
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(ethylene oxide) grafted silica nanoparticles: efficient routes of synthesis with associated colloidal stability†
Sébastien
Issa
a,
Fabrice
Cousin
*b,
Marine
Bonnevide
c,
Didier
Gigmes
a,
Jacques
Jestin
b and
Trang N. T.
Phan
 *a
*a
aAix Marseille Univ, CNRS, Institut de Chimie Radicalaire, UMR 7273-Campus Scientifique St Jérôme, Service 542, 13397 Marseille Cedex 20, France. E-mail: trang.phan@univ-amu.fr
bLaboratoire Léon Brillouin, UMR 12, Université Paris-Saclay, IRAMIS/CEA Saclay, 91191 Gif-sur-Yvette Cedex, France. E-mail: fabrice.cousin@cea.fr
cManufacture Française des Pneumatiques MICHELIN, Site de Ladoux, 23 place des Carmes Déchaux, F-63 040 Clermont-Ferrand, Cedex 9, France
First published on 14th June 2021
Abstract
In this study, poly(ethylene oxide) monomethyl ether (MPEO) of molecular weight of 5000, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and 20000 g mol−1 were grafted onto colloidal silica nanoparticles (NPs) of a 27.6 nm diameter using two distinct “grafting to” processes. The first method was based on the coupling reaction of epoxide-end capped MPEO with amine-functionalized silica NPs, while the second method was based on the condensation of triethoxysilane-terminated MPEO onto the unmodified silica NPs. The influence of PEO molecular weight, grafting process and grafting conditions (temperature, reactant concentration, reaction time) on the PEO grafting density was fully investigated. Thermogravimetric analysis (TGA) was used to determine the grafting density which ranged from 0.12 chains per nm2 using the first approach to 1.02 chains per nm2 when using the second approach. 29Si CP/MAS NMR characterization indirectly revealed that above a grafting density value of 0.3 PEO chains per nm2, a dendri-graft PEO network was built around the silica surface which was composed of PEO chains directly anchored to the silica surface and those grafted to silica NPs by intermediate of >CH–O–Si– bonds. The colloidal stability of the particles during different steps of the grafting process was characterized by small-angle X-ray scattering (SAXS). We have found that the colloidal systems are stable whatever the achieved grafting density due to the strong repulsions between the NPs, with the strength of repulsion increasing with the molecular weight of the grafted MPEO chains.
000, and 20000 g mol−1 were grafted onto colloidal silica nanoparticles (NPs) of a 27.6 nm diameter using two distinct “grafting to” processes. The first method was based on the coupling reaction of epoxide-end capped MPEO with amine-functionalized silica NPs, while the second method was based on the condensation of triethoxysilane-terminated MPEO onto the unmodified silica NPs. The influence of PEO molecular weight, grafting process and grafting conditions (temperature, reactant concentration, reaction time) on the PEO grafting density was fully investigated. Thermogravimetric analysis (TGA) was used to determine the grafting density which ranged from 0.12 chains per nm2 using the first approach to 1.02 chains per nm2 when using the second approach. 29Si CP/MAS NMR characterization indirectly revealed that above a grafting density value of 0.3 PEO chains per nm2, a dendri-graft PEO network was built around the silica surface which was composed of PEO chains directly anchored to the silica surface and those grafted to silica NPs by intermediate of >CH–O–Si– bonds. The colloidal stability of the particles during different steps of the grafting process was characterized by small-angle X-ray scattering (SAXS). We have found that the colloidal systems are stable whatever the achieved grafting density due to the strong repulsions between the NPs, with the strength of repulsion increasing with the molecular weight of the grafted MPEO chains.
Introduction
Addition of inorganic nanoparticles (NPs) to a polymer matrix is a well-known strategy for improving the various properties of polymeric materials such as mechanical strength,1–3 thermal stability,4,5 barrier properties6–8 and optical properties.9–11 Such improvements are induced not only by the physical presence of the NPs forming a connected filler network12 but also by the interactions of the polymer with the NPs and the dispersion state of the NPs within their polymer hosts.13–15 Most importantly, the last factor is a key determinant of the physical properties of the polymer–NP composites.16 However, the irreversible aggregation of NPs in polymers can occur due to the high surface to volume ratio of NPs and strong particle–particle interactions, resulting in deleterious effects on the final properties of the materials.17,18 One of the main strategies to prevent the close particle–particle surface contacts, thereby controlling the dispersion state of the NPs within the polymer matrix, is grafting chains with an analogous chemistry as the one of the polymer matrix onto the NPs.1,19–22 Previous research has demonstrated miscibility of polymer-grafted NPs in a chemically identical polymer matrix for various grafting density and the molecular weight of grafted chains and host chains.21,23,24Among different inorganic fillers, silica NPs were widely studied and used due to their well-defined ordered structure, excellent biocompatibility, thermal stability, high surface area, versatility for surface modification and relatively low cost.25–28 The covalent grafting of polymers onto the surface of silica NPs has attracted much attention in the past few decades.29–31 There are traditionally two main pathways to attach polymer chains onto NPs cores, the “grafting from” and the “grafting to” methods. The “grafting from” method consists of growing polymer chains directly from a surface functionalized with an initiator using various reversible deactivation radical polymerization techniques (RDRP).32,33 A large number of vinylic monomers including styrene,34–37 (meth)acrylates38–41 and diene monomers30,42 were grafted onto silica surfaces using this approach. In the “grafting to” method, NPs and polymer are synthesized separately and then subsequently connected together.31,43–46 While the “grafting from” method results in higher grafting densities of polymer chains, the main advantage of the “grafting to” method is the facile design and characterization of specific structures and architectures of the pre-fabricated polymer chains. Furthermore, the “grafting to” approach is more suitable for polymers synthesized by ionic polymerization which requires harsh polymerization conditions, for example poly(oxazoline),45,47 and poly(ethylene oxide)31,48 or polymers synthesized using catalyzed chain growth like polyolefins.49 However, the grafting density obtained by the “grafting to” process is generally low, particularly when the polymer chains are long. Indeed, once a first chain is grafted on the silica surface, it hinders another chain in solution approaching and anchoring to the silica surface. An increase of the molecular weight of the grafted polymer significantly reduces the access of the surface to the functional end chain and hence limiting the grafting density.
In the domain of composite colloidal particle, poly(ethylene oxide) (PEO) grafted NPs have been considered as the most effective system for reducing protein adsorption50,51 and are also used as drug delivery systems.52,53 PEO is also an attractive semi-crystalline polymer, widely used as a solid polymer electrolyte for applications in solid state lithium battery materials,54 as PEO/silica nanocomposites can increase both ionic conductivity and mechanical properties of its solid polymer electrolytes.55 As previously mentioned, linear PEO grafted silica NPs are exclusively prepared by “grafting to” method that includes two procedures, the one-spot/one-step method and the post-modification in two steps. In the one spot/one step method, Xu et al.51 prepared poly(ethylene glycol) (PEG) grafted silica by adding hydroxyl terminated PEG at the beginning of the preparation process of the silica NPs according to Stober's method in a mixture of methanol/ammonia. A similar procedure was recently reported by Akbari et al.56 for the preparation of PEG-grafted silica NPs for lowering protein adsorption applications. The first example of the two steps method was reported by Bridger and Vincent, where PEG-grafted silica NPs were synthesized via the reaction of triethoxysilane-terminated PEG with bare silica.57 Similarly, Oh et al.58 prepared low molecular weight PEG-grafted silica NPs by adding triethoxysilane functionalized PEG in an emulsion preparation process of silica particles. Feng et al.48 grafted carboxyl-terminated PEG onto commercial silica particles of 40 nm bearing alkyl-hydroxy groups by a direct esterification reaction at room temperature. Recently, Shui et al.31 reported a facile approach to enhance the grafting density of silica NPs grafted with PEG by employing a mixture of good and poor solvents. Their study demonstrated the possibility to control the grafting density (0.76–2.80 chains per nm2) of various molecular weights of epoxide terminated PEG grafted onto dry silica particles of 50 nm functionalized with amine groups by changing the ratio of an n-decane/toluene solvent mixture. These previous studies also highlighted the critical role of the solvent nature, the molecular weight of the PEG and the nature of the functional groups on the grafting of PEG onto the surface of silica particles. The common point of these reports is that they deal with dry silica particles rather than colloidal silica solutions and the molecular weight of the grafted PEO was never exceeded of 5000 g mol−1. Additionally, the dispersion states of the resulting PEG-grafted silica NPs were only sparely investigated.31,48,57
However, the access to PEO grafted silica NPs with a molecular weight higher than the critical entanglement molecular weight of PEO (Mc of 4000 g mol−1)59 is more than ever a challenge and a real need to get well-dispersed silica NPs inside a high molecular weight PEO matrix,21 a request for improving the mechanical properties of composite materials. Furthermore, the colloidal stability during different steps of grafting is also a key point to obtain an efficient dispersion of NPs when preparing silica NPs–polymer composites. It is thus important to correctly characterize the resulting PEO grafted silica NPs to ensure colloidal stability after grafting. In addition, well-dispersed PEO grafted silica NPs as reinforcing agents for PEO based electrolyte matrixes could be a promising potential candidate for lithium battery applications.
For these reasons, the aim of this research was to systematically study the effect of grafting density, PEO molecular weight and synthesis process on the colloidal stability of the resulting PEO grafted silica NPs of 27.6 nm. We used the two steps “grafting to” method, where end-functionalized poly(ethylene oxide) methyl ether (MPEO) (with molecular weights of 5000, 10000 and 20000 g mol−1) were grafted onto either amino-modified or bare silica NPs in the colloidal state in N,N-dimethyl acetamide (DMAc). The structure and crystallization behavior of the resulting PEO grafted silica NPs were deeply investigated by a combination of different complementary techniques. A careful attention has been devoted to analyze in detail the level of particles dispersion and stability by using small-angle X-rays scattering (SAXS).
Experimental
Materials
Poly(ethylene oxide) methyl ether (MPEO) (with number average molecular weight, Mn, of 5000, 10000 and 20000 g mol−1), epichlorohydrin (99%), sodium hydride (60% dispersion in mineral oil), (3-aminopropyl) triethoxysilane (APTES, 98%), anhydrous methanol (99.8%), anhydrous ethanol (99,5%) and N,N-dimethylacetamide (DMAc, ≥99%) were purchased from Aldrich, France and used as received. Ultracel® membranes with molecular weight cutoff of 10, 30, 100 kDa were obtained from Merck Millipore. All solvents and other reagents were synthesis grade and used without further purification.
Spherical SiO2 nanoparticles (Ludox TM40) were purchased from Aldrich, France initially dispersed in water at 40 wt%. They were transferred in DMAc by evaporation of the water according to the procedure described in the literature.37 The NPs diameter dispersed in DMAc was determined by SAXS to be equal to 276 Å.
000 and 20000 g mol−1. These functionals MPEO are further denoted as epoxy-MPEOx where x represents the Mn of MPEO in kg mol−1.
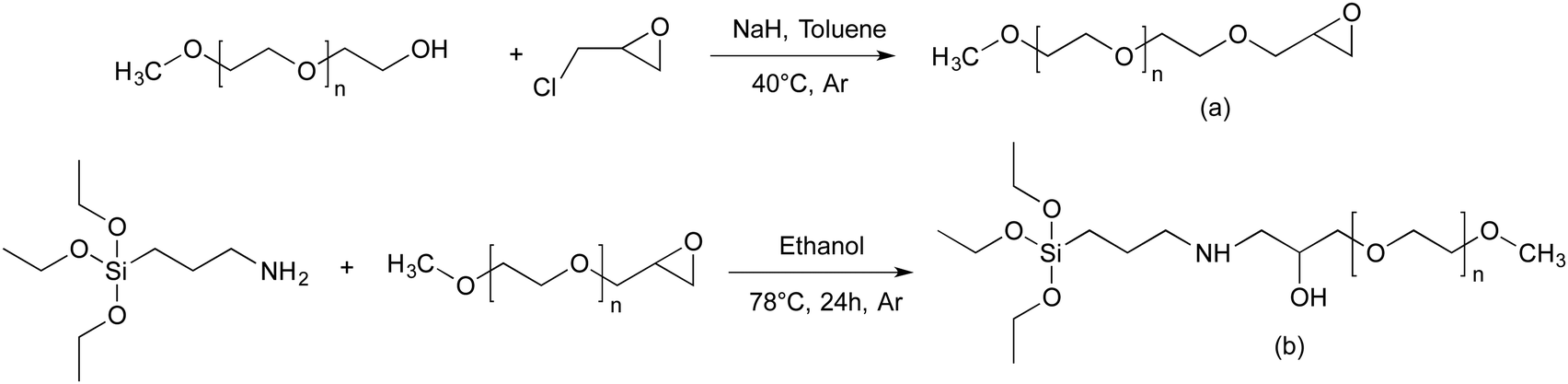 | ||
| Scheme 1 Synthesis pathway towards end functionalized methoxy-poly(ethylene oxide). (a) α-Methoxy-ω-epoxy-PEO, (b) triethoxysilane-terminated MPEO. | ||
000 and 20000 g mol−1.
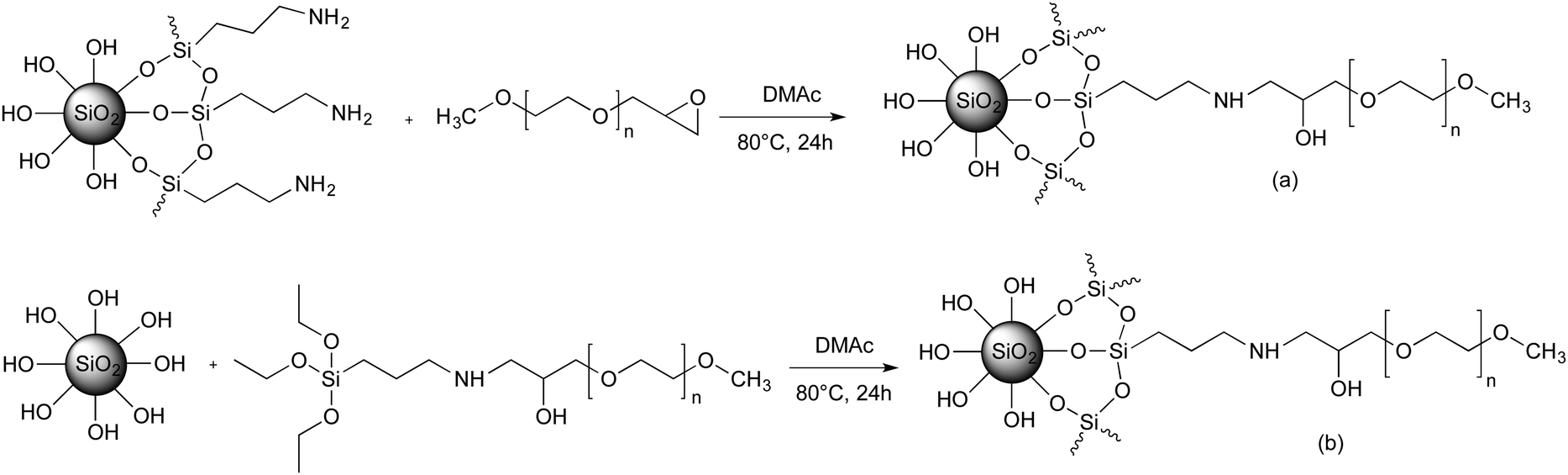 | ||
| Scheme 2 General strategy for preparing of PEO-grafted silica NPs via the “grafting to” method. (a) Coupling reaction of epoxide-terminated MPEO with amino-modified silica; (b) Coupling reaction of triethoxysilane-terminated MPEO with native silica NPs. | ||
| Entry | Sample name | σ (th) | PEO mass (g) | Weight loss between 250 °C–750 °Cb (%) | σ (exp) |
|---|---|---|---|---|---|
| All experiments were carried out with 10 g of silica NPs solution of 6 wt%. Experiments of entries 1–7 were performed with PEO of 5000 g mol−1 whereas those of entries 8–11 were performed with PEO of 10000 g mol−1. σ is the grafting density expressed as PEO chains per nm2.a Targeted σ.b Determined by TGA.c Experimental value of σ calculated by using eqn (1) from the data obtained by TGA.d Experiment carried out at 80 °C for 48 h.e Experiment carried out at 80 °C for 72 h.f The concentration of silica NPs in the resulting mixture was fixed at 1.5 wt%. |
|||||
| 1 | PEO5-SiO2_0.06 | 5 | 1.384 | 7.65 | 0.06 |
| 2 | PEO5-SiO2_0.09 | 8 | 2.222 | 10.60 | 0.09 |
| 3 | PEO5-SiO2_0.12 | 10 | 2.769 | 14.25 | 0.12 |
| 4d | PEO5-SiO2_0.17 | 10 | 2.769 | 17.70 | 0.17 |
| 5e | PEO5-SiO2_0.19 | 10 | 2.769 | 19.25 | 0.19 |
| 6 | PEO5-SiO2_0.14 | 13 | 3.600 | 15.15 | 0.14 |
| 7 | PEO5-SiO2_0.15 | 15 | 4.153 | 16.00 | 0.15 |
| 8 | PEO10-SiO2_0.03 | 2.17 | 1.205 | 7.70 | 0.03 |
| 9 | PEO10-SiO2_0.04 | 3.55 | 1.971 | 9.65 | 0.04 |
| 10f | PEO10-SiO2_0.02 | 3.55 | 1.971 | 5.60 | 0.02 |
| 11 | PEO10-SiO2_0.05 | 5 | 2.776 | 11.50 | 0.05 |
| Entry | Sample name | σ (th) | PEO mass (g) | Weight loss between 250 °C–750 °Cb (%) | σ (exp) |
|---|---|---|---|---|---|
| All experiments were carried out with 10 g of a silica NPs solution of 6 wt%. Entries 1 and 2 were performed with MPEO of 5000 g mol−1, while entries 3 – 6 were performed with MPEO of 10000 g mol−1 and the last entry (entry 7) with MPEO of 20000 g mol−1. σ is the grafting density expressed as PEO chains per nm2.a Targeted σ.b Determined by TGA.c Experimental value of σ calculated by using eqn (1) from the data obtained by TGA. | |||||
| 1 | PEO5-SiO2_0.25 | 1 | 0.697 | 22.55 | 0.25 |
| 2 | PEO5-SiO2_1.02 | 2 | 1.395 | 54.30 | 1.02 |
| 3 | PEO10-SiO2_0.025 | 0.1 | 0.139 | 5.50 | 0.025 |
| 4 | PEO10-SiO2_0.22 | 0.5 | 0.697 | 33.90 | 0.22 |
| 5 | PEO10-SiO2_0.30 | 1 | 1.395 | 41.10 | 0.30 |
| 6 | PEO10-SiO2_0.82 | 1.5 | 2.092 | 65.60 | 0.82 |
| 7 | PEO20-SiO2_0.38 | 1 | 2.790 | 63.85 | 0.38 |
Analytical techniques
All solid-state NMR spectra were obtained on a Bruker Avance-400 MHz NMR spectrometer operating at 29Si resonance frequency of 79.5 MHz. Approximatively 50 mg of samples were placed in a zirconium dioxide rotor of 4 mm outer diameter and spun at a Magic Angle Spinning rate of 10 kHz in a commercial Bruker Double Channel probe. 29Si CP/MAS experiments were performed with the Cross Polarization (CP) technique62 using a ramped 1H-pulse starting at 100% power and decreasing until 50% during the contact time (5 ms) in order to circumvent Hartmann–Hahn mismatches.63,64 To improve the resolution, a dipolar decoupling GT8 pulse sequence was applied during the acquisition time.65
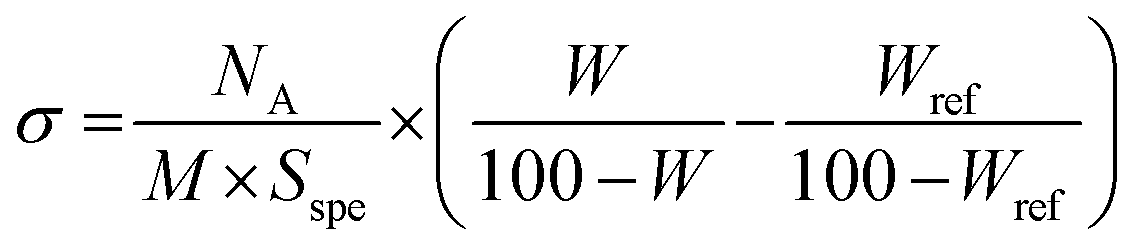 | (1) |
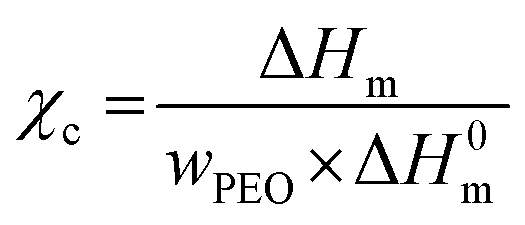 | (2) |
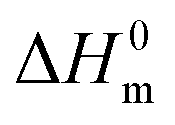 is the melting enthalpy, 195 J g−1, of a 100% crystalline PEO.66 The glass transition temperature of the PEO could not be properly measured because it is mostly crystalline.
is the melting enthalpy, 195 J g−1, of a 100% crystalline PEO.66 The glass transition temperature of the PEO could not be properly measured because it is mostly crystalline.
Results and discussion
Preparation of end-functional Methoxy-PEO
Prior proceeding to the preparation of PEO-grafted silica NPs, we need to introduce the appropriated end-functionality to the methoxy-PEO. For this purpose, α-methoxy-PEO (MPEO) was end-capped by an epoxy group at the ω-position. According to the reaction scheme reported by Van Butsele et al.60 (Scheme 1a), the ω-hydroxy end group of MPEO was deprotonated by sodium hydride and then reacted with epichlorohydrin in toluene at 40 °C. The sodium chloride byproduct formed a precipitate in toluene thereby allowing a shift in the reaction equilibrium toward the expected α-methoxy-ω-epoxy-PEO. The MPEO samples of different molecular weight (Mn = 5000, 10000 and 20000 g mol−1) were epoxidized. The 1H NMR spectrum of epoxide-terminated MPEO of 5000 g mol−1 (Fig. S1, ESI†) shows key signals corresponding to the expected structure of epoxy-MPEO. The signals at 3.57 and 3.31 ppm correspond to the methylene protons of the ethylene oxide units and the methoxy α-end group respectively (labelled a, b) whereas those at 3.08, 2.72 and 2.54 ppm were attributed to the protons of the epoxide end group (labelled c, d, e). The epoxidation yield was estimated from the relative integral value of the peaks (a) and (c). This ratio was close to 3:1, indicating a quasi-quantitative conversion of the hydroxy-end group of MPEO into an epoxy unit. The 1H NMR spectra of epoxy-MPEO10 and epoxy-MPEO20 are shown in Fig. S2 and S3 (ESI†). 1H NMR analysis indicates a quasi-quantitative epoxidation yield for the MPEO of 10000 and 20000 g mol−1.
To obtain the triethoxysilane-terminated MPEO, the α -methoxy- ω-epoxy-PEO (epoxy-MPEO) was reacted with (3-aminopropyl)triethoxysilane (APTES). The reaction between epoxide and amines is well-known for the fabrication of epoxy resin without catalyst or formation of byproducts. A large excess of APTES (15 eq. compared to epoxy-PEO) was used to shift the reaction equilibrium toward the expected triethoxysilane-terminated MPEO. The MPEO of different molecular weights (Mn = 5000, 10000 and 20000 g mol−1) were functionalized with triethoxysilane group. The 1H NMR spectrum (Fig. 1) of the triethoxysilane-terminated MPEO of 5000 g mol−1 shows key signals corresponding to the expected structure. The signal at 3.57 and 3.31 ppm correspond to methylene protons of the ethylene oxide units and the methoxy α-end group respectively (labelled a, b). The signals at 1.23 ppm and 3.78 ppm are attributed to CH3 and CH2 protons of the ethoxysilane group, (CH3–CH2–O–)3Si–, (labelled c, d) respectively. However, the proton signal of –CH2–O–Si– are overlaid with the rotation signals of the methylene protons of ethylene oxide units. The functionalization yield was estimated from the relative integral value of the peaks (a) and (c). This ratio was close to 3:9, indicating a quasi-quantitative conversion of the epoxy-end groups of the MPEO into triethoxysilane groups. The 1H NMR spectra of the triethoxysilane-terminated MPEO10 and triethoxysilane-terminated MPEO20 are shown in Fig. S4 and S5 (ESI†). 1H NMR analysis indicates that the quasi-quantitative functionalization yield was also obtained for the MPEO of 10000 and 20000 g mol−1.
 | ||
| Fig. 1 1H NMR spectrum of triethoxysilane-MPEO of 5000 g mol−1 in CDCl3. | ||
Grafting of functional MPEO onto colloidal silica NPs
All reactions were carried out in colloidal silica solutions in DMAc. The choice of DMAc as solvent for the dispersion of silica NPs and the “grating to” process was motivated by the good dispersion of silica NPs reported in the literature.37,67 It seems that such stability is governed by the polarity of the DMAc. In addition, DMAc is a good solvent for PEO. The dispersion stability of modified silica NPs in DMAc was checked at each step of the reaction by SAXS measurements. According to the protocol 1, the silica NPs were first functionalized by silanization reaction using APTES to introduce amino functionality to their surface. After the separation by ultrafiltration of the grafted aminosilane molecules from the unreacted ones, the dried amine-SiO2 NPs was characterized by TGA in order to determine the content of grafted aminosilane. For an initial targeted grafting density of 1 silane molecule per nm2, we obtained 0.4 silane molecule per nm2. This value was comparable to that reported in the literature for the same silica NPs.37 Other experiments have been investigated in order to increase the amine-SiO2 grafting density by varying the reaction time, the targeted grafting density and the reaction temperature. However, they either lead to a similar grafting density, or to an aggregation of the NPs in solution. In the second step of protocol 1, the PEO chains are attached to the surface of silica NPs by the coupling reaction between epoxide-terminated PEO and amine-SiO2 in DMAc at 80 °C. The reaction takes place without a catalyst and without the formation of by-products, and hence the colloidal stability is better preserved in particular by avoiding possible interactions with such by-products. In order to obtain the optimal reaction conditions by using epoxy-MPEO5 as a model, numerous experiments have been performed by changing the NPs concentration, the reaction time, the reaction temperature and the targeted grafting density. From this screening study, several experiments showed an aggregation of NPs during the grafting reaction or during the sample storage (less than one week), for example when the silica NPs concentration in the resulting mixture was higher than 3 wt% or when the reaction temperature was higher than 80 °C. The optimal reaction conditions while maintaining the colloidal stability were obtained for 24 h of reaction at 80 °C with a final concentration of NPs in the mixture not larger than 3 wt%. The data of these experiments are summarized in Table 1.Fig. S6 (ESI†) shows the TGA curves of native silica, amine-SiO2 and PEO grafted silica NPs sample (Table 1, entry 1). The weight loss for amine-SiO2 is mainly attributed to the decomposition of –(CH2)3–NH2 of grafted APTES whereas that of PEO–SiO2 is due to the decomposition of both APTES segments and PEO chains. The σ of the grafted PEO onto silica NPs was calculated based on eqn (1) and the results are reported on Table 1. The obtained results show that the PEO σ increases while maintaining colloidal stability when the theoretical targeted σ (Table 1, entries 1–7 for PEO of 5000 g mol−1 and entries 8–11 for PEO of 10000 g mol−1) and the reaction time (Table 1, entries 3–5) increase. One can also note that σ slightly decreases with an increase in PEO molar mass for the similar coupling conditions (Table 1, entries 1 and 11) and a decrease in the silica NP concentrations in the reaction mixture (Table 1, entries 9 and 10). Dependence of the σ with the molar mass of the grafted chains was already reported by Shui et al.31 and this value decreased from 2.0 to 0.5 PEO chains per nm2 for MPEO of 750 and 4000 g mol−1 respectively. The authors explained this difference by the fact that PEO of high Mn excludes a greater volume than the PEO of low Mn, which hinder other PEO chains approaching and anchoring to the silica surface. However, the σ reported by these authors was higher than those obtained in the present work. In our best case, the maximal σ value was 0.19 PEO chains per nm2 for MPEO of 5000 g mol−1. This difference can be explained by several factors such as the use of bigger particle size (50 nm instead of 27.6 nm in our study), working with dried silica instead of colloidal silica, and the use of a longer amine spacer (N-(2-aminoethyl)-3-aminopropylmethyldimethoxy-silane instead of APTES) and smaller PEO chains. The limiting point to increase the PEO σ was the low grafting density of amine molecules onto the silica surface in the first step. Indeed, only 0.4 amine molecules per nm2 were grafted versus 2.14 amines per nm2 in the work of Shui et al.31 However, the coupling yield, by comparing the ratio of PEO σ and amine σ, was high in our study, 37.5% (Table 1, entry 7) versus 23.4% for the same reaction time of 24 h.
Despite our efforts, increasing the grafting density of high Mn PEO was unachieved by approach 1 (Table 1, entries 9–11). For this reason, another strategy was developed (protocol 2) which is based on the organosilane condensation of triethoxysilane-terminated MPEO onto the silica NP surfaces (Scheme 2b). This strategy was first proposed by Bridger et al.57 in which the authors studied the grafting of triethoxysilane-end capped PEO chains of 18000 g mol−1 directly added to a silica dispersion in a water/methanol mixture during the later stage of its formation. They found a grafting density of 0.03 PEO chains per nm2 for silica particles of 162 nm. In the present study, three PEO of 5000, 10000 and 20000 g mol−1 were grafted onto colloidal silica NPs via condensation process carried out at 80 °C for 24 h in a mixture containing 3 wt% of silica NPs. Experimental data as well as σ determined by TGA measurements are reported in Table 2.
The results in Table 2 clearly show that the PEO σ were higher than those obtained using protocol 1. By varying the targeted σ from 0.1 to 1.5 MPEO per nm2 (Table 2, entries 3–6), one can achieve a very high grafting densities ranging from 0.025 to 0.82 PEO per nm2 for a PEO of 10000 g mol−1. On the other hand, at a same theoretical σ value fixed at 1 PEO chain per nm2 (Table 2, entries 1, 5 and 7), one can note that the experimental σ increases with the MPEO molecular weight that is contrary to the results observed in protocol 1 and those reported in the litterature.31 We explain these high grafting densities by the possibility of the condensation reaction of triethoxysilane-terminated MPEO with hydroxyl groups present on the functional PEO chain in addition to the condensation reaction of triethoxysilane-terminated MPEO with silanol functions at the silica surface (Scheme 3). The hydroxyl group was formed by the reaction of the epoxide group on the MPEO end chain with the primary amine of APTES during the ring-opening process (Scheme 1b). This hypothesis was further verified using 29Si CP/MAS NMR analysis (see “chemical and structural characterization of the grafted PEO” section).
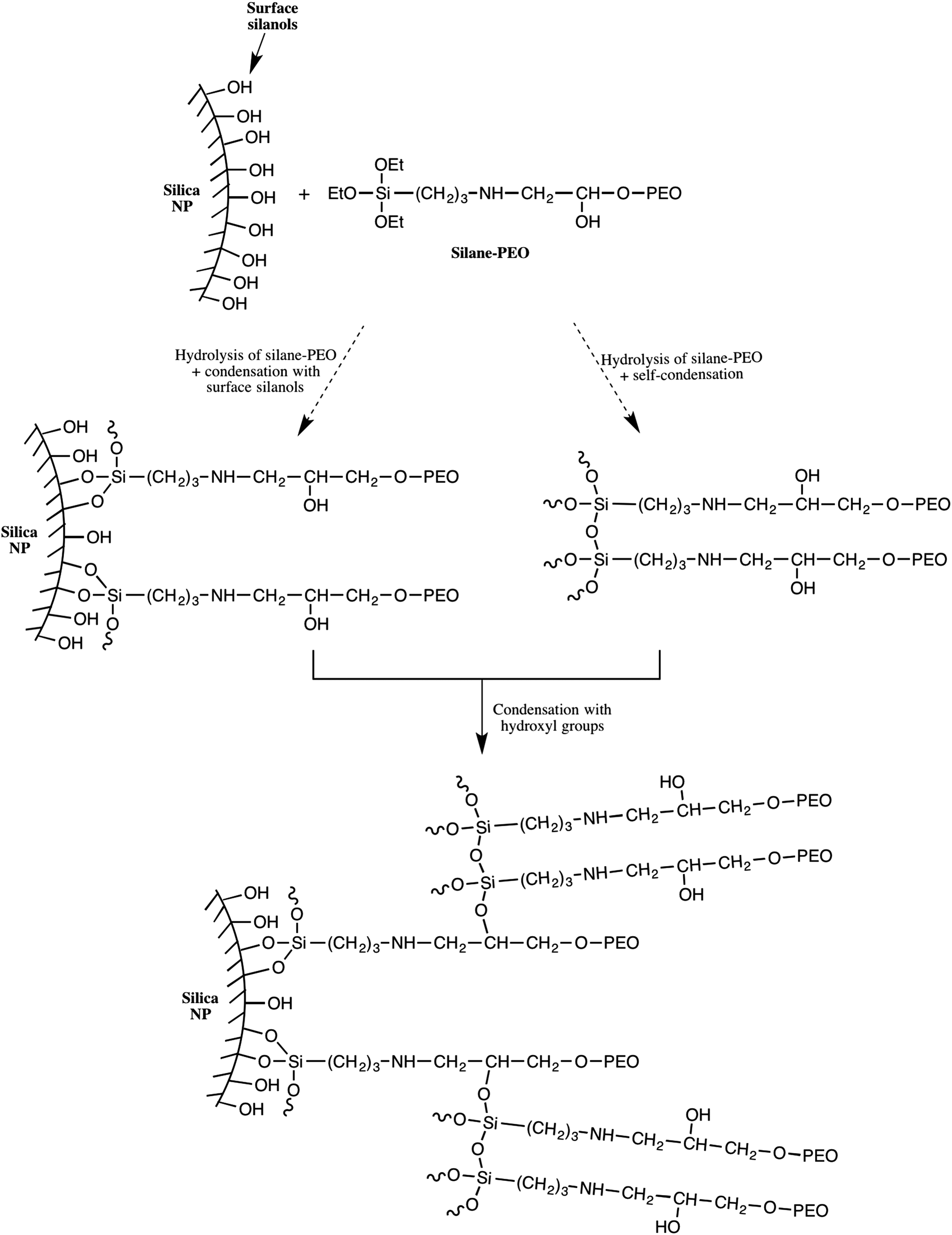 | ||
| Scheme 3 Illustration of the formation of PEO network shell around the silica nanoparticle. | ||
Chemical and structural characterizations of PEO-grafted silica NPs
The surface functionalized silica NPs were first characterized by ATR-FTIR. The spectra of unmodified silica, silane-MPEO and PEO-functionalized silica at different grafting densities are showed in Fig. 2a. In the spectrum of unmodified silica, the broad peak at 1100–1000 cm−1 is assigned to the Si–O–Si asymmetric stretching vibration and the small peak around 800 cm−1 is attributed to the asymmetric stretching vibration of Si–OH. After the grafting of PEO onto silica surface, the spectra of the functionalized silica displayed the new peaks at 1342 cm−1, 1466 cm−1 and around 2750-3000 cm−1 which are attributed to –CH2–wagging vibration, bending vibration and stretching vibration respectively. This indicates that PEO chains were successfully grafted onto silica NPs. In addition, the intensity of these peaks relating to grafted PEO increased with the grafting density of PEO chains (Fig. 2b).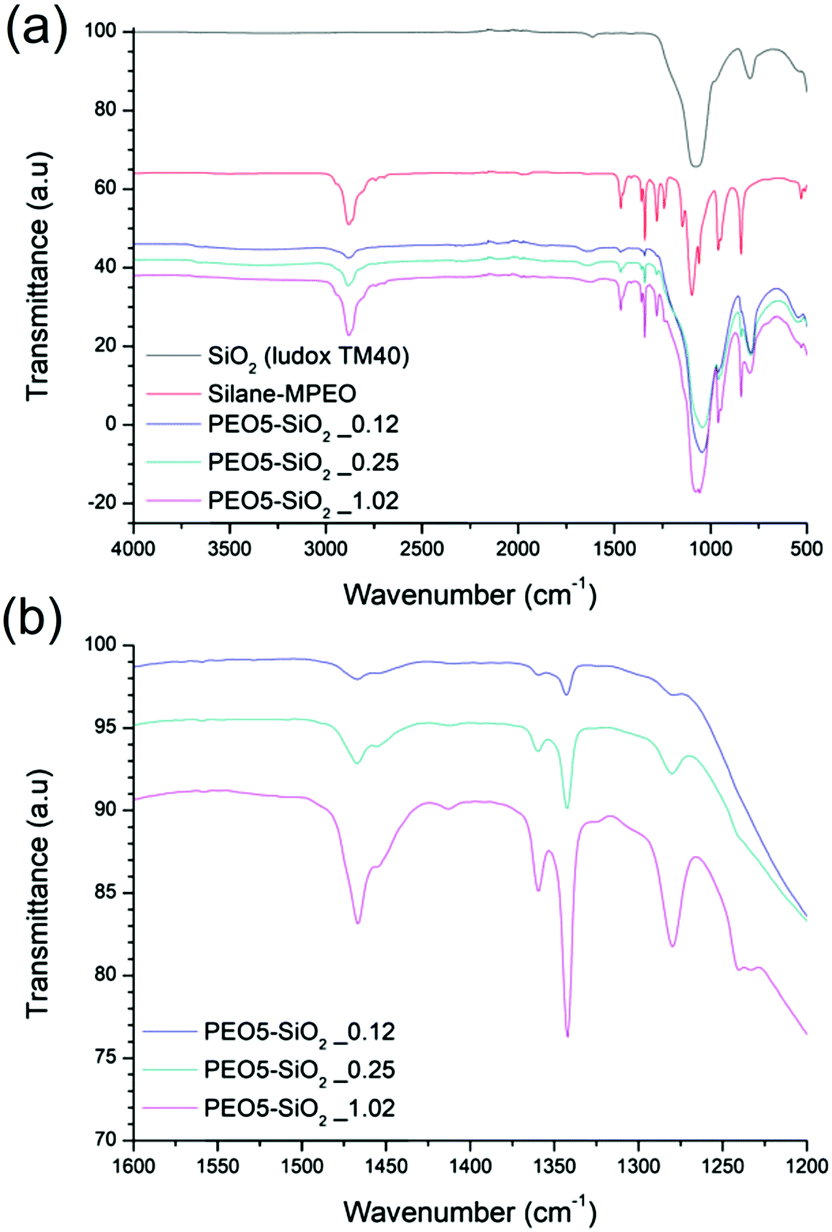 | ||
| Fig. 2 ATR-FTIR spectra of (a) unmodified silica, silane-MPEO and PEO-functionalized silica at different grafting densities and (b) spectra of PEO-functionalized silica at different grafting densities in a wavenumber rang from 1600 cm−1 to 1200 cm−1. | ||
The surface chemistry of the native and modified silica NPs was analyzed by 29Si CP/MAS NMR. As shown in Fig. 3, native silica exhibits several overlapping signals associated with Qn units: three resonances at −91.2 ppm, −101.7 ppm and −111.0 ppm are assigned to Q2, Q3 and Q4 species, respectively. The Q2, Q3 and Q4 species represent (HO)2Si(OSi)2 (geminal silanol), HOSi(OSi)3 single silanols (either isolated or vicinal) and Si(OSi)4 (fully condensed silica) species respectively.31 After the condensation reaction with APTES followed by the reaction of amine-grafted silica (NH2–SiO2) with epoxy-MPEO (Protocol 1, Scheme 2a), the 29Si MAS NMR spectra of modified-silica NPs (Fig. 3) showed a new peak around −65 ppm which is assigned to Tn centers. On these spectra, two resonances were observed at −58 ppm and −66 ppm which are attributed to R-Si(OH)(OSi)2 (T2) and R–Si–(OSi)3 (T3) centers respectively, where R represents an alkyl chain. MestReNova software was used to fit the multiple Gaussian distributions of the 29Si CP/MAS NMR spectra. Based on Gaussian deconvolution of Qn signal species, the ratio of r(Q) = (Q2 + Q3)/Q4 contributions decreased in NH2–SiO2 (Table 3, entries 1 and 2). After grafting of epoxy-MPEO, this ratio remains the same as that of NH2–SiO2 (Table 3, entry 3) since the grafting of MPEO was performed via the reaction with amine and not with silanols. The apparition of signals of T2 and T3 species and the decrease of Q2 and Q3 in favor of Q4 suggest that silanols at the silica surface were consumed in the silanization process with APTES. Since APTES was covalently grafted to silica surface therefor MPEO was too.
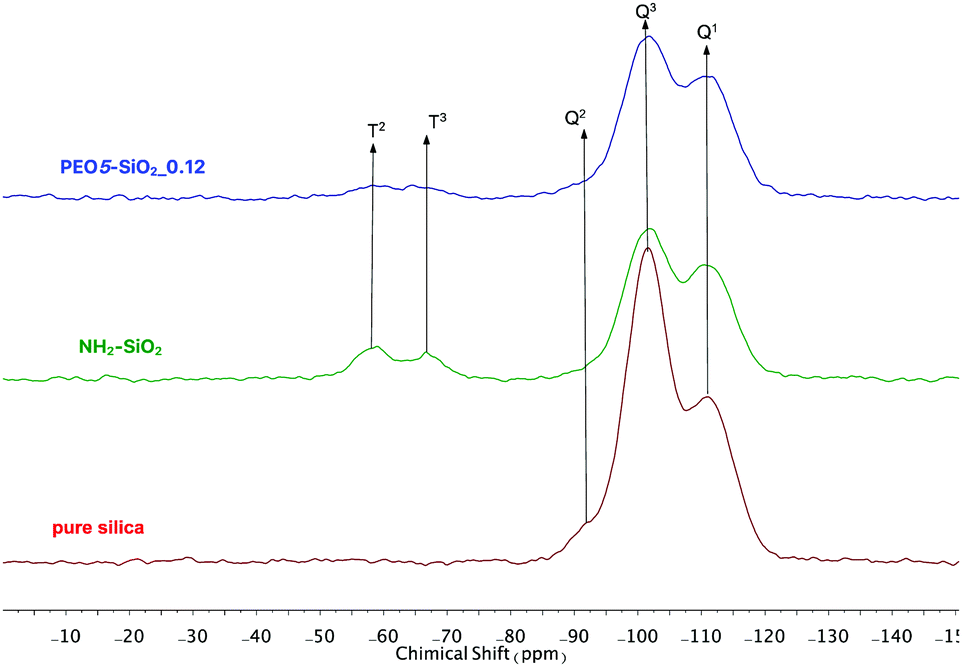 | ||
| Fig. 3 29Si CP/MAS NMR spectra of (bottom) pure silica, (middle) NH2-grafted silica and (top) PEO5-SiO2_0.12 obtained by protocol 1. | ||
| Entry | Sample name | Grating protocol | σ | r(Q) |
|---|---|---|---|---|
| 1 | Pure silica | — | 0 | 2.5 |
| 2 | NH2-SiO2 | Protocol 1 | 0.39 | 1.4 |
| 3 | PEO5-SiO2_0.12 | 0.12 | 1.4 | |
| 4 | PEO5-SiO2_0.25 | Protocol 2 | 0.25 | 1.8 |
| 5 | PEO10-SiO2_0.22 | 0.22 | 2.1 | |
| 6 | PEO10-SiO2_0.3 | 0.3 | 1.4 | |
| 7 | PEO10-SiO2_0.82 | 0.82 | 1.4 |
Analyzing the data of r(Q) ratio of all samples prepared by protocol 2 (Table 3, entries 3–7), we noted that r(Q) varied from 2.5 to 1.4 with the increase of σ and it attained a plateau at σ 0.3. This means that the conversion of Q2 and Q3 species to Q4 species by direct silanization of alkoxysilane compounds with silanols at the silica surface is limited. Indeed, once the PEO chains bind to the silica surface, they occupy a volume and hinder other chains approaching the surface of the silica NPs. Although the r(Q) value remains unchanged, we still note an increase of σ (Table 3, entry 7). These results suggest that in addition to the condensation reaction of triethoxysilane-terminated MPEO with surface silanols, there was an over-grafting of PEO chains via the condensation of triethoxysilane-terminated MPEO with hydroxyl groups present on the functional PEO chain. These grafted PEO chains were not directly anchored to the silica surface but grafted to the silica NPs through a network built by >CH–O–Si bonds and siloxane bonds (Scheme 3). The following mechanism is proposed: firstly, triethoxysilane-terminated MPEO condenses with surface silanols turning the Q2 and Q3 species into Q4 until the steric hindrance inhibits this reaction; secondly, ethoxysilane groups bearing by silane-MPEO chains condense together or with a hydroxy group on another PEO chain, which is free or already anchored to silica surface, constituting a polymer shell network around the silica NPs. The proposed structure of the PEO network around the silica NPs is schematically illustrated in Scheme 2.
0.3. This means that the conversion of Q2 and Q3 species to Q4 species by direct silanization of alkoxysilane compounds with silanols at the silica surface is limited. Indeed, once the PEO chains bind to the silica surface, they occupy a volume and hinder other chains approaching the surface of the silica NPs. Although the r(Q) value remains unchanged, we still note an increase of σ (Table 3, entry 7). These results suggest that in addition to the condensation reaction of triethoxysilane-terminated MPEO with surface silanols, there was an over-grafting of PEO chains via the condensation of triethoxysilane-terminated MPEO with hydroxyl groups present on the functional PEO chain. These grafted PEO chains were not directly anchored to the silica surface but grafted to the silica NPs through a network built by >CH–O–Si bonds and siloxane bonds (Scheme 3). The following mechanism is proposed: firstly, triethoxysilane-terminated MPEO condenses with surface silanols turning the Q2 and Q3 species into Q4 until the steric hindrance inhibits this reaction; secondly, ethoxysilane groups bearing by silane-MPEO chains condense together or with a hydroxy group on another PEO chain, which is free or already anchored to silica surface, constituting a polymer shell network around the silica NPs. The proposed structure of the PEO network around the silica NPs is schematically illustrated in Scheme 2.
Thermal properties of PEO-grafted silica NPs
The thermal transition temperatures of PEO-grafted silica were evaluated by DSC measurements. Since the content of PEO in PEO-grafted SiO2 composites is too low when σ < 0.2, it is difficult to measure properly the thermal transitions of PEO, so the DSC analysis was only performed for samples with σ value higher than 0.2 and DSC data are summarized in Table 4. Due to the low fraction of amorphous phases and small amount of sample used in DSC experiments, neither raw PEO nor PEO-grafted silica samples show a glass transition temperature. All composite samples exhibit a melting peak of the PEO chains attached to silica NPs, which was at a temperature lower than that of the corresponding raw PEO. The thermograms obtained for the melting of PEO in the PEO5-SiO2 and PEO10-SiO2 series are given in Fig. S7 and S8 (ESI†) respectively. The evolution of the PEO melting peaks in these figures clearly shows that both the melting temperature and the degree of the crystallinity of grafted PEO chains are depressed compared to the corresponding value of the parent homopolymer. The same results are also obtained for grafted PEO chains of 20000 g mol−1 (Table 4, entry 6). Similar trends have been reported in the literature.48,68 Feng et al. attributed this phenomenon to a decrease of the mobility of the grafted PEO chains which is hindered by the proximity with the silica surface.48 The proximity with the rigid silica phase strongly disturbed the PEO chains aligning together to form ordered regions and hence the formation of spherulites. As a consequence, there are less spherulites or faulty spherulites and thus the grafted PEO chains melt at a lower temperature with a smaller quantity of heat enthalpy compared to raw PEO.
| Entry | Sample name | T m (°C) | ΔHm (J g−1) | w PEO | χ c (%) |
|---|---|---|---|---|---|
|
a Weight fraction of PEO in PEO-grafted silica NPs composites determined by TGA.
b
,c,dcommercialized PEO of 5000, 10000 and 20000 g mol−1 respectively. |
|||||
| 1 | PEO5-SiO2_0.25 | 39.2 | 28.0 | 0.23 | 61.9 |
| 2 | PEO5-SiO2_1.02 | 46.9 | 70.6 | 0.55 | 65.5 |
| 3 | PEO10-SiO2_0.22 | 44.2 | 40.3 | 0.34 | 60.3 |
| 4 | PEO10-SiO2_0.30 | 50.9 | 55.9 | 0.42 | 69.1 |
| 5 | PEO10-SiO2_0.82 | 54.6 | 100.5 | 0.66 | 78.1 |
| 6 | PEO20-SiO2_0.38 | 55.9 | 92.7 | 0.64 | 74.2 |
| 7 | PEO5b | 57.8 | 174.4 | 1 | 89.4 |
| 8 | PEO10c | 60.9 | 154.7 | 1 | 79.3 |
| 9 | PEO20d | 58.7 | 149.6 | 1 | 76.7 |
Fig. S7 and S8 (ESI†) and Table 4 show that the values of Tm and crystallinity degree increase with the grafting density. This dependence has been systematically investigated by Wen et al.68 They found that the crystallization temperature of grafted PEO, hindered by confinement effects, decreases gradually as grafting density decreases. This trend is more pronounced with the grafted PEO chains of low Mn because they are more confined and more difficult to stretch out on SiO2–NH2 surfaces. In our materials, in parallel to the confinement effects, it was suggested that beyond a σ value of 0.3 chain per nm2, grafted PEO chains are not directly anchored to the silica surface but connected together via a >CH–O–Si and siloxane bonds network as schematically represented in Scheme 2 and described in previous section. These PEO chains are more mobile than those directly attached to silica surface, as a result, the values of Tm and χc increase with σ.
State dispersion of modified-silica NPs
SAXS analysis was used to evaluate the dispersion of modified silica NPs including amine-grafted silica NPs and PEO-grafted silica NPs in solution after their synthesis. The SAXS curve of the NH2-SiO2 solution in DMAc is presented in Fig. 4. The scattering curve displays four main features: (i) at very low q values (q < 0.01 Å−1), the scattering intensity decreases when q decreases, showing that the isothermal osmotic compressibility (χT) of the system is very weak; (ii) at q* ∼ 0.01 Å−1, it shows a strong correlation peak; (iii) at intermediate q values (0.01 Å−1 < q < 0.1 Å−1), it shows three oscillations which are characteristic of the form factor of spheres; (iv) at the largest q, it decays like q−4. The q−4 decay at large q is characteristic of the Porod law and demonstrates that one probes a sharp interface with the solvent at local scale. The thickness of the aminopropyl layer is indeed too small to give a signicant contribution to the scattering. | ||
| Fig. 4 (a) SAXS measurement of amine-grafted silica NPs (NH2–SiO2) in DMAC at Φ = 0.0345 (empty symbols) and its theoretical scattering intensity for one particle (red line). (b) Structure factor (S(q)) obtained by SAXS for the NH2–SiO2 colloidal solution. | ||
Since the nanoparticles are centrosymmetric objects, the scattering intensity I(q) is given by eqn (3).
| I(q) = Φ(ρSiO2 − ρDMAc)VP(q)S(q) | (3) |
At large q, S(q)q→∞ = 1 by principle, the scattered intensity can thus be fitted by a pure form factor P(q) in the q-range. This has been done by the form factor of spheres with a lognormal distribution with a radius R0 = 135 Å and polydispersity Đ = 0.12, using the SASfit software, which gives a mean radius Rmean of 138 Å, where  . Such model enables to fit perfectly the experimental curve for q > 0.02 Å−1 (Fig. 4a). The values of R0 and Đ correspond to those provided by the manufacturer.
. Such model enables to fit perfectly the experimental curve for q > 0.02 Å−1 (Fig. 4a). The values of R0 and Đ correspond to those provided by the manufacturer.
The structure factor S(q) can then be extracted from such form factor P(q) using eqn (3) (Fig. 4b). The isothermal osmotic compressibility χT(S(q)q→0) tends to a value of 0.55 that demonstrates that the potential interaction between nanoparticles is dominated by strong repulsions. These repulsions are presumably of electrostatic origin as DMAc is a polar solvent (εr = 37.8) and there remains some charged silanol groups at the surface, which ensure the good colloidal stability to the system. The large correlation peak at q* = 0.0096 Å−1 corresponds to the mean distance between nanoparticles dmean of 654 Å (2π/q*). Owing to the repulsions, the nanoparticles must be homogeneously distributed within the solution that would correspond to a dmean of ∼(2R0) (π/6Φ)1/3. The obtained value is 683 Å with Φ = 0.0345, which is very close to the experimental one, that demonstrates the perfect dispersion of the nanoparticles.
When the nanoparticles are grafted with PEO chains, whether by protocol 1 or protocol 2, the scattering intensities of all samples show the same features as those of the NH2–SiO2 solution, in particular the Porod-like q−4 decay at large q (see Fig. 5 for the PEO5–SiO2 samples synthesized by protocol 1 with different σ and Fig. 6 for the PEOx–SiO2 synthesized by protocol 2 with different σ).
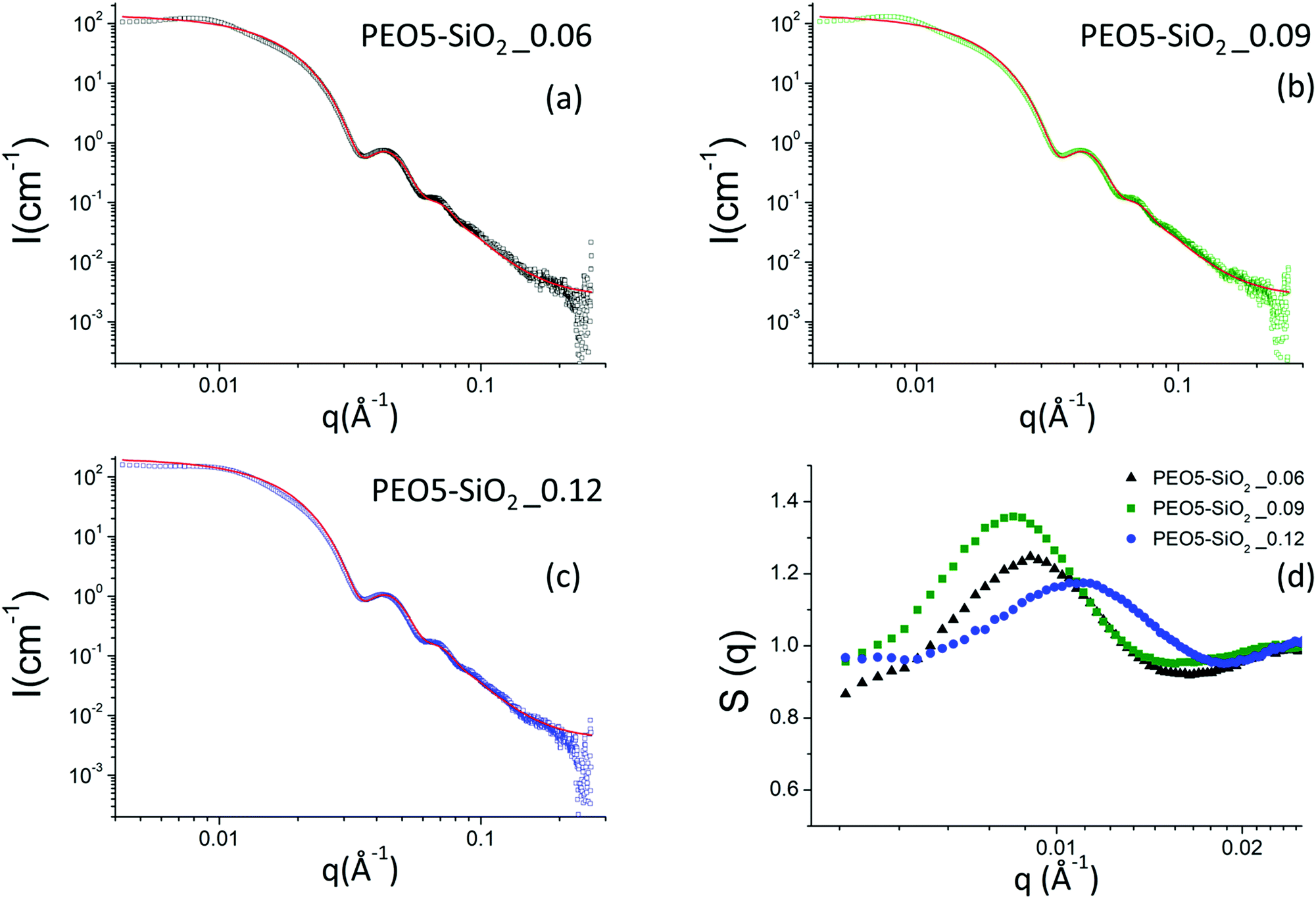 | ||
| Fig. 5 (a–c) Scattering intensities obtained by SAXS for a series of MPEO of 5000 g mol−1 grafted onto silica NPs in DMAc at different grafting densities. The continuous red line corresponds to the form factor of the silica nanoparticles. (d) Nanoparticle structure factors obtained by SAXS using the nanoparticle form factor of the NH2–SiO2 colloidal solution (Fig. 4a) for a series of MPEO of 5000 g mol−1 grafted onto silica NPs in DMAc at different grafting densities. The grafting was performed according to protocol 1 (Scheme 2a and Table 1). | ||
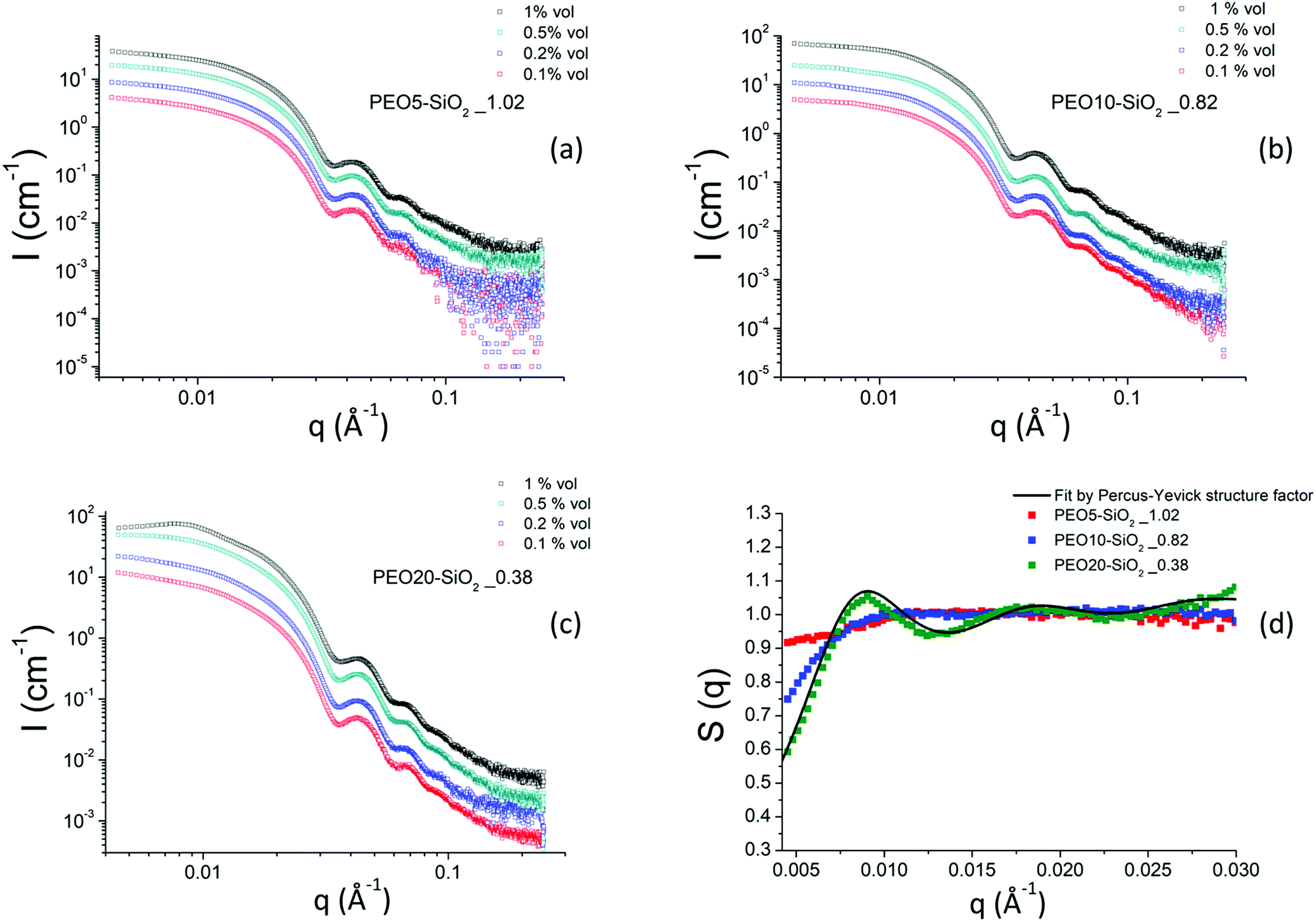 | ||
| Fig. 6 (a–c) Scattering intensities obtained by SAXS for MPEO of 5000, 10000 and 20000 g mol−1 grafted onto silica NPs in DMAc as function of nanoparticle volume fraction. (d) Nanoparticle structure factors obtained by SAXS for a series of MPEO of 5000 g mol−1, 10000 g mol−1 and 20000 g mol−1 grafted silica NPs in DMAc. The black continuous line is a fit by Percus–Yevick structure factor (see text). The grafting was performed according to protocol 2 (Scheme 2b and Table 2). | ||
However, at this local scale, one could expect to obtain another power law corresponding to the conformation of the grafted PEO chains. The contribution of these chains is however very weak compared to that of silica nanoparticles in SAXS. Indeed, since the electronic scattering length densities of the components are respectively ρSiO2 = 18.9 1010 cm−2, ρDMAc = 8.8 1010 cm−2 and ρPEO= 11.2 × 1010 cm−2, the SiO2 core of the nanoparticles are much more contrasted than the PEO coronas since (ρSiO2 − ρDMAc)2/(ρSPEO − ρDMAc) = 18, assuming a dry PEO corona. A solvation of the corona by DMAc would obviously strongly enhance such a ratio. Moreover, the volume of the corona is expected to be lower than those of the core due to the low molar mass of PEO chains. The presence of the PEO chains however slightly influences the scattered intensity at intermediate q in the regime where the oscillations for the form factor is marked. When the content of the grafted chains is low, e.g. in the samples grafted with MPEO of 5000 g mol−1 using protocol 1, the calculated form factor derived from the NH2–SiO2 sample matches the oscillations of the experimental scattered intensity (Fig. 5). In this regime, the contribution of the PEO chains to the form factor is so negligible that the structure factor of the solutions can be extracted from the division of the scattered intensity with such a form factor (by eqn (3)). On the contrary, when the number of grafted chains is high, e.g., for all samples obtained by protocol 2, the presence of the corona has a direct influence on the form factor of the nanoparticles. Indeed, it appears that the oscillations of the experimental scattered intensity are shifted with respect to those of the form factor derived from the NH2–SiO2 solution, revealing the presence of the corona. By principle, a fit of the scattered intensity in the q-regime would give access to the structural parameters of the corona (thickness and solvation). However, the contrast of such corona for X-rays remains however too low compared to those of the silica core to provide a meaningful fit as it is difficult to decouple the respective influence of thickness and solvation on the signal.
The scattering intensities of the PEO5–SiO2 synthesized by protocol 1 with three different grafting densities are represented in Fig. 5a–c and all show the characteristic correlations peaks of repulsive systems at low q. The corresponding structure factors are presented in Fig. 5d. One can note that the volume fraction of the sample solutions was not exactly the same and varies from one solution to another.
All samples show an isothermal osmotic compressibility χT lower than 1 and exhibit a strong correlation peak around 0.01 Å−1 (Fig. 5d). There are thus repulsive interactions between nanoparticles and therefore good colloidal stability. The χT of these functionalized silica samples tend to a value ranging between 0.8 and 0.9. Contrary to the case of NH2–SiO2 solutions, the potential interactions between the PEO-grafted silica nanoparticles are dominated by strong repulsions which are due to the steric hindrance of grafted PEO and not originated from the remaining charges of surface silanol groups. Indeed, given the size of grafted MPEO chains, we suppose that they mask the surface charge of silica NPs. The values of the large correlation peaks (q*), the volume fraction (Φ) and the calculated dmean are reported in the Table S1 (ESI†). The calculated distance between the NPs with the structure factor (2π/q*) are of the order of magnitude of the distance calculated with the volume fraction (2R0) (π/6Φ)1/3.
Fig. 6 shows the scattered intensities of the PEO-grafted silica NPs colloidal solutions synthesized using approach 2 for three PEO grafted chains of 5000, 10000 and 20000 g mol−1 respectively. We show in this figure only the results of colloidal solutions corresponding to the highest grafted density reached for a given molar mass at various volume fractions. Other samples with lower grafting densities were also measured by SAXS and their scattered intensity are presented in the ESI† in Fig. S9.
The behavior of the scattered intensities at the low q of the most concentrated samples give a first indication of the sign and strength of interactions in the system. For the samples grafted with the MPEO of 5000 g mol−1, it shows a plateau close to a classical Guinier plateau, i.e. a pure form factor, revealing that the systems behave almost like a non-interacting system. For the samples grafted with the MPEO of 10000 g mol−1, a small correlation peak is visible along with a scattering intensity that decays when going towards q = 0, suggesting weak repulsions. Finally, for the samples grafted with the MPEO of 20000 g mol−1, there exists a strong correlation peak at low q and a strong decay when going towards q = 0, showing the presence of strong repulsions. In order to go further in the analysis, we have derived the structure factor for the 3 solutions at the highest grafting density for each molecular weight. To this aim, we have obtained the form factor corresponding to each grafting density from the most diluted samples. Indeed, for solutions at low volume fraction, the osmotic pressure can be developed by a Virial expansion limited to the second order and the isothermal compressibility becomes:
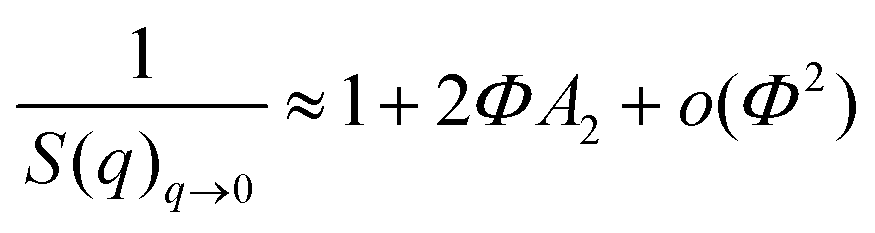
In practice, we have considered that the regime of dilution where the form factor is reached when I(q)/Φ is constant for two samples at different volume fractions (Fig. S10, ESI†). This has enabled us to obtain the structure factors of samples for the three molecular weights of MPEO chains at the same volume fraction in silica (Φsil ∼ 0.01) (Fig. 6d). These structure factors confirm the trends of a qualitative description of the scattered intensities: the systems are repulsive in all cases and an increase of the molecular weight of the grafted chains increase the strength of repulsions. χT indeed tends towards ∼0.9 for the PEO5–SiO2_1.02, revealing moderate repulsions for such sample, and then strongly decreases when increasing chains length towards respective values of ∼0.65 for PEO10–SiO2_0.82 and ∼0.4 for PEO20–SiO2_0.38. It is thus likely that the steric hindrance between particles increase with the chain length of the grafted chains, suggesting an increase of corona thickness. Moreover, for the sample PEO20–SiO2_0.38, the structure factors show a strong correlation peak at q* = 0.009 Å−1 and its second order at 2q* at 0.018 Å−1. In order to get an insight of the magnitude order of the corona thickness, we have fitted such structure factor with the well-known Percus−Yevick structure factor describing hard-sphere systems,69,70 that is well suited for spherical objects interacting through steric repulsions. In this model, the two fitting parameters are the radius of the hard sphere RHS, fitted here with a value of 336 Å, and the sphere volume fraction ΦHS, fitted here with a value of 0.115 (Fig. 6d). RHS is the sum of the silica core radius (138 Å) and of the thickness of the PEO corona, even if this latter is almost invisible by SAXS with respect to the core for contrast reasons. This provides an overall thickness of the corona of 198 Å. The obtained ΦHS is consistent with the silica volume fraction (∼0.01). The thickness is around twice the gyration radius of a 20000 g mol−1 chain, considering that DMAC is a good solvent for PEO. This thickness is much lower than those estimated from scaling laws for grafted brush in a good solvent (∼900 Å for σ = 0.38).71 This shows that such value of σ = 0.38, obtained by TGA and that takes into account all the chains within the corona, is much larger than the σ of the chains that are really grafted on the silica surface. It thus indirectly confirms that the corona is a dendri-graft PEO network.
Conclusions
In this work, we presented a detailed study of PEO-grafted hybrid materials where the PEO chains were grafted onto silica NPs of 27.6 nm using two approaches of the “grafting to” method. The efficiency of the grafting and the control of the colloidal stability of the NPs during different steps of the process were optimized by tuning synthesis parameters and have been proven by the use of different characterization techniques, namely, TGA, ATR-FTIR, solid NMR and SAXS. We demonstrated for the first time the possibility to graft a high amount of high molecular weight of PEO chains (Mn = 5000, 10000 and 20000 g mol−1) onto the colloidal silica NPs while keeping their stability throughout the process and beyond. According to SAXS analysis, the stability of the studied colloidal systems, whatever the achieved grafting density is due to the strong repulsions between NPs and this trend increases with the molecular weight of the PEO chains. On the other hand, for hybrid samples with high grafting density (σ > 0.3 PEO nm−2), 29Si solid NMR measurements indirectly revealed that PEO chains were grafted to silica NPs by two types of binding. There are the PEO chains directly anchored to the silica surface and the other ones that are grafted to silica NPs by intermediate of siloxane and >CH–O–Si– bonds. The latter are formed by hydrolysis/condensation of ethoxysilane groups (CH3–CH2–O–Si–) together and/or with hydroxyl group of the functional PEO chain, constituting an external layer of PEO around the silica NPs (Scheme 3). This result is supported by the data obtained in DSC measurements where the melting temperature of PEO and its crystallinity degree increase with σ. Indeed, the PEO chains grafted to the silica NPs via C–O–Si and siloxane bonds are more mobile than those directly anchored to silica surface and hence, the thermodynamic properties of these PEO chains are less affected.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was undertaken within the French ANR program under the contract SELPHy, No. ANR-17-CE05-0032-01. The authors thank for the funding.References
- C. Chevigny, F. Dalmas, E. Di Cola, D. Gigmes, D. Bertin, F. Boue and J. Jestin, Macromolecules, 2011, 44, 122–133 Search PubMed.
- K. Mishra, D. Gidley and R. P. Singh, Eur. Polym. J., 2019, 116, 283–290 Search PubMed.
- F. Ondreas, P. Lepcio, M. Zboncak, K. Zarybnicka, L. E. Govaert and J. Jancar, Macromolecules, 2019, 52, 6250–6259 Search PubMed.
- J. W. Gilman, Appl. Clay Sci., 1999, 15, 31–49 Search PubMed.
- S. H. Qin, Q. F. Li, M. He, H. J. Shao, J. Yu, J. B. Guo, K. Zhang and W. Yan, Polym. Eng. Sci., 2014, 54, 2911–2917 Search PubMed.
- L. N. Sridhar, R. K. Gupta and M. Bhardwaj, Ind. Eng. Chem. Res., 2006, 45, 8282–8289 Search PubMed.
- Y. B. Cui, S. Kumar, B. R. Kona and D. van Houcke, RSC Adv., 2015, 5, 63669–63690 Search PubMed.
- C. Wolf, H. Angellier-Coussy, N. Gontard, F. Doghieri and V. Guillard, J. Memb. Sci, 2018, 556, 393–418 Search PubMed.
- A. Dang, S. Ojha, C. M. Hui, C. Mahoney, K. Matyjaszewski and M. R. Bockstaller, Langmuir, 2014, 30, 14434–14442 Search PubMed.
- C. L. Wetteland and H. N. Liu, J. Biomed. Mater. Res., Part A, 2018, 106, 2692–2707 Search PubMed.
- O. Sakhno, P. Yezhov, V. Hryn, V. Rudenko and T. Smirnova, Polymers, 2020, 12, 1–21 Search PubMed.
- D. Zhao, S. F. Ge, E. Senses, P. Akcora, J. Jestin and S. K. Kumar, Macromolecules, 2015, 48, 5433–5438 Search PubMed.
- T. Wei, K. L. Jin and J. M. Torkelson, Polymer, 2019, 176, 38–50 Search PubMed.
- A. M. Jimenez, D. Zhao, K. Misquitta, J. Jestin and S. K. Kumar, ACS Macro Lett., 2019, 8, 166–171 Search PubMed.
- S. Li, Z. Y. Zhang, G. Y. Hou, J. Liu, Y. Y. Gao, P. Coates and L. Q. Zhang, Phys. Chem. Chem. Phys., 2019, 21, 11785–11796 Search PubMed.
- F. Hussain, M. Hojjati, M. Okamoto and R. E. Gorga, J. Compos. Mater., 2006, 40, 1511–1575 Search PubMed.
- M. A. Ashraf, W. X. Peng, Y. Zare and K. Y. Rhee, Nanoscale Res. Lett., 2018, 13, 214 Search PubMed.
- S. Mishra and N. G. Shimpi, J. Appl. Polym. Sci., 2007, 104, 2018–2026 Search PubMed.
- M. Bonnevide, A. M. Jimenez, D. Dhara, T. N. T. Phan, N. Malicki, Z. M. Abbas, B. Benicewicz, S. K. Kumar, M. Couty, D. Gigmes and J. Jestin, Macromolecules, 2019, 52, 7638–7645 Search PubMed.
- S. Srivastava, P. Agarwal and L. A. Archer, Langmuir, 2012, 28, 6276–6281 Search PubMed.
- S. K. Kumar, N. Jouault, B. Benicewicz and T. Neely, Macromolecules, 2013, 46, 3199–3214 Search PubMed.
- R. Hasegawa, Y. Aoki and M. Doi, Macromolecules, 1996, 29, 6656–6662 Search PubMed.
- A. S. Robbes, F. Cousin, F. Meneau, F. Dalmas, R. Schweins, D. Gigmes and J. Jestin, Macromolecules, 2012, 45, 9220–9231 Search PubMed.
- D. Maillard, S. K. Kumar, A. Rungta, B. C. Benicewicz and R. E. Prud'homme, Nano Lett., 2011, 11, 4569–4573 Search PubMed.
- Colloidal silica: fundamentals and applications, ed. E. H. Bergna and O. W. Roberts, CRS Press, 2005 Search PubMed.
- The chemistry of silica: solubility, polymerization, colloid and surface properties, and biochemistry of silica, ed. K. G. Iler, John Wiley & Sons, Inc., 1979 Search PubMed.
- R. Ciriminna, A. Fidalgo, V. Pandarus, F. Beland, L. M. Ilharco and M. Pagliaro, Chem. Rev., 2013, 113, 6592–6620 Search PubMed.
- D. W. Lee and B. R. Yoo, J. Ind. Eng. Chem., 2016, 38, 1–12 Search PubMed.
- R. Francis, N. Joy, E. P. Aparna and R. Vijayan, Polym. Rev., 2014, 54, 268–347 Search PubMed.
- M. M. Khani, Z. M. Abbas and B. C. Benicewicz, J. Polym. Sci. Part A: Polym. Chem., 2017, 55, 1493–1501 Search PubMed.
- Y. D. Shui, Y. L. Su, X. Kuang, W. W. Zhao, Y. L. Cai and D. J. Wang, Polym. Int., 2017, 66, 1395–1401 Search PubMed.
- Controlled Radical Polymerization at and from Solid Surfaces, ed. P. Vana, Spinger, 2016 Search PubMed.
- L. Wu, U. Glebe and A. Boker, Polym. Chem., 2015, 6, 5143–5184 Search PubMed.
- C. H. Liu and C. Y. Pan, Polymer, 2007, 48, 3679–3685 Search PubMed.
- M. N. Tchoul, M. Dalton, L. S. Tan, H. C. Dong, C. M. Hui, K. Matyjaszewski and R. A. Vaia, Polymer, 2012, 53, 79–86 Search PubMed.
- L. Wu, U. Glebe and A. Boker, Macromol. Rapid Commun., 2017, 38, 1600475 Search PubMed.
- C. Chevigny, D. Gigmes, D. Bertin, J. Jestin and F. Boue, Soft Matter, 2009, 5, 3741–3753 Search PubMed.
- P. Chmielarz, J. J. Yan, P. Krys, Y. Wang, Z. Y. Wang, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2017, 50, 4151–4159 Search PubMed.
- K. Ohno, Y. Ma, Y. Huang, C. Mori, Y. Yahata, Y. Tsujii, T. Maschmeyer, J. Moraes and S. Perrier, Macromolecules, 2011, 44, 8944–8953 Search PubMed.
- K. Ueno, A. Inaba, T. Ueki, M. Kondoh and M. Watanabe, Langmuir, 2010, 26, 18031–18038 Search PubMed.
- C. Deleuze, M. H. Delville, V. Pellerin, C. Derail and L. Billon, Macromolecules, 2009, 42, 5303–5309 Search PubMed.
- M. Bonnevide, T. N. T. Phan, N. Malicki, S. K. Kumar, M. Couty, D. Gigmes and J. Jestin, Polymer, 2020, 190, 122190 Search PubMed.
- S. S. Balamurugan, E. Soto-Cantu, R. Cueto and P. S. Russo, Macromolecules, 2010, 43, 62–70 Search PubMed.
- R. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217–6223 Search PubMed.
- G. Bissadi and R. Weberskirch, Polym. Chem., 2016, 7, 1271–1280 Search PubMed.
- S. Ghasemi and S. Karim, Colloid Polym. Sci., 2018, 296, 1323–1332 Search PubMed.
- J. P. Gann and M. Yan, Langmuir, 2008, 24, 5319–5323 Search PubMed.
- L. B. Feng, Y. L. Wang, N. Wang and Y. X. Ma, Polym. Bull., 2009, 63, 313–327 Search PubMed.
- Z. C. Peng, Q. Li, H. Y. Li and Y. L. Hu, Ind. Eng. Chem. Res., 2017, 56, 5892–5898 Search PubMed.
- P. F. Holmes, E. P. K. Currie, J. C. Thies, H. C. van der Mei, H. J. Busscher and W. Norde, J. Biomed. Mater. Res., Part A, 2009, 91A, 824–833 Search PubMed.
- H. Xu, F. Yan, E. E. Monson and R. Kopelman, J. Biomed. Mater. Res., Part A, 2003, 66A, 870–879 Search PubMed.
- T. Andreani, A. L. R. de Souza, C. P. Kiill, E. N. Lorenzon, J. F. Fangueiro, A. C. Calpena, M. V. Chaud, M. L. Garcia, M. P. D. Gremiao, A. M. Silva and E. B. Souto, Int. J. Pharm., 2014, 473, 627–635 Search PubMed.
- C. L. Lay, H. Q. Liu, D. C. Wu and Y. Liu, Chem. – Eur. J., 2010, 16, 3001–3004 Search PubMed.
- M. Armand, Solid State Ionics, 1983, 9-10, 745–754 Search PubMed.
- Z. Jia, W. Yuan, H. Zhao, H. Y. Hu and G. L. Baker, RSC Adv., 2014, 4, 41087–41098 Search PubMed.
- A. Akbari, R. Yegani and B. Pourabbas, Colloids Surf., A, 2015, 484, 206–215 Search PubMed.
- K. Bridger and B. Vincent, Eur. Polym. J., 1980, 16, 1017–1021 Search PubMed.
- C. Oh, C. Do Ki, J. Y. Chang and S. G. Oh, Mater. Lett., 2005, 59, 929–933 Search PubMed.
- B. J. Anderson and C. F. Zukoski, Langmuir, 2010, 26, 8709–8720 Search PubMed.
- K. V. Butsele, F. Stoffelbach, R. Jérôme and C. Jérôme, Macromolecules, 2006, 39, 5652–5656 Search PubMed.
- A. L. Li, H. Shen, H. H. Ren, C. Wang, D. C. Wu, R. A. Martin and D. Qiu, J. Mater. Chem. B, 2015, 3, 1379–1390 Search PubMed.
- J. Schaefer and O. E. Stejskal, J. Am. Chem. Soc., 1976, 98, 1031–1032 Search PubMed.
- O. B. Peersen, X. L. Wu, I. Kustanovich and S. O. Smith, J. Magn. Reson. Ser. A, 1993, 104, 334–339 Search PubMed.
- R. L. Cook, C. H. Langford, R. Yamdagni and C. M. Preston, Anal. Chem., 1996, 68, 3979–3986 Search PubMed.
- G. Gerbaud, F. Ziarelli and S. Caldarelli, Chem. Phys. Lett., 2003, 377, 1–5 Search PubMed.
- L. O. Griffin, Physical constants of linerar homopolymers, Springer Verlag: Berlin, 1986 Search PubMed.
- C. Chevigny, D. Gigmes, D. Bertin, R. Schweins, J. Jestin and F. Boue, Polym. Chem., 2011, 2, 567–571 Search PubMed.
- X. N. Wen, Y. L. Su, Y. D. Shui, W. W. Zhao, A. J. Muller and D. J. Wang, Macromolecules, 2019, 52, 1505–1516 Search PubMed.
- J. K. Percus and G. J. Yevick, Phys. Rev. A: At., Mol., Opt. Phys., 1958, 110, 1–13 Search PubMed.
- F. Cousin, in Jdn 21 – Neutrons and Materials for Energy, ed. M. Ceretti, W. Paulus, M. H. Mathon and C. Ritter, 2015, vol. 104 Search PubMed.
- S. Alexander, J. Phys., 1977, 38, 983–987 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of functional MPEO, TGA curves of unmodified and modified silica, DSC thermograms of raw MPEO and grafted PEO, SAXS curves of different PEO-grafted silica NPs solutions and table of the values of NPs mean distances. See DOI: 10.1039/d1sm00678a |
| This journal is © The Royal Society of Chemistry 2021 |