
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen inhibition of free-radical polymerization is the dominant mechanism behind the “mold effect” on hydrogels†
Rok
Simič
 ,
Joydeb
Mandal
,
Kaihuan
Zhang
and
Nicholas D.
Spencer
*
,
Joydeb
Mandal
,
Kaihuan
Zhang
and
Nicholas D.
Spencer
*
Laboratory for Surface Science and Technology, Department of Materials, ETH Zürich, Switzerland. E-mail: nspencer@ethz.ch
First published on 8th June 2021
Abstract
Hydrogel surfaces are of great importance in numerous applications ranging from cell-growth studies and hydrogel-patch adhesion to catheter coatings and contact lenses. A common method to control the structure and mechanical/tribological properties of hydrogel surfaces is by synthesizing them in various mold materials, whose influence has been widely ascribed to their hydrophobicity. In this work, we examine possible mechanisms for this “mold effect” on the surface of hydrogels during polymerization. Our results for polyacrylamide gels clearly rule out the effect of mold hydrophobicity as well as any thermal-gradient effects during synthesis. We show unequivocally that oxygen diffuses out of certain molding materials and into the reaction mixture, thereby inhibiting free-radical polymerization in the vicinity of the molding interface. Removal of oxygen from the system results in homogeneously cross-linked hydrogel surfaces, irrespective of the substrate material used. Moreover, by varying the amount of oxygen at the surface of the polymerizing solutions using a permeable membrane we are able to tailor the surface structures and mechanical properties of PAAm, PEGDA and HEMA hydrogels in a controlled manner.
Introduction
Hydrogels consist of a cross-linked polymer network and a large amount of water, which provides them with unique properties. Due to their structural similarities to biological tissues, they are often used as scaffolds for cell growth and engineering,1,2 in regenerative medicine3 and as adhesive transdermal drug-delivery patches.4 The significant water component can lead to effective lubrication properties, which has prompted applications in catheter coatings and contact lenses.5,6 In most applications, the surface structure of the hydrogels is of key importance, giving hydrogels adhesive or highly lubricious properties. Although modification of hydrogel surface properties can be achieved by various physical or chemical approaches, one of the most straightforward ways is to synthesize hydrogels by free-radical polymerization against different molding materials, which are known to influence the surface structure of the produced hydrogel.7,8Gong and coworkers first reported the heterogeneous polymerization of various hydrogels made from water-soluble vinyl monomers against a number of polymeric surfaces.7 The heterogeneous polymerization led to a gradient hydrogel structure, in which the polymer network at the surface was loosely cross-linked and less dense than in the bulk. They demonstrated the effect for monomers including acrylamide, acrylic acid and the sodium salt of styrene sulfonate, while the tested molding surfaces included polytetrafluoroethylene (PTFE), polypropylene (PP), polyethylene (PE), polystyrene (PS), polymethylmethacrylate (PMMA), and polyvinylchloride (PVC). Polymerization of the precursor solution against hydrophilic molds, such as glass, mica or sapphire, appeared to be homogeneous, resulting in an even hydrogel structure from the surface to the bulk, in contrast to the behavior against polymeric molding surfaces. Using electronic speckle pattern interferometry, the authors observed a clear interface in the vicinity of the polymeric molds during polymerization.7 The distance of the observed interface from the mold surface was shown to increase with decreasing surface energy of the molding material. On the other hand, performing the polymerization in ethanol with addition of a hydrophobic monomer greatly suppressed the appearance of the interface. Although no definitive explanation was provided for the phenomenon, the authors ascribed the appearance of the interface to the difference in surface energies of the mold and the surface tension of the polymerizing solution. It was suggested that during the retraction of the interface from the mold, the monomer-depleted solution in the vicinity of the interface leads to a formation of a soft, low-density polymer network, which is full of branched, dangling polymer chains.7,9
In a recent study we used nanoindentation to measure the elastic moduli at the surface of such gels and could show that hydrogel surfaces molded against polymeric molds were indeed significantly softer compared to those molded against glass.8 The softer surface in this case was ascribed to the presence of loose, dangling chains. However, the elastic modulus of the hydrogel surfaces seemed to not be directly related to the surface energy of the mold. Although at that point we could not explain the underlying mechanism nor confirm the hypothesis of the hydrophobic effect, we could show that polymerizing solution does not chemically react with the molding surfaces, leaving the surface properties of the molds intact after the polymerization reaction was completed.8
Due to the exothermic nature of free-radical polymerization, the temperature of the polymerizing solution can rise by several tens of degrees Celsius above the surrounding temperature. Therefore, polymerization kinetics could be significantly different close to a mold with good thermal conductivity (e.g. glass) compared to a poorly heat-conducting mold (e.g. most polymeric molds). However, it has been suggested that any such effects caused by poor thermal conductivity of the polymeric substrates might be excluded, since a PTFE mold, with relatively high thermal conductivity for a polymer, led to the most heterogeneous hydrogel structure.7 Despite that, no other evidence seems to exist that would allow us to completely reject thermal effects.
It has also been suggested that oxygen, which could become entrapped between a rough hydrophobic surface and a monomer solution, could inhibit the polymerization near the interface.10 This does not explain the effect observed on smooth molding surfaces, where the diffusion of oxygen out of oxygen-permeable molds could lead to inhibition of free-radical polymerization and result in the observed gradient hydrogel structure. The phenomenon has been reported in several fields, such as sealants,11 adhesives,12 coatings,13 microfluidics14 and photolithography.15,16 Incompletely polymerized surfaces usually suffer from decreased bonding strength and poor mechanical properties, and several approaches have been proposed to avoid the negative effects.17 While the phenomenon is detrimental in certain cases, it can be beneficial in others, such as for continuous 3D printing18 or high-throughput microparticle lithography.15 The effect has also been modeled mathematically19 as well as harnessed to form wrinkle patterns.20 While oxygen inhibition is well known and has been widely studied, there are practically no studies that would rule out the other presented mechanisms and show the dominating effect of the oxygen-inhibition when synthesizing hydrogels in different molds.
Although the exact mechanism behind heterogeneous network formation has been unknown, the resulting hydrogels were shown to have orders of magnitude lower friction compared to the same hydrogels synthesized against a glass mold.8,21,22 It was suggested that the branched polymer chains of the heterogeneously polymerized gels entrap large amounts of liquid at the surface and thus enhance the hydrodynamic thickness of the solvent layer, hence reducing friction. It has also been shown for cross-linked hydrogels that increasing the amount of water in the polymer network decreases the shear stress during sliding.23 Similar principles for reducing sliding shear are used in the contact-lens industry, where manufacturers produce lenses with soft, hydrated surface layers.24 This principle also mimics the natural lubrication mechanism in the eye, which relies upon hydrated, surface-attached mucin layers.25 Although the benefits in friction reduction of such heterogeneous systems with sparse surfaces are clear, understanding their formation in the case of hydrogels would enable a higher level of control over the process and thus of the surface properties.
In order to resolve the underlying mechanism, we have examined several plausible mechanisms in this work. The hydrophobic effect was tested by using hydrophobized glass surfaces and hydrophilized PS surfaces. To directly test the thermal effects, we have performed experiments with variously thick molding materials, which substantially affects the overall heat conduction while keeping other surface properties constant. Lastly, we have performed experiments with different amounts of oxygen in the system to check whether the heterogeneous structure is in fact caused by oxygen inhibition of the free-radical polymerization. This was done by evacuation of the air in the surroundings of the mold and back-filling with nitrogen. A second approach involved synthesis of hydrogels against an oxygen-permeable membrane while regulating the amount of oxygen on the membrane's other side. The obtained results have enabled us to reject certain hypotheses and reveal the main underlying mechanism.
Experimental
Hydrogel synthesis
The polyacrylamide (PAAm) hydrogels were synthesized from a polymerizing solution consisting of 9.6 wt% of acrylamide monomer (>99%, Sigma-Aldrich, St Louis, MO, USA) and 0.4 wt% of N,N′-methylene-bis-acrylamide cross-linker (>99.5%, Sigma-Aldrich, St Louis, MO, USA) in MilliQ water. Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photo-initiator was synthesized according to the procedure described in our previous work26 and added to the solution in the amount of 0.01 wt%. Poly(ethylene glycol) (PEG) hydrogels were synthesized from 10 wt% of poly(ethylene glycol) diacrylate (PEGDA) with average molecular weight Mn = 700 Da (Sigma-Aldrich, St Louis MO, USA), dissolved in MilliQ water together with 0.01 wt% of LAP photo-initiator. Poly(hydroxyethyl methacrylate) (PHEMA) hydrogels were synthesized from an aqueous solution of 45 wt% hydroxyethyl-methacrylate (HEMA), 15 wt% N-vinylpyrrolidone (NVP), 1.2 wt% ethylene-glycol-dimethacrylate (EGDMA), all from Sigma-Aldrich, St Louis, MO, USA. In the HEMA gels, 0.1 wt% of LAP photo-initiator was used.Once poured in the desired molds, the solutions for PAAm and PEGDA gels were polymerized for 20 min under UV light with an intensity of about 1.4 mW cm−2 at a wavelength of 365 nm (Stratalinker UV Crosslinker 2400, Stratagene Corp., La Jolla, CA, USA). HEMA gels, on the other hand, were polymerized for 40 min under the same conditions. The synthesized hydrogels were demolded and immersed in ample Milli-Q water for at least 48 hours to remove unreacted species and allow swelling of the hydrogels. Some of the HEMA gels were immersed in ethanol after demolding, which exchanged with the interstitial water and allowed additional swelling of the gels.
Molding surfaces
The selected molding materials were glass Petri dishes and sterilized polystyrene (PS) Petri dishes. The roughness values Ra of the glass and the PS molds were 1 and 5 nm, respectively. Hydrophobized glass surfaces were prepared by functionalizing the glass Petri dishes with octadecyl-trichlorosilane (OTS, >95%, Acros Organics, Geel, Belgium) by chemical vapor deposition for several hours and then rinsing with toluene. Hydrophilized PS Petri dishes were prepared by immersion in sulfuric acid for several hours and then rinsing with ultrapure water. For the thermal-conductivity experiments, thin PS sheets with a thickness of 30, 50, 125 and 190 μm (GoodFellow Cambridge Limited, Huntington, England) were used as received. Glass cover slips (Menzel-Gläser, Thermo Fischer Scientific, Massachusetts, USA) with a thickness of 150 μm were used for comparison—the thermal conductivities of glass and PS are substantially different and are about 1.0 and 0.1 W m−1 K−1, respectively.27 For the oxygen-permeability study, 11 μm-thick polyethylene (PE) and polyvinylidene-chloride (PVDC) food-wrap foils were used to cover the PS Petri dishes.The same PE foils were also used to separate the polymerizing solution from the gas mixtures of air and nitrogen. In this case, the PE foils were placed in flow-cells, which were made from polymethylmethacrylate (PMMA) plates by laser cutting. The shallow rectangular polymerization vessel measured 20 mm in width and 50 mm in length, incorporating a side-wall of 5 mm in height. The base of the vessel consisted of a 11-μm PE foil membrane, which allowed oxygen diffusion. The vessel was glued to a 2-mm-high lower chamber of same lateral dimensions as the reaction vessel. The lower chamber was connected to tubes at each end, serving as inlet and outlet. The gas flow was controlled by a syringe pump and the flow rate was set to 5 ml min−1, corresponding to a mean gas velocity of 2 mm s−1 within the chamber. Four milliliters of polymerizing solution were poured into the vessel after the gas had been flowing for several minutes and the polymerization was carried out as described earlier in the text.
Contact-angle analysis
Contact angles of ultrapure water on the surfaces were measured using a drop-shape analyzer (DSA100, Krüss GmbH, Hamburg, Germany) in sessile-drop mode. The values presented in Table 1 were measured before the polymerization on different locations of the substrates to check for their homogeneity. According to our recent study, the angles should not be affected by the polymerization.8| Substrate | Contact angle (°) | Oxygen permeability28 (cm3 25 μm (m2 24 h atm)−1) |
|---|---|---|
| Glass | <5 | 0 |
| OTS-Functionalized glass | 102 ± 2 | 0 |
| PS | 85 ± 3 | 4300 |
| Acid-treated PS | 29 ± 5 | N/A |
| PE foil | 96 ± 6 | 8586 |
| PVDC foil | 76 ± 6 | 1.2–2.3 |
Nanoindentation
In the past, we have evaluated differently molded hydrogel surfaces using various characterization techniques, including infrared spectroscopy,8 neutron reflectometry,29 rheology,22 tribology8,22,29,30 as well as various indentation methods.8,22,29,30 All the obtained data correlated well with each other, and enabled us to elucidate the surface structures of differently molded hydrogels. For this work, nanoindentation was selected for identification of the different hydrogel surfaces due to its sensitivity and nanoscopic depth resolution. The nanoindentation experiments were performed with samples fully immersed in Milli-Q water using an atomic force microscope (AFM, MFP-3D™, Asylum Research, Santa Barbara, USA). For the PHEMA gels swollen in ethanol, however, the nanoindentation experiments were performed with samples fully immersed in ethanol. The indentation probe was manufactured by attaching a silica microsphere (Kromasil, Nouryon – Separation Products, Bohus, Sweden) with a radius of 11 μm to the end of a tipless, gold-coated cantilever (NSC-36, Mikromash, Bulgaria) by means of a 2-component epoxy resin adhesive (UHU GmbH, Germany). The normal spring constant of the bare cantilever k0 was determined by applying the Sader method.31 The effective spring constant of the manufactured probe was calculated as k = k0(L0/L)3 = 1.69 N m−1, where L0 and L are the distances from the base of the cantilever to its tip and to the microsphere position, respectively.32 The relation between the cantilever deflection x and the photodiode signal U, known as optical-lever sensitivity S, was calibrated by pressing the probe against a silicon wafer in water, where the wafer was assumed to be infinitely stiff. The indentation depth d was thus calculated as d = Z − x = Z − SU, where Z is the vertical piezo displacement. The force was calculated as F = kx. The contact point was determined as the last data point lying within 2σ from the zero-force line on the approach, where σ is the root-mean-square value of the signal noise away from the surface (≈20–30 pN). Indentation and retraction speeds were set to 1 μm s−1. At this speed, only a minor hysteresis between the forces during scanning in both directions was observed, indicating the insignificance of the visco- or poro-elastic contributions to the measured force. All measurements were performed at a room temperature of 25 °C ± 1 °C. Force maps of 3 × 4 force curves were obtained over an area of 40 × 40 mm2 on 3 different locations of a sample to check for reproducibility. The graphs show representative force–indentation curves, but the given elastic moduli are the average values, with errors corresponding to one standard deviation of all measured force curves.Results and discussion
Hydrophobicity
Fig. 1a shows force–indentation curves obtained on PAAm hydrogel surfaces synthesized against hydrophilic and hydrophobic (OTS-functionalized) glass surfaces, and against hydrophobic and hydrophilic (H2SO4-treated) PS surfaces. Both glass molds, hydrophilic or hydrophobic, resulted in practically identical force–indentation curves on the hydrogel surfaces. The experimental data showed very good agreement with the Hertzian contact model (see Fig. S1, ESI†), indicating a homogeneous hydrogel structure. The extracted elastic modulus was 30 kPa ± 1 kPa for both glass-molded hydrogel surfaces. In the past, the elastic moduli of similar glass-molded hydrogels were also measured using microindentation29 and macroindentation,30 which showed a good agreement with both the Hertzian and the Winkler contact models. With both these models assuming a homogenous structure, glass-molded hydrogels appear to have a homogeneous polymer density profile throughout their thickness. While hydrogels molded against both PS molds had much softer surfaces compared to the glass-molded gels, they also appeared to be unaffected by the surface treatment. The elastic modulus extracted from within the first 1.5 μm of indentation data was well below 1 kPa, indicating a very sparse and soft structure. Increasing the indentation depth resulted in material stiffening, pointing to a progressive densification of the polymer network with increasing distance from the surface. Judging from the evolution of the force–indentation curves, the estimated thickness of this softer gradient layer is in the range of 10–20 μm. The estimated thickness is in agreement with the thickness reported for equivalent hydrogels in the literature.33 It is important to note, however, that despite the differences at the surface, glass- and PS-molded gels have a similar bulk structure.30 As expected, we again confirmed that different molding materials result in hydrogels with different surface structures. However, surface treatment and thus the hydrophobicity of the molding surface appears to have no significant effect on the surface structure of the synthesized hydrogels. | ||
| Fig. 1 (a) Representative force–indentation curves for PAAm hydrogel surfaces synthesized against a hydrophilic glass, an OTS-functionalized, hydrophobic glass, a PS surface and an acid-treated, hydrophilic PS surface. (b) Coefficient of friction as a function of speed for a hydrophilic-glass-molded PAAm surface and a hydrophobic-PS-molded PAAm surface when rubbed in a reciprocating way against a flat, hydrophilic-glass-molded PAAm pin at a contact pressure of 6 kPa in water as described in our previous study.22 | ||
To present the difference in sliding behavior of these hydrogel surfaces, Fig. 1b shows the coefficients of friction of a cross-linked, glass-molded PAAm hydrogel surface and a sparse, PS-molded hydrogel surface when slid against a cross-linked hydrogel pin at 6 kPa contact pressure in water. About an order of magnitude difference in friction can be seen over a broad range of sliding speeds. This is in agreement with the general correlation of friction and indentation data, where softer, sparser hydrogel surfaces always result in lower friction.8,22,23,30,34
Thermal conductivity
Due to the exothermic nature of free-radical polymerization, the temperature of the polymerizing solution can rise significantly above that of its surroundings. Although the exact temperature increase depends on factors such as monomer concentration, the rate of the reaction and the heat-dissipation rate, the thermal conductivity of the molding material affects the temperature of the solution near the interface (see Fig. S2, ESI†). Due to the higher thermal conductivity of glass compared to PS, the solution temperature in the vicinity of a glass surface is lower than that in the vicinity of PS. The lower solution temperature at the glass surface should lead to slower reaction kinetics in that zone and thus to a more inhomogeneous structure compared to the relatively uniform temperature distribution of the PS case. However, this would cause the opposite trend to that seen for the structures shown in Fig. 1a, which makes the thermal effects unlikely to lie behind the observed differences in the hydrogel surfaces.However, to verify any thermal effects at the interface, we have performed experiments with PS foils of different thicknesses, in order to vary the total thermal conductivity of the mold while keeping the surface chemistry the same. Fig. 2 shows the results as well as a scheme of a one-sided polymerization setup for the case of PAAm hydrogels, where pure water (thermal conductivity: λ ∼ 0.6 W m−1 K−1) was used as a heat sink, separated from the polymerizing solution (heat source) by a thin glass sheet or PS foils of different thicknesses. In the limiting case of an infinitely thin barrier, the thermal conductivity of the system would equal that of the water, which is relatively similar to that of the glass. If temperature would indeed be controlling the polymerization in this case, hydrogel surfaces would get stiffer and more similar to glass-molded hydrogel surfaces with decreasing PS foil thickness. However, irrespective of the PS foil thickness, all hydrogels had similarly soft surfaces resembling that of the bulk PS mold, Fig. 2a.
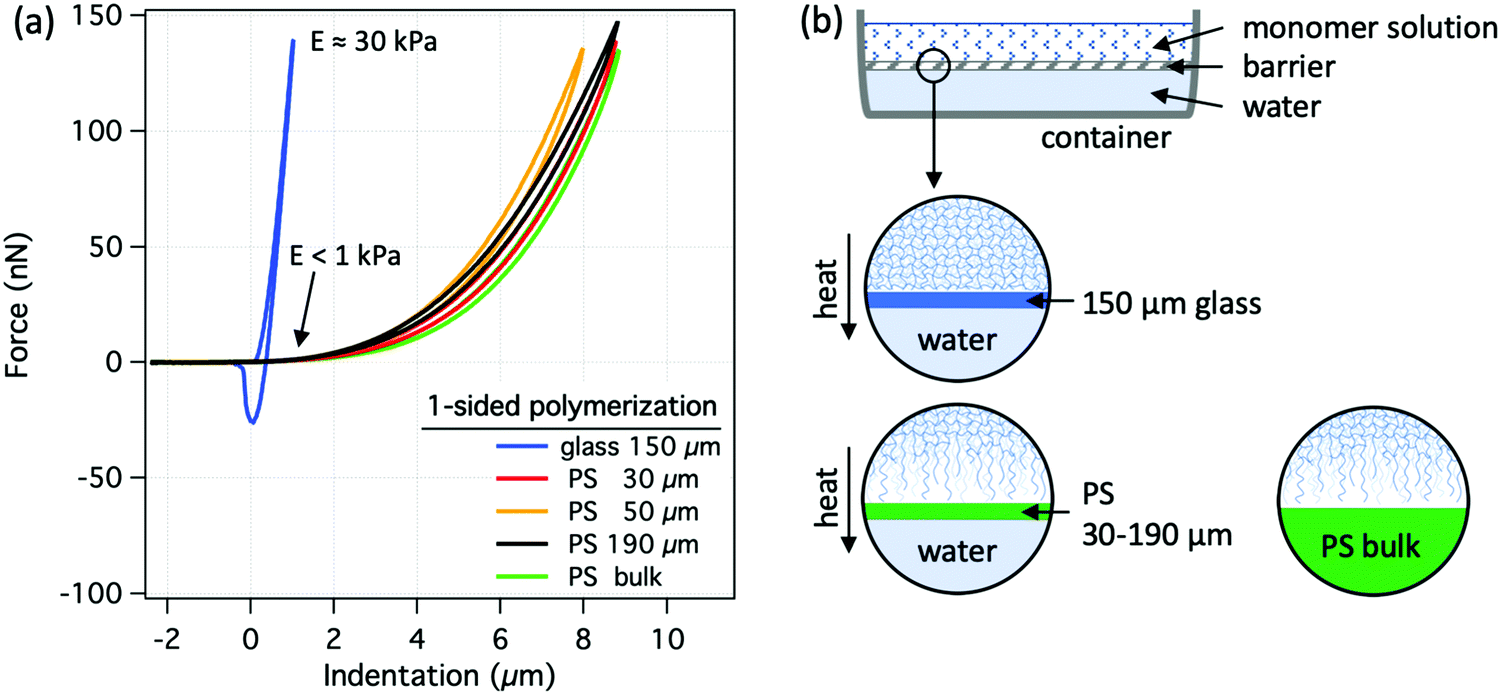 | ||
| Fig. 2 One-sided polymerization, where differently thick PS foils were used to control heat dissipation from the reaction mixture. (a) Representative force–indentation curves for variously molded PAAm hydrogel surfaces. (b) Scheme of one-sided polymerization, where the polymerizing solution was separated from pure water (good heat conductor) by a thin glass or PS barrier. | ||
A complementary experiment was performed by synthesizing hydrogels on both sides of all the above-mentioned barriers, Fig. 3. In this case, the polymerizing solution served as a heat source on both sides of the barrier, which should substantially reduce the temperature gradient at the vicinity of the barrier and thus approximate the polymerization against a bulk PS. The hydrogel surfaces formed at the two thickest PS foils were indeed sparse, soft and similar to the case with bulk PS. However, the 30 and 50 μm thick PS foils resulted in significantly stiffer hydrogel surfaces with elastic moduli in the range of 3–5 kPa within the first 1.5 μm. Besides, there was a noticeable adhesion between the silica microsphere and the two hydrogel surfaces upon retraction of the AFM probe. Both retraction force curves were phenomenologically similar to those for the cross-linked, glass-molded hydrogel surfaces, Fig. 3a. Hydrogel surfaces separated by the thin glass sheet were even stiffer and similar to the dense, cross-linked hydrogel surface synthesized against bulk glass mold, which is a good heat sink. These results indicate that heat transfer and thus the temperature cannot be responsible for the structural differences observed for the variously molded hydrogel surfaces.
 | ||
| Fig. 3 Two-sided polymerization, where differently thick PS foils were used to act as a barrier between two polymerizing solutions. No major heat dissipation was expected at the barrier–solution interface. (a) Representative force–indentation curves for PAAm hydrogels. (b) Scheme of two-sided polymerization, with either a thin glass sheet or PS foils of different thickness acting as a barrier. | ||
After ruling out the hydrophobic and the temperature effects, the observed behavior could be explained by the presence of oxygen and related inhibition of free-radical polymerization. It is known that glass is essentially impermeable to oxygen, however, the oxygen permeability of PS is about 4000 cm3 25 μm (m2 24 h atm)−1 at room temperature and 0% relative humidity.28 In the case of one-sided polymerization, oxygen could diffuse from water through the PS films and inhibit the polymerization, making all the PS-molded hydrogel surfaces sparse and soft, Fig. 2. In the case of the two-sided polymerization, the oxygen within the solution was consumed during the reaction. However, there seems to be a sufficient amount of oxygen stored within the thick (>125 μm) PS foils, which can diffuse out and inhibit the polymerization reaction at the interface. On the other hand, the lesser amount of oxygen in the two thinner (<50 μm) PS foils could only partially inhibit the reaction, resulting in cross-linked hydrogel surfaces, whose elastic moduli lay in between those of the regular glass-molded and PS-molded hydrogels, Fig. 3. Last but not least, the diffusion of oxygen out of the molding material also explains the results in Fig. 1, where surface functionalization had no effect on the surface structure, which appeared to depend solely on the “bulk” molding material.
Oxygen
To verify the hypothesis of oxygen inhibition of the polymerization reaction, we have performed two sets of experiments: (i) separating the polymerizing solution from the source of molecular oxygen (i.e. PS) by high- and low-oxygen-permeable foils, such as PE and PVDC, respectively (Fig. 4b), and (ii) synthesizing hydrogels against PS in oxygen-free environments (Fig. 5b). | ||
| Fig. 4 Using low- and high-oxygen-permeable foils to modulate oxygen transport from PS to the polymerizing solution. (a) Representative force–indentation curves for PAAm gels. Glass- and PS-molded PAAm hydrogels are shown for comparison. (b) Scheme of the experimental setup with PE- or PVDC-covered PS (oxygen source). | ||
 | ||
| Fig. 5 Hydrogels synthesized in an oxygen-free environment achieved by evacuation and subsequent back-filling with nitrogen gas. (a) Scheme of the experimental setup. (b) Representative force–indentation curves for PAAm hydrogels. Results for hydrogels molded against ambient-equilibrated glass and PS are shown for comparison. (c) Elastic modulus as a function of evacuation time of the PS mold material. | ||
Fig. 4 shows the force–indentation curves for PAAm hydrogels molded against PS, which was covered by either a permeable PE foil or by PVDC foil with low oxygen permeability. Reference curves obtained on glass- and PS-molded hydrogels are shown for comparison. Hydrogels obtained in “PE-on-PS” molds had similarly sparse surfaces as the reference PS-molded hydrogels, which is consistent with the high permeability of oxygen through the PE foil. Hydrogel surfaces made in “PVDC-on-PS” molds were much stiffer (E ∼ 16 kPa ± 0.4 kPa) compared to the PE-on-PS and PS-molded gels. Besides, the PVDC-molded gels showed adhesion characteristics similar to those of the cross-linked hydrogel surfaces. This indicates that the PVDC foil allows undisturbed polymerization of the hydrogel network, presumably due to the very low oxygen permeability of PVDC. The coefficients of friction for all four hydrogels were found to increase with the elastic modulus of surface (Fig. S3, ESI†), supporting the major role of oxygen rather than the hydrophobicity of the mold.
A more direct demonstration of oxygen inhibition of the polymerization reaction is shown in Fig. 5. Flasks containing PS molds were evacuated at 1.1–1.3 × 10−2 mbar for different times to remove the air from the system, and then subsequently back-filled with pure N2 to ambient pressure, Fig. 5a. An evacuation time of 2 hours already resulted in significantly stiffer hydrogel surfaces (E = 4 kPa ± 0.4 kPa) compared to the case with air-exposed PS, Fig. 5b. Increasing the evacuation time to 6 and to 24 hours increased the elastic modulus to 8 kPa ± 0.3 kPa and 23 kPa ± 0.5 kPa, respectively. Elastic moduli are shown in Fig. 5c as a function of evacuation time before back-filling with N2. It is therefore clear that longer evacuation times removed more oxygen from the PS molds and thus resulted in more homogeneous polymerization near the molding surface.
Controlling the amount of interfacial oxygen
Dedicated vessels were constructed to allow the synthesis of hydrogels against a permeable PE membrane, under which a mixture of molecular oxygen and nitrogen was flowed, Fig. 6a. Changing the gas composition allowed control over the amount of molecular oxygen that was diffusing through the membrane and interacting with the reaction mixture. The indentation curves for PAAm hydrogels synthesized against the PE membrane in combination with different amounts of oxygen in the gas composition are shown in Fig. 6b. It is clear that increasing the amount of oxygen monotonically decreases the elastic modulus of the hydrogel surface. The measured elastic modulus at the surface as a function of the amount of oxygen in the source gas is shown in Fig. 6c. The elastic modulus at 0% O2 was lower than that of the surface prepared against glass, presumably due to the oxygen trapped within the PE membrane or oxygen that could enter the system through the polymer tubing. Despite that, the measured indentation curve indicated a relatively dense and crosslinked hydrogel surface structure. Elastic modulus was then observed to decrease with increasing amount of oxygen with nearly inverse proportionality.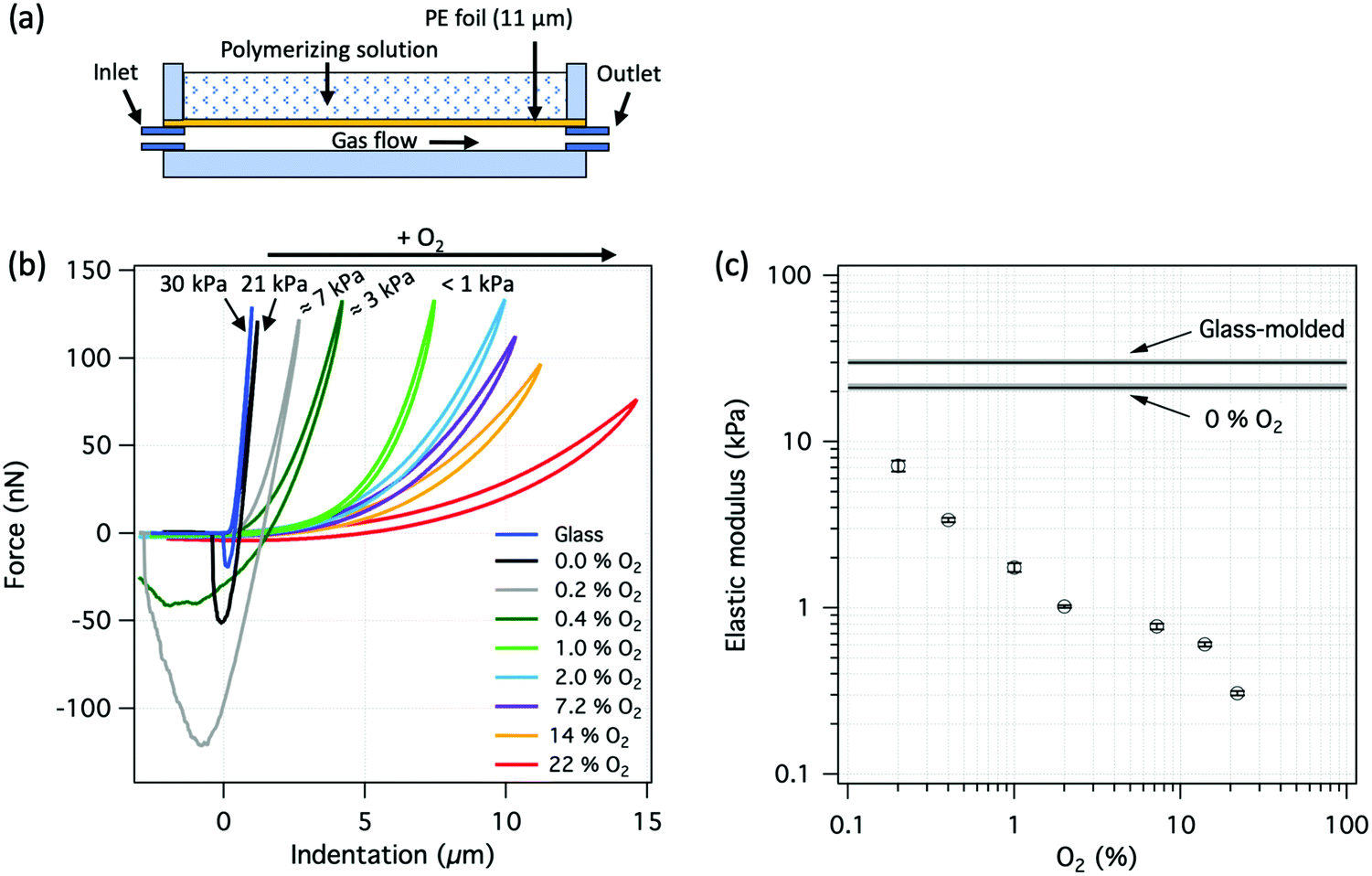 | ||
| Fig. 6 PAAm hydrogel surfaces prepared against a permeable PE foil, separating the polymerizing solution from the different gas mixtures. (a) Experimental setup. (b) Representative force–indentation curves for PAAm hydrogel surfaces prepared against a permeable PE foil.35 (c) Elastic modulus at the surface for PAAm gels as a function of oxygen in the source gas. | ||
The same principle was used for the synthesis of PEGDA and PHEMA hydrogels, Fig. 7, as examples of “crosslinker-free” and methacrylate gels, respectively. For the PEGDA hydrogels (Fig. 7a), increasing the oxygen amount had a substantial effect on the elastic modulus of the surface, which is very similar to the behavior of the PAAm hydrogels. For the PHEMA gels, the effect appeared rather limited with a moderate modulus decrease from 480 ± 10 kPa to 190 ± 10 kPa when oxygen was increased from 0% to 22%. However, immersing and swelling such PHEMA gels in ethanol, being a much better solvent for HEMA-based gels compared to water, the difference in surface modulus became more apparent. The elastic modulus at the surface of the same two gels in ethanol changed to 440 ± 30 kPa and 13 ± 1 kPa, respectively, revealing the strong effect of molecular oxygen on the polymerization also in the case of the methacrylate gels.
 | ||
| Fig. 7 Representative force–indentation curves for (a) PEGDA and (b) PHEMA hydrogel surfaces prepared in a flow-cell against a permeable PE foil, under which a gas with various amounts of oxygen was flowed. Force-indentation curves for PHEMA gels in ethanol are also shown. | ||
The results thus show that oxygen inhibition is the main mechanism behind the “mold effect”. A quantitative description using existing mathematical models19 would be an appropriate next step towards elucidating the exact relationships between the amount of supplied oxygen and the structure of the produced gradient zone. Such an understanding is key, in order to harness the phenomenon in a predictable way.
The mechanism
Based on the obtained results, oxygen inhibition appears to be the mechanism behind the formation of sparse, hydrogel surfaces with a vertical gradient in properties, when they are synthesized against oxygen-permeable surfaces. A schematic of a free-radical polymerization reaction of common hydrogels in the presence and in the absence of molecular oxygen is presented in Fig. 8 (an example of the corresponding chemical reactions is shown in the ESI,† Fig. S4). The initiator molecules first form radicals, initiating the reaction with monomers, which then propagates, converting monomers into polymer chains. The bifunctional, cross-linking monomers covalently connect the chains to form a polymer network, Fig. 8a. The presence of oxygen is known to inhibit radical polymerization by reacting with the active radicals and generating dead chain ends. The amount of oxygen present within the starting polymerizing solution is usually small, and it gets consumed evenly during the reaction without affecting the homogeneity of the network structure. However, the amount of oxygen at the interface of the solution with either air or oxygen-permeable surfaces can be sufficient to significantly hinder the polymerization reactions, resulting in the formation of a low-cross-linked, sparse hydrogel surface full of dangling, non-active chain ends, Fig. 8b. Cross-linking is more susceptible to oxygen than chain propagation, since to be effective, two radicals need to be formed on the cross-linking species. As a result, hydrogels formed in the presence of interfacial oxygen have significantly different surface properties from the equivalent hydrogels formed in its absence. The sparse, soft hydrogel surfaces can display, for example, up to an order of magnitude lower friction than cross-linked hydrogel surfaces.7,8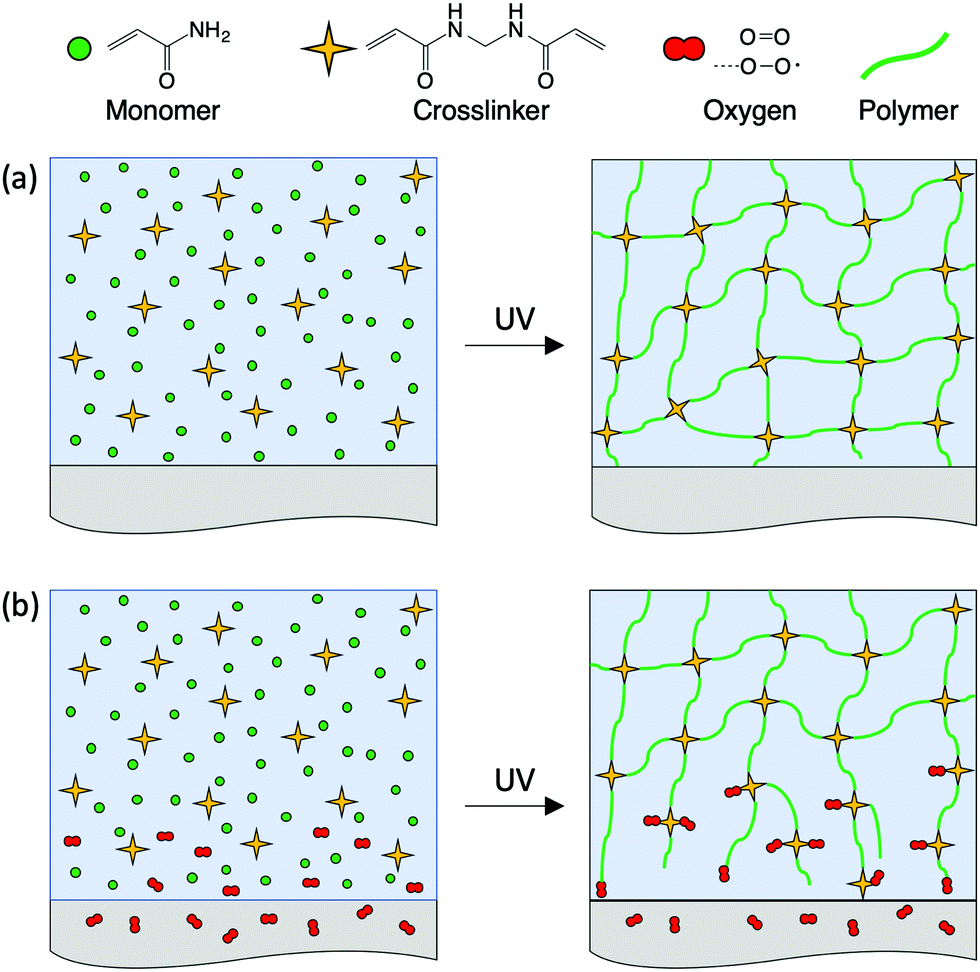 | ||
| Fig. 8 Schematic of hydrogel network formation in the absence and in the presence of oxygen at the interface during free-radical polymerization. (a) In the absence of oxygen, i.e. using oxygen-impermeable molds or in an oxygen-free environment, a hydrogel with a uniformly cross-linked surface is formed. (b) In the case of oxygen-permeable molds, oxygen inhibits the reaction near the surface, resulting in dead chain end formation, and reduced cross-linking. | ||
Conclusions
In this work we have investigated the mechanisms behind the formation of sparse hydrogel surfaces when synthesized against certain mold materials. Experiments with polyacrylamide gels showed that surface functionalization, which affects the surface energy and thus the hydrophobicity of the mold, does not affect the hydrogel surface structure in a significant way. We have also shown that the temperature dissipation during the exothermic polymerization reaction has no significant effect on the polyacrylamide hydrogel surface. Experiments with examples of an acrylate, a methacrylate, and a crosslinker-free gel showed, however, that the presence of oxygen emanating from the surface of certain mold materials was shown to inhibit polymerization, resulting in hydrogels with sparse, slippery surfaces. The source of oxygen can be the bulk of the molding material itself, which in the case of most polymeric materials entraps significant amounts of molecular oxygen. Alternatively, molecular oxygen can diffuse from an oxygen-rich medium such as a gas through a thin, oxygen-permeable membrane when used as a barrier. Therefore, synthesizing hydrogels against materials with low affinity to oxygen, against oxygen-impermeable membranes or in an oxygen-free environment results in a uniform hydrogel structure from the bulk to the surface. Alternatively, controlling the amount of oxygen arriving at the mold surface allows the structure of the hydrogel surfaces and thus their moduli and frictional behavior, to be precisely controlled.Author contributions
RS and NDS conceptualized and designed the study. RS, JM, and KZ prepared and conducted the experiments. RS collected and processed the data. All authors contributed to the interpretation of data. RS drafted the manuscript; all authors revised it critically and approved the final version of the manuscript. NDS acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 669562). The authors wish to thank Prof. W. Gregory Sawyer of the University of Florida for useful hints and fruitful discussions.References
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 Search PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 Search PubMed.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–124 Search PubMed.
- T. B. Reece, T. S. Maxey and I. L. Kron, Am. J. Surg., 2001, 182, S40–S44 Search PubMed.
- N. J. Irwin, C. P. McCoy and J. L. Trotter, Hydrogels: Design, Synthesis and Application in Drug Delivery and Regenerative Medicine, 2018, p. 89 Search PubMed.
- P. C. Nicolson and J. Vogt, Biomaterials, 2001, 22, 3273–3283 Search PubMed.
- A. Kii, J. Xu, J. P. Gong, Y. Osada and X. Zhang, J. Phys. Chem. B, 2001, 105, 4565–4571 Search PubMed.
- Y. A. Meier, K. Zhang, N. D. Spencer and R. Simic, Langmuir, 2019, 35, 15805–15812 Search PubMed.
- J. P. Gong, A. Kii, J. Xu, Y. Hattori and Y. Osada, J. Phys. Chem. B, 2001, 105, 4572–4576 Search PubMed.
- M. Peng, J. Ping Gong and Y. Osada, Chem. Rec., 2003, 3, 40–50 Search PubMed.
- I. E. Ruyter, Acta Odontol. Scand., 1981, 39, 27–32 Search PubMed.
- W. J. Finger, K.-S. Lee and W. Podszun, Dent. Mater., 1996, 12, 256–261 Search PubMed.
- C. Decker, F. Masson and R. Schwalm, JCT Res., 2004, 1, 127–136 Search PubMed.
- D. Chandra, J. A. Taylor and S. Yang, Soft Matter, 2008, 4, 979–984 Search PubMed.
- D. Dendukuri, D. C. Pregibon, J. Collins, T. A. Hatton and P. S. Doyle, Nat. Mater., 2006, 5, 365–369 Search PubMed.
- F. Di Benedetto, A. Biasco, D. Pisignano and R. Cingolani, Nanotechnology, 2005, 16, S165 Search PubMed.
- S. C. Ligon, B. Husar, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 Search PubMed.
- J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland and A. Ermoshkin, Science, 2015, 347, 1349–1352 Search PubMed.
- D. Dendukuri, P. Panda, R. Haghgooie, J. M. Kim, T. A. Hatton and P. S. Doyle, Macromolecules, 2008, 41, 8547–8556 Search PubMed.
- D. Chandra and A. J. Crosby, Adv. Mater., 2011, 23, 3441–3445 Search PubMed.
- J. P. Gong, T. Kurokawa, T. Narita, G. Kagata, Y. Osada, G. Nishimura and M. Kinjo, J. Am. Chem. Soc., 2001, 123, 5582–5583 Search PubMed.
- R. Simič, M. Yetkin, K. Zhang and N. D. Spencer, Tribol. Lett., 2020, 68, 1–12 Search PubMed.
- A. A. Pitenis and W. G. Sawyer, Tribol. Lett., 2018, 66, 113 Search PubMed.
- A. C. Dunn, J. M. Uruena, Y. Huo, S. S. Perry, T. E. Angelini and W. G. Sawyer, Tribol. Lett., 2013, 49, 371–378 Search PubMed.
- J. M. Coles, D. P. Chang and S. Zauscher, Curr. Opin. Colloid Interface Sci., 2010, 15, 406–416 Search PubMed.
- K. Zhang, R. Simic, W. Yan and N. D. Spencer, ACS Appl. Mater. Interfaces, 2019, 11, 25427–25435 Search PubMed.
- Encyclopedia of Chemical Technology, ed. H. F. Mark, D. F. Othmer, C. G. Overberger and G. T. Seaborg, Wiley Interscience, New York, 3rd edn, 1989 Search PubMed.
- Kuraray-EVAL, Technical Bulletin No. 110.
- Y. Gombert, R. Simič, F. Roncoroni, M. Dübner, T. Geue and N. D. Spencer, Adv. Mater. Interfaces, 2019, 6, 1901320 Search PubMed.
- W. Liu, R. Simič, Y. Liu and N. D. Spencer, Friction, 2020, 1–14 Search PubMed.
- J. E. Sader, J. W. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 Search PubMed.
- R. J. Cannara, M. Eglin and R. W. Carpick, Rev. Sci. Instrum., 2006, 77, 053701 Search PubMed.
- A. Chau, M. Cavanaugh, Y.-T. Chen and A. Pitenis, Exp. Mech., 2021, 1–5 Search PubMed.
- J. M. Urueña, A. A. Pitenis, R. M. Nixon, K. D. Schulze, T. E. Angelini and W. G. Sawyer, Biotribology, 2015, 1, 24–29 Search PubMed.
- R. Simič and N. D. Spencer, Tribol. Lett., 2021, 69, 86 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm00395j |
| This journal is © The Royal Society of Chemistry 2021 |