
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecorated networks of native proteins: nanomaterials with tunable mesoscopic domain size†
Ioatzin
Ríos de Anda
 *ab,
Angélique
Coutable-Pennarun
cd,
Christopher
Brasnett
a,
Stephen
Whitelam
e,
Annela
Seddon
af,
John
Russo
bg,
J. L. Ross
Anderson
dh and
C. Patrick
Royall
aijk
*ab,
Angélique
Coutable-Pennarun
cd,
Christopher
Brasnett
a,
Stephen
Whitelam
e,
Annela
Seddon
af,
John
Russo
bg,
J. L. Ross
Anderson
dh and
C. Patrick
Royall
aijk
aH.H. Wills Physics Laboratory, Tyndall Avenue, Bristol, BS8 1TL, UK. E-mail: ioatzin.riosdeanda@bristol.ac.uk
bSchool of Mathematics, University Walk, Bristol, BS8 1TW, UK
cBrisSynBio Synthetic Biology Research Centre, Life Sciences Building, Tyndall Avenue, Bristol, BS8 1TQ, UK
dSchool of Biochemistry, University of Bristol, Bristol, BS8 1TD, UK
eMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
fBristol Centre for Functional Nanomaterials, University of Bristol, Bristol, BS8 1TL, UK
gDipartimento di Fisica and CNR-ISC, Sapienza-Università di Roma, Piazzale A. Moro 2, 00185 Roma, Italy
hSchool of Cellular and Molecular Medicine, University Walk, Bristol, BS8 1TD, UK
iGulliver UMR CNRS 7083, ESPCI Paris, Université PSL, 75005 Paris, France
jSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
kCentre for Nanoscience and Quantum Information, Tyndall Avenue, Bristol, BS8 1FD, UK
First published on 30th June 2021
Abstract
Natural and artificial proteins with designer properties and functionalities offer unparalleled opportunity for functional nanoarchitectures formed through self-assembly. However, to exploit this potential we need to design the system such that assembly results in desired architecture forms while avoiding denaturation and therefore retaining protein functionality. Here we address this challenge with a model system of fluorescent proteins. By manipulating self-assembly using techniques inspired by soft matter where interactions between the components are controlled to yield the desired structure, we have developed a methodology to assemble networks of proteins of one species which we can decorate with another, whose coverage we can tune. Consequently, the interfaces between domains of each component can also be tuned, with potential applications for example in energy – or electron – transfer. Our model system of eGFP and mCherry with tuneable interactions reveals control over domain sizes in the resulting networks.
1 Introduction
Complex, hierarchical, materials with long-range ordering, comprised from building blocks at the nano- and micro-scale can be obtained by controlling self-assembly, through careful manipulation of the interactions between the assembling components.1–3 If the constituents of such structures exhibit useful optical, magnetic, electric, chemical or biological properties, then the assemblies formed hold great potential for a myriad of applications including photonics, energy transfer and storage, catalysis, drug delivery and tissue scaffolding.4–9 Perhaps the greatest source of inspiration for design and construction at the nanoscale is nature itself. The exquisite level of complexity, specificity, efficiency and sophistication of systems found in nature makes their building blocks (proteins, nucleic acids, carbohydrates and lipids) an attractive possibility to be exploited for new materials. Of these building blocks, proteins exhibit the largest diversity of structure and function and thus have the greatest potential to be exploited as the functional components for novel nanostructured materials.5–8,10–13 They are capable of carrying out structural, catalytic, transport, packaging, optical, specific recognition, electrical, information storage and metabolic functions.5,6Among synthetic materials, dispersions of colloids and nanoparticles self-assemble to a very wide variety of structures, which can be controlled by tuning the interactions between the particles.14 Similarly interactions between proteins determine the structures into which they assemble.15–20 Here we take inspiration from self-assembly of soft matter systems such as colloids in the context of protein assembly to form a 3d network of a binary protein system.
In nature, assembly of proteins into networks is often avoided, so that a condensed phase is formed only if (i) they have evolved to do so (i.e. compartmentalised structures like carboxysomes or viral capsids), if (ii) mutations lead to mis-folding or to interactions that produce aggregation, or if (iii) changes in the medium (i.e. ionic strength or significant changes in temperature or pH) occur.16,19,21,22 In the latter two scenarios, the proteins may lose their functionality. Furthermore, unlike most gel networks found in nature, here we aim to produce an architecture with a binary system in which the domain size of each species can be controlled. By incorporating multi-enzyme cascades, light harvesting arrays and electron transfer proteins, such multicomponent networks hold great potential as advanced materials for catalysis, energy transduction and small scale electronics.
While so-called bigels of two distinct unconnected, mixed or bicontinuous networks have been produced with colloids,23 (denatured) proteins24 and in simulations with patchy particles,25 here our focus is on a single network of two components, where we can control the assembly such that the domain size and hence the interfaces between domains can be tuned. In particular, we aim to self-assemble decorated protein networks formed by distinguishable protein domains, where these preserve their native structure. The fluorescent proteins enhanced green fluorescent protein (eGFP) and mCherry were chosen as the model system due to the ease of monitoring the assembly process with fluorescent microscopy.
To assemble these structures, we develop a strategy to control the interactions of each species independently, similar to Immink and collaborators.26 Here, we exploit the effects of salts on protein solutions,18,27–29 based on specific salt–protein interactions. Trivalent ions have been shown to selectively interact with the surface-exposed carboxylic groups of the acidic residues of the proteins, which in turn, leads to gelation or crystallisation.30–32 Thus, by effectively modifying the surface of the proteins, we provide the specificity required to gain control over their self-assembly at mesoscopic lengths of tens of nanometers and upwards. We also use nonspecific interactions via the addition of ammonium sulphate so that we have two methods to control the interactions between the proteins.
Since the specific interactions are controlled by trivalent ions (here Y3+) which interact with a negatively charged protein (eGFP), the non-specific protein (mCherry) is cationised such that it should be independent of the yttrium. This leads to a binary protein system with opposite charges which can then weakly associate. Therefore our strategy to assemble the binary network is to first assemble the eGFP into a backbone of the network and then to decorate this backbone with mCherry, which we assemble by addition of ammonium sulphate. Depending on the concentration of mCherry, we find different mechanisms of assembly of the mCherry which provides a means to tune the coverage of the eGFP network. As such, the strategies proposed herein represent a promising route to yield a new type of functional biomaterial, a multicomponent 3d network whose domains could have enzymatic, electron or energy transfer properties for example.
Our work is organised as follows. In Section 2 we describe the methods followed. In Section 3 we present and discuss our results. We start by showing the increment in the specificity of protein–salt interactions to gain sufficient control over the gelation process. This is followed by studying the nature and strength of the protein–salt interactions in the presence of different salt concentrations using small-angle X-ray scattering (SAXS). Once we stablished the interactions, we then proceed to assemble the desired structures, following the proposed two-step addition of native and modified proteins to different salts, along with the structure characterisation using confocal laser scanner microscopy. We then implement our strategy for different protein compositions to tune the protein domain sizes within the network and propose a mechanism for our findings. Finally, we study quantify the structure of our decorated networks via their fractal dimension. In Section 4 we summarise our conclusions.
2 Methods
2.1 Cellular culture for the expression of eGFP
Escherichia coli BL21 (DE3) competent (able to receive DNA) cells were previously transformed with the DNA plasmid-pET45b(+)-eGFP. First, a mini-culture was prepared by inoculating 100 mL of lysogeny broth (LB) of nutrients and the antibiotic carbenicillin (50 μg mL−1) with E. coli. The culture was left to grow for 16 h at 37 °C and 180 rpm. 20 mL of this culture were then used to inoculate 1 L of LB containing carbenicillin (50 μg mL−1), which was left to grow under the same previous conditions. The optical density (OD600nm) was monitored to a value of 0.5–0.6, when the production of eGFP was induced by adding 1 mM of Isopropyl β-D-1-thiogalactopyranoside (IPTG). After 1 h of induction time, the temperature was changed to 30 °C. After 16 h of incubation, the cell culture was centrifuged at 4500g for 15 min at 4 °C. The supernatant obtained was discarded and the pellet was resuspended in a lysis buffer (20 mM imidazole, 300 mM NaCl and 50 mM potassium phosphate at pH 8.0) and stored at −20 °C.332.2 Cellular culture for the expression of mCherry
Escherichia coli BL21 (DE3) competent cells were previously transformed with the DNA plasmid pBADmCherry. A mini-culture was prepared under the same conditions as for the expression of eGFP. Equally, 20 mL of said culture were used to inoculate 1 L of LB with 50 μg mL−1 carbenicillin, which was then incubated as before. In this case the OD600nm of the culture was closely monitored until it reached a value between 0.6–0.8, when mCherry expression was induced by adding L-arabinose from a 20% stock solution for a final concentration of 0.2% (w/v).34 The rest of the steps followed are the same as the ones described above for eGFP.2.3 Purification of fluorescent proteins
The purification of both proteins followed the same protocol. Cell pellets were thawed and kept on ice, broken down by lysis using 3 sonication cycles of 30 seconds (Soniprep 150 plus MSE) and centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (Sorvall SS34 rotor) at 4 °C for 30 min. The supernatant was recovered and filtered through a 0.22 μm syringe filter (Millipore) and injected to a nickel–nitrilotriacetic acid (Ni–NTA) agarose column (Qiagen) connected to an ÄKTA START purification system (GE Healthcare). The column had been previously equilibrated with the lysis buffer mentioned above.
000 rpm (Sorvall SS34 rotor) at 4 °C for 30 min. The supernatant was recovered and filtered through a 0.22 μm syringe filter (Millipore) and injected to a nickel–nitrilotriacetic acid (Ni–NTA) agarose column (Qiagen) connected to an ÄKTA START purification system (GE Healthcare). The column had been previously equilibrated with the lysis buffer mentioned above.
After the protein solution addition, the bound fluorescent proteins were washed with the same lysis buffer to elute unbound proteins. eGFP and mCherry were later eluted with a linear gradient (0–100%) of a 500 mM imidazole, 300 mM NaCl, 50 mM potassium phosphate buffer at pH 8. The recovered proteins were dialysed against dH2O for 16 h using a 10MWCo dialysis membrane. Finally, the proteins were collected and stored at −20 °C.
2.4 Concentration of fluorescent proteins
Dialysed proteins were filtered through a 0.22 μm syringe filter (Millipore) and concentrated by reducing a volume of ∼30 mL to ∼1 mL using protein 30 kDa concentrators (ThermoFisher Scientific) at 5000 rpm and 4 °C for the time required to reach the desired volume. The protein concentration was determined by measuring the absorbance at λeGFP = 488 nm and λmCherry = 587 nm, using molar extinction coefficients values of εeGFP = 56000 M−1 cm−135 and εmCherry = 72000 M−1 cm−1.36
2.5 Network formation of native and cationised eGFP and mCherry with yttrium chloride
Different yttrium chloride (YCl3, Sigma Aldrich) concentrations (1, 2, 5, 10, 50 mM) were added from stock solutions to 30 μL of 7 mg mL−1 solutions of native and cationised proteins, separately. The solutions were mixed in a vortex and analysed immediately after.2.6 Network formation of eGFP with native and cationised mCherry with ammonium sulphate
Different amounts of ammonium sulphate, (NH4)2SO4, Sigma Aldrich) were increasingly added to a 100 μL of 7 mg mL−1 total mixture of eGFP and native mCherry to reach final salt concentrations from 0.3 to 3 M. Additionally, to test its effect on modified proteins, (NH4)2SO4 was added to 50 μL of 7 mg mL−1 solutions of cationised mCherry (see section below) for a final concentration of 3 M of the salt. All samples were vortexed and analysed immediately after.2.7 Cationisation of mCherry
Protein cationisation was performed through the addition of a stock solution of hexamethylenediamine (HMDA, Sigma Aldrich) at a pH 6.0–6.5 whose concentration was 10 times that of the protein solution to 10 mL of a known concentration of native protein solution. The pH was adjusted to 6.0–6.5 with HCl 1 M. An equal concentration to the protein of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Sigma Aldrich) was added to the reaction at two different times: half of the required reagent was added after HMDA and the remaining half after ∼3 h of reaction. The pH was monitored constantly and adjusted to 6.0–6.5 as required for the first 6 h. The mixture was left stirring at room temperature overnight (∼18 h).37 Finally, the solution was filtered through a 0.22 μm syringe filter to remove any precipitates, dialysed against dH2O for 24 h and concentrated following the same procedure described before.2.8 Electrophoretic mobility measurement
We followed Roosen-Runge et al.31 to determine the effective charge of our proteins. We performed electrophoretic mobility μe measurements in a Zetasizer Nano ZS (Malvern, UK) at a detector angle of 13° and a 4 mW 633 nm laser beam. Using electrophoretic light scattering (ELS) via phase analysis light scattering (M3-PALS), the μe of the proteins is determined as an external electric field is applied. The zeta potential z for a spherical particle with radius a is then given by the equipment following: | (1) |
![[small zeta, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e126.gif) = (eζ)/(2kBT) through:31
= (eζ)/(2kBT) through:31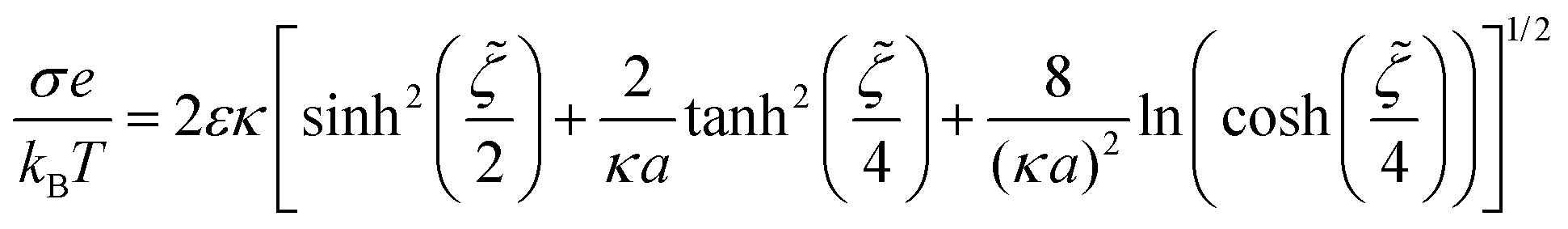 | (2) |
| Q = 4πa2σ | (3) |
As indicated by Roosen-Runge and collaborators,31 this approach assumes a spherical shape for the proteins and an isotropic ion distribution on their surfaces. This is not the case for our model proteins, and as such, the charges calculated should only be considered as effective charges suitable to understand the phenomena observed in our experiments.
All measurements were performed at 20 °C using 1 mL of 2 mg mL−1 of native and modified proteins in a NaCl 10 mM solution adjusted at a pH 7.4 with dropwise additions of either 0.1 M HCl solution or 0.1 M NaOH solution.
2.9 Size exclusion chromatography purification
Size exclusion chromatography purification was carried out for small angle X-ray scattering analysis of both proteins. eGFP and cationised mCherry were concentrated to ∼3 mL in a 25 mM Tris–base 150 mM NaCl buffer at pH 7.4. The proteins were applied to a HiLoad Superdex 75 16/600 size exclusion column using ÄKTA START purification system (GE Healthcare) pre-equilibrated with the same buffer. Protein elution was monitored at 280 nm.2.10 Small angle X-ray scattering measurements
Small angle X-ray scattering measurements were done on 25 μL of 10 mg mL−1 of both eGFP and cationised mCherry in a 25 mM Tris–base 150 mM NaCl buffer at pH 7.4, with increasing concentrations of YCl3 and (NH4)2SO4, respectively. All measurements were performed on a SAXSLAB Ganesha 300XL instrument with a wavevector q range of 0.006–0.30 Å−1. Samples were studied as prepared and no concentration corrections from potential protein aggregation were pursued. Background corrections were made with both an empty capillary, and with capillaries containing the same buffer and the corresponding salt concentrations. All the data were fitted using the SasView version 4.0 software package.382.11 Decorated network formation of native and cationised proteins
In order to form the desired decorated network, we propose a two-step condensation mechanism, where we precipitate one protein first, followed by the addition and subsequent condensation of the second protein. Our methodology is as follows: first, an eGFP backbone gel was formed by adding 5 mM YCl3 as described above to a 4 mg mL−1 solution of only eGFP. Then (NH4)2SO4 was added and dissolved for a final concentration of 3 M. Finally, 4 mg mL−1 of cationised mCherry were added to the solution, mixed for 5 min and analysed immediately. The final total protein concentration was kept at 8 mg mL−1. Three more samples following this protocol were prepared where the mass ratio of eGFP:cationised mCherry was varied to 2:1, 5:1 and 10:1.
2.12 Imaging of the protein networks
All samples were confined to capillaries with a square cross-section of 0.50 × 0.50 mm (Vitocrom) and sealed on the ends with Norland 61 optical adhesive. Confocal laser scanning microscopy Leica TCS with a white light laser emitting at 500 nm was used to study any gelation, using a NA 1.4 63× oil immersion objective, with a resolution of 0.1 μm in x and y and 0.6 μm in z. The channels used for the proteins were 488 nm for eGFP and 587 nm for native and cationised mCherry. Scans of the capillary in the z-axis were also acquired to analyse the network structure in 3d, where care was taken to assure the pixel size was equal in all axes (100 nm per pixel). Images of the binary networks were deconvolved with Huygens Professional version 15.05 (Scientific Volume Imaging, The Netherlands, http://svi.nl).2.13 Analysis of the mixing and domain sizes of eGFP and cationised mCherry in the binary networks
A 3d scan of the networks was obtained and analysed individually to measure the percentage of mixing and de-mixing of the proteins in the binary network, along with the sizes of de-mixed domains. Care was taken to obtain images with the same sizes and the intensities of the images was normalised for all of them. Then, the pixels were classified according to their intensities as yellow (mixed proteins), green (eGFP) and red (cationised mCherry). Different intensity thresholds were tested to optimise the results. The percentage of each colour was obtained to study the mixing of the proteins. Additionally, the volumes of the identified regions of individual proteins (de-mixed domains) were measured by counting the number of pixels on said domains, which were then converted to μm3 using the pixel size.2.14 Fractal dimensions of the mixing and domain sizes of eGFP and cationised mCherry in the binary networks
The fractal dimension, df, describes how the number N of domains i identified as eGFP, cationised mCherry or mixed proteins, depend on their domain size, i.e., their radius of gyration, Rig, following:39 | (4) |
 | (5) |
3 Results and discussion
We present our results and discussion in four main sections. The first section corresponds to tuning the protein–protein interactions to gain sufficient control over the assembly process using small-angle X-ray scattering (SAXS). We then proceed to assemble the desired structures following the two-step process of assembling a network of eGFP which is subsequently decorated with mCherry. In the third section, we demonstrate tuning of the protein domain sizes within the network. Finally, we obtained the fractal dimension of the decorated networks to characterise their structure.3.1 Controlling specificity of protein interactions
To suppress non-specific interactions between the salts and the proteins, we require high protein–salt specificity. To achieve this, we focus on yttrium chloride (YCl3).31 While iron chloride would in principle be an alternative, precise control over the pH during the assembly process is important and we are more confident of this with YCl3. The results are illustrated in Fig. 3a and Fig. S1a (ESI†) where eGFP forms a network readily upon the addition of Yttrium Chloride.So that the yttrium chloride only interacts with the eGFP, we cationise the mCherry, which suppresses its interaction with the metal cation due to the positive charge on the protein resulting from the cationisation. We determined the cationised mCherry (c-mCherry) ζ-potential at pH = 7.4 and calculated its effective surface charge Q31 (see Methods section for further details). We observed a charge inversion for the cationised mCherry (ζc-mCherry = +9.3 mV and Qc-mCherry = +7.51 e) from its native negative counterpart (ζmCherry = −7.0 mV and QmCherry = −5.90 e).
To test if we have effectively “blocked” the interaction between YCl3 and c-mCherry, we mixed solutions of c-mCherry against the same concentrations of salts as for the eGFP. We did not observe any assembling for the c-mCherry at any concentration of YCl3 up to 50 mM as shown in Fig. S2a (ESI†). Finally, we demonstrated assembly through non-specific interactions using ammonium sulphate,29 (NH4)2SO4. To destabilise c-mCherry, we added 3 M of ammonium sulphate to a 7 mg mL−1 solution of the protein. Images of the results obtained are shown in Fig. 3b and Fig. S2b (ESI†) where the cationised protein formed networks whilst retaining its fluorescent properties.
We thus obtained a system where eGFP forms a network through specific interactions with YCl3, whereas the cationisation of mCherry successfully modified the net charge of the protein from negative to positive and the assembly of the cationised protein with said salt was effectively avoided. However, the non-specific interactions with ammonium sulphate were not affected and c-mCherry assembled readily with this salt. We used these specific and differential interactions to yield the decorated networks.
3.2 Determination of protein–protein interactions
In order to determine the interactions in our system, we conducted small-angle X-ray scattering (SAXS) studies on protein solutions (10 mg mL−1) with different salt concentrations: eGFP with YCl3 and c-mCherry with (NH4)2SO4, all below the precipitation concentration. We chose to consider solutions below the precipitation concentration because under these conditions it is possible to relate the protein–protein interactions to the static structure factor S(q).14 Additionally, we further purified the proteins through size exclusion chromatography to eliminate any impurities that might interfere with the measurements. However, we did not filter the samples after salt addition and thus they were studied as prepared. We consider solutions of one species only for simplicity and also because each species is in solution at different stage of the assembly sequence (Fig. 1). Further details are given in the Methods section. The scattering intensity, I(q) is related to the product of the form factor P(q) and the static structure factor S(q). I(q) = ϕVprotein(Δρ)2P(q)S(q) where ϕ is the volume fraction of the proteins, Vprotein is the volume of the protein, Δρ is the difference in scattering length density between the proteins and its supporting solvent. Under certain assumptions, the structure factor is uniquely determined by the pair interaction potential between the proteins.18,40,41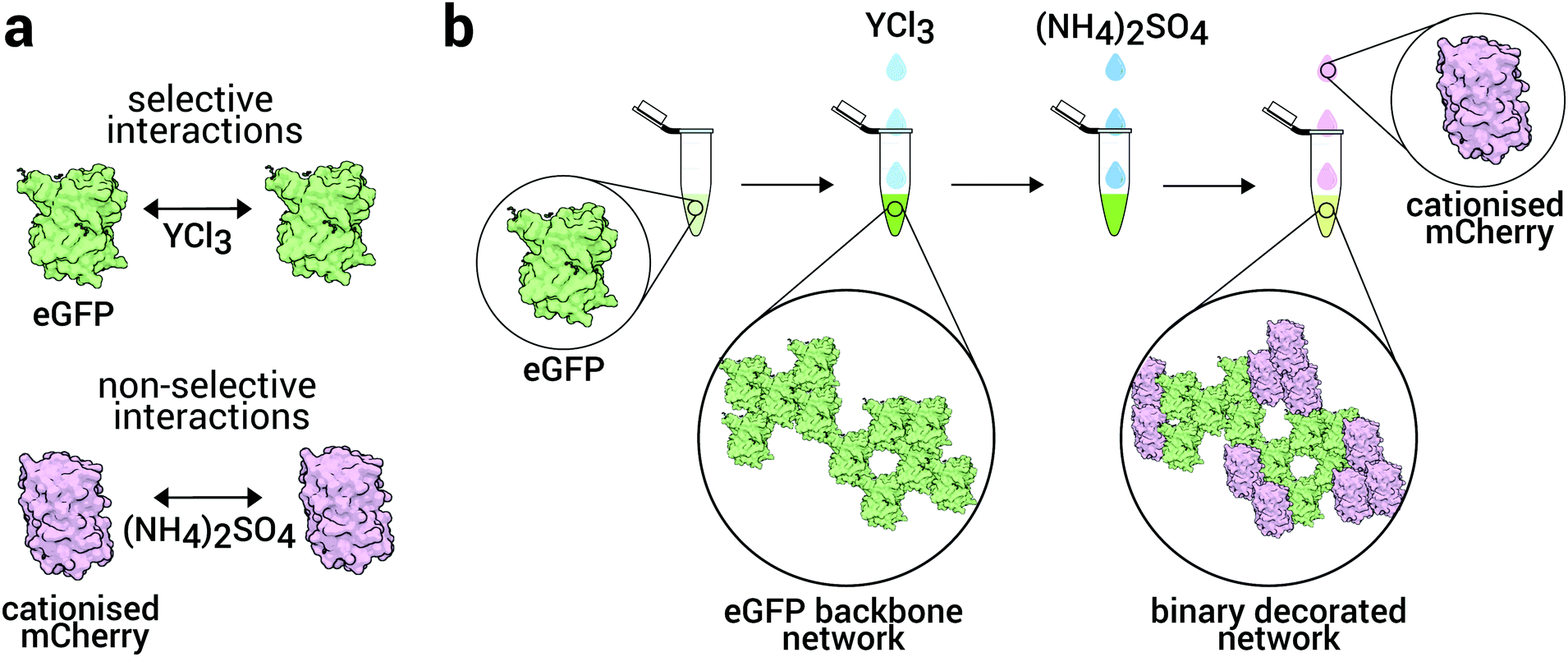 | ||
| Fig. 1 Strategy to yield decorated protein networks. (a) Specific interactions of trivalent Y3+ ions and eGFP and protein surface modification (cationisation) of mCherry are exploited to provide and control the specificity of protein–salt interactions to yield a decorated network with distinguishable protein domains. (b) Two-step design methodology, where a backbone network of eGFP is formed first, followed by its decoration with cationised mCherry domains. | ||
The scattering intensities for eGFP and c-mCherry are shown in Fig. 2a and c, respectively. It has been previously found that interactions in globular protein solutions in the presence of salts resemble those of charged particles with short-range attractive potentials.30–32,42–44 In our system, in the absence of added salt we obtain a system with electrostatic repulsions. When the salt concentration is increased, we find a reduction of the strength of the repulsive interactions due to attractions between the proteins by either forming bridges (YCl3)30–32,43 or by decreasing their solubility through non-specific interactions ((NH4)2SO4).29,45
 | ||
| Fig. 2 Determining protein–protein interactions with small angle X-ray scattering (SAXS). (a) and (c) SAXS scattering intensity with a cylinder and a Ornstein–Zernike model fitting for 10 mg mL−1 protein solutions of eGFP with different YCl3 concentrations and a cylinder fitting of cationised mCherry with various (NH4)2SO4 concentrations, respectively. (b) Evolution of the lengths of the protein dimers and small aggregates and correlation lengths, ξ of eGFP at different YCl3 concentrations. (d) Evolution of the length of cationised mCherry as (NH4)2SO4 concentration is increased. | ||
In order to determine these interactions we first fitted our SAXS data with a cylindrical form factor46 to obtain information on the dimerisation and aggregation of small numbers of proteins. The results obtained are shown in Fig. 2a and c and in Tables SI and SII (ESI†), where we found different behaviour for each protein. It is worth noting that we sought to describe the protein–salt behaviour in our gel formation strategy, thus, we did not filter any of the samples to eliminate small aggregates, nor did we perform any concentration corrections in the data to account for the possible formation and precipitation of the former.
Previous work on eGFP revealed that the protein exists in dimers.46 When no YCl3 is present, SAXS data indicates the presence of cylinders with a diameter of 30 Å and lengths of ∼80 Å, consistent with dimers as illustrated in the inset in Fig. 2a (Table SI, ESI† and Fig. 2b). As the concentration of YCl3 is increased, the length of the cylinders we fit also increases, indicating protein association. This is also shown as an upturn in S(q) at low wavevector q in Fig. 2a. To fit this q range we used the product of the cylinder form factor and the Ornstein–Zernike (OZ) form for the structure factor. We obtained the correlation length, ξ, of the density fluctuations, which we interpret as transient clusters, using S(q) ∼ [1 + (qξ)2]−1.47 The results are shown in Fig. 2b, where we found an increase in the correlation length ξ as the salt concentration is increased. However, we do obtain large errors for these fittings. We then can only interpret these results as an indication of sequential protein association, following yttrium ions bridging.30 The fitted parameter for the form factor and structure factor indicate we have charge-stabilised eGFP dimers between which the repulsion is decreased as YCl3 is added to the system as illustrated in the inset in Fig. 2a.
On the other hand, c-mCherry presents a completely different behaviour upon addition of ammonium sulphate. Our form factor fitting where no salt is present in the protein solution indicates the presence of monomers of ∼32 Å diameter and length of ∼50 Å, consistent with monomers of mCherry48 (see Table SII, ESI† and inset in Fig. 2c). The small size increase is likely due to the cationisation. For c-mCherry, blue we only observe a change of the form factor when a high salt concentration (close to the gel formation concentration of 1.6 M) is added to the system, as shown in Fig. 2d. Moreover, there is a lack of an increase in intensity at low q as observed for eGFP and the cylinder form factor alone is enough to describe c-mCherry. The upturn observed is independent of the salt concentration and is likely due to aggregates formed during sample manipulation. It is worth highlighting the big difference on the background signal at high q for salt concentrations above 0.75 M (Fig. 2c). Further investigation in this respect, showed this signal came from some protein denaturation. However, we still observed significant chromophore fluorescence of this protein even at much higher salt concentrations, as shown in the previous section. Due to the presence of salts and the formation of aggregates, we faced limitations to assess protein stability via conventional techniques such as circular dichroism, for example. Thus, to further investigate the level of protein folding of cationised mCherry, we measured its the emission fluorescence at different temperatures. This represents a sensitive metric of structural integrity as the chromophore hydrolyses when exposed to water.49 The results are shown in Fig. S3 in the ESI,† where a clear decrease of emission signal is observed as the temperature is increased, and it is lost above 80 °C. From these results we can conclude that the unfolded protein looses its fluorescence and will not contribute to the signal observed in the gels.
Thus, unlike eGFP where association and dimerisation is controlled continuously by adding YCl3, in the case of c-mCherry, we have charge-stabilised c-mCherry monomers whose attraction is only abruptly increased when a sufficiently large amount of (NH4)2SO4 is added to the solution, as illustrated in the sketch in Fig. 2b.
3.3 Two-step formation of decorated protein networks
As outlined in the introduction, we first assemble a network of eGFP via the specific interactions of the trivalent salt, and then decorate with c-mCherry via the addition of the nonspecific ammonium sulphate interactions. Building on our investigation of the interactions between the proteins discussed above, to assemble a robust eGFP backbone network, we prepared a system of eGFP at 4 mg mL−1 with a trivalent salt concentration of YCl3 at 5 mM. The resulting network is shown in Fig. 3a. We also tested the stability of this network in the presence of ammonium sulphate. The structure is shown in Fig. S4 (ESI†), where we do not observe major changes in the gel structure in comparison with the gels shown in the figure mentioned before. We proceeded to prepare an c-mCherry solution at 4 mg mL−1 and tested its assembly first in the absence of the eGFP, using a saturated ammonium sulphate concentration of 3 M to ensure full aggregation, as shown in Fig. 3b. | ||
| Fig. 3 Deconvolved confocal images of individual and decorated eGFP and cationised mCherry networks. (a) eGFP network formed through the addition of yttrium chloride. (b) Cationised mCherry network formed by adding ammonium sulphate. (c and d) Confocal images of the expected decorated gel networks with distinctive eGFP (green) and cationised mCherry (red) domains. 4 mg mL−1 of eGFP were mixed on their own with 5 mM of yttrium chloride to form a gel. 3 M of ammonium sulphate is added followed by 4 mg mL−1 of cationised mCherry at pH = 7. Inset in (c) rendering of a binary network from confocal microscopy images showing a percolating structure in 3d. Scale bars = 10 μm. | ||
We assemble the decorated network, as follows. To the eGFP network (volume 15 μl), we added ammonium sulphate and then c-mCherry solution (volume 5 μl), such that the concentrations in the final solution of (volume 20 μl), was 4 mg mL−1 for both the eGFP and mCherry, 5 mM for the YCl3 and 3 M for the (NH4)2SO4. The results of this approach are shown in Fig. 3c and d, where we can observe a backbone eGFP network decorated with c-mCherry.
3.4 Tuning the coverage of the backbone eGFP network with cationised mCherry
The eGFP network coverage exhibits clearly identifiable domains of both eGFP and c-mCherry. This indicates that they are at least predominantly composed of one of the proteins. To confirm that the structures found are indeed 3d networks in Fig. 3c inset we show a 3d rendering of confocal microscopy image data where we see percolation in all three dimensions. It is worth highlighting that the elongation in the z plane is related to the limited optical resolution.Thus, the strategy proposed successfully produced decorated networks with distinctive protein domains. However, there are still large yellow areas (overlay of green and red channels), indicating that c-mCherry deposited directly on the surface of the preexisting eGFP network which we will refer as “mixture” below. Both observations are clearer at higher magnifications (Fig. 3d).
We further confirmed that the strategy followed above to increase salt–protein specificity is indeed required to yield the decorated structures. Rather than the two-step approach of decorating a gel of eGFP and then adding mCherry, we mixed solutions of eGFP and mCherry and added several (NH4)2SO4 salt concentrations. The results are shown in Fig. S5 in the ESI.† No assembly occurs before 1 M of (NH4)2SO4, and we only start to find clusters of ∼5 μm formed solely by eGFP up to 1.5 M of (NH4)2SO4. However, we start observing co-precipitation of the proteins at a salt concentration of 1.6 M (NH4)2SO4, where the proteins are indistinguishably mixed on the lengthscales we access. This is more evident as the salt concentration increases. Full co-precipitation is observed at 3 M of (NH4)2SO4.
This coprecipitation is due to non-specific interactions between the proteins due to the addition of (NH4)2SO4. Indeed, the isoelectric point and acidic surface residues of the two proteins are similar and only a few salt (Ca2+ and SO42−) specific sites are present in eGFP, as shown in Table SIII (ESI†).50,51 One of these is a site for a sulphate group for eGFP.‡
Therefore, choosing salts with specific protein interactions and the modification of the surface of the proteins carried out above are essential for the successful decorated network formation.
Having assembled the desired architecture, we then investigated whether we could control the coverage of the eGFP network and the domain sizes of eGFP and c-mCherry. To do this, we decreased the concentration of c-mCherry to 0.5, 0.2, and 0.1 of the eGFP concentration, which was kept at 4 mg mL−1, similar to the strategy proposed by de las Heras et al. on simulations of binary mixtures of patchy spheres.25Fig. 4a shows the percentages of protein domains identified as eGFP (green), c-mCherry (red) and mixture (yellow) according to the different eGFP:c-mCherry composition. Unexpectedly, as we decreased the amount of c-mCherry, the percentage of domains identified as eGFP also decreased slightly, whereas the percentage of mixed network increased (Fig. 4a), making almost half of the network composition when the eGFP:c-mCherry ratio is 10:1. The small decrease of eGFP coverage suggests that the increase in mixing occurs mainly at expense of individual domains of c-mCherry, however, we would expect to see a more significant reduction of the regions identified as eGFP domains.
 | ||
| Fig. 4 Control over eGFP gel coverage with cationised mCherry. (a) Comparison of individual regions of eGFP (green), cationised mCherry (red) and mixture (yellow) in the binary network when the eGFP:cationised mCherry concentration ratio of the proteins is varied from 1:1, 2:1, 5:1, 10:1. (b) Comparison of the distribution of the domain volume size of individual regions of eGFP (left), cationised mCherry (centre) and mixture (right) in the decorated networks. The eGFP:cationised mCherry concentration ratio is varied from 1:1 (pink), 2:1 (purple), 5:1 (blue) and 10:1 (turquoise). (c) and (d) Cationised mCherry competition between forming clusters before their deposition (c: c-mCherry rich) or depositing directly on the pre-existing eGFP backbone (d: c-mCherrry poor). | ||
Finally we measured the sizes of the eGFP, c-mCherry and mixture domains to see if their distribution also changed with the different ratios of protein concentration tested. To carry out this analysis, the neighbours of pixels identified as a particular domain type were counted and the number of pixels per domain was obtained. The resulting probability distribution functions are shown in Fig. 4b, where the left panel shows the size distribution of eGFP domains, the centre one corresponds to c-mCherry and the right panel pertains to the domains of mixed proteins.
The plots show that as the concentration of c-mCherry is decreased, the amount of small domain sizes also decreases for eGFP and mixed protein regions. Such reduction in domain sizes is more evident at 5:1 and 10:1 protein ratios (pink and turquoise colours, respectively in Fig. 4b). However, c-mCherry shows a different behaviour, with a general reduction of the amount of protein domains consistent with the previous analysis. A reduction of the sizes of eGFP and mixed protein domains, but not of eGFP gel coverage, might be a consequence of non-homogenous and more scattered deposition and mixing of c-mCherry as its concentration is reduced. Thus, the sizes of protein domains and the protein mixing can be manipulated by modifying the composition of the system.
3.5 Two mechanisms for network decoration
This unexpected behaviour of increasing protein mixing by c-mCherry upon decreasing the concentration suggests that more than one mechanism of c-mCherry assembly onto the network is at play. We propose the following.• If the amount of regions identified as c-mCherry increases or remains the same as the quantity of this protein is reduced, there may be a preference for aggregation between like proteins (eGFP–eGFP and c-mCherry–c-mCherry). This might occur because at high concentrations c-mCherry is more likely to encounter more protein of its kind in solution. As a result, before depositing on the surface of the pre-existing eGFP gel, c-mCherry will aggregate with itself, forming small clusters, as illustrated in Fig. 4c (c-mCherry rich).
• On the other hand if the protein mixing increases as less c-mCherry is added, then the likelihood of c-mCherry to come in contact with eGFP increases since there is now less c-mCherry available in solution, and thus the precipitating c-mCherry will deposit directly on the surface of the pre-existing eGFP gel. This mixing and deposition is non-homogenous, leading to small domain sizes of both eGFP and mixed protein [Fig. 4d (c-mCherry poor)].
We believe that the c-mCherry coverage as a function of reducing the quantity added may be due to a competition between these two different extreme scenarios arising from varying the concentration of c-mCherry solely. At high c-mCherry concentrations, like-protein encounters dominate with individual domains of c-mCherry forming, prior to decorating the eGFP network. However, as we decrease the concentration of c-mCherry, then eGFP–c-mCherry encounters seem to dominate and the latter preferentially mixes with the eGFP gel, leading to smaller domain sizes of individual proteins.
3.6 Gel fractal dimensions
Since our gels are constituted by individual and mixed domains of two proteins, we can describe their structure through the connectivity of the eGFP, c-mCherry and mixed clusters via their fractal dimension, df.52–54 As discussed in the previous section, the size distribution of domains changes according to the different protein concentrations used, thus, df can also give information about how the overall gel structure might be altered by changing the amount of protein. The values obtained for the df of each domain are plotted in Fig. 5b according to the eGFP:c-mCherry ratio tested. All the values obtained lay within a range of 2.2–2.6, consisting with df ∼ 2.5 characteristic of percolating clusters, where the structure expands in 3d without filling all the available space.52,53,55 Additionally, this value of df is closer to gels pertaining to the reaction-limited cluster aggregation (RLCA) regime (df ∼ 2). This classification is based on the kinetics of cluster aggregation, and for the case of RLCA, the formation rate is limited by the probability of particles bonding upon collision.56 These results further support our picture of the two mechanisms at play for the network decoration, where the preferential aggregation of c-mCherry with other c-mCherry in solution or with the pre-existing eGFP gel depends on the likelihood of the protein finding one or the other first. Despite of the changes in gel composition, we do not observe significant variation on the df for any of the different protein ratios. This might be due to the fact that the initial gel consisted of only eGFP and the deposition of c-mCherry and/or protein mixing occurred on top of it. Since the concentration of eGFP remained constant, so did the overall structure of the gel.
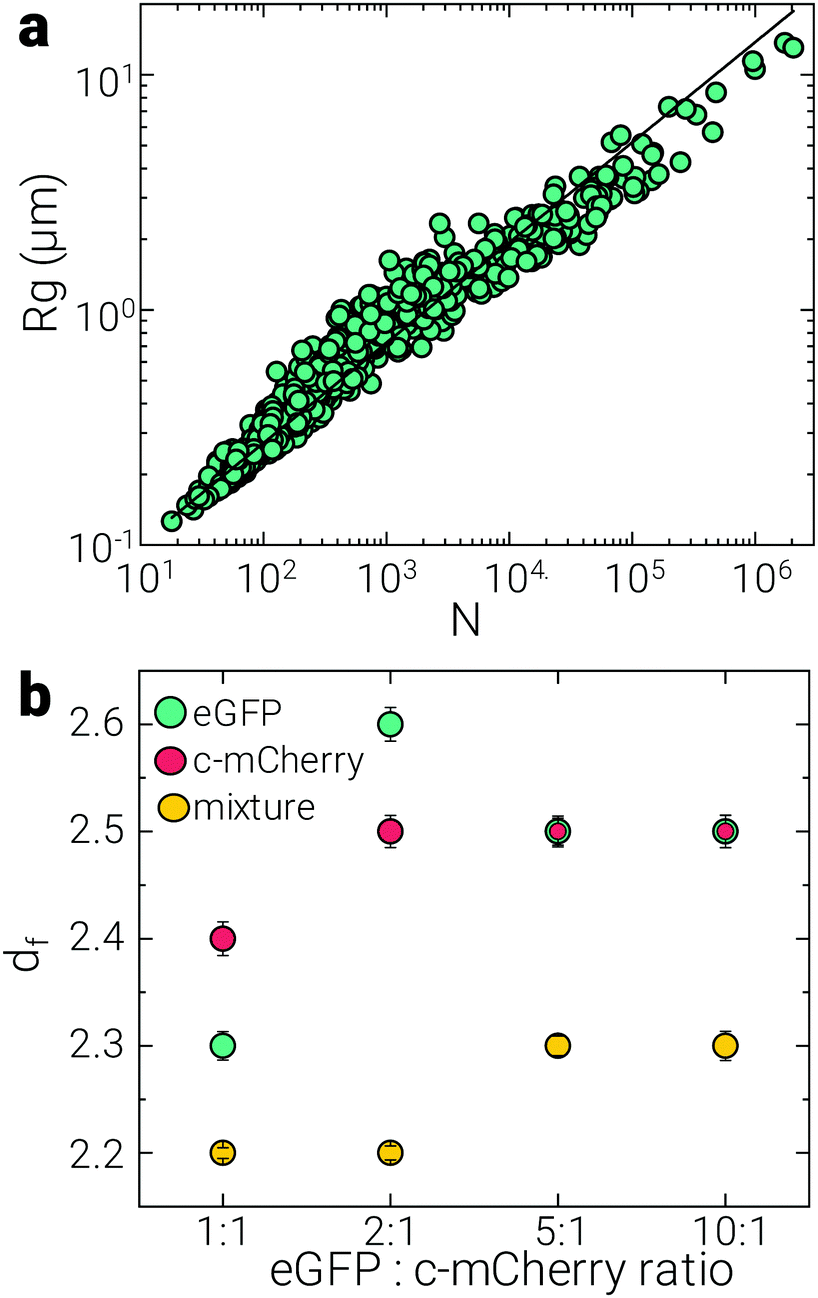 | ||
| Fig. 5 Gel structural analysis. (a) Radius of gyration of eGFP clusters as a function of the number of eGFP domains shown to illustrate the calculation of the fractal dimension, df (b) df of eGFP (green), cationised mCherry (red) and mixture (yellow) domains according to the different eGFP–cationised mCherry gel compositions. Note: for eGFP–cationised mCherry ratios 5:1 and 10:1, eGFP and cationised mCherry domains have the same df = 2.5. The latter have been plotted in a smaller size for clarity. | ||
4 Conclusions
By carefully controlling interactions in a binary protein system, we have assembled a nanoarchitecture with tunable domain sizes. In particular we have used specific interactions between a trivalent ion (Y3+) to assemble a backbone network of eGFP. In the second step of assembly, we add another protein mCherry, which has been cationised such that it does not interact with the trivalent ion. In order that the mCherry binds to the eGFP network, we use a nonspecific salt, ammonium sulphate. Our assembly approach leads to mesoscopic domains of mCherry on the eGFP network. By tuning the composition of the two proteins, we find that the domain size can be tuned, and we deduce that this is due to competing assembly mechanisms of the mCherry onto the network: at low mCherry concentrations, mCherry–eGFP encounters dominate, leading to more protein mixing and smaller domain sizes. Increasing the mCherry concentration leads to mCherry–mCherry encounters dominating instead, such that aggregates of mCherry form prior to decorating the eGFP network and decreasing mixing.An interesting possibility for the future would be to develop computational approaches similar to the ground breaking work on the crystallisation of rubredoxin by Fusco et al.57 This would help to shed further light into the protein–salt interactions as well as the self-assembly and network decoration mechanisms at play. However, in making analogy with colloidal systems, we believe that the networks formed here are consistent with spinodal gelation,53,58 pertaining to systems where strong attractions, without significant directional dependence drive gelation, as has been observed previously in lyzozyme gels.59 In this sense, these are non-equilibrium structures and on long timescales could perhaps condense further.54
Our work emphasises the complexity of protein–salt interactions and highlights the need for further studies to understand better the specific interactions involved to obtain the desired structures. To this end, we have investigated selective control of protein–protein interactions, fundamental to realising the goal of tunable multi-protein architectures at mesoscopic lengthscales. Additionally, we have shown the structures formed are networks and determined the protein–protein interactions with small angle X-ray scattering and found behaviour consistent with theory.18
By using fluorescent proteins to monitor the protein stability, we provide some evidence that the structure may be preserved during the self-assembly process, since only folded proteins will contribute to the fluorescence corresponding to their fluorophore observed in the gels. As such, binary mixtures of proteins with enzymatic, charge-carrying, antibiotic and light-harvesting abilities can be designed de novo and assembled into micron-sized porous networks through the methodology developed herein.
Compartmentalisation and/or immobilisation of enzymes allows for the combination of complex multi-step reactions and mimics the cascade metabolic pathways found in nature. Additionally, the enzyme close proximity resulting from it, might improve synergistic interactions between enzymes involved in sequential reactions.60,61 For applications, immobilisation also improves enzyme operational, thermal, mechanical and storage stability.62 Efforts to immobilise single or mixtures of enzymes have been obtained before in so called cross-linked enzyme aggregates (CLEAs), where the proteins are mixed together in solution, precipitated and crosslinked.60–62 In these aggregates the protein catalytic activity is comparable with their counterparts in solution.60 In contrast to CLEAs, our approach offers a better control over the assembly and the interactions of binary protein structures. In addition, the porous structure on our gels, might also facilitate the flux of substrates and products, and might help with the recovery of the final compounds of interest.
Moreover, the proteins involved in our decorated networks do not require compatibility or specificity on their mutual interactions. Additionally, group-specific cross-linking of the decorated networks can provide further stability and will allow the elimination of the precipitating salts for future applications. Finally our methodology is straightforward, cheap, versatile and scalable, and hence, it opens the door for new strategies to produce a novel class of innovative functional biomaterials beyond the fluorescent proteins we have used here to demonstrate the method. For instance, a similar approach with enzymes can yield advanced materials where sequential multi-enzyme catalysis can be incorporated; light harvesting arrays can be obtained for energy transduction; and electron transfer proteins can form small scale electronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Nicholas Wood, Yushi Yang and Levke Ortlieb for providing the code for image analysis. The authors are grateful for very enriching discussions with Peter Schurtenberger and Bob Evans, and to Jennifer McManus for a critical reading of the manuscript. The work of IRdA and JR was funded by a Leverhulme Trust Philip Leverhulme Prize. IRdA was supported by a doctoral scholarship from CONACyT. JR acknowledges support from the European Research Council Grant DLV-75918. IRdA, AC-P and CPR gratefully acknowledge the ERC Grant agreement no. 617266 “NANOPRS” for financial support and Engineering and Physical Sciences Research Council (EP/H022333/1). The Ganesha X-ray scattering apparatus used for this research was purchased under EPSRC Grant “Atoms to Applications” (EP/K035746/1). This work benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547.Notes and references
- C. N. Lam and B. D. Olsen, Soft Matter, 2013, 9, 2393 RSC.
- C. S. Thomas, M. J. Glassman and B. D. Olsen, ACS Nano, 2011, 5, 5697 CrossRef CAS PubMed.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189 CrossRef CAS PubMed.
- X. Zongwei, W. Liyang, F. Fengzhou, F. Yongqi and Z. Yin, Curr. Nanosci., 2016, 12, 725 CrossRef.
- Y. Bai, Q. Luo and J. Liu, Chem. Soc. Rev., 2016, 45, 2756 RSC.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571 CrossRef CAS PubMed.
- N. Kobayashi, K. Inano, K. Sasahara, T. Sato, K. Miyazawa, T. Fukuma, M. H. Hecht, C. Song, K. Murata and R. Arai, ACS Synth. Biol., 2018, 7, 1381 CrossRef CAS PubMed.
- L. Yang, A. Liu, S. Cao, R. M. Putri, P. Jonkheijm and J. J. L. M. Cornelissen, Chem. – Eur. J., 2016, 22, 15570 CrossRef CAS PubMed.
- W. M. Park and J. A. Champion, J. Am. Chem. Soc., 2014, 136, 17906 CrossRef CAS PubMed.
- T. Vo-Dinh, in Protein Nanotechnology, ed. T. Vo-Dinh, Humana Press, Totowa, NJ, 2005, pp. 1–13 Search PubMed.
- D. W. Watkins, J. M. X. Jenkins, K. J. Grayson, N. Wood, J. W. Steventon, K. K. Le Vay, M. I. Goodwin, A. S. Mullen, H. J. Bailey, M. P. Crump, F. MacMillan, A. J. Mulholland, G. Cameron, R. B. Sessions, S. Mann and J. L. R. Anderson, Nat. Commun., 2017, 8, 358 CrossRef PubMed.
- A. J. Simon, Y. Zhou, V. Ramasubramani, J. Glaser, A. Pothukuchy, J. Gollihar, J. C. Gerberich, J. C. Leggere, B. R. Morrow, C. Jung, S. C. Glotzer, D. W. Taylor and A. D. Ellington, Nat. Chem., 2019, 11, 204 CrossRef CAS PubMed.
- A. Ciach, J. Pekalski and W. T. Góźdź, Soft Matter, 2013, 9, 6301 RSC.
- M. Doi, Soft Matter Physics, Oxford Univ. Press, 2013 Search PubMed.
- A. C. Dumetz, A. M. Chockla, E. W. Kaler and A. M. Lenhoff, Biophys. J., 2008, 94, 570 CrossRef CAS PubMed.
- J. J. McManus, P. Charbonneau, E. Zaccarelli and N. Asherie, Curr. Opin. Colloid Interface Sci., 2016, 22, 73 CrossRef CAS.
- E. van der Linden and A. Foegeding, Gelation: Principles, models and applications to proteins, in Modern Biopolymer Science, ed. S. Kasapis, J. B. Norton and I. T. Ubbink, Elsevier, 2009, ch. 2, p. 29 Search PubMed.
- F. Zhang, M. W. A. Skoda, R. M. J. Jacobs, R. A. Martin, C. M. Martin and F. Schreiber, J. Phys. Chem. B, 2007, 111, 251 CrossRef CAS PubMed.
- Y. Wang, A. Lomakin, T. Hideshima, J. P. Laubach, O. Ogun, P. G. Richardson, N. C. Munshi, K. C. Anderson and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13359 CrossRef CAS PubMed.
- R. P. Sear, J. Phys.: Condens. Matter, 2007, 19, 033101 CrossRef.
- H. Wang, X. Yan, H. Aigner, A. Bracher, N. D. Nguyen, W. Y. Hee, B. M. Long, G. D. Price, F. U. Hartl and M. Hayer-Hartl, Nature, 2019, 566, 131 CrossRef CAS PubMed.
- A. Šarić, Y. C. Chebaro, T. P. J. Knowles and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17869 CrossRef PubMed.
- L. Di Michele, F. Varrato, J. Kotar, S. H. Nathan, G. Foffi and E. Eiser, Nat. Commun., 2013, 4, 2007 CrossRef PubMed.
- A. Blumlein and J. J. McManus, J. Mater. Chem. B, 2015, 3, 3429 RSC.
- D. de las Heras, J. M. Tavares and M. M. Telo da Gama, Soft Matter, 2012, 8, 1785 RSC.
- J. N. Immink, J. J. E. Maris, J. J. Crassous, J. Stenhammar and P. Schurtenberger, ACS Nano, 2019, 13, 3292 CrossRef CAS PubMed.
- A. H. Elcock and J. A. McCammon, Biophys. J., 2001, 80, 613 CrossRef CAS PubMed.
- K. C. Duong-Ly, S. B. Gabelli and J. Lorsch, Chapter Seven – Salting out of Proteins Using Ammonium Sulfate Precipitation, Academic Press, 2014, vol. 541, p. 85 Search PubMed.
- H. I. Okur, J. Hladlková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997 CrossRef CAS PubMed.
- F. Zhang, G. Zocher, A. Sauter, T. Stehle and F. Schreiber, J. Appl. Crystallogr., 2011, 44, 755 CrossRef CAS.
- F. Roosen-Runge, B. S. Heck, F. Zhang, O. Kohlbacher and F. Schreiber, J. Phys. Chem. B, 2013, 117, 5777 CrossRef CAS PubMed.
- F. Zhang, F. Roosen-Runge, A. Sauter, M. Wolf, R. M. J. Jacobs and F. Schreiber, Pure Appl. Chem., 2014, 86, 191 CAS.
- T.-Y. D. Tang, D. Cecchi, G. Fracasso, D. Accardi, A. Coutable-Pennarun, S. S. Mansy, A. W. Perriman, J. L. R. Anderson and S. Mann, ACS Synth. Biol., 2018, 7, 339 CrossRef CAS PubMed.
- R. Schleif, Trends Genet., 2000, 16, 559 CrossRef CAS PubMed.
- M. Kaishima, J. Ishii, T. Matsuno, N. Fukuda and A. Kondo, Sci. Rep., 2016, 6, 35932 CrossRef CAS PubMed.
- Y. Li, A. M. Sierra, H.-w. Ai and R. E. Campbell, Photochem. Photobiol., 2007, 84, 111 Search PubMed.
- R. C. Deller, B. M. Carter, I. Zampetakis, F. Scarpa and A. W. Perriman, Biochem. Biophys. Res. Commun., 2018, 495, 1055 CrossRef CAS PubMed.
- SasView https://www.sasview.org.
- D. Kroll and S. Croll, Polymer, 2017, 115, 154 CrossRef CAS.
- M. Wolf, F. Roosen-Runge, F. Zhang, R. Roth, M. W. A. Skoda, R. M. J. Jacobs, M. Sztucki and F. Schreiber, J. Mol. Liq., 2014, 200, 20 CrossRef CAS.
- P. Singh, A. Roche, C. F. van der Walle, S. Uddin, J. Du, J. Warwicker, A. Pluen and R. Curtis, Mol. Pharmaceutics, 2019, 16, 4775 CrossRef CAS PubMed.
- A. Stradner, H. Sedgwick, F. Cardinaux, W.-C. Poon, S. Egelhaaf and P. Schurtenberger, Nature, 2004, 432, 492 CrossRef CAS PubMed.
- F. Zhang, S. Weggler, M. J. Ziller, L. Ianeselli, B. S. Heck, A. Hildebrandt, O. Kohlbacher, M. W. A. Skoda, R. M. J. Jacobs and F. Schreiber, Proteins: Struct., Funct., Bioinf., 2010, 78, 3450 CrossRef CAS PubMed.
- R. Piazza and M. Pierno, J. Phys.: Condens. Matter, 2000, 12, A443 CrossRef CAS.
- P. T. Wingfield, Curr. Protoc. Protein Sci., 2001 DOI:10.1002/0471140864.psa03fs13.
- D. P. Myatt, L. Hatter, S. E. Rogers, A. E. Terry and L. A. Clifton, Biomed. Spectrosc. Imaging, 2017, 6, 123 CAS.
- J.-P. Hansen and I. Macdonald, Theory of Simple Liquids, Academic, London, 1976 Search PubMed.
- A. Huang, H. Yao and B. D. Olsen, Soft Matter, 2019, 15, 7350 RSC.
- J. Zhou, A. E. Panaitiu and G. Grigoryan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1059 CrossRef CAS PubMed.
- J. A. J. Arpino, P. J. Rizkallah and D. D. Jones, PLoS One, 2012, 7, e47132 CrossRef CAS PubMed.
- X. Shu, N. C. Shaner, C. A. Yarbrough, R. Y. Tsien and S. J. Remington, Biochemistry, 2006, 45, 9639 CrossRef CAS PubMed.
- M. Lattuada, H. Wu, A. Hasmy and M. Morbidelli, Langmuir, 2003, 19, 6312 CrossRef CAS.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- C. P. Royall, M. A. Faers, S. L. Fussell and J. E. Hallett, 2021, arXiv: 2104.04090.
- A. S. Balankin and B. E. Elizarraraz, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 056314 CrossRef PubMed.
- R. C. Ball, D. A. Weitz, T. A. Witten and F. Leyvraz, Phys. Rev. Lett., 1987, 58, 274 CrossRef PubMed.
- D. Fusco, J. J. Headd, A. De Simone, J. Wang and P. Charbonneau, Soft Matter, 2014, 10, 290 RSC.
- P. J. Lu, E. Zaccarelli, F. Ciulla, A. B. Schofield, F. Sciortino and D. A. Weitz, Nature, 2008, 453, 499 CrossRef CAS PubMed.
- F. Cardinaux, A. Stradner, P. Schurtenberger, F. Sciortino and E. Zaccarelli, Europhys. Lett., 2007, 77, 48004 CrossRef.
- L. T. Nguyen and K.-L. Yang, Enzyme Microb. Technol., 2017, 100, 52 CrossRef CAS PubMed.
- A. Bhattacharya and B. I. Pletschke, J. Mol. Catal. B: Enzym., 2015, 115, 140 CrossRef CAS.
- S. Rehman, H. N. Bhatti, M. Bilal and M. Asgher, Int. J. Biol. Macromol., 2016, 91, 1161 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm02269a |
| ‡ However the observation of the presence of these specific sites is related to the crystallisation process followed to determine the crystal structure in the PDB,50 and there may well be similar sites on mCherry. |
| This journal is © The Royal Society of Chemistry 2021 |