
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-inspired incorporation of phenylalanine enhances ionic selectivity in layer-by-layer deposited polyelectrolyte films†
Stephen J.
Percival
 *,
Sara
Russo
,
Chad
Priest
,
Ryan C.
Hill‡
,
James A.
Ohlhausen
,
Leo J.
Small
,
Susan B.
Rempe
and
Erik D.
Spoerke
*
*,
Sara
Russo
,
Chad
Priest
,
Ryan C.
Hill‡
,
James A.
Ohlhausen
,
Leo J.
Small
,
Susan B.
Rempe
and
Erik D.
Spoerke
*
Sandia National Laboratories, PO Box 5800, MS 1411, Albuquerque, NM 87185, USA. E-mail: sperciv@sandia.gov; edspoer@sandia.gov
First published on 13th May 2021
Abstract
The addition of a common amino acid, phenylalanine, to a Layer-by-Layer (LbL) deposited polyelectrolyte (PE) film on a nanoporous membrane can increase its ionic selectivity over a PE film without the added amino acid. The addition of phenylalanine is inspired by detailed knowledge of the structure of the channelrhodopsins family of protein ion channels, where phenylalanine plays an instrumental role in facilitating sodium ion transport. The normally deposited and crosslinked PE films increase the cationic selectivity of a support membrane in a controllable manner where higher selectivity is achieved with thicker PE coatings, which in turn also increases the ionic resistance of the membrane. The increased ionic selectivity is desired while the increased resistance is not. We show that through incorporation of phenylalanine during the LbL deposition process, in solutions of NaCl with concentrations ranging from 0.1 to 100 mM, the ionic selectivity can be increased independently of the membrane resistance. Specifically, the addition is shown to increase the cationic transference of the PE films from 81.4% to 86.4%, an increase on par with PE films that are nearly triple the thickness while exhibiting much lower resistance compared to the thicker coatings, where the phenylalanine incorporated PE films display an area specific resistance of 1.81 Ω cm2 in 100 mM NaCl while much thicker PE membranes show a higher resistance of 2.75 Ω cm2 in the same 100 mM NaCl solution.
Introduction
Controlling ionic transport across separator membranes is highly desirable for applications ranging from chemical separations to energy storage. Nanoporous membranes offer a convenient platform with which to control ion transport.1 The ion transport through the nanopores can be tuned by controlling the surface charge on the pore wall.2–5 A variety of unique and responsive chemistries have been demonstrated to alter the surface charge, and resulting ion transport, through a nanoporous membrane.6–12 Altering surface charge can be achieved by simply grafting a charged polymer, or polyelectrolyte (PE), to the surface of the pore.4,12–14 Many methods are used to modify pore surfaces, including both chemical and physical deposition techniques.Layer-by-Layer (LbL) deposition of PE offers a scalable, inexpensive and facile method by which to impart many desirable properties to a range of substrate materials. Deposition through the LbL process is a simple yet powerful technique where alternating layers of different materials are deposited in a controlled manner.15–20 The bottom-up approach of LbL assembly has been leveraged using different materials to create many active and passive functional materials, including polymer/clay anti-corrosion coatings,21–27 fire-retardant coatings,28,29 nanoparticle electrocatalysts30,31 and ion selective membranes.32–36 LbL deposition of PEs offers an attractive method for modification of nanoporous membranes, which can be used to tune the ion transport properties of nanoporous membranes for various applications, ranging from water purification to energy storage and chemical separations/synthesis.
During the LbL deposition process of a PE coating, an anionic polymer and a cationic polymer are sequentially deposited on a substrate, forming one “bilayer” (BL).37 These polymers are deposited from dilute aqueous solutions of the respective polymers and the substrate (or membrane) is simply dip-coated from one solution to the other to form the PE films.35,37–39 However, additional post coating functionalization or crosslinking can further be applied to tailor the coating properties.32,35,40–43 This post deposition processing has been shown to be a crucial step for many systems geared towards a specific application where crosslinking can make a film more ion selective32,35 but can also be controlled to make a film more impermeable41–43 if desired for a given application. Surface functionalization can make a membrane capture selective proteins44 or anitbodies45 for isolation and purification.
Besides post-coating crosslinking, the films’ properties can also be tailored by incorporation of different constituents. Such alteration can come in the form of nanoparticles,46,47 functional molecule filled nanocontainers48,49 or small functional molecules.16,50–52 Here, we seek inspiration from biological systems, such as ion channels, aiming to incorporate a functional amino acid to help improve ion-selectivity of our PE coatings. Addition of amino acids to different polymer systems has been demonstrated to change the properties or chemical interactions of the polymers selectively. These amino acid functionalities have been reported to affect silver complexation in olefin transport membranes,53 pH-dependent ion transport,54 improved desalination membrane permeability,55 and anti-fouling properties56,57 of membranes. However, most of these processes simply grafts the amino acid to the surfaces of the polymer or support membrane. Few integrate the functional amino acid into the bulk of the film where it may have a more substantial effect, similar to biological protein ion channels. The presence of the molecules in the bulk could affect the local energy environments akin to how the local chemistry present in an ion permeation pathway of a biological pore controls selective ion permeation in ion channels.
Biological ion channel proteins form pores, often sub-nm in diameter, that span cellular membranes and control ion transport across those membranes.58 Membrane transport proteins have inspired,59–61 or been incorporated into,62,63 synthetic membranes to gain high selectivity and low resistance for the transport of ions or small molecules. Specific amino acids from each protein interact with ions, directly or indirectly,64 and account for the channel transport properties. As an example, the side chain phenyl group of phenylalanine (Phe) and tyrosine, along with amino acids containing carboxylates and polar functional groups, line the ion permeation pathway of channelrhodopsin (ChR) proteins, shown in Fig. 1 in the computed open-state conformation (allowing ion transport through the protein). That chemical composition facilitates the transport of sodium and other cations across cellular membranes, while rejecting anions.65 Since the ChR family of proteins are also light sensitive, they are well known in the field of optogenetics, where light is applied to control cells (e.g., neurons) that have been modified genetically to express ChR channels, and as such, have been extensively studied and characterized.66–69 Here, we utilize ChR as inspiration for increasing the cationic selectivity of layer-by-layer deposited polyelectrolyte films. Specifically, the prevalence of Phe in the ion channel, which lines the permeation pathway, encouraged its incorporation into ion selective membranes. While not forming the exact structured protein ion channel that inspired the use of Phe, we hypothesized that we could achieve a higher cation selectivity from a type of ion selective membranes by simply including the amino acids in the LbL polyelectrolyte film deposition.
 | ||
| Fig. 1 Snapshot from molecular simulations of channelrhodopsin cation channel protein, C1C2, in the computed open state. Amino acids associated with the ion permeation pathway are color-coded (pink – phenylalanine (PHE), green – glutamic acid (GLU), blue – asparagine (ASN), red – aspartic acid (ASP), orange – tyrosine (TYR)). Ion permeation occurs between helices I, II, and VII, represented by sodium ion (purple sphere) at positions along the permeation pathway. Light sensitivity is given by the retinal molecule (black), shown here for reference. | ||
In this work, LbL deposited PE coatings of polyethyleneimine (PEI) and polyacrylic acid (PAA) are modified by the simple addition of amino acids to the dip solutions. Phe was the amino acid used as an additive in the coating process because Phe contains a hydrophobic phenyl ring, which has been shown in previous studies to be an essential amino acid in ion channel proteins, such as channelrhodopsin.70 Recent computational studies of the free energy along the channelrhodopsin C1C2 ion permeation pathway indicate that binding sites that include Phe stabilize the sodium ion by an amount similar to sodium hydration free energy.71 We show that the Phe is effectively incorporated into the self-assembled PE films. It is important to note that while this work is inspired by observations of Phe in biological ion channels, we do not believe the simple incorporation of the phenylalanine into the self-assembled PE film means that it will behave as if it is in the protein ion channel which has a very specific structure that leads to highly efficient sodium ion transport. Specifically, incorporation of Phe into the film increases the ionic selectivity while maintaining a relatively low membrane resistance. In contrast, the same increase in ionic selectivity can be achieved by coating more PE layers onto the membranes leading to thicker coatings, which in turn increase the resistance of the membranes. These results are a key demonstration of how understanding of protein ion channel structures can be used for informed engineering of inexpensive ion selective membranes.
Experimental
Membrane synthesis
All chemicals were purchased and used without further purification. Sodium chloride (NaCl, ACS reagent grade >99.0%, Sigma-Aldrich) poly(acrylic acid) (PAA, 35 wt% in H2O, average Mw = ∼100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich), polyethylenimine (PEI, branched, average Mw = ∼25000, Sigma Aldrich), glutaraldehyde (GA, 25% solution in H2O, Sigma-Aldrich), D-phenylalanine (>98.0%, Sigma-Aldrich), and sodium hydroxide (NaOH, >98%, Fisher Scientific). Aqueous solutions were made in 18.2 MΩ cm DI water.
000, Sigma-Aldrich), polyethylenimine (PEI, branched, average Mw = ∼25000, Sigma Aldrich), glutaraldehyde (GA, 25% solution in H2O, Sigma-Aldrich), D-phenylalanine (>98.0%, Sigma-Aldrich), and sodium hydroxide (NaOH, >98%, Fisher Scientific). Aqueous solutions were made in 18.2 MΩ cm DI water.
Track-etched nanoporous polycarbonate (PC) support membranes (0.05 μm pore, 6 × 108 pores per cm2, 25 mm or 90 mm diameter, Sterlitech Corporation) were first sonicated in water to ensure the pores were fully filled with water and then treated to remove a thin unwanted polyvinyl pyrrolidone (PVP) layer. The membranes were etched in a 4.5 M NaOH solution for 5 minutes, followed by rinsing in DI water. Then they were treated with UV-ozone (UVO-Cleaner model 144A, Jelight Company Inc.) for 10 minutes on each side, while slightly moist to prevent the membranes from curling up during the process. Immediately thereafter, the membranes were immersed in the 0.1 wt% PEI solution (pH = 10.4) for 5 minutes, rinsed in DI water, then immersed in the 0.07 wt% PAA solution (pH = 3.2) for 5 minutes. In these solutions the phenylalanine had a slightly different charge state based on the pH and the respective pKa of the amino acid functional groups. In the PAA solution (pH = 3.2) the phenylalanine had a protonated primary amine and deprotonated carboxylic acid. In the PEI solution (pH = 10.4) the phenylalanine was negatively charged with the carboxylic acid deprotonated. This constituted the 1st BL of the LbL assembly process. Each rinsing step consisted of two large volumes of DI water where the membrane was placed first in one DI water container for 1 minute and then the second DI water container for an additional minute, before going to the next polymer solution. The positively charged PEI was coated onto the membrane first because the PC membrane is negatively charged in solution and thus motivates laying down the oppositely charged polymer first. Subsequent BLs were assembled by dipping in the polymer solutions for only 1 minute each. For the dip solutions containing amino acid additives, the concentration of the phenylalanine was 1 mM in the unchanged concentrations of PEI and PAA solutions (amino acid was added to both polymer solutions for dipping). The phenylalanine is not expected to leach out of the self-assembling film during the coating process due to the strong electrostatic interactions between the charged molecules, typically known as electrostatic crosslinking.72 The self-assembled PE and PE with incorporated phenylalanine (referred to as PE + Phe from this point on) BLs were then crosslinked with glutaraldehyde. The membranes were immersed in a 25% glutaraldehyde solution for 24 hours and then washed with copious amounts of DI water. The 25 mm diameter membranes were tested as made after crosslinking. From the 90 mm diameter membranes, daughter membranes 25 mm in diameter were punched out for all subsequent testing. All membranes tested were immersed in 0.1 mM NaCl solution for 72 hours before testing.
SEM characterization
Scanning electron microscopy (SEM) was performed using a Zeiss GeminiSEM 500 at 2–5 kV accelerating voltage and 3–5 mm working distance. A 1–3 nm layer of Au–Pd was sputtered onto the samples to minimize the effects of sample charging. Cross-sections of PC membranes were obtained by a freeze-fracture method where the membranes were frozen at cryogenic temperatures in water, forming an ice block which could then be snapped in two. The fractured membrane was warmed to room temperature to release it from the ice and dried under nitrogen before interrogation in SEM. Film thicknesses were measured across multiple points along the membrane cross-section (both top on bottom) and averaged together to obtain the thickness of the LbL deposited film.Electrochemical measurements
Ionic selectivity measurements were performed following a potentiometric method described in depth elsewhere.9,35 Briefly, each membrane was sealed in a U-shaped cell equipped with Luggin probes positioned 1 cm from the membrane, and a Ag/AgCl wire placed in each Luggin probe. Both sides were filled with 0.1 mM aqueous NaCl and the potential, measured by HP 34401A multimeter, was allowed to equilibrate to 0 ± 2 mV. The concentration of aqueous NaCl on the left side of the membrane was then fixed at 0.1 mM, while the concentration on the right side was varied from 0.1 mM to 1 M (all NaCl concentrations were sequentially diluted from a 1 M stock solution). At each concentration, the voltage was allowed to stabilize (equilibrated for 45 minutes). Then the cell was rinsed and equilibrated to 0.1 mM NaCl, after which the sides were switched, with a constant 0.1 mM aqueous NaCl on the right side of the cell. Before use, all solutions were allowed to equilibrate with laboratory atmosphere, stabilizing at pH = 6.0. Ionic selectivity measurements were performed in triplicate on different membranes.Membrane area specific resistance
The area specific resistance (ASR) of the bare PC membranes and the coated PE membranes was measured using a Solartron Analytical Modulab following a previously developed procedure.73 The membranes were first soaked in aqueous NaCl solutions of 100 mM, 1 M, and 3 M concentrations for at least 3 days to allow the solution within the coating to equilibrate with the bulk solution. For each concentration, the membranes were then placed between two 316 stainless steel electrodes within a Teflon® Swagelok® cell. To measure the ASR of the membranes, impedance spectroscopy was performed at 10 mV RMS AC and 0 V DC with respect to the open circuit potential and over 1 MHz to 1 Hz. The cell resistance was defined as the point at which the complex impedance curve crossed the real impedance axis (when the imaginary component was zero). This measurement was repeated with 2, 3, and 4 stacked membranes. The individual membrane resistance was then deduced from the slope of the line of cell resistance vs. number of membranes. The ASR of the membrane was then calculated using the membrane cross-sectional area.Time of flight – secondary ion mass spectroscopy (ToF-SIMS)
Time-of-flight secondary ion mass spectrometry (ToF-SIMS), (Ion-TOF ToF.SIMS 5, Ion-TOF Gmbh, www.iontof.com), was employed to interrogate the surface of each film. A 25 kV Bi3++ primary ion probe in high-current, bunched mode was rastered over 3 random 100 × 100 μm2 area locations on each film. Spectra were created by integrating 40 scans consisting of a random-rastered 128 × 128 pixel “image” with 1 shot per pixel over each area. Charge compensation was not needed. Separate positive and negative secondary ion spectra were acquired at each location. The resulting spectra were calibrated using standard aliphatic hydrocarbon peaks.Molecular simulations
Interactions between Na+ ions and components of the polyelectrolyte membrane were studied by electronic structure calculations utilizing the Gaussian 09 software package.74 Lowest energy molecular structures were obtained at the B3LYP level of density functional theory (DFT) with the aug-cc-pVDZ basis set. Analytical normal mode frequency analysis was added to predict reaction free energies.75 An implicit polarizable continuum model (PCM), with dielectric value of 78 and default boundaries, was used to model aqueous solution.76 To validate the methods, aqueous phase hydration free energy of Na+ was computed using quasi-chemical theory77 and a cluster of Na+ with four waters, [Na(H2O)4]+, following procedures described earlier.78,79 We limit our analysis to Na+, reserving studies of selectivity among other cations like K+ for future work. Note that prior work by Stevens & Rempe suggest that the smaller Na+ would bind more strongly than K+ to carboxylates that are freely accessible for binding. A four-coordinate cluster was chosen for analysis because it represents the most probable cluster size observed in ab initio molecular dynamics simulations of Na+ in liquid water.79 The molecular electronic potential (MEP), and atomic charges fitted to reproduce the MEP, were computed at the same level of theory and with the same basis sets. An isolated charged iso-butyrate (IB−) was utilized to model a simple monomer of PAA, and a single charged phenylalanine (Phe−) was used to model Phe embedded in the polyelectrolyte membrane. Phe was modeled in an unprotonated state to represent Phe− crosslinked with glutaraldehyde, which reduces the proton charge density of the Phe amine group. Although 4–5 waters also ligate carboxylate groups in ab initio molecular dynamics simulations of formate ion in aqueous solution,80 hydration of these groups in IB− and Phe− were treated implicitly with the PCM model. The reason for that choice is to focus the current studies on the exchange of sodium ion's ligating waters for direct interactions with components of the LbL coatings (X− = IB− or Phe−), as seen in eqn (1).[Na(H2O)4]+ + X− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [Na(X)(H2O)4−n] + nH2O [Na(X)(H2O)4−n] + nH2O | (1) |
Results and discussion
Membrane LbL coating
The LbL deposition of the PE thin films was accomplished by sequentially dipping the nanoporous PC into solutions containing the desired cationic and anionic polymers, as shown in Scheme 1. During this dipping process, the first polymer, in this case the cationic PEI, will coat the membrane. Dipping is followed by rinsing to remove weakly adhered excess polymers from the membrane. The membrane is then dipped into the 2nd polymer solution, where the anionic polymer, PAA in this case, will self-assemble onto the first layer of PEI, again followed by rinsing. A complete BL is formed after dipping into each solution. In the case of using the amino acid additive, 1 mM Phe was dissolved into each solution (both PEI and PAA) to ensure it was incorporated throughout the PE coating. This process could be repeated over and over to build up many BLs, 10 being the most studied here. Phenylalanine was added to both solutions to promote homogeneous incorporation of Phe into the film. The coated membranes were all further chemically crosslinked using glutaraldehyde which reacts with the primary amines of the PEI and Phe. This covalent crosslinking not only increases the ionic selectivity (by reducing the cationic charge density of the film by reacting with the amines35), but also ensures the PE films were chemically stable against high salt concentrations when testing. | ||
| Scheme 1 Schematic of the Layer-by-Layer (LbL) deposition process where polymer layers are deposited onto the nanoporous membrane from sequential dipping in solutions containing the cationic and anionic polymers each with added amino acid additives, in this case phenylalanine. Film deposits on both sides of membrane despite being shown growing on one side only for clarity. | ||
Membrane characterization
Time of Flight Secondary Ion Mass Spectroscopy (ToF-SIMS) was used to confirm the presence of Phe in the PE film and to quantify the amount of Phe deposited. A calibration curve was made using ToF-SIMS where a known amount of Phe was mixed with PEI and PAA solutions and both polymer solutions drop cast onto a silicon wafer and allowed to dry (no crosslinking step was performed on these samples). This ensured a known amount of Phe was incorporated into the dried polymer film eliminating the possibility that the amount of Phe was lower than expected due to preferential adsorption of polymers to the substrate surface. Concentrations of Phe used in the calibration curve samples were from 0 mM to 2 mM in increments of 0.5 mM. Test samples, similar to the PC membranes, were made by dip coating silicon wafers with 5 BLs of PE using the same conditions that were used for the membranes, these samples were quickly analyzed using ToF-SIMS after making them.Fig. 2 shows the results of interest from the ToF-SIMS analysis. We have identified a peak at m/z = 166.08 which is attributed to Phe with an additional hydrogen (Phe + H) associated with it (making it a positively charged ion). The peak located at 166.13 m/z, next to the Phe peak, is attributed to background hydrocarbon contamination (probably of the form C11H18O+) and is present on all samples (even the control sample with no added Phe). The Phe peak, located at 166.08 m/z increases with increasing amounts of Phe added to the drop cast samples. The dip coated sample also shows this peak, clearly indicating that Phe is present in the sample, though the intensity of the peak appears smaller as compared to the drop cast sample with 1 mM Phe added to the solution (same concentration as in the dip coating solutions). The smaller size of the 166.08 peak from the dip coated sample compared to the drop cast sample indicates that not all of the dissolved Phe was incorporated into the film during the self-assembly of the BLs. The drop cast control samples were used to create a calibration curve of the normalized m/z 166.08 peak counts as a function of Phe concentration in the solution. To compare the different sample spectra, the data was normalized to a specific peak associated with PAA in each of the ToF-SIMS spectra (i.e. each spectrum was normalized to its own PAA peak before comparison). This reference peak was located at m/z = 117 and is attributed to a sodium adduct of the PAA monomer unit (C3H3O2Na2+). From this semi-quantitative calibration curve (semi-quantitative because the control samples and experimental samples were not coated on the silicon wafers in an equivalent manner, drop cast vs. dip coat) we can calculate the dip coated 5 BL PE + Phe sample had the same amount of Phe as a control sample would have if ∼0.24 mM Phe was added to the solution before drop casting.
 | ||
| Fig. 2 ToF-SIMS data showing the peak at m/z = 166.08 for a 5 BL PE + Phe dip coated sample and a control containing the PE and 1 mM phenylalanine drop cast onto a silicon wafer. This peak is attributed to the phenylalanine plus a proton (Phe + H+), making it positively charged. The peaks have been normalized to their respective m/z = 117 peak in each ToF-SIMS spectrum, which is attributed to the PAA fragment. | ||
To gain further understanding of the self-assembled PE coating structure and thickness, cross-sectional analysis was performed. Fig. 3A shows the cross section of a 10 BL PE sample. The cross section shows the PC membrane at the bottom of the SEM image, as evidenced by the porous cross-section, with the PE coating on top (indicated by the brackets). There is a clear interface between the porous PC membrane and the self-assembled PE coating, but a demarcation (red dashed line) is included for easy identification. Measuring the thickness of the PE at different points in the SEM cross sections, the 5 BL PE membrane was found to be 1.23 ± 0.19 μm thick and the 5 BL PE + Phe had a similar thickness of 1.27 ± 0.37 μm. The 10 BL PE film, however, was much thicker, at 2.71 ± 0.44 μm. Similar thickness results for PE bilayers were seen in previous reports,35,81 where thick polymer films were also deposited from PEI and PAA solutions. From the SEM cross-sectional analysis we cannot identify any obvious structural changes between the polyelectrolyte coatings with and without added phenylalanine. These measured films were largely uniform on the top and the bottom of the support membrane as the LbL coating deposits on both sides of the membrane at the same rate. Additionally, the pores were filled with the deposited polyelectrolyte, which is consistent with previous studies of these LbL PE films.35
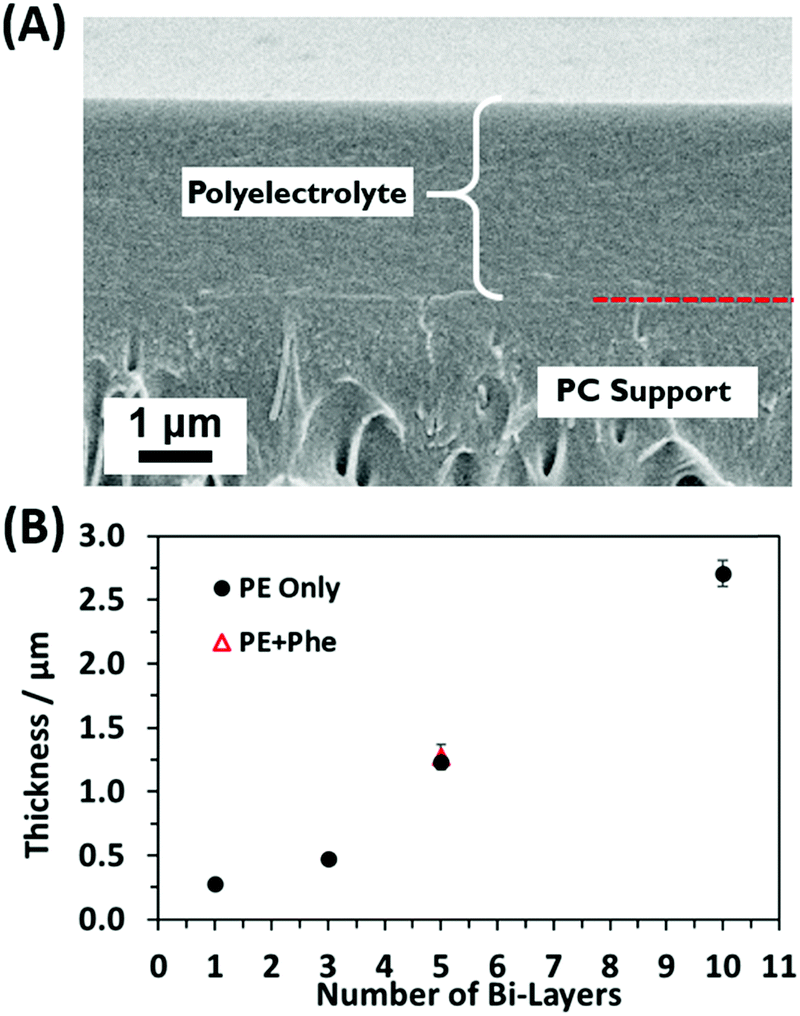 | ||
| Fig. 3 (A) SEM image of the cross section from a 10 BL PE film on the PC membrane support. The red dashed line denotes the interface between the PE coating and the PC support. (B) Plot showing the linearly increasing PE thickness with number of coated BLs. | ||
Electrochemical measurements
The thickness of the PE coating is predicted to affect the membrane ASR. To investigate what effect the coating thickness has on the resulting resistances of the membranes, the resistance of stacks of membranes cut from the same mother membrane were measured via impedance spectroscopy as detailed in the experimental section. The membranes were stacked and measured to obtain an average resistance per membrane, thereby eliminating contributions to resistance from the cables and fixturing. Example plots of the total resistance vs. the number of membranes stacked together for bare PC and coated PE membranes (5 BL PE and 10 BL PE) in 1 M NaCl solution are plotted in Fig. 4A. The slope of the resulting best fit line is a measure of the resistance increase per membrane which can then be used to calculate the ASR using the known area of the membranes (all membranes were 0.712 cm2). All the resistance vs. number of stacked membranes plots show an offset that arises from the test cables and test assembly and is not involved in the calculation of the average specific resistance of a membrane. | ||
| Fig. 4 (A) Total resistance of stacks of membranes in 1 M NaCl. The slope of the line was used to calculate the Area Specific Resistance (ASR). (B) Resulting ASR of the bare PC, 5 and 10 BL PE only, and 5 BL PE + Phe in 0.1, 1 and 3 M NaCl. | ||
The resistances for the different membranes, including the 5 BL PE + Phe, are plotted at different salt concentrations in Fig. 4B. As expected, the bare PC membranes have the lowest ASR. Obviously, this is due to the pores being open and allowing facile, less selective movement of ions through the membrane. However, the addition of ion selective PE film increases the resistance for the 5 BL PE and again, further increases for the 10 BL PE. The increase in the overall resistance of the membrane is attributed to the presence of, and the increase in thickness of the PE (going from 5 to 10 BLs). Both 5 BL PE and 5 BL PE + Phe have similar ASRs, which are higher than the bare PC membranes, at all salt concentrations. These coated membranes approach the ASR of the bare membranes at higher NaCl concentrations. Finally, the 10 BL PE coated membranes have the highest ASR of the tested membranes, with an ASR approximately double the 5 BL coated membranes at every concentration. This increased ASR corresponds with the increased PE thickness and number of coated PE BLs which are coating the PC membrane. The presence of the PE in the pores and on the face of the membrane was expected to increase the ASR, compared to bare PC, as it physically decreases the concentration of mobile charge carriers in the nanopores. As the NaCl concentration decreases, however, the PE serves as an ionic buffer in the nanpores and on the membrane faces. The ASR of the different membranes are summarized in Table S1 in the ESI.† The change in ASR with decreasing NaCl concentration from 3 M to 0.1 M (Table S1, ESI†) is observed to be dependent on the coating. The 5 BL PE membrane ASR changes by 4.15× (1.87/0.45), and the bare PC membrane changes by 4.64× (1.21/0.26). The addition of Phe further reduces the concentration dependence, with the ASR of 5 BL PE + Phe changing only 2.97× (1.81/0.61). Despite having the highest ASR, the 10 BL PE coated membranes showed the lowest concentration dependence with a 2.08× (2.75/1.32) change from 3 M to 0.1 M NaCl concentration, in line with previous discussion.
Ultimately, the membranes’ selectivity for charged ionic species dissolved in solution is the focus of this study. To determine the ionic selectivity of the membranes, each type of membrane was exposed to solutions with different NaCl concentrations placed on opposite sides of the membrane. When exposed to these conditions, a permselective membrane will begin to preferentially allow some ions to diffuse from a region of high concentration to low concentration. Depending on the membrane's polyelectrolyte charge density, the membrane will equilibrate with the ions partitioned at the membrane/solution interfaces at different levels.82–84 This ion partitioning will lead to a buildup of charge at the interface and the ion transport in the membrane will stop (in the absence of an applied electric field).82–84 The buildup of charge at the membrane interface, which is a reflection of the membrane's selectivity, will set up an electrical potential difference on the membrane, which can be measured.84 The influence of the PE layers on the ionic selectivity was determined from the resulting transmembrane voltage. The membrane voltage, Vm, for a 1:1 monovalent salt can be described by a modified version of the Nernst equation shown in eqn (2).1,9,35,84
 | (2) |
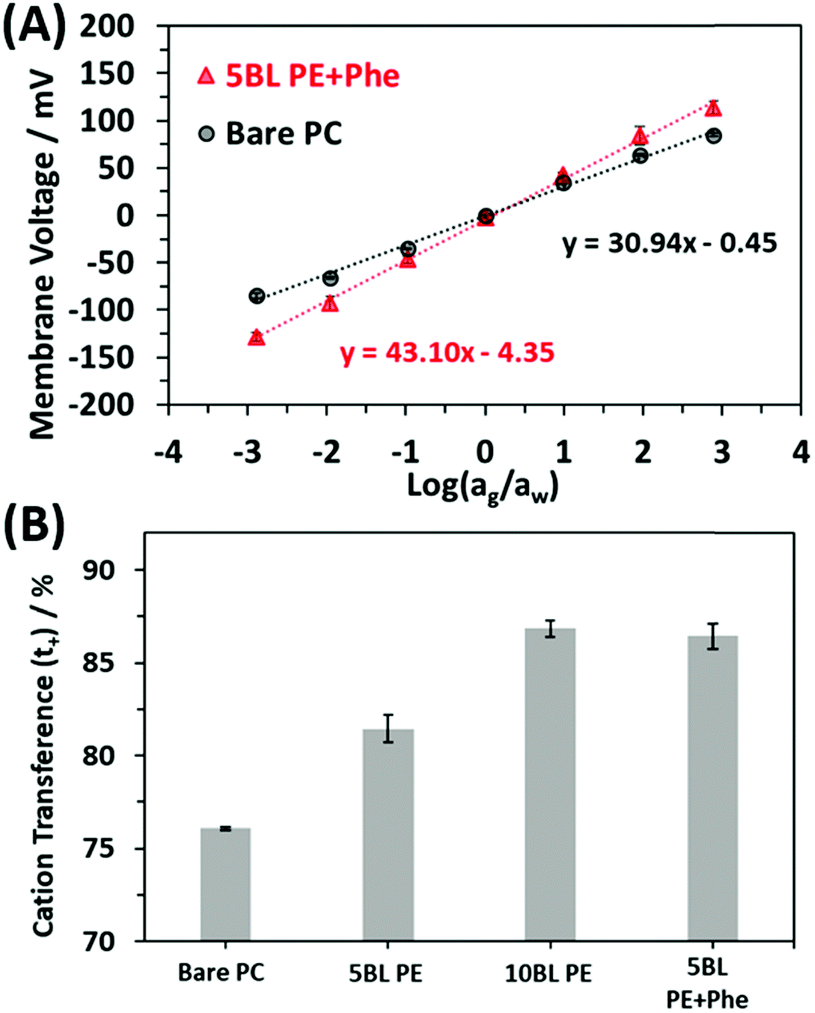 | ||
| Fig. 5 (A) Plots showing the averaged resulting membrane voltage curves as a function of salt concentration on either side of the membrane where the coefficient of determination (R2 value) was 0.998 for the 5 BL PE + Phe and 0.995 for the Bare PC membrane (number of membrane responses averaged together is n = 4 for bare PC and n = 6 for 5BL PE + Phe). (B) Cation transference numbers for bare PC, 5 BL PE, 10 BL PE and 5 BL PE + Phe membranes. | ||
The bar graph in Fig. 5B shows the resulting cation selectivity for each of the samples tested. The selectivity values are also presented in Table 1 for comparison. All the coated samples have a higher selectivity than the bare PC, as expected. Additionally, the selectivity of the PE only coatings increases as more is deposited, where 10 BL PE has a cation transference (t+) of 86.8% which is higher than that of 5 BL PE, 81.4%. However, the 5 BL PE + Phe shows a significant increase in transference as compared to the 5 BL PE despite having the same thickness of PE material. In fact, the incorporation of Phe increases the cation transference to 86.4% which is on par with the 10 BL PE. This effect is akin to doubling the number of pure PE BLs applied to the membrane as seen by comparing the 10 BL PE to the 5 BL PE but does not increase the actual thickness of the membrane coating (Fig. 3) and does not increase the resistance of the membrane (Fig. 4). This is an important finding for these membranes as it has been reported that ionic selectivity and conductivity are typically bound by a tradeoff relationship, i.e. increasing ion selectivity will lead to a decrease in conductivity of a membrane and vice versa.86
| Slope of line/mV | (t+ − t−)/% | t +/% | |
|---|---|---|---|
| Bare PC | 30.9 ± 0.1 | 52.2 ± 0.2 | 76.1 ± 0.1 |
| 5 BL PE | 37.2 ± 0.9 | 62.9 ± 1.5 | 81.4 ± 0.7 |
| 10 BL PE | 43.6 ± 0.6 | 73.7 ± 0.9 | 86.8 ± 0.5 |
| 5 BL PE + Phe | 43.1 ± 0.8 | 72.8 ± 1.4 | 86.4 ± 0.7 |
Computational analysis
When comparing the effect of the added amino acid, Phe incorporation induced a significant effect on the ion-selectivity of the coatings. These results indicate that the Phe presence in the film is an important aspect in the interaction with solvated cations in the membrane, offering parallels to the influence of Phe seen in the protein ion channel, channelrhodopsin, discussed previously. However, the orientation and nature of structural integration of the Phe molecules into the PE film is not known and likely plays a substantial role in the increase in selectivity. To investigate how the Phe in the film may be influencing the selectivity of the membrane, computational analysis was used to determine relative interaction energies of the ions and Phe in the film.Earlier analysis of Na+ binding free energy along the ion permeation pathway of channelrhodopsin C1C2 shows that the protein stabilizes Na+ by an amount roughly equal throughout the full permeation pathway, with many small wells and barriers. This stabilization through the whole of the permeation pathway (in the protein ion channel) is due to the complex interaction of all the amino acid constituents working together. Solvation free energy within the channel is expected to match Na hydration free energy.79 Direct interactions between Na+ and functional groups from Phe and other amino acids account for the stabilization in the protein, and such a profile promotes rapid Na transport.79
A snapshot of the open state channelrhodopsin protein, C1C2, is shown in Fig. 1 and was previously discussed. A sodium ion (Na+) trajectory through the polypeptide maps the ion's trajectory through the protein.71 In native environments, Na+ enters from the extracellular side of the membrane and traverses to the intracellular side. As Na+ transits through C1C2, it interacts with several amino acids. The permeation path mainly contains glutamate (Glu), asparagine (Asn), and multiple Phe. The carboxylate-bearing amino acid side chains of glutamic and aspartic acids are considered most important to selectivity of cations over anions,65,87 but Phe also may play a substantial role.
A notable difference between the channelrhodopsin protein and the system under study here is that Phe backbone atoms, including those from the carboxylate group, may interact with the ions during permeation through the synthetic membrane. Negatively charged carboxylate groups will naturally attract Na+. To investigate the roles of carboxylates from PAA compared with Phe on stabilizing cations, we used electronic structure calculations to compute enthalpy changes for Na+ binding to IB− (analog to the PAA repeat unit) and Phe−. A computed molecular electrostatic potential (MEP) map emphasizes the contrasting electronic states of molecules used to model the PAA film, with and without incorporation of Phe (Phe and IB− molecules) as seen in Fig. 6. These molecular electrostatic potentials visually illustrate the most negatively charged regions are localized around the carboxylate groups, as expected. Compared to oxygen atoms of water, the carboxylate oxygens are more negatively charged, with those from Phe− more negative than IB− (Fig. 6).
 | ||
| Fig. 6 Molecular electronic potential representation for (A) IB− (B) PHE− and (C) H2O. Scale bar is provided below to distinguish negative sites (red) and positive sites (blue). Spheres are color coded with carbon shown as gray, hydrogen as white and oxygen as red. | ||
We found the lowest energy structures for Na+ ions binding to a single charged Phe− and a charged IB− (chosen as a model for a simple monomer of PAA). Studies on Na+ binding to the carboxylate groups of both molecules (Phe− & IB−) resulted in the most favorable binding energies relative to other potential binding sites, as anticipated from the electrostatic profiles (Fig. 6). To better represent the experimental system, we treated Na+ in complex with its preferred number of contacting water molecules, seen in Fig. 7A, determined earlier by ab initio molecular dynamics simulations of Na+ in liquid water.79 Those contacting waters occupy a volume within 3 Å from the ion. In the reactions studied, the [Na(H2O)4]+ complex exchanges from one to four waters with one of the substrates to achieve the most stable new coordinating complex (with either IB− or Phe−). Our modeled complexes change Na+ coordination number, depending on the number of waters exchanged. As illustrated in Fig. 7, Na+ expands its coordination to 5 when one water is exchanged. In this particular case, Na(Phe−) and Na(IB−) are complexed in a bidentate binding mode, along with three coordinating water molecules to complete the five-fold coordination structure (Fig. 7B and C). In the other cases studied, Na+ also interacts with carboxylates of Phe− and IB− in bidentate mode (see Fig. S2 of ESI†). With exchange of 2 ligating waters of Na+ for an LbL component, a four-fold coordination structure is maintained. With exchange of 3 ligating waters, a three-fold coordination structure forms. With exchange of all four ligating waters, a two-fold coordination complex forms with Na+ interacting with the carboxylate group in bidentate binding mode (Fig. 7D and E).
 | ||
| Fig. 7 Cluster models for (A) hydrated Na+ ion with 4 water ligands, (B) Na+ with IB− exchanged for 2 water ligands, (C) Na+ with Phe− exchanged for 2 water ligands, (D) Na+ with IB− exchanged for 4 water ligands, and (E) Na+ with Phe− exchanged for 4 water ligands. Red dashed lines represent distances between Na+ and directly coordinating ligands. These distances are 2.33 Å on average for water (A), 2.32 Å for IB− and water (B), 2.33 Å for Phe− and water (C), 2.19 Å for IB− only (D), and 2.19 Å for Phe− only (E). Spheres are color coded with carbon shown as gray, hydrogen as white, oxygen as red, nitrogen as blue and sodium as purple. | ||
The models provided in Fig. 7 and ESI,† Fig. S2 allow for the calculation of reaction free energy. We first cross-checked our computational methods by comparing our calculated hydration free energy of sodium ion with experimentally estimated values. Our calculated hydration value of −101.7 kcal mol−1 compares well with earlier calculations78 and an experimental estimate of −90.8 kcal mol−1.88 The calculated free energy differences for exchange of one ligating water ([Na(H2O)4]+) with IB− and Phe− (ΔGgas) illustrate that Phe− has a slightly higher propensity for exchange interaction with [Na(H2O)4]+ in the gas phase (Table 2). This same small favoring of Phe− over IB− occurs for exchange of two, three, and four waters, but the most favorable reaction free energy occurs for exchange of one water (see Tables S2 and S3 in the ESI†). However, this difference does not account for the large difference in selectivity unveiled by the experiments. Therefore, we investigated the exchange of these reaction in the dielectric of water by applying the implicit polarizable continuum model (PCM). In the dielectric continuum, the differences in reaction free energy ΔGaq are more pronounced (see Tables S2 and S3 in the ESI†). For the most favorable reaction, Phe− shows a more stable energy of −77.94 kcal mol−1 compared to −47.46 kcal mol−1 for IB− when all four waters of Na+ ion are exchanged (Table 2). The difference in free energy strongly favors exchange of sodium's ligating waters for Phe− over IB− by approximately 29 kcal mol−1. This indicates that the Na+ ion preferentially associates with Phe− over the PAA carboxylic acids, which may aid in the initial Na+ ion insertion into the film, i.e. going from fully solvated in the aqueous solution to transitioning into the polyelectrolyte film, thus aiding transport and increasing selectivity. Transport likely involves the binding of many Na+ to multiple binding sites, and their collective movement, but that topic will be the subject of future work.
| Reactions | ΔGgas/kcal mol−1 | ΔGaq/kcal mol−1 |
|---|---|---|
| [Na(H2O)4]+ + IB− [Na(IB)(H2O)3] + 1H2O |
−87.16 | −31.55 |
| [Na(H2O)4]+ + Phe− [Na(Phe)(H2O)3] + 1H2O |
−90.64 | −61.25 |
| [Na(H2O)4]+ + IB− [Na(IB)] + 4H2O |
−77.42 | −47.46 |
| [Na(H2O)4]+ + Phe− [Na(Phe)] + 4H2O |
−80.94 | −77.94 |
This study demonstrates how biological ion conducting proteins can inform the engineering of new, more efficient ion exchange membranes, where the selectivity of LbL deposited PE films can be increased by the addition of Phe to the dip coating solutions. The increase in selectivity was shown to be enhanced due to interactions of Na+ with Phe, a result consistent with predictions from the DFT calculations of channelrhodopsin. The increase in selectivity could be a result of possible structural changes, such as phenyl ring stacking between phenylalanine molecules, forming more complex structures in the polyelectrolyte, or preferential adsorption of the phenyl ring on the surface of the polycarbonate within the pores, or even membrane swelling. While the co-deposition and self-assembly of the Phe with the PE polymers may preferentially form structures in the coating that are beneficial for cation transport, we have no evidence of this at this time and more work needs to be done to confirm possible structural changes.
Conclusion
Inspired by biological protein ion channels, the amino acid Phe was incorporated into PE coatings on nanoporous separator membranes to tailor transmembrane ion transport. The inclusion of this small molecule amino acid was shown to increase the ionic selectivity of a LbL deposited PE coating. The PE films incorporated with Phe had consistently higher cationic selectivity than similar thickness films made without Phe, suggesting that a unique chemical interaction attributed to Phe is responsible. The added amino acid, in fact, increased the selectivity of the PE film to rival that of a film that was more than twice as thick but without the penalty of an increased membrane resistance. The increased selectivity was attributed to the stabilization of the Na+ ion in the film as determined from the computational analysis of ion binding free energies. This study provides insights into how bio-inspired structure–function relationships can inform tailored performance of new ionic materials.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Sara Dickens for recording SEM micrographs. This work was supported by the Laboratory Directed Research and Development (LDRD) program at Sandia National Laboratories and by the Energy Storage Program, managed by Dr Imre Gyuk, through the U.S. Department of Energy's Office of Electricity. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.References
- C. R. Martin, M. Nishizawa, K. Jirage, M. S. Kang and S. B. Lee, Adv. Mater., 2001, 13, 1351–1362 Search PubMed.
- H. Daiguji, Chem. Soc. Rev., 2010, 39, 901–911 Search PubMed.
- W. Guo, Y. Tian and L. Jiang, Acc. Chem. Res., 2013, 46, 2834–2846 Search PubMed.
- L. H. Yeh, M. Zhang, S. Qian, J. P. Hsu and S. Tseng, J. Phys. Chem. C, 2012, 116, 8672–8677 Search PubMed.
- F. M. Gilles, M. Tagliazucchi, O. Azzaroni and I. Szleifer, J. Phys. Chem. C, 2016, 120, 4789–4798 Search PubMed.
- F. Buyukserin, P. Kohli, M. O. Wirtz and C. R. Martin, Small, 2007, 3, 266–270 Search PubMed.
- J. Elbert, F. Krohm, C. Ruttiger, S. Kienle, H. Didzoleit, B. N. Balzer, T. Hugel, B. Stuhn, M. Gallei and A. Brunsen, Adv. Funct. Mater., 2014, 24, 1591–1601 Search PubMed.
- T. Liu, C. Y. Bao, H. Y. Wang, Y. Lin, H. J. Jia and L. Y. Zhu, Chem. Commun., 2013, 49, 10311–10313 Search PubMed.
- L. J. Small, D. R. Wheeler and E. D. Spoerke, Nanoscale, 2015, 7, 16909–16920 Search PubMed.
- I. Vlassiouk, C. D. Park, S. A. Vail, D. Gust and S. Smirnov, Nano Lett., 2006, 6, 1013–1017 Search PubMed.
- Q. Q. Zhang, Z. Y. Hu, Z. Y. Liu, J. Zhai and L. Jiang, Adv. Funct. Mater., 2014, 24, 424–431 Search PubMed.
- T. W. Lin, J. P. Hsu, C. Y. Lin and S. Tseng, J. Phys. Chem. C, 2019, 123, 12437–12443 Search PubMed.
- Y. X. Zhao, J. M. Janot, E. Balanzat and S. Balme, Langmuir, 2017, 33, 3484–3490 Search PubMed.
- C. Cheng, A. Yaroshchuk and M. L. Bruening, Langmuir, 2013, 29, 1885–1892 Search PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 Search PubMed.
- G. Decher, M. Eckle, J. Schmitt and B. Struth, Curr. Opin. Colloid Interface Sci., 1998, 3, 32–39 Search PubMed.
- G. Decher, in Multilayer Thin Films: Sequential Assembly of Nanocomposite Materials, ed. G. Decher and J. B. Schlenoff, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2nd edn, 2012, vol. 1, ch. 1, pp. 1–19 Search PubMed.
- J. J. Richardson, M. Bjornmalm and F. Caruso, Science, 2015, 348, aaa2491 Search PubMed.
- A. Ostendorf, C. Cramer, G. Decher and M. Schonhoff, J. Phys. Chem. C, 2015, 119, 9543–9549 Search PubMed.
- E. Sorrenti, V. Ball, D. Del Frari, C. Arnoult, V. Toniazzo and D. Ruch, J. Phys. Chem. C, 2011, 115, 8248–8259 Search PubMed.
- S. J. Percival, M. A. Melia, C. A. Alexander, D. W. Nelson, E. J. Schindelholz and E. D. Spoerke, Surf. Coat. Technol., 2020, 383, 125228 Search PubMed.
- E. J. Schindelholz, E. D. Spoerke, H. D. Nguyen, J. C. Grunlan, S. Qin and D. C. Bufford, ACS Appl. Mater. Interfaces, 2018, 10, 21799–21803 Search PubMed.
- M. A. Melia, S. J. Percival, S. Qin, E. Barrick, E. Spoerke, J. Grunlan and E. J. Schindelholz, Prog. Org. Coat., 2020, 140, 105489 Search PubMed.
- F. Fan, C. Y. Zhou, X. Wang and J. Szpunar, ACS Appl. Mater. Interfaces, 2015, 7, 27271–27278 Search PubMed.
- J. H. Dai, D. M. Sullivan and M. L. Bruening, Ind. Eng. Chem. Res., 2000, 39, 3528–3535 Search PubMed.
- T. R. Farhat and J. B. Schlenoff, Electrochem. Solid-State Lett., 2002, 5, B13–B15 Search PubMed.
- D. V. Andreeva, D. Fix, H. Mohwald and D. G. Shchukin, J. Mater. Chem., 2008, 18, 1738–1740 Search PubMed.
- T. Guin, M. Krecker, A. Milhorn, D. A. Hagen, B. Stevens and J. C. Grunlan, Adv. Mater. Interfaces, 2015, 2, 1500214 Search PubMed.
- K. M. Holder, R. J. Smith and J. C. Grunlan, J. Mater. Sci., 2017, 52, 12923–12959 Search PubMed.
- C. Han, S. J. Percival and B. Zhang, Langmuir, 2016, 32, 8783–8792 Search PubMed.
- M. Yaqub, S. Imar, F. Laffir, G. Armstrong and T. McCormac, ACS Appl. Mater. Interfaces, 2015, 7, 1046–1056 Search PubMed.
- J. L. Stair, J. J. Harris and M. L. Bruening, Chem. Mater., 2001, 13, 2641–2648 Search PubMed.
- J. J. Harris, J. L. Stair and M. L. Bruening, Chem. Mater., 2000, 12, 1941–1946 Search PubMed.
- J. A. Armstrong, E. E. L. Bernal, A. Yaroshchuk and M. L. Bruening, Langmuir, 2013, 29, 10287–10296 Search PubMed.
- S. J. Percival, L. J. Small, E. D. Spoerke and S. B. Rempe, RSC Adv., 2018, 8, 32992–32999 Search PubMed.
- L. Yang, C. Tang, M. Ahmad, A. Yaroshchuk and M. L. Bruening, ACS Appl. Mater. Interfaces, 2018, 10, 44134–44143 Search PubMed.
- J. J. Richardson, J. W. Cui, M. Bjornmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 Search PubMed.
- C. Y. Cho, F. Xiang, K. L. Wallace and J. C. Grunlan, Macromolecules, 2015, 48, 5723–5729 Search PubMed.
- D. A. Hagen, B. Foster, B. Stevens and J. C. Grunlan, ACS Macro Lett., 2014, 3, 663–666 Search PubMed.
- J. H. Dai, A. W. Jensen, D. K. Mohanty, J. Erndt and M. L. Bruening, Langmuir, 2001, 17, 931–937 Search PubMed.
- A. M. Lehaf, M. D. Moussallem and J. B. Schlenoff, Langmuir, 2011, 27, 4756–4763 Search PubMed.
- Y. H. Yang, L. Bolling, M. Haile and J. C. Grunlan, RSC Adv., 2012, 2, 12355–12363 Search PubMed.
- J. J. Harris, P. M. DeRose and M. L. Bruening, J. Am. Chem. Soc., 1999, 121, 1978–1979 Search PubMed.
- H. Feroz, J. Meisenhelter, G. Jokhadze, M. Bruening and M. Kumar, Biotechnol. Prog., 2019, 35, e2859 Search PubMed.
- W. J. Liu, A. L. Bennett, W. J. Ning, H. Y. Tan, J. D. Berwanger, X. Q. Zeng and M. L. Bruening, Anal. Chem., 2018, 90, 12161–12167 Search PubMed.
- D. V. Golubenko, R. R. Shaydullin and A. B. Yaroslavtsev, Colloid Polym. Sci., 2019, 297, 741–748 Search PubMed.
- S. Bhattacharjee and M. L. Bruening, Langmuir, 2008, 24, 2916–2920 Search PubMed.
- D. G. Shchukin and H. Mohwald, Small, 2007, 3, 926–943 Search PubMed.
- D. G. Shchukin, M. Zheludkevich, K. Yasakau, S. Lamaka, M. G. S. Ferreira and H. Mohwald, Adv. Mater., 2006, 18, 1672–1678 Search PubMed.
- D. Yoo, A. P. Wu, J. Lee and M. F. Rubner, Synth. Met., 1997, 85, 1425–1426 Search PubMed.
- W. F. Paxton, P. T. McAninch, K. E. Achyuthan, S. H. R. Shin and H. L. Monteith, Colloids Surf., B, 2017, 159, 268–276 Search PubMed.
- I. M. Henderson, H. A. Quintana, J. A. Martinez and W. F. Paxton, Chem. Mater., 2015, 27, 4808–4813 Search PubMed.
- S. W. Kang, J. H. Kim, J. Won, K. Char and Y. S. Kang, J. Membr. Sci., 2005, 248, 201–206 Search PubMed.
- M. Maeda, M. Kimura, Y. Hareyama and S. Inoue, J. Am. Chem. Soc., 1984, 106, 250–251 Search PubMed.
- I. M. Kolangare, A. M. Isloor, Inamuddin, A. M. Asiri and A. F. Ismail, Environ. Chem. Lett., 2019, 17, 1053–1059 Search PubMed.
- A. Nguyen, S. Azari and L. D. Zou, Desalination, 2013, 312, 82–87 Search PubMed.
- Y. F. Liu, C. Xu, B. B. Xie, W. H. Hu, Y. Li and C. Yao, J. Coat. Technol. Res., 2018, 15, 403–414 Search PubMed.
- B. Hille, Ionic Channels of Excitable Membranes, Sinauer Associates, Sunderland, MA, 3rd edn, 2001 Search PubMed.
- R. T. Cygan, C. J. Brinker, M. D. Nyman, K. Leung and S. B. Rempe, MRS Bull., 2008, 33, 42–47 Search PubMed.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, eaab0530 Search PubMed.
- Y. Q. Fu, Y. B. Jiang, D. Dunphy, H. F. Xiong, E. Coker, S. Chou, H. X. Zhang, J. M. Vanegas, J. G. Croissant, J. L. Cecchi, S. B. Rempe and C. J. Brinker, Nat. Commun., 2018, 9, 990 Search PubMed.
- C. Y. Tang, Y. Zhao, R. Wang, C. Helix-Nielsen and A. G. Fane, Desalination, 2013, 308, 34–40 Search PubMed.
- C. Helix-Nielsen, Membranes, 2018, 8, 44 Search PubMed.
- S. Varma, D. M. Rogers, L. R. Pratt and S. B. Rempe, J. Gen. Physiol., 2011, 137, 479–488 Search PubMed.
- K. Deisseroth and P. Hegemann, Science, 2017, 357, eaan5544 Search PubMed.
- G. Nagel, D. Ollig, M. Fuhrmann, S. Kateriya, A. M. Mustl, E. Bamberg and P. Hegemann, Science, 2002, 296, 2395–2398 Search PubMed.
- G. Nagel, T. Szellas, W. Huhn, S. Kateriya, N. Adeishvili, P. Berthold, D. Ollig, P. Hegemann and E. Bamberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13940–13945 Search PubMed.
- E. S. Boyden, F. Zhang, E. Bamberg, G. Nagel and K. Deisseroth, Nat. Neurosci., 2005, 8, 1263–1268 Search PubMed.
- T. Bruegmann, D. Malan, M. Hesse, T. Beiert, C. J. Fuegemann, B. K. Fleischmann and P. Sasse, Nat. Methods, 2010, 7, U897–U845 Search PubMed.
- H. E. Kato, F. Zhang, O. Yizhar, C. Ramakrishnan, T. Nishizawa, K. Hirata, J. Ito, Y. Aita, T. Tsukazaki, S. Hayashi, P. Hegemann, A. D. Maturana, R. Ishitani, K. Deisseroth and O. Nureki, Nature, 2012, 482, U369–U115 Search PubMed.
- C. Priest, M. VanGordon, C. S. Rempe, M. I. Chaudhari, M. Stevens and S. B. Rempe, in Computing the Potential of Mean Force Profiles for Ion Permeation through Channelrhodopsin Chimera, C1C2, ed. R. E. Dempski, SpringerUS, 1st edn, 2020 DOI:10.1007/978-1-0716-0830-2.
- J. A. Jaber and J. B. Schlenoff, J. Am. Chem. Soc., 2006, 128, 2940–2947 Search PubMed.
- N. S. Hudak, L. J. Small, H. D. Pratt and T. M. Anderson, J. Electrochem. Soc., 2015, 162, A2188–A2194 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- S. B. Rempe and H. Jonsson, Chem. Educ., 1998, 3, 17 Search PubMed.
- F. Lipparini and B. Mennucci, J. Chem. Phys., 2016, 144, 160901 Search PubMed.
- L. R. Pratt and S. B. Rempe, in Quasi-chemical theory and implicit solvent models for simulations, ed. L. R. Pratt and G. Hummer, 1999, vol. 492, pp. 172–201 Search PubMed.
- M. J. Stevens and S. L. B. Rempe, J. Phys. Chem. B, 2016, 120, 12519–12530 Search PubMed.
- M. I. Chaudhari, J. M. Vanegas, L. R. Pratt, A. Muralidharan and S. B. Rempe, in Hydration Mimicry by Membrane Ion Channels, ed. M. A. Johnson and T. J. Martinez, 2020, vol. 71, pp. 461–484 Search PubMed.
- K. Leung and S. B. Rempe, J. Am. Chem. Soc., 2004, 126, 344–351 Search PubMed.
- Y. H. Yang, M. Haile, Y. T. Park, F. A. Malek and J. C. Grunlan, Macromolecules, 2011, 44, 1450–1459 Search PubMed.
- J. Kamcev, M. Galizia, F. M. Benedetti, E. S. Jang, D. R. Paul, B. D. Freeman and G. S. Manning, Phys. Chem. Chem. Phys., 2016, 18, 6021–6031 Search PubMed.
- J. Kamcev, D. R. Paul, G. S. Manning and B. D. Freeman, ACS Appl. Mater. Interfaces, 2017, 9, 4044–4056 Search PubMed.
- G. M. Geise, H. J. Cassady, D. R. Paul, B. E. Logan and M. A. Hickner, Phys. Chem. Chem. Phys., 2014, 16, 21673–21681 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 86th edn, 2005 Search PubMed.
- H. Q. Fan and N. Y. Yip, J. Membr. Sci., 2019, 573, 668–681 Search PubMed.
- M. R. VanGordon, G. Gyawali, S. W. Rick and S. B. Rempe, Biophys. J., 2017, 112, 943–952 Search PubMed.
- Y. Marcus, Biophys. Chem., 1994, 51, 111–127 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Plot of the normalized peak heights from the ToF-SIMS calibration curve, table listing the ASR for the different membranes, structures of the ion complexes considered in computational studies and tables listing the change in Gibbs free energy of sodium ion interactions with phenylalanine and iso-butyrate. See DOI: 10.1039/d1sm00134e |
| ‡ Current address: Department of Chemical and Materials Engineering, University of Kentucky, Lexington, KY, 40506-0046. |
| This journal is © The Royal Society of Chemistry 2021 |