
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular gelation controlled by an iodine clock†
Solenn
Riedel
a,
Thomas
Schweizer
b,
Katrina
Smith-Mannschott
a,
Eric R.
Dufresne
 a and
Guido
Panzarasa
*ac
a and
Guido
Panzarasa
*ac
aLaboratory of Soft and Living Materials, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 1-5/10, Zürich 8093, Switzerland. E-mail: guidop@ethz.ch
bLaboratory of Soft Materials, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 1-5/10, Zürich 8093, Switzerland
cWood Materials Science, Institute for Building Materials, ETH Zürich, Stefano-Franscini-Platz 3, Zürich 8093, Switzerland
First published on 27th January 2021
Abstract
Programming supramolecular assembly in the time domain is a fundamental aspect of the design of biomimetic materials. We achieved the time-controlled sol–gel transition of a poly(vinyl alcohol)–iodine supramolecular complex by generating iodine in situ with a clock reaction. We demonstrate that both the gelation time and the mechanical properties of the resulting hydrogel can be tuned by properly selecting the clock parameters or through competitive iodine complexation.
Synthetic living materials, indistinguishable in form and function from their biological counterparts, are a major goal of contemporary materials science.1 Recent progress in systems chemistry2,3 is catalyzing efforts toward their practical realization. In this regard, the design of time-programmable supramolecular self-assembly and gelation by means of far-from-equilibrium chemical networks is an especially promising approach.4–7
The sol–gel transition, also known as gelation, occurs in a broad class of soft and biological materials, from colloidal suspensions to protein solutions. Gels are an integral part of biological systems, from the cytoskeleton to the extracellular matrix.8 Control of gelation is central to both healthy biological functions9–11 as well as pathological states.12 Gels have countless technological applications: from food to energy, from the environment to biomedicine.13–15 Robust approaches to control gelation are necessary to the production of printable,16 injectable17 gels. However, tuning the time of sol–gel transition without compromising the structure and properties of the final gel remains challenging.18
In supramolecular hydrogels, the constitutive elements (small molecules or polymers) are held together by non-covalent interactions (e.g. hydrogen bonding, π–π stacking, van der Waals forces), giving them outstanding stimuli-responsive behavior.19–23 Supramolecular hydrogels can be prepared by a variety of approaches, including: electrostatic interaction and coacervation, complex formation with metal ions, and host–guest interactions.22,24 Often, however, these approaches require the synthesis of specially designed building blocks, limiting their practical relevance.
We previously demonstrated how pH-clocks, reaction networks able to generate pH changes after an initial lagtime, can be used to achieve the autonomous programming of colloidal materials.25–29 The usefulness of chemical clocks to control gelation in time, which can lead to increased gel homogeneity, has been demonstrated already. However, the majority of these approaches rely on pH-triggered interactions, e.g. electrostatic coupling. These pH changes are produced either by slow acid generators, such as δ-gluconolactone,30,31 or, most often, by enzymatic systems, such as the urease-urea.32–39
Here, we explore gelation by supramolecular complexation, applying an “iodine clock” for controlling the time of sol–gel transition and the mechanical properties of a poly(vinyl alcohol) (PVA)–iodine‡ supramolecular complex. By generating iodine autonomously in situ, we achieved the delayed formation of an homogeneous supramolecular polymeric hydrogel without an external trigger, and were able to investigate the gelation dynamics rheologically.
Many polymers, both natural (e.g. amylose, cyclodextrins) and artificial (e.g. poly(vinyl alcohol) PVA), can form supramolecular complexes with iodine.40 The relevance of such complexes, apart from fundamental interest in unraveling their structure,41 ranges from analytical applications,42,43 to thermoelectrochemical cells,44 from antimicrobial agents45 to advanced photovoltaics.46
Poly(vinyl alcohol) PVA is a water-soluble, industrially relevant polymer, cheap and widely available with different molecular weights, stereoregularity and composition (e.g. degree of acetylation). It is non-toxic, biocompatible, and useful for a variety of biomedical applications.47 Its supramolecular complexes with iodine (often prepared by directly mixing, or subliming, iodine in the polymer matrix) have been studied for decades.48–52 PVA is thus a perfect playground for investigating time-controlled supramolecular gelation.
An “iodine clock” is a network of chemical reactions in which iodine is produced after an induction time. For our investigations we chose the iodide–persulfate–thiosulfate (IPT) reaction, a reliable substrate-depletive iodine clock with a well-established mechanism.53–55 In the IPT system, iodine is generated by the reaction of sodium iodide NaI with ammonium persulfate (NH4)2S2O8, but is immediately reduced back to iodide by sodium thiosulfate Na2S2O3. As a result, iodine can accumulate only after thiosulfate has been completely consumed (Fig. 1A). As shown in Fig. 1B for a typical experiment with our IPT–PVA system ([NaI] = 50 mM, [Na2S2O3] = 40 mM, [(NH4)2S2O8] = 200 mM, 5% PVA 130 kDa), after an initial time lag, the appearance and progressive accumulation of iodine are marked respectively by a sudden change of the reaction mixture from colorless to yellow, and then to dark orange. Once the iodine concentration has reached a certain threshold, the colour of the mixture shifts from dark orange to green-brown, indicating that the blue charge-transfer PVA–iodine complex started to form. Eventually, a dark blue gel is obtained (see also Movie S1, ESI†). According to literature,56,57 PVA forms channel inclusion complexes, very similar to those formed by amylose,58 in which linear tri- or poly-iodides are wrapped inside PVA helices. If the concentration of the PVA is high enough, as in our conditions, helices can then associate via intermolecular bonds giving rise to a gel (Fig. S1, ESI†).
 | ||
| Fig. 1 (A) Mechanism of the iodide–persulfate–thiosulfate–poly(vinyl alcohol) (IPT–PVA) system. (B) Photographic sequence of a typical ITP–PVA system over time, showing the color changes and the resulting gel (conditions: [NaI] = 50 mM, [Na2S2O3] = 40 mM, [(NH4)2S2O8] = 200 mM, 5% PVA 130 kDa). | ||
Thanks to the initial lagtime, we could investigate every phase of the gelation process by rheological measurements.59 In Fig. 2A, the dynamic time sweep plot shows the evolution of the elastic G′ and dissipative G′′ shear moduli as a function of time for a representative IPT–PVA system. Initially G′′ dominates over G′, a typical sol behavior. The clock then “goes off”, iodine accumulates in the solution, and the PVA–iodine complex starts to form and associate. The resulting microstructure resists the deformation induced as strain is applied by the rheometer. This resistance manifests as a steep increase in both moduli, especially G′, which intersect in what is usually taken as the gelation time tgel (Fig. 2A, inset). A control experiment showed that the applied oscillating strain did not influence the final microstructure responsible for the mechanical properties of the resulting gel (Fig. S2, ESI†).
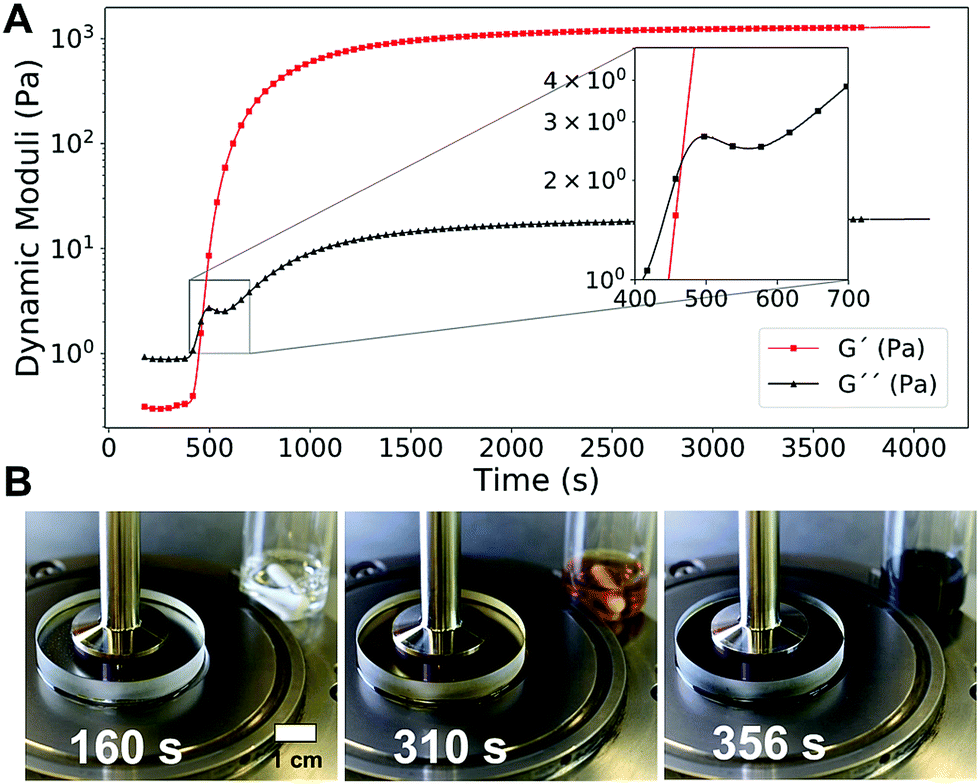 | ||
| Fig. 2 (A) Dynamic time sweep curve (ω = 10 rad s−1, γ0 = 10%) showing the evolution of the elastic G′ and dissipative G′′ shear moduli during the formation of a typical PVA–iodine gel ([NaI] = 50 mM, [Na2S2O3] = 40 mM, [(NH4)2S2O8] = 200 mM, 5% PVA 130 kDa). (B) A transparent rheometer plate allowed to monitor the rheological properties of the PVA–IPT system together with its color evolution (see also Movie S2, ESI†). | ||
Using a transparent rheometer plate, we could directly relate the color evolution with the change in rheological properties, confirming that the blue color of the PVA–iodine complex appears well before the gel formation (Fig. 2B and Movie S2, ESI†). This result suggests that a critical iodine concentration must be reached for the gel to form.
By monitoring the gelation with consecutive frequency sweep tests, and analysing them as suggested by H. Winter and F. Chambon,61 we could show that the sample at the crossover point is behaving like a critical gel with frequency-independent equality of G′ and G′′ (ESI,† Fig. S3). We also studied the evolution of the gel stiffness over long times (tens of hours) by acoustic resonance spectroscopy (ARS), a non-destructive, impedance-based method62 (ESI,† Fig. S4). The results are in accordance with the rheology data. However, ARS data suggest that the PVA–IPT gel is still evolving several hours after its formation, a detail which could not be appreciated from rheology alone. The reasons for such behavior are not yet clear and could deserve dedicated investigations.
Tuning the molecular weight of PVA (ESI,† Fig. S5) and the parameters of the iodine clock allows to change the dynamics of the IPT–PVA system as well as the mechanical properties of the resulting gels.
We started by investigating the effect of iodide concentration. Increasing [NaI] from 25 to 100 mM resulted in faster gelation rates (from 1.1 × 10−3 s−1 to 4.2 × 10−3 s−1) and increased storage moduli G′, from 0.1 kPa to 6.8 kPa (Fig. 3A–C and Fig. S6, ESI†). Increasing the amount of iodide fed to the system results in an increased iodine concentration, strengthening the elastic matrix of the gel (more crosslinks) and decreasing the clock lagtime (faster consumption of thiosulfate), which in turn gives a linearly increasing rate of gelation.
 | ||
| Fig. 3 Effect of iodide concentration (A–C), thiosulfate concentration (D–F), and of competitive iodine complexation (G–I) on the rheological dynamics (A, D and G) and gelation rate (B, E and H) of the IPT–PVA system, and on the mechanical properties of the resulting PVA–iodine gel (C, F and I). For the sake of clarity, we show only the evolution of G′, together with the Cao fit60 used to determine the final value of G′. The complete curves as well as a more detailed description of the fit are available in the ESI† (Fig. S6–S8 and eqn (S1)). | ||
Conversely, by tuning the concentration of thiosulfate Na2S2O3 for a fixed [NaI], the rate of gelation decreased linearly for increasing [Na2S2O3], leaving almost unchanged the gel's mechanical properties (Fig. 3D–F and Fig. S7, ESI†). Thiosulfate rapidly transforms iodine back to iodide, thus increasing the lagtime. Since under the applied conditions persulfate (NH4)2S2O8 is in excess, all the iodide is eventually oxidized to iodine, regardless of the Na2S2O3 concentration. As a result, the amount of iodine available for complexation is independent from thiosulfate, and the final gel modulus is the same.
We then explored the impact of competitive iodine complexation on the IPT–PVA system. Cyclodextrins are well-known to form host–guest complexes with many different species, including iodine.63,64 We chose methyl-β-cyclodextrin (mβCD) thanks to its excellent water solubility and good iodine complexation properties. Methyl-β-cyclodextrin was substituted to thiosulfate, resulting in a new system iodide–persulfate–mβCD.
As shown in Fig. 3G–I and Fig. S8 (ESI†), the higher the concentration of mβCD the longer the lagtime until gel formation, with a linearly decreasing rate of gelation – a result similar to that obtained with thiosulfate. However, a significant difference is observed for the elastic modulus G′, which dramatically decreases for increasing [mβCD] while it was almost unaffected by thiosulfate. These results can be explained by taking into account the dramatically different mechanisms of iodine scavenging for thiosulfate and methyl-β-cyclodextrin. Thiosulfate converts iodine into iodide, but after it has been completely decomposed, iodine can accumulate in the system and form the complex with PVA. On the other hand, mβCD, which forms a complex with iodine more readily than PVA, sequesters it quickly after its formation. Only after the methyl-β-cyclodextrin has been saturated, the iodine can interact with PVA (Fig. S9, ESI†). The mechanical properties of the resulting PVA–iodine gels are thus dictated by the amount of iodine available, which decreases by increasing the concentration of mβCD.
To summarize: increasing the iodide concentration increases the amount of iodine available for complexation with PVA, increasing both the gelation rate and the mechanical modulus of the gel; conversely, increasing the concentration of thiosulfate decreases the gelation rate but, since it does not change the final iodine concentration, leaves the gel's mechanical modulus unaffected. Methyl-β-cyclodextrin, by competing with PVA to complex with iodine, decreases both the gelation rate and the mechanical modulus of the gel. Thus, by tuning these three parameters [NaI], [Na2S2O3], and [mβCD], it is possible to tailor both the gelation rate and the mechanical properties of the resulting PVA–iodine gel.
Regardless of its method of formation, the PVA–iodine gel is a supramolecular polymeric hydrogel, held together by non-covalent interactions. Thus, removing the iodine, either by decomposition or sequestration, will result in gel disappearance. Fig. 4 demonstrates this concept by showing what happens to identical pieces of PVA–iodine gels when they are left immersed in relatively concentrated (0.1 M) solutions of thiosulfate (Fig. 4A) or methyl-β-cyclodextrin (Fig. 4B). Pure water (Fig. 4C) is also used as a control (the solubility of iodine in water is very limited, around 0.29 g L−1 at 20 °C).65
 | ||
| Fig. 4 Dissolution of a typical PVA–iodine gel ([NaI] = 50 mM) in the presence of (A) an iodine-decomposing agent (sodium thiosulfate), or (B) an iodine-sequestering agent (methyl-β-cyclodextrin). In water (C) the gel is stable for long times. In (A) the solution is colorless as iodine is transformed into iodide, while in (B) the yellow color is due to the mβCD–iodine complex. In (C) the faint yellow color is due to a small amount of free iodine leached from the gel. Regardless of the approach used, the PVA–iodine gel dissolves through surface erosion, as shown schematically in (A, inset). The length of the scale bar is 1 cm. | ||
As depicted in the inset of Fig. 4A, the gel dissolves by surface erosion. Depending on the type and concentration of the iodine-removing agent, it is possible to adjust the time required to achieve a partial or complete gel dissolution.
Our PVA–iodine gel may find applications in fields as diverse as biomedicine (e.g. as a controlled iodine release system for antimicrobial wound dressings) to energy generation (e.g. as an active element in (thermo)electrochemical cells).
Conclusions
We demonstrated the control of sol–gel transition of a supramolecular poly(vinyl alcohol)–iodine complex orchestrated by a chemical clock as an in situ source of iodine.We investigated the effects of key parameters, such as the iodine clock composition, the presence of competitive iodine complexation, and the molecular weight of PVA, on the gelation time and the mechanical properties of the resulting gels. Additional factors, which are known to influence the physical characteristics (including the color) of PVA–iodine complexes, such as the stereoregularity and degree of acetylation of PVA, could be investigated as well, further widening the tunability of this system. Since the colloidal properties of PVA in aqueous solution can be affected by certain salts (e.g. sulfate66), this could also be an interesting direction for further research.
Our results further suggest the vast future opportunities for the application of chemical clocks to materials science, and in particular for the understanding and design of time-controlled supramolecular self-assembly systems.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. W. Tibbitt and R. Langer, Acc. Chem. Res., 2017, 50, 508–513 CrossRef CAS.
- H. W. H. Van Roekel, B. J. H. M. Rosier, L. H. H. Meijer, P. A. J. Hilbers, A. J. Markvoort, W. T. S. Huck and T. F. A. De Greef, Chem. Soc. Rev., 2015, 44, 7465–7483 RSC.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS.
- M. M. Lerch, A. Grinthal and J. Aizenberg, Adv. Mater., 2020, 32, 1905554 CrossRef CAS.
- A. Walther, Adv. Mater., 2020, 32, 1905111 CrossRef CAS.
- L. Heinen and A. Walther, Soft Matter, 2015, 11, 7857–7866 RSC.
- J. Prost, F. Jülicher and J. F. Joanny, Nat. Phys., 2015, 11, 111–117 Search PubMed.
- J. A. Riback, C. D. Katanski, J. L. Kear-Scott, E. V. Pilipenko, A. E. Rojek, T. R. Sosnick and D. A. Drummond, Cell, 2017, 168, 1028–1040.e19 CrossRef CAS.
- S. Kroschwald and S. Alberti, Cell, 2017, 168, 947–948 CrossRef CAS.
- L. W. Janson, J. Kolega and D. L. Taylor, J. Cell Biol., 1991, 114, 1005–1015 CrossRef CAS.
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS.
- G. R. Hendrickson, M. H. Smith, A. B. South and L. A. Lyon, Adv. Funct. Mater., 2010, 20, 1697–1712 CrossRef CAS.
- C. Echeverria, S. Fernandes, M. Godinho, J. Borges and P. Soares, Gels, 2018, 4, 54 CrossRef.
- D. Klinger and K. Landfester, Polymer, 2012, 53, 5209–5231 CrossRef CAS.
- Y. Dong, S. Wang, Y. Ke, L. Ding, X. Zeng, S. Magdassi and Y. Long, Adv. Mater. Technol., 2020, 5, 2000034 CrossRef CAS.
- K. H. Bae, L. S. Wang and M. Kurisawa, Biodegradable hydrogels: P. and challenges, J. Mater. Chem. B, 2013, 1, 5371–5388 RSC.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 2002759 CrossRef CAS.
- R. Eelkema and A. Pich, Adv. Mater., 2020, 32, 1906012 CrossRef CAS.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937–954 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- G. Panzarasa, A. Osypova, A. Sicher, A. Bruinink and E. R. Dufresne, Soft Matter, 2018, 14, 6415–6418 RSC.
- G. Panzarasa, T. Sai, A. L. Torzynski, K. Smith-Mannschott and E. R. Dufresne, Mol. Syst. Des. Eng., 2020, 5, 445–448 RSC.
- G. Panzarasa, A. L. Torzynski, T. Sai, K. Smith-Mannschott and E. R. Dufresne, Soft Matter, 2020, 16, 591–594 RSC.
- C. C. M. Sproncken, B. Gumi-Audenis, G. Panzarasa and I. K. Voets, ChemSystemsChem, 2020, 2, e2000005 CrossRef.
- G. Panzarasa and E. R. Dufresne, Chimia, 2020, 74, 612 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506–6521 CAS.
- E. Jee, T. Bánsági, A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 47–50 RSC.
- S. Panja, K. Boháčová, B. Dietrich and D. J. Adams, Nanoscale, 2020, 12, 12840–12848 RSC.
- S. Mondal, D. Podder, S. K. Nandi, S. Roy Chowdhury and D. Haldar, Soft Matter, 2020, 16, 10115–10121 RSC.
- S. Panja and D. J. Adams, Chem. Commun., 2019, 55, 10154–10157 RSC.
- S. Panja, C. Patterson and D. J. Adams, Macromol. Rapid Commun., 2019, 40, 1900251 CrossRef.
- L. Heinen, T. Heuser, A. Steinschulte and A. Walther, Nano Lett., 2017, 17, 4989–4995 CrossRef CAS.
- A. Chenite, S. Gori, M. Shive, E. Desrosiers and M. D. Buschmann, Carbohydr. Polym., 2006, 64, 419–424 CrossRef CAS.
- S. Moulay, J. Polym. Eng., 2013, 33, 389–443 CAS.
- S. Madhu, H. A. Evans, V. V. T. Doan-Nguyen, J. G. Labram, G. Wu, M. L. Chabinyc, R. Seshadri and F. Wudl, Angew. Chem., Int. Ed., 2016, 55, 8032–8035 CrossRef CAS.
- J. J. Colin and H. F. Gaultier de Clauby, Ann. Chim. Phys., 1814, 90, 87–100 Search PubMed.
- F. Stromeyer, Ann. Phys., 1815, 49, 146–153 CrossRef.
- H. Zhou, T. Yamada and N. Kimizuka, Sustainable Energy Fuels, 2018, 2, 472–478 RSC.
- H.-U. Schenck, P. Simak and E. Haedicke, J. Pharm. Sci., 1979, 68, 1505–1509 CrossRef CAS.
- W. S. Yang, B. W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS.
- C. C. DeMerlis and D. R. Schoneker, Food Chem. Toxicol., 2003, 41, 319–326 CrossRef CAS.
- H. Yang and F. Horii, Polymer, 2008, 49, 785–791 CrossRef CAS.
- T. Takahama, S. M. Saharin and K. Tashiro, Polymer, 2016, 99, 566–579 CrossRef CAS.
- K. Tashiro, H. Kitai, S. M. Saharin, A. Shimazu and T. Itou, Macromolecules, 2015, 48, 2138–2148 CrossRef CAS.
- N. N. Bozhko, V. P. Stolyarov, N. N. Baranov, E. B. Bablyuk and V. G. Nazarov, Inorg. Mater. Appl. Res., 2010, 1, 329–334 CrossRef.
- M. Keizo, Adv. Polym. Sci., 1993, 91–129 Search PubMed.
- P. C. Moews and R. H. Petrucci, J. Chem. Educ., 1964, 41, 549–551 CrossRef CAS.
- A. E. Burgess and J. C. Davidson, J. Chem. Educ., 2012, 89, 814–816 CrossRef CAS.
- T. S. Rao and S. I. Mali, Z. Naturforsch., A: Phys., Phys. Chem., Kosmophys., 1974, 29, 141–144 CAS.
- M. M. Zwick, J. Appl. Polym. Chem., 1965, 9, 2393–2424 CrossRef CAS.
- J. G. Pritchard and D. A. Akintola, Talanta, 1972, 19, 877–888 CrossRef CAS.
- M. M. Zwick, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 1642–1644 CrossRef CAS.
- G. M. Kavanagh and S. B. Ross-Murphy, Prog. Polym. Sci., 1998, 23, 533–562 CrossRef CAS.
- X. J. Cao, H. Z. Cummins and J. F. Morris, Soft Matter, 2010, 6, 5425–5433 RSC.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- J. M. Ide, Rev. Sci. Instrum., 1935, 6, 296–298 CrossRef.
- G. Panzarasa and E. R. Dufresne, Chaos, 2019, 29, 071102 CrossRef.
- J. W. Minns and A. Khan, J. Phys. Chem. A, 2002, 106, 6421–6425 CrossRef CAS.
- H. Hartley and N. P. Campbell, J. Chem. Soc., Trans., 1908, 93, 741–745 RSC.
- B. Filova, L. Musilova, A. Mracek, M. L. Ramos, L. M. P. Veríssimo, A. J. M. Valente and A. C. F. Ribeiro, J. Mol. Liq., 2020, 304, 112728 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete experimental details, additional figures and experimental movies. See DOI: 10.1039/d0sm02285c |
| ‡ In our experimental conditions the formation of water-soluble triiodide I3− (and possibly of other poly(iodide) species) should be preferred over the formation of elemental iodine I2, as shown by the mechanism in Fig. 1A. However, for the sake of simplicity, throughout the manuscript we refer to triiodide and poly(iodide) species collectively as to “iodine”. |
| This journal is © The Royal Society of Chemistry 2021 |