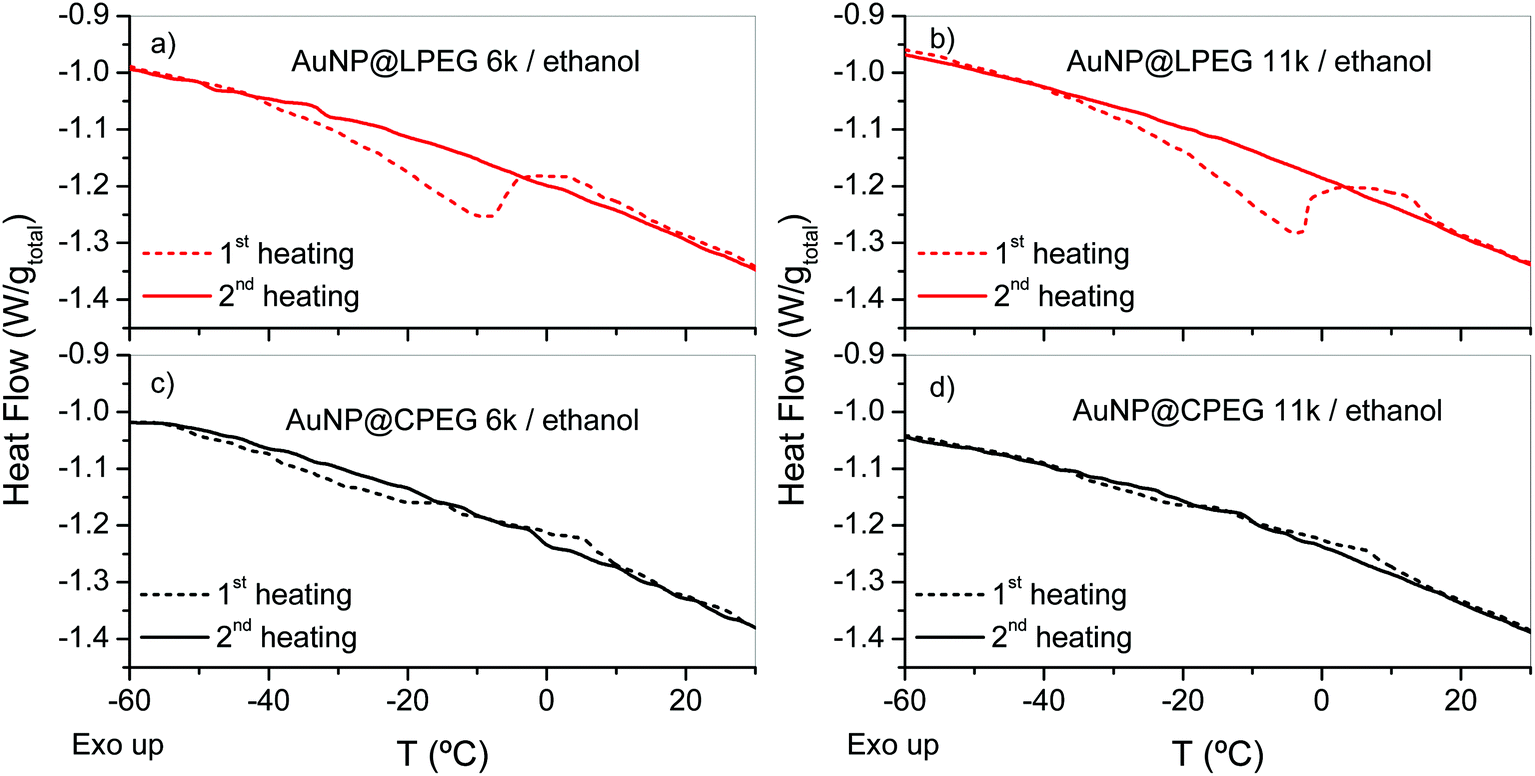 | ||
| Fig. 8 Heating DSC scans (30 °C min−1) of AuNP@PEG in ethanol. 1st heating scan of samples cooled at −25 °C in a freezer for 5 days and immediately transferred to the DSC platform at −150 °C. Then, samples were cooled to −150 °C at 30 °C min−1 and a 2nd heating scan was recorded at 30 °C min−1. (a and b) AuNP@LPEG of Mn = 6 and 11 kg mol−1. (c and d) AuNP@CPEG of Mn = 6 and 11 kg mol−1. | ||
The sedimentation of AuNP@LPEG in ethanol at −25 °C and the redispersion at room temperature can be undoubtedly ascribed to the PEG brush behavior in ethanol. We hypothesize that upon cooling, monomer–monomer interactions are favored over monomer–ethanol interactions causing polymer desolvation and chain collapse. To evaluate our hypothesis, we recorded 1H NMR data of AuNP@CPEG 6k and AuNP@LPEG 6k in ethanol-d6 as a function of decreasing temperature from 25 °C to −30 °C (Fig. 9). We observed significant peak broadening and downfield shift of PEG CH2 in both samples as a result of a change in chain conformation and slowdown of the overall molecular motion. This change occurs relatively fast (just the time needed to go from one temperature to other, in approximately 10–15 min) and reverses while warming back up to 25 °C. The peak intensity (relative to that at 25 °C) decreases and the difference in the full width at half maximum (ΔFWHM) with respect to that at 25 °C increases in both samples in a similar way up to −20 °C (Fig. 9c and 9d). Then, the CPEG brushes exhibited a sudden increment of ΔFWHM from −20 to −30 °C, accompanied by a slight decrease of peak intensity, suggesting that the cyclic brushes attain a relatively high collapsed conformation at these temperatures with respect to that exhibited at 25 °C. This increment of ΔFWHM is smoother in the linear brushes suggesting gradual chain collapse upon cooling. By storing the samples used for NMR analysis at −25 °C, similar results to the previous ones were obtained (Fig. S7, ESI†), even when they contained a higher [Au°] concentration and deuterated ethanol where isotopic effects could eventually occur (e.g. changes in the PEG solubility).28
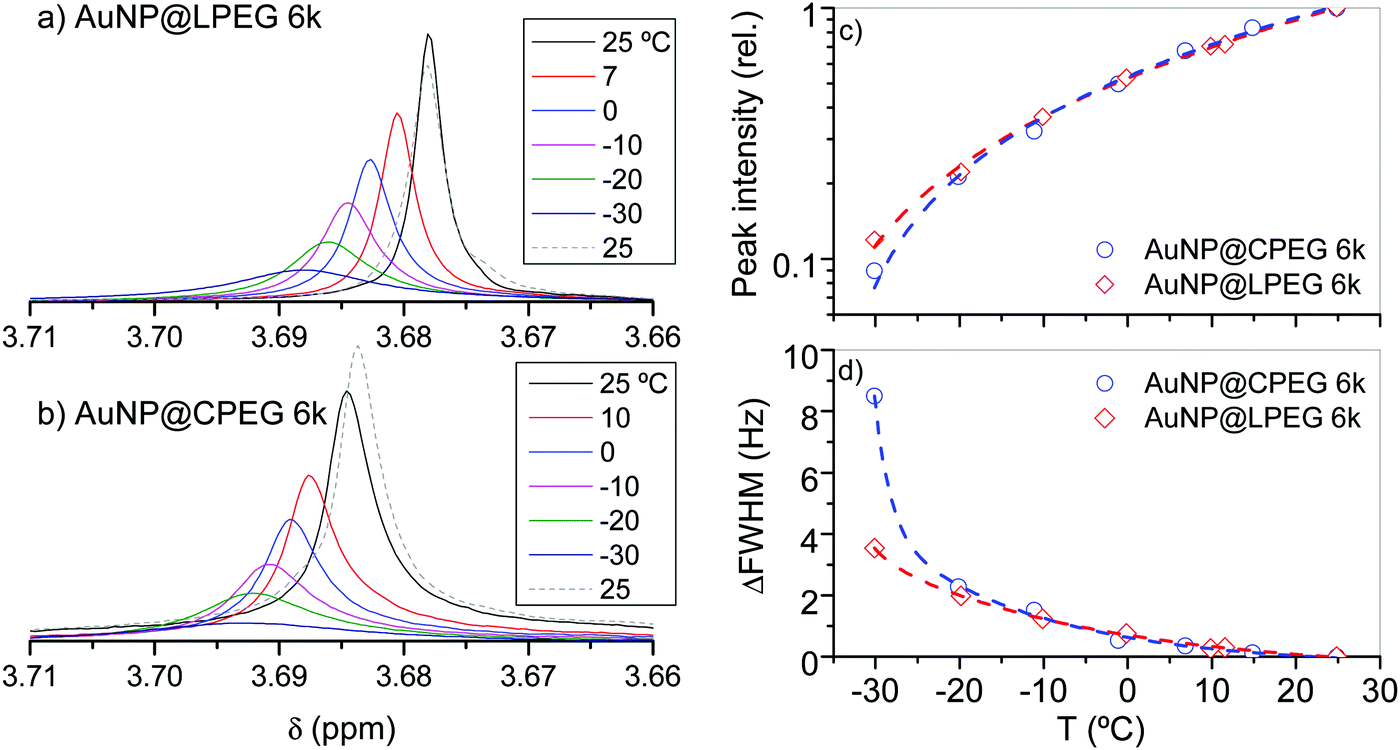 | ||
| Fig. 9 1H NMR of (a) AuNP@LPEG 6k and (b) AuNP@CPEG 6k registered upon cooling in ethanol-d6. [Au°] = 3 mM. The methyl group signals of residual protonated ethanol at 1.14 ppm were used as a reference. (c) Peak intensity relative to that at 25 °C. (d) ΔFWHM = FWHM(T) − FWHM(25 °C). | ||
The experimental evidence points to the fact that the collapsed CPEG brushes endow stability to AuNPs in ethanol at −25 °C. In contrast, the system containing LPEG brushes sediments slowly in such conditions. We propose the following mechanism, which involves three states (Fig. 10). In the first state, the polymer brushes are in good solvent conditions at room temperature. The polymer chains are extended providing good colloidal stability to the nanoparticles. Upon cooling, minute-scale brush collapse occurs with entropy and enthalpy loss due to deviations of polymer conformation from a random coil, while retaining colloidal stability. This brush collapse is reversible upon heating. Then, during isothermal annealing at −25 °C for several hours, at high enough NP concentration the frequent collisions of NPs favor the interpenetration of linear chain brushes of adjacent NPs (Fig. 10a). This process occurs with further entropy loss of the random coil conformation induced by the compression ejected by adjacent NPs. The incorporation of new nanoparticles into the aggregates leads to their sedimentation. Importantly, even after several months, the aggregated nanoparticles at the bottom of the flask conserve the initial red color, suggesting the absence of plasmon coupling. This scenario is quite different from a typical salt-induced aggregation of PEG-coated gold nanoparticles,15 where dehydration of brushes causes polymer shell collapse, making insufficient steric repulsions to overcome van der Waals attractions.
 | ||
| Fig. 10 Scheme of a plausible mechanism for the sedimentation of AuNP@LPEG in ethanol at low temperatures (a), while AuNP@CPEG remains stable (b). | ||
Interpenetration of linear chain brushes has already been suggested in linear PEO brushes of 2700 and 5500 g mol−1 molecular weight attached to iron oxide NPs of 7.3 nm of diameter.29 The authors attributed the irreversible thermoresponsible behavior observed for this system to the irreversible kinetic trapping caused by the interdigitation of PEO chains on adjacent NPs. In fact, chain interpenetration is an important effect for many instances of the physical properties of brushes, as it controls entanglements, and entanglements slow the polymer dynamics.30 Theoretical studies on the compression and subsequent extension of two main chain liquid crystalline polymer brushes (very high interaction between neighboring chains) facing towards one another showed that upon compression, the brushes interpenetrated with each other.31 Once the chains underwent extension the polymers stayed “glued” together, remaining in the same structure as the compressed brushes.
The resistance of our collapsed cyclic PEO brushes to interpenetrate at −25 °C would explain why AuNP@CPEG remains stable for a long period of time (Fig. 10b). Chain interpenetration is unlikely in cyclic brushes due to steric constraints as predicted theoretically32 and demonstrated experimentally,33 where the reduced friction between two TiO2 surfaces grafted with cyclic polyoxazolines led to the impenetrability of cyclic brushes upon high compression forces.33
When the AuNP@PEG systems are warmed up to room temperature, State I is achieved quickly (in minutes) in both topologies. The interactions between polymer brushes and solvent are favorable again allowing the chains to extend and disentangle. Then, the nanoparticles return to the colloidal state. Furthermore, overcoming the entropic penalty resulting from recovering the random coil configuration would explain the huge heat effect observed on heating (see Fig. 8 panels a and b), otherwise not justified only considering the heat of melting and heat of solution of LPEG in ethanol.27
The UV-Vis data in Fig. 3 and DSC data in Fig. 8 indicate that some changes also occur in AuNP@CPEG but in minimum amounts, and that there is no effect of molecular weight in the range investigated. For similar grafting densities of linear and cyclic brushes (0.4 and 0.3 chains per nm2 for each pair of PEO brushes, Table 1), the latter have to accommodate in less space near the particle surface causing increased concentration of polymer segments. Therefore, if chain interpenetration occurs in cyclic brushes, it must occur only in the very external part of the polymer shell.
We could also speculate that polymer crystallization might eventually occur in Stage II based on the ability of neat PEG to form crystal phases with ethanol. However, this is difficult to detect experimentally with standard laboratory techniques. Our attempts to detect crystals of PEG brushes in ethanol by X-ray techniques were unsuccessful due to the predominance of AuNP scattering in AuNP@PEG samples. In the absence of solvent, we were able to detect crystallization in CPEG and LPEG brushes by fast scanning calorimetry (FSC) on dry AuNP@PEG 11k samples. This technique measures micrometer-scale samples and works at extremely high cooling rates (≥1000 K s−1). This allows generating, for instance, fully amorphous bulk PEG samples22 which is otherwise not possible at the standard rates of conventional DSC (<40 K min−1) due to the unavoidable crystallization of PEG. FSC data (Fig. S8, ESI†) showed that in dry AuNP@PEG 11k samples, both CPEG and LPEG brushes, are able to crystallize even at such high cooling rate (1000 K min−1) in stark contrast to bulk PEG samples, whose crystallization is avoided in these conditions.22 Furthermore, we observe no significant differences in the crystallization behavior of CPEG and LPEG brushes that could explain the sedimentation of AuNPs. This result supports the hypothesis that the phase separation observed in AuNP@LPEG at low temperatures bears little or no relation to polymer crystallization.
Conclusions
We have reported on the high stability of AuNPs modified with cyclic PEG brushes of Mn = 6 and 11 kg mol−1 in ethanol at −25 °C for, at least, four months. These results are in contrast to those obtained with analogous linear PEG brushes inducing the nanoparticle sedimentation at −25 °C overnight, and then, their redispersion at room temperature. Sedimentation is independent of the PEO molecular weight for brushes of similar grafting density. These results indicate that the topology of PEG brushes plays an important role on the colloidal stability of AuNPs.We provide a plausible explanation for the cooling-induced sedimentation of AuNP@LPEG in ethanol. We postulate that the collapsed polymer brushes at low temperature start to interpenetrate at certain interparticle distances. This mechanism is only favorable in linear chains. Steric constraints in cyclic polymer brushes do not allow sufficient chain interpenetration, leaving the nanoparticles dispersed and stable in ethanol. Upon warming back to room temperature, the polymer brushes recover their initial extended dimensions. The linear brushes disentangle and the nanoparticles recover their colloidal stability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank J. I. Miranda for NMR experiments and both, J. I. Miranda and A. Moreno for helpful discussions. We also gratefully acknowledge support from the Spanish Ministry “Ministerio de Ciencia, Innovación y Universidades” (PGC2018-094548-B-I00, MICINN/FEDER, UE, and PID2019-111772RB-I00), Basque Government (IT-1175-19 and PIBA 2018-34) and Diputación Foral de Guipúzcoa (RED 2018).References
- J. J. Giner-Casares, M. Henriksen-Lacey, M. Coronado-Puchau and L. M. Liz-Marzán, Mater. Today, 2016, 19, 19–28 CrossRef CAS.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2020, 120, 464–525 CrossRef CAS PubMed.
- R. A. Potyrailo, Chem. Soc. Rev., 2017, 46, 5311–5346 RSC.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- V. Chiozzi and F. Rossi, Nanoscale Adv., 2020, 2, 5090–5105 RSC.
- Q. Dai, C. Walkey and W. C. W. Chan, Angew. Chem., Int. Ed., 2014, 53, 5093–5096 CrossRef CAS.
- H. Zhang, W. Wang, S. Mallapragada, A. Travesset and D. Vaknin, Nanoscale, 2017, 9, 164–171 RSC.
- M. A. Schroer, F. Lehmkühler, J. Möller, H. Lange, G. Grübel and F. Schulz, J. Phys. Chem. Lett., 2018, 9, 4720–4724 CrossRef CAS.
- A. B. Serrano-Montes, D. Jimenez de Aberasturi, J. Langer, J. J. Giner-Casares, L. Scarabelli, A. Herrero and L. M. Liz-Marzán, Langmuir, 2015, 31, 9205–9213 CrossRef CAS PubMed.
- M. Liu, W.-C. Law, A. Kopwitthaya, X. Liu, M. T. Swihart and P. N. Prasad, Chem. Commun., 2013, 49, 9350–9352 RSC.
- A. M. Alkilany, A. I. B. Yaseen, J. Park, J. R. Eller and C. J. Murphy, RSC Adv., 2014, 4, 52676–52679 RSC.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, 2003 Search PubMed.
- T. A. Grünewald, A. Lassenberger, P. D. J. van Oostrum, H. Rennhofer, R. Zirbs, B. Capone, I. Vonderhaid, H. Amenitsch, H. C. Lichtenegger and E. Reimhult, Chem. Mater., 2015, 27, 4763–4771 CrossRef PubMed.
- R. Iida, H. Mitomo, Y. Matsuo, K. Niikura and K. Ijiro, J. Phys. Chem. C, 2016, 120, 15846–15854 CrossRef CAS.
- M. A. Aboudzadeh, A. Iturrospe, A. Arbe, M. Grzelczak and F. Barroso-Bujans, ACS Macro Lett., 2020, 9, 1604–1610 CrossRef CAS.
- Y. Wang, J. E. Q. Quinsaat, T. Ono, M. Maeki, M. Tokeshi, T. Isono, K. Tajima, T. Satoh, S.-I. Sato, Y. Miura and T. Yamamoto, Nat. Commun., 2020, 11, 6089 CrossRef CAS.
- Y. Wang, W. Qin and D. Qiu, Langmuir, 2014, 30, 5170–5175 CrossRef CAS PubMed.
- A. P. Hill, C. Kunstmann-Olsen, M. P. Grzelczak and M. Brust, Chem. – Eur. J., 2018, 24, 3151–3155 CrossRef CAS PubMed.
- G. Morgese, B. Shirmardi Shaghasemi, V. Causin, M. Zenobi-Wong, S. N. Ramakrishna, E. Reimhult and E. M. Benetti, Angew. Chem., Int. Ed., 2017, 56, 4507–4511 CrossRef CAS.
- D. L. Ho, B. Hammouda, S. R. Kline and W.-R. Chen, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 557–564 CrossRef CAS.
- I. Ojea-Jiménez, N. G. Bastús and V. Puntes, J. Phys. Chem. C, 2011, 115, 15752–15757 CrossRef.
- M. A. Aboudzadeh, M. Dolz, X. Monnier, E. González de San Román, D. Cangialosi, M. Grzelczak and F. Barroso-Bujans, Polym. Chem., 2019, 10, 6495–6504 RSC.
- X.-P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- Y. Long, R. A. Shanks and Z. H. Stachurski, Prog. Polym. Sci., 1995, 20, 651–701 CrossRef CAS.
- G. Zardalidis, J. Mars, J. Allgaier, M. Mezger, D. Richter and G. Floudas, Soft Matter, 2016, 12, 8124–8134 RSC.
- E. Reimhult, M. Schroffenegger and A. Lassenberger, Langmuir, 2019, 35, 7092–7104 CrossRef CAS.
- C. Wohlfarth, CRC Handbook of Enthalpy Data of Polymer-Solvent Systems, 2006 Search PubMed.
- G. Yuan and B. Hammouda, Polymer, 2019, 166, 178–183 CrossRef CAS.
- T. Gillich, C. Acikgöz, L. Isa, A. D. Schlüter, N. D. Spencer and M. Textor, ACS Nano, 2013, 7, 316–329 CrossRef CAS PubMed.
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS PubMed.
- V. M. Amoskov, T. M. Birshtein and V. A. Pryamitsyn, Macromolecules, 1998, 31, 3720–3730 CrossRef CAS.
- A. Erbas and J. Paturej, Soft Matter, 2015, 11, 3139–3148 RSC.
- M. Divandari, L. Trachsel, W. Yan, J.-G. Rosenboom, N. D. Spencer, M. Zenobi-Wong, G. Morgese, S. N. Ramakrishna and E. M. Benetti, ACS Macro Lett., 2018, 7, 1455–1460 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures of NIR, DSC, and FSC data, and pictures of the AuNP@PEG dispersions and CPEG in ethanol. See DOI: 10.1039/d1sm00720c |
| This journal is © The Royal Society of Chemistry 2021 |