
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure evolution during phase separation in spin-coated ethylcellulose/hydroxypropylcellulose films
Pierre
Carmona
 *ab,
Magnus
Röding
ac,
Aila
Särkkä
c,
Christian
von Corswant
d,
Eva
Olsson
b and
Niklas
Lorén
*ab
*ab,
Magnus
Röding
ac,
Aila
Särkkä
c,
Christian
von Corswant
d,
Eva
Olsson
b and
Niklas
Lorén
*ab
aUnit Product Design, Department Agriculture and Food, Division Bioeconomy and Health, RISE Research Institute of Sweden, Gothenburg, Sweden. E-mail: pierre.carmona@ri.se; niklas.loren@ri.se
bDivision Nano-and BioPhysics, Department of Physics, Chalmers University of Technology, Gothenburg, Sweden
cDepartment of Mathematical Sciences, Chalmers University of Technology and Gothenburg University Gothenburg, Gothenburg, Sweden
dOral Product Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Gothenburg, Sweden
First published on 12th March 2021
Abstract
Porous phase-separated films made of ethylcellulose (EC) and hydroxypropylcellulose (HPC) are commonly used for controlled drug release. The structure of these thin films is controlling the drug transport from the core to the surrounding liquids in the stomach or intestine. However, detailed understanding of the time evolution of these porous structures as they are formed remains elusive. In this work, spin-coating, a widely applied technique for making thin uniform polymer films, was used to mimic the industrial manufacturing process. The focus of this work was on understanding the structure evolution of phase-separated spin-coated EC/HPC films. The structure evolution was determined using confocal laser scanning microscopy (CLSM) and image analysis. In particular, we determined the influence of spin-coating parameters and EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HPC ratio on the final phase-separated structure and the film thickness. The film thickness was determined by profilometry and it influences the ethanol solvent evaporation rate and thereby the phase separation kinetics. The spin speed was varied between 1000 and 10000 rpm and the ratio of EC:HPC in the polymer blend was varied between 78:22 wt% and 40:60 wt%. The obtained CLSM micrographs showed phase separated structures, typical for the spinodal decomposition phase separation mechanism. By using confocal laser scanning microscopy combined with Fourier image analysis, we could extract the characteristic length scale of the phase-separated final structure. Varying spin speed and EC:HPC ratio gave us precise control over the characteristic length scale and the thickness of the film. The results showed that the characteristic length scale increases with decreasing spin speed and with increasing HPC ratio. The thickness of the spin-coated film decreases with increasing spin speed. It was found that the relation between film thickness and spin speed followed the Meyerhofer equation with an exponent close to 0.5. Furthermore, good correlations between thickness and spin speed were found for the compositions 22 wt% HPC, 30 wt% HPC and 45 wt% HPC. These findings give a good basis for understanding the mechanisms responsible for the morphology development and increase the possibilities to tailor thin EC/HPC film structures.
:HPC ratio on the final phase-separated structure and the film thickness. The film thickness was determined by profilometry and it influences the ethanol solvent evaporation rate and thereby the phase separation kinetics. The spin speed was varied between 1000 and 10000 rpm and the ratio of EC:HPC in the polymer blend was varied between 78:22 wt% and 40:60 wt%. The obtained CLSM micrographs showed phase separated structures, typical for the spinodal decomposition phase separation mechanism. By using confocal laser scanning microscopy combined with Fourier image analysis, we could extract the characteristic length scale of the phase-separated final structure. Varying spin speed and EC:HPC ratio gave us precise control over the characteristic length scale and the thickness of the film. The results showed that the characteristic length scale increases with decreasing spin speed and with increasing HPC ratio. The thickness of the spin-coated film decreases with increasing spin speed. It was found that the relation between film thickness and spin speed followed the Meyerhofer equation with an exponent close to 0.5. Furthermore, good correlations between thickness and spin speed were found for the compositions 22 wt% HPC, 30 wt% HPC and 45 wt% HPC. These findings give a good basis for understanding the mechanisms responsible for the morphology development and increase the possibilities to tailor thin EC/HPC film structures.
1 Introduction
For decades, solid oral dosage forms have been the most popular way of drug administration.1 One way to make such forms is to produce a capsule that contains many small pellets. The microstructure of the pellet coating controls the mass transport of the drug from the core to the surrounding liquid in the stomach or intestine. By controlling the manufacturing and formulation, and therefore the coating structure, the drug release rate can be tailored.2 Coatings made of phase-separated films are known to be very effective for controlling mass transport,3 but the kinetics and mechanisms of the formation of the film structure are not completely understood.The use of the two cellulose derivatives ethyl cellulose (EC, water-insoluble) and hydroxypropyl cellulose (HPC, water-soluble) is well established in the pharmaceutical industry.4 Mixed with a common solvent e.g. ethanol, EC and HPC form a one-phase polymer blend at low polymer concentration. Then, as the solvent evaporates, the mixture becomes incompatible and starts to phase separate. Under certain process conditions and ratios of EC and HPC, a phase separated structure is formed. Due to the gradual viscosity increase as the solvent evaporates, the phase-separated structure eventually becomes kinetically trapped. When the dried film comes in contact with water, the HPC dissolves and leaches out, leaving a porous network through which the drug can diffuse. The different steps in the film formation and the drug release are illustrated in Fig. 1. The 3D structure of the porous network dictates the drug transport rate.5,6 By controlling the kinetics of the phase separation and the film structure, it is possible to tailor the drug transport rate.
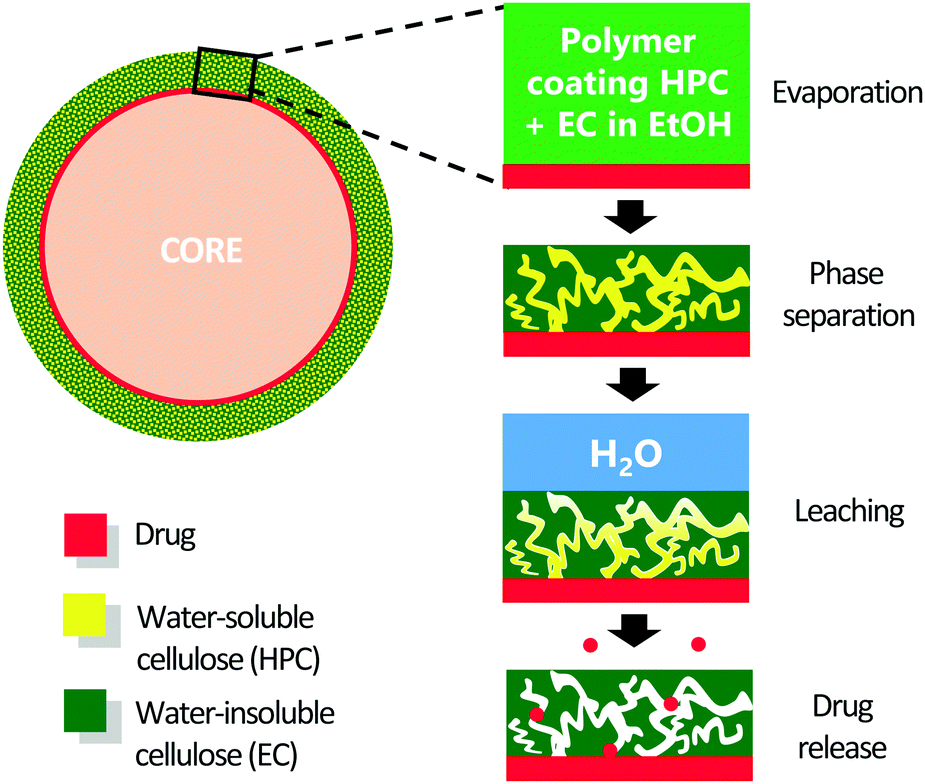 | ||
| Fig. 1 Formation of a porous film made of EC and HPC at the surface of a pharmaceutical pellet used for drug delivery. From top to bottom the different steps: ethanol evaporation, phase separation and kinetic trapping, HPC leaching, and drug release. | ||
The industrial coating of drug-containing pellets with thin phase-separated films is often made batch-wise in a fluidized bed. One challenge in this process is to relate the final properties of the polymer coating to the parameters of the industrial production of the pellets. During spraying, small droplets containing EC, HPC, and ethanol hit the pellet and form a coating. At the pellet surface, the new droplets are mixed with the existing coating. During manufacturing, the pellets are passing through the spraying zone in the fluidized bed several times. Thus, the coating layer is built up during a sequence of passages. Many mechanisms are involved in the formation of the phase-separated films on the pellets, and they are competing in a complex and time-dependent manner.7 Examples of involved mechanisms are phase separation, kinetic trapping, solvent evaporation, rewetting, and mixing.
It is challenging to follow the multilayer formation and the phase separation occuring in a fluidized bed. One technique with which phase separation during solvent evaporation in a thin film is much easier to follow and understand is spin-coating. Therefore, spin-coating is selected in this work to mimic the structure formation occuring in a fluidized bed. Spin-coating is a broadly used technique for producing uniform, thin polymer films.8–10 There has been an increasing interest in this technique due to the possibility of obtaining a broad range of different morphologies and hence the ability to tailor the structure.9 The spin-coated, phase-separated structure depends on the transition from a one-phase state of two immiscible polymers in a common solvent to a two-phase state during the evaporation of the solvent.11 It has been demonstrated experimentally that the final morphology of the phase-separated film depends on a wide range of factors including properties of the polymers (molecular weight,12 solubility,13 surface tension, and viscosity) of the blended solution (solid content,14 solvent properties,13 blend ratio), and properties of the substrate (chemistry,13 roughness, size and shape). Along with the polymer and substrate features, the final morphology also depends on the spin-coating parameters such as spin speed,15 acceleration, spin time, and the surrounding atmosphere.16 Toolan et al.17 recently developed an in situ method for studying the phase separation of PS/PMMA systems (polystyrene/poly(methyl-methacrylate)) synchronizing stroboscopic LED illumination with a highly sensitive electron-multiplying charge coupled device camera (EMCCD) connected to an optical microscope. They proposed a two-step mechanism for the phase separation during spin-coating: a hydrodynamic thinning (flow dominated step) followed by a solvent thinning (solvent evaporation dominated) step. They determined the influence of the spin-coating parameters on the final morphology of the film. Furthermore, a relationship between characteristic length scale and thickness has been demonstrated for thin films made of synthetic polymers.9,18–20 However, information about this relationship in mixed biopolymer systems such as mixtures of EC and HPC is lacking.
To study and characterize the phase-separated structure of thin polymer films, different techniques have been used, including atomic force microscopy (AFM),13,21–23 electron microscopy,9 grazing incidence ultrasmall-angle scattering (GIUSAXS),21 X-ray scattering,24 and small angle light scattering.10,17 Image analysis, especifically the 2D fast Fourier transform (2D-FFT) has been widely used to characterize the structure of polymer and metal systems at the microscopic scale and in particular to determine the characteristic length scale of phase-separated structures.5,8,9,25–29 The 3D structure of EC/HPC films has been characterized in high resolution using focused ion beam scanning electron microscopy (FIB-SEM).30–32 Porosity, pore size distribution, pore shape and connectivity have been determined from the FIB-SEM data using 3D image analysis.32 Furthermore, confocal laser scanning microscopy (CLSM) has been used to characterize EC/HPC film structure at the micrometer level.6
EC and HPC can be dissolved in ethanol. During ethanol evaporation, the polymer blend concentration increases, and the mixture moves into the incompatible region of the phase diagram and starts to phase separate. The phase separation results in an enrichment of the polymers in two separate phases, called segregative phase separation.33 When the evaporation of ethanol is complete, a kinetically trapped, phase-separated film of EC/HPC remains. The mixture undergoes phase separation either through the spinodal decomposition mechanism34,35 or the nucleation and growth mechanism.36,37 Depending on the composition (ratio EC:HPC), the phase volumes, and the shape of the phase diagram, different morphologies are obtained. There are three main cases: (i) the mixture phase separates through nucleation and growth resulting in a discontinuous structure with inclusions having different sizes that appear at different times; (ii) a discontinuous structure is formed by spinodal decomposition and the inclusions have similar size and appear at the same time; (iii) a bicontinuous structure is formed by spinodal decomposition.38 For temperature-induced phase separation, the characteristic length scale and the formed morphologies are mainly determined by the coarsening kinetics and the composition (phase volumes).39,40 In the case of solvent-quenched systems, several studies have shown that faster evaporation lead to smaller features in the final dried structures.9,13,41 The characteristic length scale of the phase separated structure not only depends on the coarsening time but also on the time-dependent depth of the solvent quench.40,42 During solvent evaporation, the depth of the solvent quench changes continuously, which will influence the characteristic length scale. Previous works have shown that the structure of EC and HPC in the film and its properties depend on numerous parameters such as the ratio EC:HPC, the molecular weight distribution of EC and HPC,43,44 and the processing parameters (temperature, gas flow, spray rate).45 Differences were observed in the microstructure, in water and solute permeability, in drug release rate and release rate of HPC. It was found that a higher molecular weight of HPC results in a lower permeability.46 Marucci et al.45 studied permeability and leaching of EC/HPC films with different compositions. They discovered the presence of a percolation onset at about 22 wt% HPC. When the HPC ratio exceeded 22 wt%, both the amount of HPC leached out and the water permeability of the films increased linearly with the HPC ratio up to a certain concentration of HPC.3,43,45,47,48
Several studies have been conducted on the phase separation of EC/HPC systems.44–46,49 In this work, we studied the spin-coated EC/HPC thin film with CLSM, Fourier image analysis, and profilometry. To the best of our knowledge this combination has never been used to study the EC/HPC fim structure. CLSM and Fourier image analysis have previously been used to evaluate structure evolution of mixed biopolymer systems of whey protein isolate and gellan gum.50 CLSM was chosen because it facilitates 3D imaging of the structure and provides for differentiating EC and HPC using selective fluorescence labelling. In addition, CLSM is a non-invasive technique which does not need a long and difficult sample preparation and enables characterization of the bulk structure inside a material. CLSM was used together with Fourier image analysis to quantify the changes in characteristic length scale as a function of the composition of the polymer blend and the spin speed.
The overall purpose of this work was to understand the mechanisms controlling the structure evolution during phase separation within a thin film formed by solvent evaporation. Here, spin-coating has been used to mimic the industrial process that involves deposition and rapid drying of a thin layer of EC/HPC on a pellet surface. Spin-coating was used in this article to produce thin polymer films with a well-controlled final phase-separated structure of the films. This opens up for tailoring the final structure and hence the drug release. New information regarding the influence of composition and spin-coating parameters on the final phase-separated structure of thin EC/HPC films is provided in this work.
This paper is structured as follows (i) the dependence of the characteristic length scale of the phase-separated structure with spin speed and composition is shown, (ii) these results are correlated to rheology and profilometry measurements, and (iii) the validity of the established spin curves and the relationship between characteristic length scale and thickness are discussed.
2 Material and methods
2.1 Solution preparation
Solutions with 4 different compositions of HPC (Klucel Pharm HPC, grade LF, Ashland Inc, Covington, Kentucky, USA) with a mean molecular weight of 95000 Daltons51 and EC (Ethocel Standard Premium, viscosity 10 cps, Dow Cellulosics, Dow Chemical Company, Midland, Michigan, USA) with a mean molecular weight of 30000 Daltons52 were prepared. For the microscopic analysis, 6 wt% of polymer blend was mixed in a solution of 2 mM Na-fluorescein (CAS. 518-47-8, Sigma Aldrich, St. Louis, Missouri, USA) in ethanol (CAS. 64-17-5 AnalR NORMAPUR® 96%, VWR Chemical, Radnor, Pennsylvania, USA). The EC/HPC polymer ratios studied were 22, 30, 45 and 60 wt% of HPC in the polymer blend. The compositions were chosen according to previous studies performed on the same system.45,47 Fluorescein was chosen as a fluorescent tracer because it exhibits a strong affinity to HPC.43
For the viscosity measurements, 16 solutions were prepared with the following compositions: 22, 30, 45, and 60 wt% HPC and 4 ratios of solvent: 94, 90, 85 and 80 wt% of ethanol (96%). Ethanol was added to the polymer blend and the solutions were stirred overnight.
2.2 Sample preparation by spin-coating
A spin-coater (WS-650MZ-23NPP, Laurell Technologies, North Wales, Pennsylvania, USA) was used to spin-coat the EC/HPC solutions. The volume of solution was optimized and fixed at 250 μL. Fig. 2 shows the different steps of the spin-coating process. The polymer solution was deposed on the surface of a 25 × 75 × 1 mm3 glass microscope slide (SuperFrost Ground 90°, Thermo Scientific, Waltham, Massachusetts, USA). The microscopic observation was performed in the rotational center area of the microscope glass slide. The spin coater bowl was equipped with a nitrogen purge which ensures a nitrogen environment while coating. The temperature was held constant at 23 °C in the spin-coating chamber. The drying takes place during spin-coating. The spin-coating parameters were optimized, and it was found that the structure evolution was kinetically trapped after about 15 s at 1000 rpm. Therefore, the spin time was fixed at 45 s to ensure that the film was totally dried after spinning. The acceleration ratio, i.e. the ratio between the acceleration speed and the spin speed, was fixed at 1:1 (e.g. for a spin speed of 3000 rpm, the acceleration was 3000 rpm per s). The morphologies of the films for 1000, 2000, 3000, 4000, 5000, 6000, 8000 and 10000 rpm were studied. For each parameter combination, the experiment was performed in triplicate.
 | ||
| Fig. 2 Illustration of the spin-coating process. (a) Deposition of the polymer solution. (b) Spinning, dominated by flow. (c) Spinning dominated by solvent evaporation. (d) The film is dried, and the phase separation has occurred. | ||
2.3 Thickness measurement by profilometry
To determine the thickness of the spin-coated film, a stylus profilometer (Alpha-Step D-100 KLA Tencor, Milpitas, CA, USA) was utilized. Stylus profilometers use a contact probe to detect the surface. The diamond stylus is physically moving along the surface in order to measure the surface height. Prior to the measurement, the film surface was scratched with a razor blade. The stylus slides across the film and uses the difference in height from the film surface to the glass surface (inside of a scratch) to determine the film thickness with a precision of 1 nm. For each experiment, measurements on 6 areas of each microscope slide were performed in duplicate. The measurements were performed with stylus speed 0.03 mm s−1, stylus force 0.03 N, and using averaging of 16 lines per passage.2.4 Confocal scanning laser microscopy
The structure of the spin-coated thin film was determined at the micrometer level using CLSM (Leica TCS SP5, Leica, Wetzlar, Germany). A Leica 100×/1.4 PL APO oil objective was used for the characterization. A 488 nm argon laser was used for imaging. The signal emitted in the interval 500–600 nm was recorded (with a peak expected at 515 nm for maximum fluorescein emission). The lateral resolution at full-width half-maximum (FWHM) was about 130 nm and the axial resolution (FWHM) was about 330 nm. Digital zooms between 1× and 12× (the field of view (FOV) was 155 and 13 μm, respectively) were used, depending on the size of the structure observed. The images were recorded with 1024 × 1024 pixels with a scanning rate of 400 Hz and 6-line averaging. In the micrographs, the bright phase corresponds to the HPC phase and the dark phase to the EC phase. On each slide, micrographs at 4 spots with different distances with respect to the center of the slide were taken. The procedure was repeated in triplicate.2.5 Image analysis using 2D fast Fourier transform
It was found that the EC/HPC system in this work phase-separated through spinodal decomposition.46 During spinodal decomposition and solvent evaporation, a typical periodicity or characteristic length scale is unique for each structure and describes the stage of phase separation. The structure evolution during spinodal decomposition and solvent evaporation has been modelled and simulated using a combination of Flory-Huggins and Cahn–Hilliard–Cock theories.53 We study the characteristic length scale L(t) to determine the influence of spin speed and polymer blend composition on the dried film morphology. In order to extract information from the micrographs, we perform image analysis using the two-dimensional fast Fourier transform (2D-FFT). We perform image analysis in the following fashion, the steps of which are also illustrated in Fig. 3. Fig. 3a shows a CLSM micrograph of a phase separated EC/HPC film with EC as the dark phase and HPC as the bright phase. First, a background subtraction was performed by smoothing the original image with a 2D Gaussian smoothing kernel with high standard deviation (σ = 100) and subtracting the smoothed image from the original. Second, the resulting image was smoothed with another Gaussian filter (σ = 2) for noise reduction (see Fig. 3a). Third, the Fourier transform of the image was computed. Assuming that the image grayscale intensity in pixel (r1,r2) is f(r1,r2), the discrete, two-dimensional Fourier transform of an image with N × N pixels (here N = 1024) is computed by
 | ||
| Fig. 3 Determination of the characteristic length scale with 2D Fourier Transform. (a) CLSM micrograph of HPC 45 wt% spin-coated for 45 s at 3000 rpm, the dark phase is EC and the bright phase is HPC. (b) Power spectrum and representation of the radius. (c) Radial distribution: the wavenumber value at the peak corresponds to L(t) = 1.7 μm. | ||
Fourth, the so-called power spectrum or Fourier space spectrum P(k1,k2) is calculated from the Fourier transform by
| P(k1,k2) = |F(k1,k2)|2 |
Finally, an estimate of the characteristic length scale L(t) was obtained as

2.6 Viscosity measurements
To determine the zero-shear viscosity of the stock solutions (6 wt% polymer blends in 94 wt% of ethanol) during evaporation, experiments on different EC/HPC mixtures were conducted on a rotational rheometer (ARES-G2, Texas Instruments, Dallas, Texas, USA). Four compositions, EC:HPC 78:22; 70:30; 55:45; 40:60 wt%, and 4 ratios of solvent, 94, 90, 85 and 80 wt% of ethanol, were studied. A logarithmic flow sweep was performed in triplicate with a bob and cup geometry: 30 mm diameter cup and 27.7 diameter bob. The system was closed to prevent evaporation. After pouring 25 mL of solution in the cup, the sample was conditioned at 23 °C with a soak time of 30 s, a pre-shear of 20 s at 0.1 s−1 and an equilibration time of 10 s. The logarithmic flow sweep was performed in triplicate with a shear rate from 0.01 to 100 s−1 with 10 points/decade. A similar approach was used to determine the viscosity of EC/HPC solutions by Andersson et al..44 The zero-shear viscosity was extrapolated from the rheology data.54 The specific viscosity at zero shear rate was calculated through the equation:55
3 Result and discussion
3.1 The effect of spin speed on the phase-separated structure
One of our aims was to determine the effect of the spin-coating parameters on the phase-separated structure. The spin time and the acceleration ratio were fixed, and the effect of spin speed on the final film structure was investigated. Fig. 4a shows CLSM micrographs of spin-coated films at different spin speeds: 1000, 2000, 5000 and 8000 rpm at magnification 500× (FOV 31 μm) for the composition 30 wt% HPC. We reiterate that the bright phase corresponds to HPC phase and the dark phase to EC phase. The micrographs as shown in Fig. 4a were taken in the centre of the microscope slide, corresponding to the centre of rotation during spinning. Fig. 4b shows the characteristic length scale obtained with Fourier image analysis versus the spin speed. | ||
| Fig. 4 (a) CLSM micrographs of spin-coated thin films. The HPC phase is bright and the EC phase is dark. (b) Influence of spin speed on average characteristic length scale L(t) of the phase-separated structure. The error bars represent the standard deviation. For reading purposes, the data for HPC 22wt% and HPC 45wt% were intentionally shifted on the x-axis with −100 rpm and +100 rpm, respectively. | ||
In Fig. 4a, all CLSM micrographs show structures typical for spinodal decomposition. The domain size of the phase-separated pattern is decreasing with increasing spin speed for the composition 30 wt% HPC. In Fig. 4b, for the three compositions 22, 30 and 45 wt% HPC, the characteristic length scale of the phase-separated structure decreases with increasing spin speed. For example, for HPC 30 wt%, L(t)1000 = 2.5 μm and L(t)8000 = 1.1 μm. This suggests that the phase separation (spinodal decomposition followed by coarsening) is stopped in an earlier stage of coarsening at higher spin speed. One possible explanation is that at higher spin speed the evaporation is faster, meaning that a deeper solvent quench depth is reached faster. The deeper solvent quench depth will influence the characteristic length scale and the time for coarsening. The experimental observation is supported by Birnie et al.56 who determined the ethanol evaporation rate for spin speeds in the range 500–3000 rpm. They found that the ethanol evaporation rate varies with the square root of the spin speed.
For all investigated spin speeds above 1000 rpm, the structures are remarkably homogenous in size and shape throughout the microscope slide. At 1000 rpm, the structure variability is noticeably larger as displayed by the larger error bars, indicating an inhomogeneity induced by the spin-coating process. Above 1000 rpm, it is likely that the spin speed is high enough to obtain homogeneous deposition over the substrate, so that solvent evaporation takes place in the same way all over the sample. Approximately at 4000 rpm and above, the characteristic length scale reaches a plateau at L(t) ∼ 1.1 μm. Thus, increasing the spin speed above 4000 rpm has little effect on the final morphology for 22, 30 and 45 wt% HPC.
3.2 Relation between thickness and spin speed
Our purpose here was to determine if there was a correlation between thickness and spin speed. In Fig. 5, spin curves are plotted for all compositions in the polymer blend, i.e. the average thickness of the films h is plotted against the spin speed ω. The spin curves were fitted with a power law and the values of the exponent n were determined. | ||
| Fig. 5 Spin curves: average thickness plotted versus spin speed for all composition EC/HPC. R2 is the coefficient of determination (i.e. goodness of fit) of the fitted power law regression and the error bars represent the standard deviation. | ||
Fig. 5 shows that the phase-separated polymer film is thinner when the spin speed is higher, e.g. hHPC30(1000 rpm) = 1.5 μm and hHPC30(8000 rpm) = 0.5 μm. This can be explained by the fact that the increase of centrifugal forces leads to more material being spread out in all directions and hence to a thinner film. There is also a small tendency that the films become thicker when increasing the HPC ratio. A possible explanation could be that the viscosity increases with increasing HPC ratio resulting in a thicker film (see section 3.3 for further elaboration).
Meyerhofer developed a mathematical model for spin-coated films that relates the film thickness to spin speed, initial viscosity and evaporation rate.57 In particular, it was shown that the thickness h decreases with increasing spin speed ω according to the relation h ∝ kω−0.5, with a constant k that depends on the viscosity, density and concentration of the solution. The derivation of the model is built upon a two-step thinning during spin-coating. Meyerhofer proposed that during the first step, the thinning is dominated by the fluid being spun off (outflow) and that during the second step, the thinning is dominated by evaporation (see Fig. 2). At the transition point, the outflow and evaporation contributions are equal, and the thickness can be estimated analytically. This two-step thinning during spin-coating was used to explain the structure of the phase-separated polymer system.10,11,16,23 In addition, the Meyerhofer model also assumes that the evaporation rate is uniform, the thickness is constant throughout the film, and that changes of concentration in the z direction (through the film) can be neglected.
In this work, the obtained exponents are −0.54, −0.55, −0.55, and −0.51 for 22, 30, 45, and 60 wt% HPC, respectively. The fits in Fig. 5 show that the assumptions made by Meyerhofer leading to the relation h ∝ kω−0.5 are consistent with the experimental data obtained on spin-coated EC/HPC mixtures. For example, it was observed that the thickness is constant throughout the film and that the structure is homogenous in the film in the z direction. The Meyerhofer model provides an understanding of the thickness variations when changing spin-coating process parameters and that it is likely that a two-step thinning mechanism, hydrodynamic thinning followed by evaporation thinning, is active during the spin-coating of the EC/HPC films.
3.3 Viscosity of EC/HPC solutions
Rheology experiments were conducted to determine the viscosity of the polymer solutions prior to spinning. The measured samples contained 22, 30, 45 and 60 wt% HPC. Furthermore, solutions with different decreasing solvent content were investigated to evaluate the increase of viscosity during ethanol evaporation because this will influence coarsening rate and kinetic trapping of the phase separated structure. Fig. 6a shows the zero-shear viscosity versus the ratio of HPC in the polymer blend at 6 wt% total polymer concentration in the stock solutions for the four compositions (22, 30, 45, 60 wt% HPC). The value for the viscosity at zero-shear rate was approximated from the viscosity at the Newtonian plateau.54,58Fig. 6b shows the zero-shear specific viscosity versus the polymer concentration. The choice of the specific viscosity in this plot was made in order to correlate the slope with previous values in the literature. | ||
| Fig. 6 (a) Zero-shear viscosity versus the HPC ratio in the polymer blend. The solutions were the stock solution with 6 wt% polymer in 94 wt% ethanol. The error bars represent the standard deviation. (b) Zero-shear specific viscosity as a function of polymer concentration. | ||
In Fig. 6a, the viscosity of EC/HPC solutions increases linearly with the increase of the HPC ratio in the polymer blend. This result can be explained by the average molecular weight values of HPC and EC used. The molecular weight of HPC is considerably higher than the molecular weight of EC (see the Material and Methods section). The entanglements between the polymer chains are higher when increasing the HPC ratio, resulting in an increase of viscosity.55 This explains why solutions with higher HPC content exhibit higher viscosity. Consequently, and due to slower spinning off at high viscosity, the films obtained by spin-coating are thicker with higher amount of HPC (Fig. 5).
Results in Fig. 6b show that the viscosity is increasing significantly with increased evaporation of ethanol in the solution (i.e. increasing polymer concentration). The logarithmic plot of the zero-shear specific viscosity versus the polymer concentration was fitted with a power law. The determined exponents were 4.4 and 4.6 for 30 and 45 wt% HPC, respectively. Bercea et al.59 studied the viscosity of HPC solution from the same supplier (Klucel) with molecular weights in the range 80–1050 kDa. They plotted the specific viscosity as a function of c[η], where c is the polymer concentration and [η] is the intrinsic viscosity calculated from the Mark–Houwink dependence. In the entangled regime,55 Bercea et al.59 found a slope of 4. By estimating the intrinsic viscosity of our system with the HPC intrinsic viscosity reported for HPC in aqueous solutions at 25 °C,60c[η] was calculated and the specific viscosity plotted versus c[η]. The slope found exhibits nearly the same exponents: 4.4 for 30 wt% HPC and 4.6 for 45 wt% HPC. Thus, the exponents found in this work conform with the ones reported earlier for HPC solutions.
The plot in Fig. 6b can be used to estimate the rate of viscosity increase during evaporation. The rheological data help us understand kinetic trapping: with ethanol evaporation, the viscosity increases drastically, trapping the structure during the phase separation process.40 For both compositions, 30 and 45 wt% HPC, the viscosity increases at a similar rate: exponents of 4.4 for HPC 30 wt% and 4.6 for HPC 45 wt%. This observation might explain the results shown in Fig. 4b that the 3 different HPC ratios reach the same plateau of characteristic length scale around 4000 rpm. The increase of viscosity with the increase of the HPC ratio that is shown in Fig. 6a is not so pronounced compared to the high increase of viscosity during evaporation shown in Fig. 6b. Thus, the main parameters responsible for the kinetic trapping are the total polymer concentration and the ethanol evaporation rate.
3.4 Correlation between characteristic length scale and thickness
In Fig. 7, the characteristic length scale of the phase-separated structure (y-axis) obtained by Fourier image analysis (m ± sd) as a function of the average film thickness (m ± sd) (x-axis) is plotted for 3 compositions, 22, 30 and 45 wt% HPC. The thickness was obtained by profilometry. | ||
| Fig. 7 Average characteristic length scale versus average thickness for (a) EC:HPC 78:22 wt%. (b) EC:HPC 70:30 wt%. (c) EC:HPC 55:45 wt%. The error bars represent the standard deviation. Equations and coefficients of determination for the linear regressions are displayed. | ||
In Fig. 7, for all 3 compositions, the characteristic length scale of the structure increases with the thickness of the film. These results correspond well to the results found for other spin-coated phase-separated synthetic polymer systems.9,19,20 When the film is thinner, the evaporation is faster (due to the higher surface-to-volume ratio) and the phase separation is trapped in an earlier stage, leading to a smaller L(t). Fig. 7 shows a linear relationship between L(t) and the thickness fitted using linear regression. Values of the coefficient of determination (R2) obtained for the 3 plots are close to 1, showing that a linear model describes the relationship very well. However, the origin of this dependency is unclear since many different mechanisms can influence the structure evolution. The depth and the rate of the solvent quench will impact the characteristic length scale, the time for coarsening and the rate of coarsening. The coarsening is also dependent on the viscosity increase during the solvent evaporation. All these mechanisms will influence the structure evolution and more research is therefore needed to understand the background to the relationship between the L(t) and the film thickness.
Fig. 7 also shows that there is no clear trend concerning the slope dL/dh between the different compositions: 0.96, 1.62, 0.85 for HPC 22, 30, 45 wt%, respectively. The largest value of thickness and characteristic length scale for the 3 graphs correspond to the spin speed 1000 rpm. Because the thickness distribution and the structure are more heterogeneous at this spin speed than at the other measurement points, this spin speed might be considered as an outlier. Ignoring the possible outlier, the slope dL/dh would be 0.77, 1.25 and 1.13 for 22 wt% HPC, 30 wt% HPC and 45 wt% HPC, respectively. In addition, the R2 coefficients are 0.84, 0.93, 0.95 for 22 wt% HPC, 30 wt% HPC and 45 wt% HPC, respectively. Consequently, even if the possible outliers are excluded from the regression, there is no clear trend in the slopes as a function of composition.
It can be noticed that for the whole set of experiments, the characteristic length scale of the phase-separated structure is larger than the film thickness. Hence it is not unreasonable to speculate that the transition from a thick film to a thinner film also influences the coarsening of the structure and that there might be a transition from a 3D coarsening to a nearly 2D coarsening. It would therefore be very interesting to investigate the detailed structure evolution and the coarsening mechanisms in a future work.
4 Conclusions
It was found that the combination of confocal laser scanning microscopy, image analysis, rheology and profilometry is powerful to investigate spin-coated phase-separated films made of EC/HPC. The spin-coating technique showed good control and reproducibility of the final structures. The spin-coating has made it possible to investigate the phase separation as it might occur in the industrial fluidized bed. This information will help to control the formed coating structure and its release properties. Fourier image analysis was developed and optimized to determine the characteristic length scale of the phase-separated structure. The effect of spin-coater parameters on the final structure of the phase-separated spin-coated EC/HPC films, as a function of composition (ratio EC:HPC) was determined. The results showed that for the spin speed, the final characteristic length scale decreases with increasing spin speed. In addition, the film thickness decreases with increasing spin speed. Our finding on the relationship between film thickness and length scale can help understand the structure obtained on the industrial pellets. Spin curves were established and an exponent close to −0.5 was found for all compositions. Finally, strong correlation between thickness and spin speed was found for 22 wt% HPC, 30 wt% HPC and 45 wt% HPC.
Information about the phase separation kinetics will provide further understanding of the processes controlling the phase-separated structure of thin EC/HPC films. Thus, it would be interesting to perform in situ experiments with CLSM to investigate the kinetics of phase separation of spin-coated films in the future.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The Swedish Foundation for Strategic Research (SSF grant FID16-0013) is gratefully acknowledged for the funding. Christian Müller and Sandra Hutmark at the Department of Chemistry, Chalmers University of Technology, are acknowledged for access to lab facilities (spin-coater and profilometer). Anna Olsson from AstraZeneca and Cecilia Fager from Chalmers are acknowledged for valuable discussions on phase-separated EC/HPC films.References
- H. Gupta, D. Bhandari and A. Sharma, Recent Pat. Drug Delivery Formulation, 2009, 3, 162–173 CrossRef CAS PubMed.
- G. Tiwari, R. Tiwari, B. Sriwastawa, L. Bhati, S. Pandey, P. Pandey and S. K. Bannerjee, Int. J. Pharm. Invest., 2012, 2, 2–11 CrossRef PubMed.
- M. Marucci, G. Ragnarsson, C. von Corswant, A. Welinder, A. Jarke, F. Iselau and A. Axelsson, Int. J. Pharm., 2011, 411, 43–48 CrossRef CAS PubMed.
- P. Sakellariou and R. C. Rowe, Prog. Polym. Sci., 1995, 20, 889–942 CrossRef CAS.
- S. Wassén, R. Bordes, T. Gebäck, D. Bernin, E. Schuster, N. Lorén and A.-M. Hermansson, Soft Matter, 2014, 10, 8276–8287 RSC.
- T. Gebäck, M. Marucci, C. Boissier, J. Arnehed and A. Heintz, J. Phys. Chem. B, 2015, 119, 5220–5227 CrossRef.
- R. Vehring, Pharm. Res., 2008, 25, 999–1022 CrossRef CAS PubMed.
- P. D. Fowler, C. Ruscher, J. D. McGraw, J. A. Forrest and K. Dalnoki-Veress, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 90 CrossRef PubMed.
- J. J. van Franeker, D. Westhoff, M. Turbiez, M. M. Wienk, V. Schmidt and R. A. J. Janssen, Adv. Funct. Mater., 2015, 25, 855–863 CrossRef CAS.
- S. Y. Heriot and R. A. Jones, Nat. Mater., 2005, 4, 782–786 CrossRef CAS.
- D. T. W. Toolan, E. U. Haq, A. Dunbar, S. Ebbens, N. Clarke, P. D. Topham and J. R. Howse, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 875–881 CrossRef CAS.
- X. Li, R. Xing, Y. Zhang, Y. Han and L. An, Polymer, 2004, 45, 1637–1646 CrossRef CAS.
- S. Walheim, M. Böltau, J. Mlynek, K. Georg and U. Steiner, Macromolecules, 1997, 30, 4995–5003 CrossRef CAS.
- A. D. Dunbar, P. Mokarian-Tabari, A. J. Parnell, S. J. Martin, M. W. Skoda and R. A. Jones, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 31, 369–375 CrossRef CAS PubMed.
- L. Fang, M. Wei, C. Barry and J. Mead, Macromolecules, 2010, 43, 9747–9753 CrossRef CAS.
- P. Mokarian-Tabari, M. Geoghegan, J. R. Howse, S. Y. Heriot, R. L. Thompson and R. A. L. Jones, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 33, 283–289 CrossRef CAS PubMed.
- D. T. W. Toolan and J. R. Howse, J. Mater. Chem. C, 2013, 1, 603–616 RSC.
- C. Schaefer, PhD thesis, Eindhoven University of Technology, 2016.
- E. L. Williams, S. Gorelik, I. Phang, M. Bosman, C. Vijila, G. S. Subramanian, P. Sonar, J. Hobley, S. P. Singh, H. Matsuzaki, A. Furube and R. Katoh, RSC Adv., 2013, 3, 20113–20124 RSC.
- P. Muller-Buschbaum and M. Stamm, Colloid Polym. Sci., 2001, 279, 376–381 CrossRef CAS.
- R. Meier, M. A. Ruderer, A. Diethert, G. Kaune, V. Korstgens, S. V. Roth and P. Muller-Buschbaum, J. Phys. Chem. B, 2011, 115, 2899–2909 CrossRef CAS PubMed.
- C. Huang, Doktors der Naturwissenschaften, Technischen Universität Darmstadt, 2015 Search PubMed.
- S. Ebbens, R. Hodgkinson, A. J. Parnell, A. Dunbar, S. J. Martin, P. D. Topham, N. Clarke and J. R. Howse, ACS Nano, 2011, 5, 5124–5131 CrossRef CAS PubMed.
- P. Müller-Buschbaum, J. S. Gutmann, M. Wolkenhauer, J. Kraus, M. Stamm, D. Smilgies and W. Petry, Macromolecules, 2001, 34, 1369–1375 CrossRef.
- N. Lorén, M. Langton and A.-M. Hermansson, J. Chem. Phys., 2002, 116, 10536–10546 CrossRef.
- C. E. Ayres, B. S. Jha, H. Meredith, J. R. Bowman, G. L. Bowlin, S. C. Henderson and D. G. Simpson, J. Biomater. Sci., Polym. Ed., 2008, 19, 603–621 CrossRef CAS PubMed.
- T. Fujita and M. Chen, Jpn. J. Appl. Phys., 2008, 47, 1161 CrossRef CAS.
- J. S. Gutmann and P. Muller-Buschbaum, Faraday Discuss., 1999, 112, 285–297 RSC.
- J. Zhu, R. Balieu, X. Lu and N. Kringos, Mater. Des., 2018, 137, 164–175 CrossRef CAS.
- C. Fager, S. Barman, M. Röding, A. Olsson, N. Lorén, C. von Corswant, D. Bolin, H. Rootzen and E. Olsson, Int. J. Pharm., 2020, 587, 119622 CrossRef CAS PubMed.
- C. Fager, M. Röding, A. Olsson, N. Lorén, C. von Corswant, A. Sarkka and E. Olsson, Microsc. Microanal., 2020, 26, 837–845 CrossRef CAS PubMed.
- M. Röding, C. Fager, A. Olsson, C. von Corswant, E. Olsson and N. Lorén, J. Microsc., 2021, 281, 76–86 CrossRef PubMed.
- L. Picullel, K. Bergfeldt and S. Nilsson, presented in part at the Meeting, Biopolymer mixtures, Nottingham, 1994.
- R. A. L. Jones and R. W. Richards, Polymers at Surfaces and Interfaces, Cambridge University Press, Cambridge, UK, 1999 Search PubMed.
- J. W. Cahn, J. Chem. Phys., 1965, 42, 93–99 CrossRef CAS.
- J. W. Gibbs, The Scientific Papers of J. Willard Gibbs, Dover, New York, 1961 Search PubMed.
- N. C. Das, J. Microsc., 2014, 253, 198–203 CrossRef CAS.
- H. Tanaka and T. Nishi, Phys. Rev. Lett., 1985, 55, 1102–1105 CrossRef CAS PubMed.
- L. H. Sperling and Introduction to Physical Polymer Science, 4th ed., 2006 Search PubMed.
- C. Schaefer, J. J. Michels and P. Van Der Schoot, Macromolecules, 2016, 49, 6858–6870 CrossRef CAS.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS.
- C. Schaefer, P. van der Schoot and J. J. Michels, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 91, 022602 CrossRef CAS PubMed.
- H. Andersson, M. Marucci, J. Hjärtstam, M. Stading, C. von Corswant and A. Larsson, Eur. J. Pharm. Biopharm., 2009, 83, 301–306 Search PubMed.
- H. Andersson, J. Hjartstam, M. Stading, C. von Corswant and A. Larsson, Eur. J. Pharm. Sci., 2013, 48, 240–248 CrossRef CAS PubMed.
- M. Marucci, J. Hjartstam, G. Ragnarsson, F. Iselau and A. Axelsson, J. Controlled Release, 2009, 136, 206–212 CrossRef CAS PubMed.
- H. Andersson, H. Habel, A. Olsson, S. Sandhagen, C. von Corswant, J. Hjartstam, M. Persson, M. Stading and A. Larsson, Int. J. Pharm., 2016, 511, 223–235 CrossRef CAS.
- C. Fager, Licentiate thesis Licentiate of engineering, Chalmers University of Technology, 2018 Search PubMed.
- A. Jarke, Master thesis, Uppsala University, 2009.
- J. Deprez, Master thesis, Ghent University, 2016.
- S. Wassén, N. Lorén, K. van Bemmel, E. Schuster, E. Rondeau and A.-M. Hermansson, Soft Matter, 2013, 9, 2738–2749 RSC.
- Ashland and Klucel hydroxypropylcellulose, Physical and chemical properties, 2017 Search PubMed.
- ETHOCEL DowCellulosics, Ethylcellulose Polymers Technical Handbook, 2005 Search PubMed.
- J. J. Michels, ChemPhysChem, 2011, 12, 342–348 CrossRef CAS PubMed.
- H. Barnes, Handbook of Elementary Rheology, University of Wales, 2000 Search PubMed.
- P. G. De Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca and London, 1979 Search PubMed.
- D. P. Birnie and M. Manley, Phys. Fluids, 1997, 9, 870–875 CrossRef CAS.
- D. Meyerhofer, J. Appl. Phys., 1978, 49, 3993–3997 CrossRef.
- M. Shaw and W. Macknight, Introduction to Polymer Viscoelasticity, Wiley, 2005 Search PubMed.
- M. Bercea and P. Navard, Cellul. Chem. Technol., 2018, 52, 603–608 CAS.
- D. J. Goodwin, D. R. Picout, S. B. Ross-Murphy, S. J. Holland, L. G. Martini and M. J. Lawrence, Carbohydr. Polym., 2011, 83, 843–851 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |