
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Membrane-coated 3D architectures for bottom-up synthetic biology†
Hiromune
Eto
 a,
Henri G.
Franquelim
a,
Michael
Heymann
ab and
Petra
Schwille
*a
a,
Henri G.
Franquelim
a,
Michael
Heymann
ab and
Petra
Schwille
*a
aDepartment for Cellular and Molecular Biophysics, Max Planck Institute for Biochemistry, Am Klopferspitz 18, D-82152, Martinsried, Germany. E-mail: schwille@biochem.mpg.de
bDepartment of Intelligent Biointegrative Systems, Institute of Biomaterials and Biomolecular Systems, University of Stuttgart, Pfaffenwaldring 57, 70569, Stuttgart, Germany
First published on 23rd March 2021
Abstract
One of the great challenges of bottom-up synthetic biology is to recreate the cellular geometry and surface functionality required for biological reactions. Of particular interest are lipid membrane interfaces where many protein functions take place. However, cellular 3D geometries are often complex, and custom-shaping stable lipid membranes on relevant spatial scales in the micrometer range has been hard to accomplish reproducibly. Here, we use two-photon direct laser writing to 3D print microenvironments with length scales relevant to cellular processes and reactions. We formed lipid bilayers on the surfaces of these printed structures, and we evaluated multiple combinatorial scenarios, where physiologically relevant membrane compositions were generated on several different polymer surfaces. Functional dynamic protein systems were reconstituted in vitro and their self-organization was observed in response to the 3D geometry. This method proves very useful to template biological membranes with an additional spatial dimension, and thus allows a better understanding of protein function in relation to the complex morphology of cells and organelles.
Introduction
Many protein functions take place at lipid interfaces; and often, geometrical features are key in regulating this interaction. For example, membrane curvature,1,2 compartmentalisation into droplets and vesicles,3,4 and formation of domains that pattern lipid surfaces5,6 all govern pathways by influencing protein localization and behaviour. Consequently, engineering membrane geometry to mimic cellular features, in order to faithfully reconstruct biological functionality in vitro, is one of the most fundamental challenges in bottom-up synthetic biology.Over the past years, many techniques have been developed to generate artificial membranes and modulate their geometry. They often start from spherical membrane mimics, such as water-in-oil droplets and liposomes,7 which can be produced in large numbers and feature cell-sized volumes with phospholipid mono- or bilayer interfaces. By varying membrane curvature or surface tension through buffer exchange or membrane-transforming agents, the geometry of these delicate free-standing vesicles can be deformed into rod- or tubular-like shapes, mimicking certain cellular geometries.4,8,9 However, the resulting morphologies are either ill-controlled or require sophisticated techniques that are technically challenging. In particular, since non-spherical shapes are rarely in equilibrium, they tend to be temporally unstable and hard to control. Hence, the variety of shapes that free-standing vesicle membranes can adapt to is limited. Other bilayer systems, such as black lipid membranes10–12 and other emulsion transfer methods13 also suffer from this limitation; their membrane topology and topography cannot be easily varied beyond their equilibrium state.
In this regard, supported lipid membranes on substrates provide an attractive alternative, featuring high stability of membranes and facile preparation methods.14 They can be formed on planar surfaces, but their geometry can also be varied by forming them on patterned substrates15–19 or on the surfaces of microfabricated compartments.20,21 Here, the range and size of possible geometries depends on the underlying pattern or the shape of the supporting material, which allows much greater flexibility. However, accurately recreating cellular geometries often requires complex 3D features, which conventional 2D patterning methods cannot achieve.
In order to explore more complex 3D structures for membrane sculpting, we turned to two-photon direct laser writing (2PP-DLW) – a light-initiated 3D printing technology, based on photoresist polymerisation upon the quasi-simultaneous absorption of 2 photons. Owing to the higher definition of the 2-photon point spread function, structures can be printed with sub-micron resolution,22 and custom-shaped 3D microenvironments can be accurately fabricated with length scales relevant to cellular processes. 2PP-DLW has been successfully employed in optics,23 microfluidics,24 soft robotics25–27 and biological applications,26,28–30 and a variety of photoresists and post-modification methods have been developed to further increase the range of chemical functionalities for the different applications.31–33
To form supported lipid membranes on materials suitable for 3D lithography, Ormocomp, pentaerythritol triacrylate (PETA) and trimethylolpropane ethoxylate triacrylate (TPETA) are of particular interest. They are well established polymer systems in bio-applications because they have low cytotoxicity, are easy to handle, and show minimal autofluorescence with good transmittance of light for imaging.31,32,34 They are chemically and mechanically stable, with low swelling unlike typical hydrogels,35 and their elastic moduli are typical for stiff polymers (Ormocomp ∼1 GPa, PETA ∼3 GPa and TPETA ∼10 MPa).36,37 Their chemical compositions are also ideal for manipulating membranes: PETA includes an exposed hydroxyl group that aid in lipid membrane formation, whilst TPETA has a PEG-like chain that passivates against membrane fusion. Ormocomp is accessible to a wide range of available post-modification methods,29,38 including silanization, that can render the surface favourable for membrane formation. Therefore, these materials are ideal candidates to fabricate 3D-lipid bilayer architectures.
In this paper, we introduce a new method for generating custom-shaped membrane morphologies based on 2PP-DLW. We specifically tailor 3D printed structures that are templates for biomimetic lipid membranes – different lipid compositions can be deposited, based on charge interaction between lipids and support structures. We apply our membrane architectures to investigate the influence of particular spatial features on in vitro reconstituted protein systems. These include a membrane attached minimal actin cortex and the pattern forming MinDE protein system from E. coli, which displays dynamic self-organization in response to 3D spatial patterning. Although 2D structuring of membrane surfaces has previously been shown to alter the obtained patterns,39 adding another spatial dimension will bring us an important step closer towards studying true biomimetic functionality.
Experimental
Fabrication of 3D microstructures
To enhance the adhesion of the microscaffolds to the glass substrate surface for pentaerythritol triacrylate (PETA, Sigma-Aldrich) and trimethylolpropane ethoxylate triacrylate (TPETA, Sigma-Aldrich, Mn ∼ 692, Sigma-Aldrich), glass coverslips (22 mm × 22 mm, borosilicate, Menzel) were initially treated with O2 plasma (Zepto, Diener Electronic) (power 30%, pressure 0.3 mbar for 1 min), then immersed in 3-(trimethoxysilyl)propyl methacrylate (Sigma-Aldrich, ≥98%) (0.1 mL in 20 mL ethanol) overnight, rinsed in water, and dried with nitrogen. At this point, the coverslips should be slightly hydrophobic – a visual inspection of the contact angle of a small water droplet with the surface (>45°) should suffice. For Ormocomp (Microchem, USA), similarly plasma treated coverslips were spin-coated with OrmoPrime (Microchem, USA) for 40 s at 6000 rpm and then baked on a hotplate for 5 min at 150 °C.For lipid assays, plane surfaces of Ormocomp and PETA were spin-coated (60 s at 6000 rpm) and UV exposed (FormCure, Formlabs) for 15 min at room temperature. As TPETA requires either an overnight exposure, or cures more efficiently with O2-free atmosphere – a slab of PDMS was gently pressed down on a drop of photoresist and exposed to UV for 15 min at RT.
For the Min assays, treated coverslips were further spin-coated with a drop of Ormocomp and UV exposed, so that the glass surface is also coated with Ormocomp (thickness ∼10 μm).
For 3D printing, scaffold geometries were designed in Solidworks (Dassault Systèmes, France), and print parameters then specified in Describe (Nanoscribe GmbH, Germany) and fabricated on a commercial DLW system (Photonic Professional, Nanoscribe GmbH, Germany) with a Zeiss LCI “Plan-Neofluar” 25×/0.8 objective, corr-ring set on oil immersion. The photoresist was drop-cast onto the coverslip and the 3D microscaffolds were printed with an oil immersion (Carl Zeiss Immersol 518F). After exposure, the structures were developed and rinsed with isopropyl alcohol (for PETA and TPETA) or OrmoDev (Microchem, USA) and then isopropyl alcohol (for Ormocomp). The structures were kept submerged in isopropyl alcohol (IPA) and dried using a critical point dryer (Leica EM CPD300). This was because drying the beam structures directly in air would lead to them collapsing due to surface tension – for larger, sturdier structures, drying in air would be sufficient.
For composite printing, PETA was dropcast on spin-coated TPETA. The grids were printed at 80% laser power and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μm s−1. The sample was then developed and rinsed with isopropyl alcohol and dried in a stream of air.
000 μm s−1. The sample was then developed and rinsed with isopropyl alcohol and dried in a stream of air.
Scanning electron microscopy
Samples were first sputter-coated with platinum/palladium on a high-resolution automatic sputter coater (Cressington 208HR) at 20 mA and 0.1 mbar Argon for 3 × 20 s. The thickness of the applied coatings was measured with a built-in thickness controller to be 2.0 nm. They were then imaged using TESCAN MIRA3 FESEM operating at an accelerating voltage of 10 kV in SE mode.Homogeneous bilayer formation by small unilamellar vesicles (SUV) fusion
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA), unless otherwise stated. Small unilamellar vesicles (SUVs) composed of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) for neutral lipid membranes, DOPC/DOPG (1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)) for negatively charged membranes, or DOPC/DOTAP (1,2-dioleoyl-3-trimethylammonium-propane) for positively charged membranes, containing Atto655-DOPE (Atto-TEC GmbH, Siegen, Germany) in 7:3:0.005 molar ratio. Lipids dissolved in chloroform were dried under a nitrogen stream, and vials were placed in a desiccator to remove residual chloroform for at least 2 h. Afterwards, lipids were rehydrated in a buffer to a concentration of 4 mg mL−1. One of two types of buffer was used, depending on the required conditions. pH Buffer (0.1 M Na2CO3 and 0.1 M Na2HCO3 mixed in 6:4 volume ratio, pH 10.1) was prepared to provide basic conditions for deprotonation of hydroxyl groups. Buffer M (25 mM Tris–HCl, 150 mM KCl, 5 mM MgCl2, pH 7.5) was used for APTES-treated Ormocomp structures. SUVs were then generated by sonication in a water bath (model 1510; Branson) until the solution appeared clear.
SUVs were added to a chamber, assembled from a silicone isolator chamber (Thermo Fisher Scientific, 0717104) pressed on to the coverslip, at a concentration of 0.5 mg mL−1 diluted in the buffer of choice. After 3 min incubation at 37 °C on a heating block, the supported lipid bilayer (SLB) was gently washed with a total of 600 μL buffer S (25 mM Tris–HCl, 150 mM KCl, pH 7.5) to remove excess vesicles.
For the silanisation treatment of Ormocomp, the samples were first plasma treated with argon plasma (Model 950 Advanced Plasma System, Gatan), and then incubated in (3-aminopropyl)triethoxysilane (Sigma-Aldrich) at a dilution of 0.1 mL in 25 mL ethanol for 5 min. The samples were then rinsed in IPA and water. For delicate structures, the chamber was assembled with the sample still submerged in water so that the structures did not collapse under surface tension during drying. The water was exchanged with buffer, and the SUV concentration was adjusted to 0.5 mg mL−1.
Lipid patch formation by giant unilamellar vesicle (GUV) fusion
Giant unilamellar vesicles (GUVs) composed either of DOPC (neutral), DOPC/DOPG (negative) or DOPC/DOTAP (positive) mixtures with Atto655-DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) in 8:2:0.0005 molar ratio were produced by electroformation in polytetrafluoroethylene chambers with platinum electrodes 4 mm apart, as described previously.40,41 Briefly, 6 μL of the lipid mixture (2 mg mL−1 in chloroform) was spread onto two platinum wires and dried in a desiccator for 30 min. The chamber was then filled with 370 μL of a 250 mOsm kg−1 aqueous solution of sucrose. An AC electric field of 2 V (RMS) was applied at a frequency of 10 Hz for 1.5 h, followed by 2 Hz for 45 min. The obtained solution was diluted 1:10 in volume in 250 mOsm kg−1 sucrose and further diluted 1:7 in volume in their respective buffers. The vesicle solution was then deposited directly onto the polymer surface, and left for several minutes until the vesicles sank to the bottom and came into contact with the surface.
Fluorescence microscopy
FRAP experiments on plane surfaces were done on Nikon Eclipse Ti2 in a TIRF (totalinternal reflection fluorescence) set-up using a Nikon Apo TIRF 60×/1.4 oil immersion objective. We used the 640 nm diode laser lines to image Atto655-DOPE. FRAP experiments on patterned membrane patches were done on a Zeiss LSM780 confocal laser scanning microscope using a C-Apochromat 40×/1.20 water-immersion objective (Carl Zeiss AG, Oberkochen, Germany). Atto655-DOPE was excited by the 633 nm He–Ne laser.Likewise, F-actin filament and MinDE self-organisation assays were imaged on a Zeiss LSM780 confocal laser scanning microscope using a Zeiss C-Apochromat 40×/1.20 water-immersion objective (Carl Zeiss AG, Oberkochen, Germany). Alexa488 was excited using the 488 nm argon laser, Alexa568 on the 561 nm DPSS laser and Atto655-DOPE using the 633 nm He–Ne laser. Obtained images were processed using Fiji.42
4D images were taken with Nikon Eclipse Ti in a spinning disk confocal set-up, with the Nikon Plan Fluor 20×/0.75 water immersion objective (both Nikon GmbH, Düsseldorf, Germany). We used the 490 and 640 nm diode laser lines to image GFP and Atto655-DOPE, respectively. We then used a custom written script to reconstruct the 4D images. Briefly, the images were first processed by successive filters: Gaussian 3D Blur (x = 3, y = 3, z = 10; Subtract Background (rolling ball radius = 20, sliding paraboloid); Median 3D (x = 3, y = 3, z = 1). The 3D timelapse images from different fluorescence channels were then merged into one composite color hyperstack, and visualized using 3D Viewer (Display as = Volume, Color = None, Threshold = 0, Resampling factor = 2).
Fluorescence recovery after photobleaching (FRAP)
For FRAP on plane surfaces, a circular spot of 5 μm diameter was illuminated with full laser power for 260 ms, and the average fluorescence intensity was recorded every 3 s over 300 s. For FRAP on patterned membrane patches, the defined regions of interest were illuminated with full laser power for 0.82 s (50 iterations on the Zen Black software (Carl Zeiss AG, Oberkochen, Germany)), and the average fluorescence intensity was recorded every second for 100 s for the 2.5 μm diameter circular region, and every 3 s for 300 s for the entire square patch. Measurements were taken at room temperature (23 °C). Intensity in a second area of the same dimensions was recorded in the same field of view, in order to correct for the intensity drift. The intensity trace was fit to the equation derived by Soumpasis.43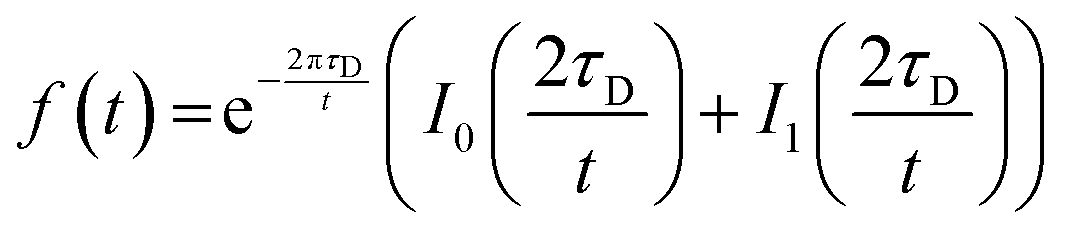 | (1) |
Here, τD is the characteristic timescale for diffusion, and t is the time. f(t) is the normalized fluorescence, and I0 and I1 are modified Bessel functions. The diffusion timescale for a bleached spot of radius r is τD = r2/4D with D the diffusion coefficient. A custom script written in Python was used for curve fitting and data analysis.
Atomic force microscopy
AFM was performed on a JPK Instruments Nanowizard III BioAFM mounted on a Zeiss LSM510 Meta laser scanning confocal microscope (Jena, Germany). AFM measurements were taken after locating the membrane patch by confocal microscopy. Cantilevers (BL-AC40TS-C2, Biolever Mini, Olympus) were used for the quantitative imaging (QI) mode, with typical spring constants of 0.09–0.1 N m−1. Setpoint force was set to 200–250 pN, acquisition speed to 61.1 μm s−1, Z-length to 110 nm and image resolution to 256 × 256 pixels. Data was analyzed using JPK data processing software Version 5.1.4 (JPK Instruments).F-Actin filament assays
Actin filaments were prepared according to the published protocol.44,45 Briefly, rabbit skeletal muscle actin monomers (Molecular Probes) and biotinylated rabbit actin monomers (tebu-bio, Cytoskeleton Inc.) were mixed in a 5:1 (actin:biotin-actin) ratio. Polymerization of the mixture (39.6 μM) was induced in Buffer F containing 50 mM KCl, 2 mM MgCl2, 1 mM DTT, 1 mM ATP, 10 mM Tris–HCl buffer (pH 7.5). The biotinylated actin filaments were labelled and stabilized with Alexa-Fluor 647 Phalloidin (Molecular Probes) according to the manufacturer protocol.
We formed the SLB as described previously, with DOPC/DOTAP (1,2-dioleoyl-3-trimethylammonium-propane), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) and Atto655-DOPE (Atto-TEC GmbH, Siegen, Germany) mixed in 6.75:2.75:0.5:0.0005 molar ratio. Streptavidin Alexa 568-conjugate (Thermo Fischer Scientific) was subsequently added at 10 nM concentration and incubated for at least 30 min. After washing away the excess streptavidin with Buffer F, F-actin filaments were added at a final concentration of 200 nM. Filaments were incubated for at least 1 h at room temperature, and the unattached filaments gently washed away with Buffer F before imaging.
MinDE self-organisation assays
MinDE dynamics were reconstituted as previously described.46 Briefly, a mixture of 1 μM MinD (doped with 30% EGFP-MinD), 2 μM MinE, and 5 mM ATP (from 100 mM ATP stock in 100 mM MgCl2, pH 7.5) in buffer M was first prepared. The mixture was then loaded onto the chambers to a final concentration of 0.5 μM MinD (doped with 30% EGFP-MinD), 1 μM MinE, and 2.5 mM ATP (i.e., to half the concentration) and incubated for at least 30 min at room temperature (23 °C) before imaging.2. Results and discussion
2.1. 3D laser lithography of microstructures
Engineering functional lipid-coated 3D geometries before a possible in vitro reconstitution of membrane proteins involves three main steps: (1) microfabrication of the 3D structures, (2) surface modification of the polymers to specifically tune lipid–polymer interactions, and (3) lipid deposition to prepare supported lipid membranes (schematic shown in Fig. 1A). In the microfabrication step, we performed a 4-dimensional parameter sweep (laser power, scan speed, z-slicing and xy-hatching), and analysed the outcome for quality by scanning electron microscopy (SEM). All these parameters contribute to the deposited energy density, which regulates the degree of polymerisation. The slicing and hatching parameters with respect to the polymerisation voxel (determined by the optical set up, chemical composition, laser power and scan speed47) also affect the structure quality. For example, a very coarse hatching with a small polymerisation voxel could lead to rough surface quality as well as underexposure.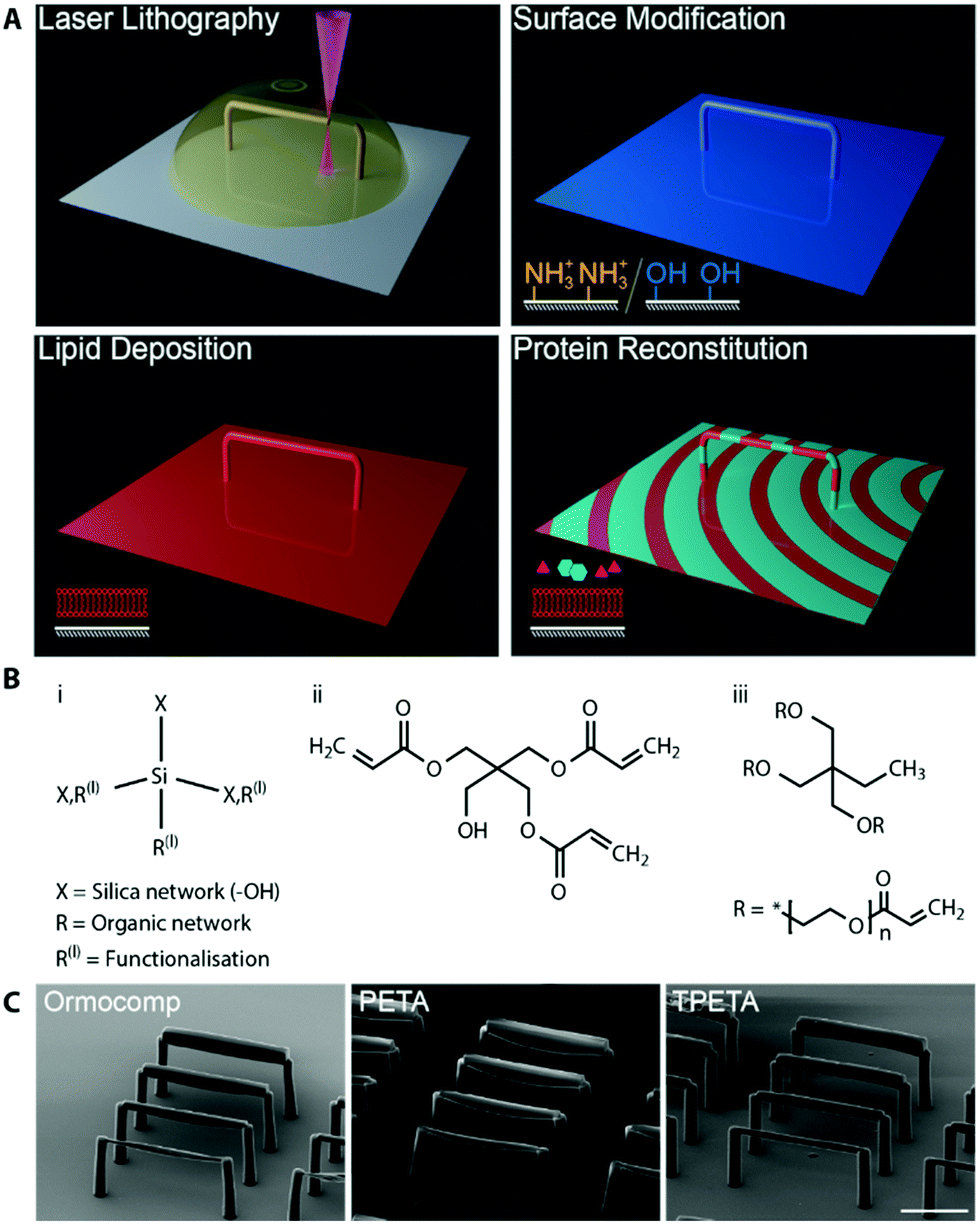 | ||
| Fig. 1 (A) Schematic the of work flow. Microstructures are printed by two-photon lithography. The microstructure surface is then modified to promote lipid membrane fusion, and coated by a supported lipid membrane. Afterwards, purified proteins are reconstituted in vitro. (B) Chemical structures of (i) Ormocomp and (ii) PETA, which allow SLB formation, and (iii) TPETA, which passivates against SLB formation. (C) scanning electron microscopy images of beam structures printed with (left to right) Ormocomp, PETA and TPETA at their near-optimum settings (see Fig. S1 for a detailed parameter sweep, ESI†). Scale bar 50 μm. | ||
We printed rod-like structures (length 100 μm, and widths 2–10 μm), as a minimal complexity design to first test out our printing parameters, (see Fig. S1 for parameter sweep, ESI†), and we subsequently used them in our in vitro reconstitution experiments. No stable structure could be printed below 2 μm width. For each of the resists (chemical structures shown in Fig. 1B), we found that the optimum writing conditions were: Ormocomp (power 80% speed 10000 μm slicing 0.1 μm hatching 0.5 μm), PETA (power 80% speed 10000 μm s−1 slicing 0.3 μm hatching 0.1 μm) and TPETA (power 100% speed 10000 μm s−1 slicing 0.3 μm hatching 0.1 μm), all with XY hatching at 90° (perpendicular) to the axis of the rods (Fig. 1C). We found that the dynamic range (range of parameters from where polymerisation starts to occur up to where resist explodes due to overexposure) of Ormocomp is narrow compared to PETA and TPETA. The narrow range of Ormocomp is consistent with previously reported results,48 and could potentially be broadened by use of additional photoinitiators.
We also found that hatching at 90° (perpendicular) to the axis of the beams provided much better results as compared to hatching at 0° (parallel) to the axis (Fig. S2, ESI†). For structures with high xy aspect ratios (length much longer compared to width), the time taken between adjacent hatchlines is much shorter with perpendicular hatching. Each hatchline is immediately stabilised by the adjacent hatchline before it has time to deform under gravity or drift, which resulted in much more stable layers of print being formed.
2.2 Surface modification of polymers for membrane fusion
After the fabrication, we went on to coat the surfaces of our printed structures with lipid membranes. Supported lipid membranes can be formed by vesicle fusion,46 whereby an attractive interaction between the lipid and the surface promotes vesicles to rupture and spread on the surface. A more detailed understanding of this process is extremely complex, as it involves a large set of parameters on both the surface and the membrane, including higher order molecular interactions, charge density on both membrane and polymer surface, as well as surface roughness.49,50 However, we can gain qualitative insights into the probable lipid behaviour by considering the charge interaction between the lipid headgroups and the functional groups on the polymer surface.51 Thus, by considering the molecular structures, we formulated lipid–polymer combinations that would form supported lipid bilayers (SLBs).PETA has hydroxyl groups which can be deprotonated under alkaline conditions (Fig. 1B). The presence of negative charges on the surface promotes fusion of positively charged lipid vesicles via direct electrostatic interaction. Ormocomp is a commercially available polymer consisting of an organic/inorganic (silica) hybrid network, in which hydroxyl groups are also present. However, the detailed chemical composition is undisclosed. Similar to PETA, these hydroxyl groups can also be deprotonated at high pH, and thus likely a useful support for membranes with an overall positive charge. In order to support negative or neutrally charged lipids, deprotonated hydroxyl groups on PETA and Ormocomp can often be bridged by positively charged divalent ions such as calcium, which is also commonly used to support negatively charged lipids on glass.52
In addition to the native properties of the polymers, there is a range of well-documented post-functionalisation methods for Ormocomp.32,38 The presence of the inorganic backbone in its chemical structure means that many treatments can be done similarly to treating glass or silicon dioxide. It can first be plasma-treated, which exposes more hydroxyl groups. This can again be used to support positively charged membranes or be bridged by divalent ions to support negative or neutral lipids. Ormocomp can further be silanized, by which positively charged amine groups are incorporated on the surface of the material by (3-aminopropyl)triethoxysilane (APTES) treatment.53 Hence, silanization allows negatively charged vesicles to fuse with the surface. Note that oxygen plasma cannot be used with Ormocomp, since it reacts with the inorganic component in the polymer network and forms porous structures.54 Hence, we have several possibilities to manipulate charge interactions that would favour fusion with vesicles of the desired lipid type with PETA and Ormocomp (Fig. 2A).
 | ||
| Fig. 2 Lipid interactions with polymer surfaces. (A) Functional groups on the polymer surface promotes membrane fusion with positive or negative lipid headgroups. (B) Homogeneous lipid membranes were formed by SUV fusion. Fluidity of the SLB was measured by FRAP. Time lapse images of FRAP, and the associated intensity trace, performed on Ormocomp with APTES treatment with DOPC:DOPG lipids doped with 0.005 mol% Atto655-DOPE. Scale bar 5 μm (C) Bilayer verification on Ormocomp. Membrane patches were prepared by fusing GUVs to the polymer surface. The membrane height (4.7 ± 0.7 nm) was measured by atomic force microscopy. Scale bar 20 μm, colour bar range 0–40 nm. | ||
TPETA, on the other hand, has PEG-like chains in their structure, which is a known passivation agent against molecular adhesion and adsorption,32 and can also prevent vesicle fusion. This passivating property of TPETA, when combined with the lipophilic property PETA and Ormocomp, could allow us to print composite structures, where we can selectively target membrane supported regions. Thus, based on simple chemistry, we evaluated multiple combinatorial scenarios to generate 3D membrane geometries with desired membrane affinity.
These charge-based considerations do not only apply to the materials investigated here, but could also be used as starting points for forming membranes on any newly developed material. As more and more photoresins are being developed for 3D printing, materials with improved physical and chemical properties would significantly widen the scope of our biological research. For example, soft materials such as shape-shifting hydrogels26 and protein-based hydrogels55–57 would allow us to create flexible and deformable structures, potentially enabling us to investigate how cytoskeletal proteins deform membranes. Materials with better optical properties are also crucial. With fluorescence-based, dynamic imaging of biological samples, the resolution and the image quality of membrane processes rely heavily on refractive index matching between the sample and the surrounding medium. Materials such as CYTOP58 and MyPolymer59 have similar refractive indices to water, which would allow super-resolution imaging, and this would give us the chance to probe biological samples at a higher level of detail.
2.3. Supported lipid membrane formation on polymer surfaces
Next, we tested how well the different combinations of polymer and surface modifications support lipid membranes of different charges. We generated small unilamellar vesicles for vesicle fusion at a high concentration of vesicles (with the amount of lipid much greater than needed to completely cover all surfaces) and deposited them on planar, spin-coated polymer surfaces. Subsequently, we checked for homogeneous, fluid bilayers indicative of continuous membranes with no defects by fluorescence recovery after photobleaching (FRAP) of labelled lipids (Fig. 2B). Diffusion coefficients are summarised in Table 1, and immobile fractions in Table S1 (ESI†).| Negative/neutral charged lipids | |||
|---|---|---|---|
| Material | Surface modifications | DOPC/DOPG | DOPC |
| Ormocomp | Plasma + APTES | 0.6 ± 0.1 | 0.7 ± 0.4 |
| Plasma + pH buffer + Ca2+/Mg2+ | 0.6 ± 0.4 | 0.7 ± 0.3 | |
| pH buffer + Ca/Mg | 0.2 ± 0.05 | 0.1 ± 0.03 | |
| PETA | pH buffer + Ca/Mg | 0.006 ± 0.0004 | 0.05 ± 0.003 |
| Positive charged lipids | ||
|---|---|---|
| Material | Surface modifications | DOTAP |
| Ormocomp | Plasma + pH buffer | 2.3 ± 1.3 |
| pH buffer | 0.3 ± 0.03 | |
| PETA | pH buffer | 1.1 ± 0.1 |
We found that for negative and neutrally charged lipids, Ormocomp with plasma treatment and silanization, or with divalent ion bridging, resulted in membranes with the highest fluidity (0.6 ± 0.4 μm2 s−1), comparable to those of membranes on the commonly employed substrate, glass (0.9 ± 0.4 μm2 s−1).60,61 Even though the fluidity of supported bilayers is typically slower than those of free-standing bilayers (which can be up to 20 μm2 s−1),62 the support does not significantly interfere with biological functions of many membrane proteins, as shown in previous studies.16,63 Untreated Ormocomp or PETA with divalent ion bridging showed very little fluorescence recovery in the timescale of our experiments, indicative of immobile membranes. For positively charged lipids, Ormocomp with plasma treatment, as well as untreated PETA, formed fluid bilayers (Fig. S3, ESI†), whilst untreated Ormocomp formed immobile membranes. On TPETA, supported lipid membranes were not formed, indicated by the lack of fluorescence on the polymer surface after vesicle deposition (Fig. S3, ESI†).
Having determined the fluidity of the respective membranes, we confirmed whether the membrane was indeed a lipid bilayer and not a lipid monolayer, as for the case of some other polymer-supported membranes.20 For this, we prepared giant unilamellar vesicles (GUVs), sized typically 5–20 μm, which form large membrane patches when they fuse onto the surface. These patches were individually imaged, by both fluorescence microscopy and atomic force microscopy, which allowed us to simultaneously verify the presence of the membrane patch, and also to directly measure the membrane height.
We measured the height of the bilayer patch for Ormocomp (4.7 ± 0.7 nm, Fig. 2C) and PETA (4.7 ± 0.9 nm, Fig. S4, ESI†). The error was calculated as a standard deviation from 3 cross-sections taken from 2 independent bilayer patches (total 6 cross-sections). Typical lipid membranes with di-oleyl acyl-chained lipids have an approximate thickness of 5 nm in height.64 Both measurements are therefore well within the height range of bilayers. When GUVs were deposited on TPETA, GUVs did not rupture and remained intact, which was confirmed by confocal microscopy (Fig. S5, ESI†).
We conclude that, on PETA and Ormocomp, lipid bilayers of different charged lipids can be formed, with membrane fluidity comparable to those formed on glass. In contrast, TPETA does not support membrane formation, and passivates against membrane fusion.
The processes we employed here to investigate membrane formation can be applied to other material, surface modification and lipid combinations that are more application specific. In many cases, synthetic lipids are suitable mimics for natural lipids – for example, most prokaryotic cell membranes are negatively charged, and therefore, many bacterial protein systems can be reconstituted on membranes containing DOPG.65–67 A wider variety of lipid species, such as PIP2 or DOPS, could be investigated to further extend the range of protein systems to be recruited. Membranes could also be formed on polymer cushions, e.g., polyethylene glycol (PEG)-silanes, and this would allow the in vitro reconstitution of transmembrane proteins.68,69 Such investigations would support us to further improve the biomimicry of our fabricated microenvironment.31,70
2.4. Lipid patterning by composite printing
One of the most attractive features of engineering microenvironments is the ability to spatially position molecules to mimic the spatial order and organisation within a living cell. Having found the polymer–lipid composition combinations that either allow or passivate against vesicle fusion, we printed composite structures that allowed selective membrane deposition. For these structures, we utilised PETA and TPETA (without post-modifications), where untreated PETA supports positively charged membranes in alkaline conditions, whereas TPETA passivates against them. We designed 10 μm grids consisting of PETA, with 10 μm spacing, that were printed on top of a planar TPETA surface.We deposited labelled lipids (DOPC:DOTAP 7:3 molar ratio doped with 5 mol% biotinylated lipid and 0.005 mol% Atto655-DOPE) on these structures and observed lipid–dye fluorescence only in the 10 μm squares with 10 μm spacing, indicating that membranes formed only on the PETA surface (Fig. 3).
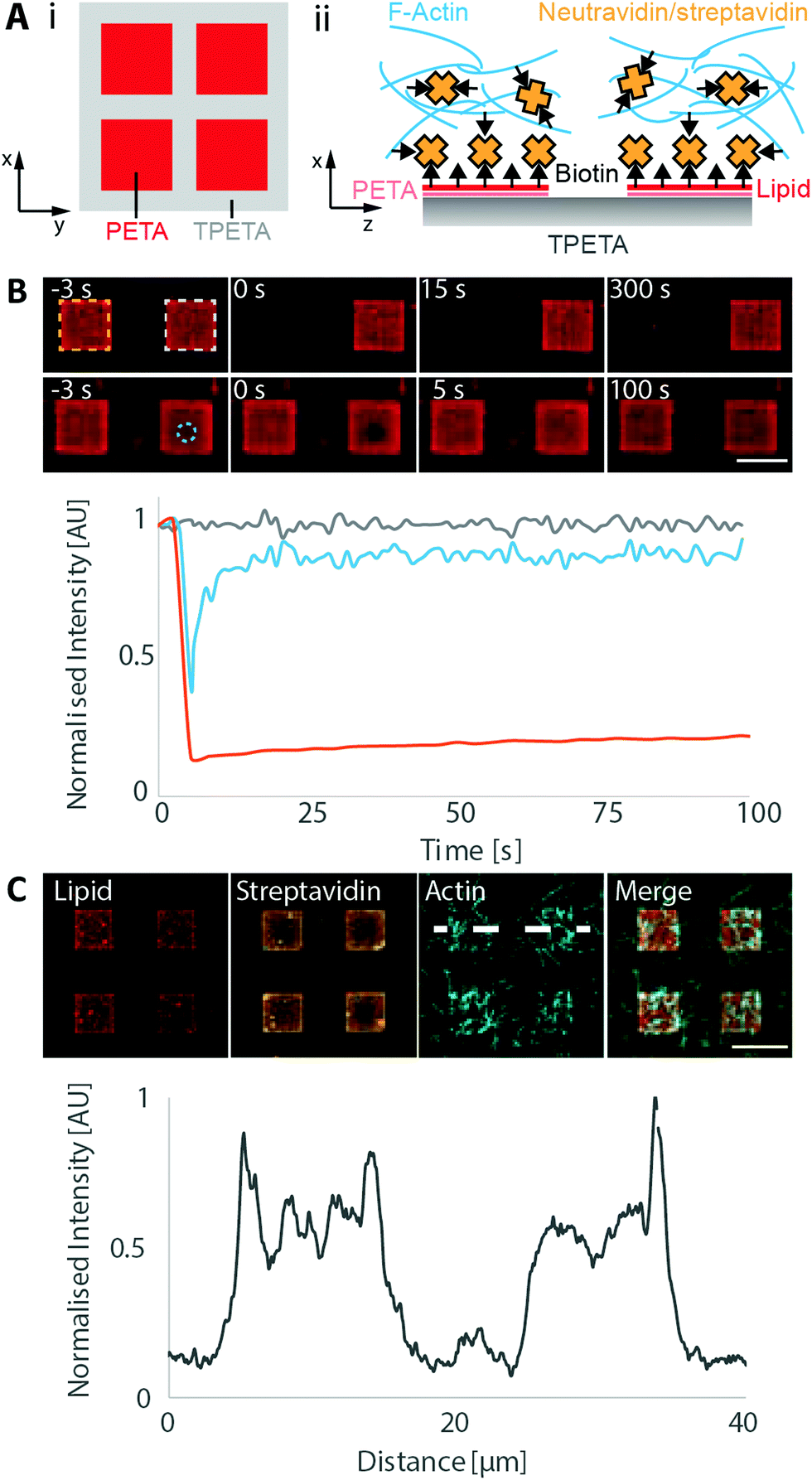 | ||
| Fig. 3 Lipid patterning by composite printing. (A) Schematics of (i) PETA grids printed on TPETA surface and (ii) in vitro reconstitution of actin filaments on patterned lipids. (B) FRAP experiment on membrane patches. Membranes with labelled lipids (DOPC:DOTAP 7:3 molar ratio doped with 5 mol% biotinylated lipid and 0.005 mol% Atto655-DOPE) were formed on square-patterned PETA patches, where an entire square (orange, top row) and a 2.5 μm diameter circular region (blue, bottom row) were subsequently bleached. Fluorescence intensity vs. time from these regions are plotted with: bleached square (orange), unbleached square (grey) and circular region (blue). (C) Confocal images of patterned lipids and actin filaments after incubation with streptavidin. Its associated intensity trace along the indicated line is plotted. Scale bar 10 μm. | ||
To assess the mobility of the lipids on these membrane patches, we first bleached a single, entire square. This square did not recover in fluorescence in the timescale of our experiments (5 min), showing that the individual membrane patches are indeed disconnected (Fig. 3B). We also bleached a smaller circular region of 2.5 μm diameter within a membrane patch, and the fluorescence recovered with a diffusion coefficient of 1.3 ± 0.8 μm2 s−1, which is similar to the values obtained on plane surfaces.
To demonstrate the utility of selective membrane deposition for targeting specific molecules onto the membranes, we used our previously developed minimal actin cortex.44 In this setup, biotinylated lipids in the membrane are bound by streptavidin that in turn recruit stabilised and biotinylated actin filaments. We found that the streptavidin was selectively recruited to the lipid patches and that the actin filaments accumulated in this area (Fig. 3C). We also performed a control experiment, where the membranes were not incubated with streptavidin before the deposition of actin filaments (Fig. S6, ESI†). We observed that a significantly reduced number of actin filaments were localized on the membranous patches. We can still see a slightly increased intensity on the membrane, as compared to the surrounding regions, which can be attributed to the non-specific binding of actin filaments with positively charged lipids due to electrostatic forces.71,72
Lastly, we performed a control on a planar PETA surface, where streptavidin was reconstituted with and without the formation of the lipid bilayer. As expected, we found that streptavidin was detected on the surface only with the presence of the lipids, excluding the possibility that they are simply adsorbing on the PETA surface (Fig. S7, ESI†), but are selectively recruited to the membranes by biotin–streptavidin coupling.
These results demonstrate that positive lipids can be patterned by composite printing of PETA and TPETA, which can further be employed to spatially pattern proteins.
2.5. Dynamic protein patterns on 3D microstructures
One of the major methodological advantages of 3D printing is that we can produce full 3D shapes that are not simply extrusions of 2D patterns. This added dimensionality gives us the opportunity to engineer and systematically vary the surface to volume ratio. This is a crucial parameter for reaction–diffusion processes that involve membranes for confinement and as catalytic surfaces, as peripheral membrane proteins attach to the lipid surface, and detach and diffuse in the bulk volume. In order to further explore this capability of our technique, we investigated the E. coli MinDE system, which has been shown in vivo to be a spatial regulator that positions the division ring to midcell,73 and forms dynamic patterns when reconstituted on supported lipid membranes in vitro.63,74Pattern formation of the E. coli MinDE system involves two proteins: MinD and MinE. MinD forms a dimer in the presence of ATP, upon which it attaches cooperatively to the membrane (autocatalytic attachment). At higher local concentrations on the membrane, it recruits MinE, which stimulates MinD's ATPase activity that triggers its detachment from the membrane (catalytic detachment). This forms the basis for a reaction–diffusion mechanism, where the two proteins cycle between the bulk volume and the lipid surface, and is dynamically regulated by biochemical reactions between them. The patterns are therefore strongly dependent on geometry with different surface to volume ratios. On planar surfaces with (quasi-) infinite bulk volume,63 Min proteins form travelling waves; on patterned (finite) surfaces with infinite bulk volume they produce directional waves;39 and in confined, cylindrical geometry, they perform pole-to-pole oscillations3 that are also seen in vivo.73
In order to investigate MinDE's geometry sensitivity in more detail, we devised a selection of 3D lipid architectures. As MinDE require negatively charged membranes for self-organization,65 we printed structures with Ormocomp and silanized them. A previous study on 2D patterned bilayers showed that MinDE waves switch from directional waves on long strips of membranes to more chaotic, spiral patterns when the strip width increased.39 We wanted to observe whether MinDE patterns would respond in a similar fashion in 3D. Therefore, we first designed rod-like structures (Fig. 4B), and increased the width in the mid-section of the rod from 10 to 50 μm, thereby providing larger surface area for the available volume.
 | ||
| Fig. 4 MinDE dynamic self-organisation on 3D structures. (A) Schematic of MinDE molecular mechanism. (B) SEM images of printed structures. (C) Confocal images of dynamic patterns on the structures (from left to right, 10 μm, 20 μm, 50 μm widths), seen from the bottom of the structures. Images from the top and their 4D reconstructions are shown in Fig. S8 and Movie 1 (ESI†). (D) Kymograph of pattern taken along the axis of rods. Scale bar 20 μm horizontal, 4 min vertical. | ||
When we reconstituted the MinDE proteins in vitro, they formed traveling wave patterns on the structures with qualitatively different behaviours. For the 10 μm rod, waves travelled directionally along the axis, assimilating 1D waves. As we increased the width, we found that at 20 μm, the pattern resembled the spiral/chaotic patterns often seen on planar surface assays (Fig. 4C, D and Fig. S8, Movie 1, ESI†). The kymographs taken along the axis of the rods also reveal that the waves travel unidirectionally along the rod for narrow widths; whereas for the 20 μm case, the waves travel outwards from the centre of the spiral. Other wave properties are similar to previously reported experiments: the measured period (1–2 min) and wavelength (∼40 μm) compare well with previous studies on bilayers (period 0.6–2 min, wavelength 50–110 μm).74 Since the width of the 10 μm rod is much smaller than the typical wavelength of the waves, the waves are spatially confined to travel along the long axis. This effect has previously been seen on 2D-patterned membranes,3,21,39 as well as in vivo by moulding bacteria into shapes.75
In order to demonstrate the complexity of membrane morphology that could be obtained with this method, we designed a single spiral, a multi-spiral and a fractal tree that we coated with lipids. We then reconstituted MinDE proteins in vitro to show that the membranes retain functionality even at such complex shapes. To capture the full 4D information resulting from the Min dynamics on the 3D structures, we turned to spinning disk confocal microscopy. The fast frame rates (typically <100 ms, compared to ∼1 s for a confocal laser scanning microscope) allowed us to record 3D Z-stacks (50–60 slices at 2–3 μm intervals) at sufficient temporal resolution (10 s) (Fig. 5, Movie 2, ESI†). On both the single and the multi-spiral structures, the waves travel linearly on each of the rods, similarly to the horizontal rod. The waves on the fractal tree become more chaotic, due to the high density of branches that complexify the bulk-to-surface ratio, but an overall directionality of waves travelling from the central stem to the outside branches can still be visually confirmed.
 | ||
| Fig. 5 Visualization of MinDE dynamic patterns on complex structures (Movie 2, ESI†). (A) SEM images of printed structures. (B) 3D view and (C) Top view of Min patterns on structures. Lipids are visualized in red, and MinD in cyan. The patterns were imaged as a 3D time lapse on a spinning disk confocal microscope, and processed using a custom written Fiji script. Scale bar 100 μm. | ||
With the toolbox developed here, we can now explore much more complex 3D geometries, which will allow us to investigate this behaviour in more detail. For example, MinDE do not only form traveling waves, but exhibits many more patterns such as quasi-stationary patterns76 that so far have only been observed in quasi-infinite plane and bulk volume assays. Recent theoretical analysis of the MinDE system has reported that the pattern formation of MinDE heavily depends on the surface to volume coupling, suggestive of even richer and partly unexplored MinDE pattern formation.77,78 In addition to their role as the spatial indicators of the E. coli midcell, MinDE waves have also been shown to position and transport biologically unrelated membrane-bound molecules by non-specific interactions.67 The directionality of MinDE waves on rods demonstrated here could be exploited to specifically guide MinDE waves and thereby transport arbitrary molecules on membrane surfaces to a desired location. Taken together, this promises fascinating further developments in many directions: the practical toolbox introduced here, theoretical modelling of Min dynamics in three dimensions, as well as the computational tools that allow 4D image analysis,79–81 which are all required to investigate these questions further.
Together with the recent advances in cell-free protein reconstitution in vitro, the 3D printed structures demonstrated here can be used to template biological membranes and significantly expands the toolkit for bottom-up synthetic biology. Although self-assembly and self-organization of functional biomolecules have been shown to partly reproduce subcellular structures, the morphological complexity of cells and organelles can still be much more efficiently addressed by additionally employing cutting-edge microengineering technology. Membranous structures like the endoplasmic reticulum, or the invaginations in the mitochondria are systems where the membrane area per reaction volume is carefully and actively regulated, and such structures can be the target of a systematic investigation. Our work can be considered a starting point for the further 3D engineering of lipid–polymer interactions using multiple lipid types, materials with greater deformability and compositional complexity, and by miniaturisation of printed materials. Biochemical investigations in such biomimetic 3D geometries shed new insights into molecular mechanisms of proteins and open up new applications in bioengineering, with an added dimensionality to the conventional 2D fabrication techniques.
Conclusions
In this work, we have introduced a novel method to fabricate microenvironments for investigations of protein–lipid interactions modulated by geometry. In order to obtain structures at biologically relevant length scales, we employed 2-photon direct laser writing to 3D print micron-sized structures from PETA, TPETA and Ormocomp. We showed that supported lipid membranes can be formed on PETA and Ormocomp, whilst TPETA passivates. By utilising these properties and incorporating them as a composite structure, membranes as well as membrane-associated proteins can be spatially patterned. Engineering surface to volume ratios on the micrometer scale, our method enables fabrication of true 3D geometries to study the dependence of protein self-organization through reaction-diffusion, in particular the MinDE system from E. coli, on membrane morphology, as visualised through 4D image analysis. We recapitulate the striking transition from regular to more turbid wave fronts as soon as membrane dimensions exceed the scale of the wavelength in a particular direction, indicating how tightly this protein system is adapted to a specific spatial setting within rod-like cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Enrico Lemma, Mark Hippler and Martin Bastmeyer (Karlsruhe Institute of Technology, Germany) for their helpful discussions on 2-photon photoresist optimization. We also thank Allen Liu (University of Michigan, USA) as well as Beatrice Ramm (Princeton University, USA) for insightful comments on the manuscript. We also thank the Biochemistry Core Facility of the Max Planck Institute of Biochemistry for assistance with protein purification, and the Imaging Core Facility of the same institution for assistance on the 4D image visualisation. The authors recognize financial support from Max Planck Bristol Centre for Minimal Biology and the Max Planck Society (MPG), as well as support from Center for Nanosciences, Munich.References
- S. Hussain, C. N. Wivagg, P. Szwedziak, F. Wong, K. Schaefer, T. Izoré, L. D. Renner, M. J. Holmes, Y. Sun, A. W. Bisson-Filho, S. Walker, A. Amir, J. Löwe and E. C. Garner, eLife, 2018, 7, 1–45 Search PubMed.
- A. A. Bridges, M. S. Jentzsch, P. W. Oakes, P. Occhipinti and A. S. Gladfelter, J. Cell Biol., 2016, 213, 23–32 CrossRef CAS PubMed.
- K. Zieske and P. Schwille, Angew. Chem., Int. Ed., 2013, 52, 459–462 CrossRef CAS PubMed.
- F. Fanalista, A. Birnie, R. Maan, F. Burla, K. Charles, G. Pawlik, S. Deshpande, G. H. Koenderink, M. Dogterom and C. Dekker, ACS Nano, 2019, 13, 5439–5450 CrossRef CAS PubMed.
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 CrossRef CAS PubMed.
- E. Sezgin, I. Levental, M. Grzybek, G. Schwarzmann, V. Mueller, A. Honigmann, V. N. Belov, C. Eggeling, Ü. Coskun, K. Simons and P. Schwille, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1777–1784 CrossRef CAS PubMed.
- M. Dezi, A. Di Cicco, P. Bassereau and D. Levy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7276–7281 CrossRef CAS PubMed.
- J. Korlach, C. Reichle, T. Müller, T. Schnelle and W. W. Webb, Biophys. J., 2005, 89, 554–562 CrossRef CAS PubMed.
- A. Yamada, S. Lee, P. Bassereau and C. N. Baroud, Soft Matter, 2014, 10, 5878–5885 RSC.
- S. Demarche, K. Sugihara, T. Zambelli, L. Tiefenauer and J. Vörös, Analyst, 2011, 136, 1077–1089 RSC.
- S. Kalsi, A. M. Powl, B. A. Wallace, H. Morgan and M. R. R. De Planque, Biophys. J., 2014, 106, 1650–1659 CrossRef CAS PubMed.
- T. Masters, W. Engl, Z. L. Weng, B. Arasi, N. Gauthier and V. Viasnoff, PLoS One, 2012, 7(8), e44261 CrossRef CAS PubMed.
- R. Watanabe, N. Soga, D. Fujita, K. V. Tabata, L. Yamauchi, S. Hyeon Kim, D. Asanuma, M. Kamiya, Y. Urano, H. Suga and H. Noji, Nat. Commun., 2014, 5, ncomms5519 CrossRef PubMed.
- F. J. Morera, G. Vargas, C. González, E. Rosenmann and R. Latorre, Methods in Membrane Lipids, 2007, vol. 400(1–2), pp. 571–585 Search PubMed.
- K. Zieske, J. Schweizer and P. Schwille, FEBS Lett., 2014, 588, 2545–2549 CrossRef CAS PubMed.
- S. D. Hansen, W. Y. C. Huang, Y. K. Lee, P. Bieling, S. M. Christensen and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15013–15022 CrossRef CAS PubMed.
- J. T. Groves, N. Ulman, P. S. Cremer and S. G. Boxer, Langmuir, 1998, 14, 3347–3350 CrossRef CAS.
- H. Nakashima, K. Furukawa, Y. Kashimura, K. Sumitomo, Y. Shinozaki and K. Torimitsu, Langmuir, 2010, 26, 12716–12721 CrossRef CAS PubMed.
- P. Heo, S. Ramakrishnan, J. Coleman, J. E. Rothman, J. B. Fleury and F. Pincet, Small, 2019, 15, 1–13 CrossRef PubMed.
- H. Eto, N. Soga, H. G. Franquelim, P. Glock, A. Khmelinskaia, L. Kai, M. Heymann, H. Noji and P. Schwille, ACS Appl. Mater. Interfaces, 2019, 11, 21372–21380 CrossRef CAS PubMed.
- Y. Caspi and C. Dekker, eLife, 2016, 5, 1–27 CrossRef PubMed.
- M. G. Guney and G. K. Fedder, J. Micromech. Microeng., 2016, 26(10), 105011 CrossRef.
- M. Kadic, G. W. Milton, M. van Hecke and M. Wegener, Nat. Rev. Phys., 2019, 1, 198–210 CrossRef.
- J. Knoška, L. Adriano, S. Awel, K. R. Beyerlein, O. Yefanov, D. Oberthuer, G. E. Peña Murillo, N. Roth, I. Sarrou, P. Villanueva-Perez, M. O. Wiedorn, F. Wilde, S. Bajt, H. N. Chapman and M. Heymann, Nat. Commun., 2020, 11, 1–12 Search PubMed.
- U. Bozuyuk, O. Yasa, I. C. Yasa, H. Ceylan, S. Kizilel and M. Sitti, ACS Nano, 2018, 12(9), 9617–9625 CrossRef CAS PubMed.
- M. Hippler, E. Blasco, J. Qu, M. Tanaka, M. Wegener and M. Bastmeyer, Nat. Commun., 2019, 1–8 CAS.
- C. de Marco, C. C. J. Alcântara, S. Kim, F. Briatico, A. Kadioglu, G. de Bernardis, X. Chen, C. Marano, B. J. Nelson and S. Pané, Adv. Mater. Technol., 2019, 4, 1–7 Search PubMed.
- F. Klein, T. Striebel, J. Fischer, Z. Jiang, C. M. Franz, G. Von Freymann, M. Wegener and M. Bastmeyer, Adv. Mater., 2010, 22, 868–871 CrossRef CAS PubMed.
- B. Richter, T. Pauloehrl, J. Kaschke, D. Fichtner, J. Fischer, A. M. Greiner, D. Wedlich, M. Wegener, G. Delaittre, C. Barner-Kowollik and M. Bastmeyer, Adv. Mater., 2013, 25, 6117–6122 CrossRef CAS PubMed.
- M. Hippler, E. D. Lemma, S. Bertels, E. Blasco, C. Barner-Kowollik, M. Wegener and M. Bastmeyer, Adv. Mater., 2019, 31(26), 1808110 CrossRef PubMed.
- C. Barner-Kowollik, M. Bastmeyer, E. Blasco, G. Delaittre, P. Müller, B. Richter and M. Wegener, Angew. Chem., Int. Ed., 2017, 56, 15828–15845 CrossRef CAS PubMed.
- B. Richter, V. Hahn, S. Bertels, T. K. Claus, M. Wegener, G. Delaittre, C. Barner-Kowollik and M. Bastmeyer, Adv. Mater., 2017, 29(5), 1604342 CrossRef PubMed.
- H. Ceylan, I. C. Yasa and M. Sitti, Adv. Mater., 2017, 29, 1605072 CrossRef PubMed.
- F. Klein, B. Richter, T. Striebel, C. M. Franz, G. Von Freymann, M. Wegener and M. Bastmeyer, Adv. Mater., 2011, 23, 1341–1345 CrossRef CAS PubMed.
- O. Okay, General Properties of Hydrogels, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 6 Search PubMed.
- C. Schizas and D. Karalekas, J. Mech. Behav. Biomed. Mater., 2011, 4, 99–106 CrossRef CAS PubMed.
- M. Hippler, K. Weißenbruch, K. Richler, E. D. Lemma, M. Nakahata, B. Richter, C. Barner-kowollik, Y. Takashima, A. Harada, E. Blasco, M. Wegener, M. Tanaka and M. Bastmeyer.
- F. R. Flohr, M. Shtil and R. Müller, Surface Functionalization Strategy for Bone Biomimetic Osteocyte Models, Eur. Cells Mater., 2016, 32(5), 76 Search PubMed.
- J. Schweizer, M. Loose, M. Bonny, K. Kruse, I. Monch and P. Schwille, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15283–15288 CrossRef CAS PubMed.
- M. I. Angelova and D. S. Dimitrov, Faraday Discuss. Chem. Soc., 1986, 81, 303–311 RSC.
- A. J. García-Sáez, D. C. Carrer and P. Schwille, in Liposomes: Methods and Protocols: Biological Membrane Models, ed. V. Weissig, Humana Press, Totowa, NJ, 2010, Volume 2, pp. 493–508 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- D. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef CAS.
- S. K. Vogel, 2016, pp. 213–223.
- S. K. Vogel, F. Heinemann, G. Chwastek and P. Schwille, Cytoskeleton, 2013, 70, 706–717 CrossRef CAS PubMed.
- B. Ramm, P. Glock and P. Schwille, J. Vis. Exp., 2018, 137, e58139 Search PubMed.
- D. Serien and S. Takeuchi, Appl. Phys. Lett., 2015, 107, 10–15 CrossRef.
- E. Harnisch, M. Russew, J. Klein, N. König, H. Crailsheim and R. Schmitt, Opt. Mater. Express, 2015, 5, 456 CrossRef.
- A. Poursoroush, M. M. Sperotto and M. Laradji, J. Chem. Phys., 2017, 146, 1–10 CrossRef PubMed.
- S. Moradi, A. Nowroozi and M. Shahlaei, RSC Adv., 2019, 9, 4644–4658 RSC.
- K. Göpfrich, B. Haller, O. Staufer, Y. Dreher, U. Mersdorf, I. Platzman and J. P. Spatz, ACS Synth. Biol., 2019, 8, 937–947 CrossRef PubMed.
- B. Seantier and B. Kasemo, Langmuir, 2009, 25, 5767–5772 CrossRef CAS PubMed.
- M. Zhu, M. Lerum and W. Chen, Langmuir, 2012, 28, 416–423 CrossRef CAS PubMed.
- S. Aura, V. Jokinen, M. Laitinen, T. Sajavaara and S. Franssila, J. Micromech. Microeng., 2011, 21(12), 125003 CrossRef.
- C. L. Lay, Y. H. Lee, M. R. Lee, I. Y. Phang and X. Y. Ling, ACS Appl. Mater. Interfaces, 2016, 8, 8145–8153 CrossRef CAS PubMed.
- H. Jia, T. Litschel, M. Heymann, H. Eto, H. G. Franquelim and P. Schwille, Small, 2020, 1906259, 1–10 Search PubMed.
- D. Serien and S. Takeuchi, ACS Biomater. Sci. Eng., 2017, 3, 487–494 CrossRef CAS PubMed.
- A. Nakahara, Y. Shirasaki, K. Kawai, O. Ohara, J. Mizuno and S. Shoji, Microelectron. Eng., 2011, 88, 1817–1820 CrossRef CAS.
- C. Niederauer, P. Blumhardt, J. Mücksch, M. Heymann, A. Lambacher and P. Schwille, Opt. Express, 2018, 26, 20492 CrossRef CAS PubMed.
- S. Chiantia, J. Ries, N. Kahya and P. Schwille, ChemPhysChem, 2006, 7, 2409–2418 CrossRef CAS PubMed.
- L. K. Tamm and H. M. McConnell, Biophys. J., 1985, 47, 105–113 CrossRef CAS PubMed.
- A. Sonnleitner, G. J. Schuetz and T. Schmidt, Biophys. J., 1999, 77, 2638–2642 CrossRef CAS PubMed.
- M. Loose, E. Fischer-Friedrich, J. Ries, K. Kruse and P. Schwille, Science, 2008, 320, 789–792 CrossRef CAS PubMed.
- S. J. Attwood, Y. Choi and Z. Leonenko, Int. J. Mol. Sci., 2013, 14, 3514–3539 CrossRef CAS PubMed.
- A. G. Vecchiarelli, M. Li, M. Mizuuchi and K. Mizuuchi, Mol. Microbiol., 2014, 93, 453–463 CrossRef CAS PubMed.
- K. Zieske and P. Schwille, eLife, 2014, 3, 1–19 CrossRef PubMed.
- B. Ramm, P. Glock, J. Mücksch, P. Blumhardt, D. A. García-Soriano, M. Heymann and P. Schwille, Nat. Commun., 2018, 9(1), 3942 CrossRef PubMed.
- E. K. Sinner and W. Knoll, Curr. Opin. Chem. Biol., 2001, 5, 705–711 CrossRef CAS PubMed.
- J. Andersson and I. Köper, Membranes, 2016, 6(2), 30 CrossRef PubMed.
- H. Ceylan, I. C. Yasa and M. Sitti, Adv. Mater., 2017, 29(9), 1–7 CrossRef PubMed.
- A. Laliberte and C. Gicquaud, J. Cell Biol., 1988, 106, 1221–1227 CrossRef CAS PubMed.
- C. F. E. Schroer, L. Baldauf, L. van Buren, T. A. Wassenaar, M. N. Melo, G. H. Koenderink and S. J. Marrink, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5861–5872 CrossRef CAS PubMed.
- D. M. Raskin and P. A. J. de Boer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4971–4976 CrossRef CAS PubMed.
- B. Ramm, T. Heermann and P. Schwille, Cell. Mol. Life Sci., 2019, 76, 4245–4273 CrossRef CAS PubMed.
- F. Wu, B. G. C. Van Schie, J. E. Keymer and C. Dekker, Nat. Nanotechnol., 2015, 10(8), 719–726 CrossRef CAS PubMed.
- P. Glock, B. Ramm, T. Heermann, S. Kretschmer, J. Schweizer, J. Mu and P. Schwille, ACS Synth. Biol., 2019, 8(1), 148–157 CrossRef CAS PubMed.
- L. Wettmann, M. Bonny and K. Kruse, PLoS One, 2018, 13, 1–20 CrossRef PubMed.
- F. Brauns, G. Pawlik, J. Halatek, J. W. J. Kerssemakers, E. Frey and C. Dekker, bioRxiv, 2020, 2020.03.01.971952.
- H. Peng, Z. Ruan, F. Long, J. H. Simpson and E. W. Myers, Nat. Biotechnol., 2010, 28, 348–353 CrossRef CAS PubMed.
- S. Daetwyler, U. Gunther, C. D. Modes, K. Harrington and J. Huisken, Dev., 2019, 146(6) DOI:10.1242/dev.173757.
- A. Spark, A. Kitching, D. Esteban-Ferrer, A. Handa, A. R. Carr, L. M. Needham, A. Ponjavic, A. M. Santos, J. McColl, C. Leterrier, S. J. Davis, R. Henriques and S. F. Lee, Nat. Methods, 2020, 17, 1097–1099 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm00112d |
| This journal is © The Royal Society of Chemistry 2021 |