
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplementing Zn2+ ion and pH-value control into artificial mussel glue proteins by abstracting a His-rich domain from preCollagen†
Sandra
Arias
 a,
Shahrouz
Amini
b,
Jana M.
Krüger
a,
Lukas D.
Bangert
a and
Hans G.
Börner
*a
a,
Shahrouz
Amini
b,
Jana M.
Krüger
a,
Lukas D.
Bangert
a and
Hans G.
Börner
*a
aHumboldt-Universität zu Berlin, Department of Chemistry, Laboratory for Organic Synthesis of Functional Systems, Brook-Taylor-Str. 2, Berlin D-12489, Germany. E-mail: h.boerner@HU-Berlin.de
bMax Planck Institute of Colloids and Interfaces, Department of Biomaterials, Potsdam 14424, Germany
First published on 5th February 2021
Abstract
A His-rich domain of preCollagen-D found in byssal threads is derivatized with Cys and Dopa flanks to allow for mussel-inspired polymerization. Artificial mussel glue proteins are accessed that combine cysteinyldopa for adhesion with sequences for pH or Zn2+ induced β-sheet formation. The artificial constructs show strong adsorption to Al2O3, the resulting coatings tolerate hypersaline conditions and cohesion is improved by activating the β-sheet formation, that enhances E-modulus up to 60%.
Since decades the adhesion apparatus of marine mussels and related biochemical processes provide a source of inspiration, challenges and nanotechnology solutions across diverse disciplines.1–7 The bioadhesion process employs a mould deposition of a set of purpose adapted mussel foot proteins (mfps).2,8,9 In those mfps, the L-3,4-dihydroxyphenylalanine (Dopa) residues present catechol moieties that are considered to be key for adhesion10,11 and cohesion.12–14 This made Dopa and synthetic catechol analogues to constitute the basis of a rich family of mussel-inspired polymers.15–18
Awareness is rising, that in mfps the adsorption properties of Dopa are strongly modulated by neighbouring residues. Thus, not only Dopa, but also the Dopa sequence environment is essential to constitute the complexity of the bioadhesion functions.11,19
Abstracting these underlying concepts advent mussel-inspired polymers beyond Dopa-bearing instant adhesives. Such adhesives promise complex functions, which reach from enzymatically activated adhesion, to structurally defined adhesive sites, material specific adhesives, self-healing properties or activatable cohesion control mechanisms.18–27 Zhong and co-workers demonstrated on a protein level the possibilities by fusing the mfp3&5 that constitute the adhesive interface in the mussel glue apparatus with Amyloid Aβ, recognized for β-sheet formation.28 Mimicking functional aspects of these complex fusion proteins with artificial mussel glue proteins of reduced complexity would bridge the gap between cost-intensive proteins and synthetic Dopa-bearing polymers.29 Recently, a tyrosinase activated polymerization of mfp1 consensus decapeptide derivative was reported to broaden sequence complexity in mussel-glue inspired polymers.27,30 The sequence derivative contained Cys and Tyr residues, where Tyr could be enzymatically oxidized with AbPPO4 tyrosinase.31 This yields Dopa-quinones to which thiols of the Cys residues add by Michael-addition leading to cysteinyldopa connectivities in each repeat unit. To implement pH activatable cohesion control mechanisms, β-sheet forming [VT]n depsi-peptides with Tyr and Cys flanks were polymerized in a tyrosinase activated manner.27 The resulting artificial mussel glue proteins show fast and strong adsorption onto different surfaces with notable adhesion energies and proved to be resistant against hypersaline solutions of 4.2 M NaCl as found in the water of the Dead Sea. A pH triggered increase of the E-modulus was found as a result of activating internal β-sheet formation. Besides the enzyme induced polymerization strategy, a chemical activation route improves ease of accessibility, decouples the sequence from meeting tyrosinase substrate constrains and even enables the use of commodity monomer plattforms.32,33
While the adhesive plug of mussels with its remarkable adhesive interface is frequently in the focus of mussel-inspired glues, the byssus thread also exhibits exceptional material properties such as self-healing coatings, high-toughness fibres, and modulus matching.34–37 These properties are linked to rapid self-assembly of protein building blocks into gradient architectures and the collagenous protein derivative preCol is one of the proteins that constitute the core of the byssal threads.2 Harrington et al. investigated the role of the His-rich preCols to coordinate metal ions for inducing secondary structure transitions and constituting dynamic bonds for self-healing properties.38
Here we expand a histidine-rich-domain (HRD) from the preCol-D protein of the mussel byssus thread39 with Dopa and Cys residues to enable the mussel-inspired polymerization via the straightforward chemically activated route. The resulting artificial mussel glue proteins combine cysteinyldopa connectivities as Dopa derivatives at each repeat unit to mediate adsorption with a switchable β-sheet domain that can be triggered by either pH 4 → 7 or Zn2+ ions for cohesion control.
The sequence AVAHAHAHAHASAGANGRARAHARA (HRD) used in this study, originates from the N-terminal domain of preCol-D, a protein that is found in the distal region of byssal threads in the Mytilus species.39 The exact sequence section and length relate to a repetitive [HA]n motif, which was investigated previously.38 Harrington et al. proved the pH sensitivity and capability to coordinate metal ions e.g. Zn2+ or Cu2+. Both pH changes or the presence of certain ions induced in HRD a structural transition from statistical chain segment conformation to β-sheets, suggesting that HRD might play a role in the self-assembly process of the hierarchical structure in byssal threads.38
To polymerize the HRD and obtain an artificial mussel glue protein that might resemble preCol-D in aspects of triggered secondary structure formation, the HRD was extended N-terminally with Dopa (Y*) and C-terminally with Cys via a GG-spacer. The resulting 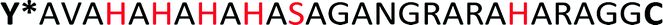 (Y*-HRD-C) was synthesized by solid-phase peptide synthesis, employing Fmoc-Dopa(acetonide)-OH as building block. After liberating Y*-HRD-C from the support, MALDI-TOF mass spectrometry confirmed the molecular structure (ESI,† Section S4).
(Y*-HRD-C) was synthesized by solid-phase peptide synthesis, employing Fmoc-Dopa(acetonide)-OH as building block. After liberating Y*-HRD-C from the support, MALDI-TOF mass spectrometry confirmed the molecular structure (ESI,† Section S4).
Y*-HRD-C is readily soluble in water or buffer in a pH window of 3–8 and chemical activation is feasible by oxidizing the Dopa residues to the corresponding Dopa-quinones (Fig. 1). By following the optimized conditions as described previously for the polymerization of a set of minimal Y*KC motifs,32 1.5 equiv. of NaIO4 lead to the formation of poly(Y*-HRD-C) under aqueous conditions. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis confirmed the polymer formation within 1 h by showing a broad polymer band in the apparent molecular weight range of 15–25 kDa (ESI,† Fig. S3). Consistent with the recently described chemically activated polymerization of tripeptides,32 the polymer formation was fast under the given conditions (room temperature, pH 7) and polymer growth was completed within the first hour as afterwards no significantly changes could be found in the SDS-PAGE (ESI,† Fig. S3). The polymerization proceeds efficiently in the pH window of 5.5–7.0, reaching the highest molecular weights at pH 7 (ESI,† Fig. S3). Below pH 5.5 and above pH 7 the polymer formation was more limited. This was expected, as under acidic conditions the protonation equilibrium shifts thiols toward reduced nucleophilicity and basic environment induces β-sheet formation that leads to aggregation of the HRD peptides.40 The isolated poly(Y*-HRD-C) was analysed by gel permeation chromatography (GPC), confirming the formation of soluble polymers with Mn,app = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 Da and Đ = 1.4 (ESI,† Fig. S4). MALDI-TOF-MS shows multimers up to tetramers and confirmed the expected repeat unit mass (ESI,† Fig. S5). 1H-NMR provided evidence for the formation poly(Y*-HRD-C), but resonances of cysteinyldopa superimpose with His side chains (ESI,† Fig. S6). Recent in-depth studies on the mussel-inspired polymerization of several different peptide sequences, provided clear analytical evidence for the formation of cysteinyldopa linkages and excluded alternative reaction pathways via didopa or lysinyldopa.27,30,32 Considering this, it appears to be valid to assume that Y*-HRD-C polymerizes via the cysteinyldopa pathway, too.
700 Da and Đ = 1.4 (ESI,† Fig. S4). MALDI-TOF-MS shows multimers up to tetramers and confirmed the expected repeat unit mass (ESI,† Fig. S5). 1H-NMR provided evidence for the formation poly(Y*-HRD-C), but resonances of cysteinyldopa superimpose with His side chains (ESI,† Fig. S6). Recent in-depth studies on the mussel-inspired polymerization of several different peptide sequences, provided clear analytical evidence for the formation of cysteinyldopa linkages and excluded alternative reaction pathways via didopa or lysinyldopa.27,30,32 Considering this, it appears to be valid to assume that Y*-HRD-C polymerizes via the cysteinyldopa pathway, too.
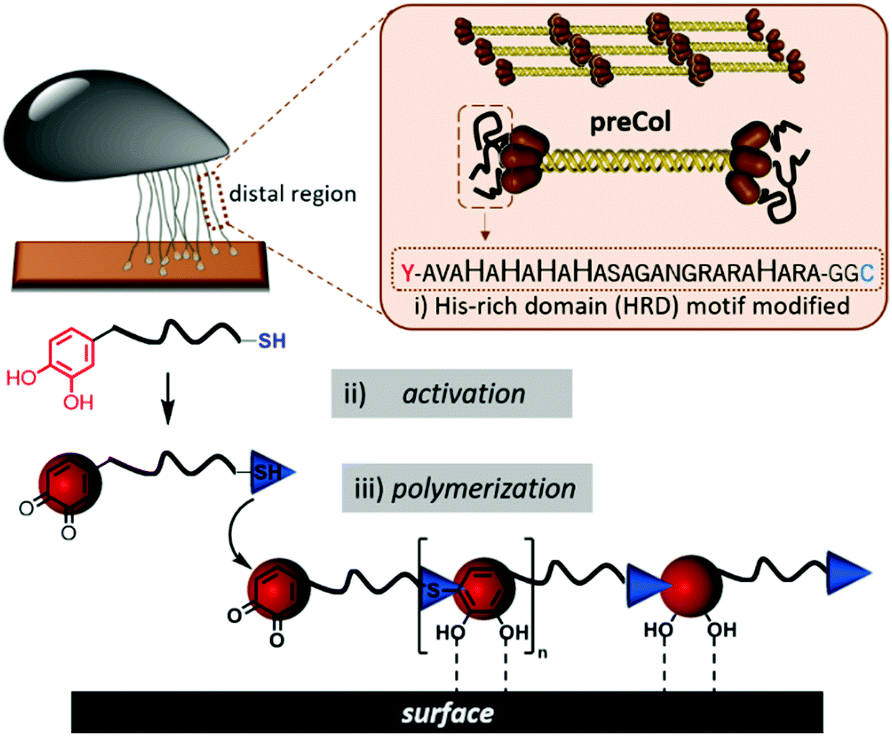 | ||
| Fig. 1 Illustration of the chemically activated mussel-inspired polymerization. Abstraction of the functional HRD peptide from preCol proteins and implementing of Dopa and Cys flanks for polymerization (i). Chemical activation with NaIO4 (ii) leads to polymerization (iii) of the HRD-peptide-based unimer to form the artificial mussel foot protein. | ||
The sequence of the poly(Y*-HRD-C) meets largely the hydrophobic–hydrophilic repeat pattern41 and matches well the β-strand periodicity.42 Moreover, with pKa = 6.8 of side chain for His a pH dependent β-sheet formation was described for HRD43 and might be anticipated for the polymeric construct as well. Infrared spectroscopy (IR) suggested the presence of β-sheet secondary structures for both unimer and poly(Y*-HRD-C) in the dry state. The IR spectra of the peptide and the polymer exhibit vibration bands in the amide I region at 1623 cm−1 and 1646 cm−1 which can be assigned to β-strands (Fig. S7, ESI†).44
By circular dichroism (CD) spectroscopy, secondary structures in solution at different pH-values were investigated. Where the Y*-HRD-C unimer adopts at pH 4–7 a statistical chain segment conformation, the poly(Y*-HRD-C) shows a pH triggered transition in secondary structure (Fig. 2 and ESI,† Fig. S8). The CD spectrum of the polymer exhibits at pH 4.0 cotton effects at (−)190 nm, indicating random coils. However, shifting the pH to 7.0 makes cotton effects at (−)223 nm evident, which are typical for β-sheets. It should be noted, that those CD spectra still indicate partial random coil structures. This was expected, as the cysteinyldopa linkages act as structural defects that cause intrinsically unstructured regions, which cannot integrate into β-sheets (Fig. 2). The pH responsive transition in secondary structure from random coil to β-sheet can be rationalized by the histidine rich face of the alternating sequence. The imidazole side chains of His residues are protonated at pH 4, improving solubility and preventing the peptide from adopting an all-trans conformation due to Coulomb repulsion. At pH 7 the His residues are mostly not protonated, enabling the segment to stretch out into an all-trans β-strand. Apparently, the conformational flexibility of the non-polymerized Y*-HRD-C peptide is not sufficiently reduced in solution to permit a structural transition, whereas the entropic penalty in the poly(Y*-HRD-C) is expected to be lower to enable the secondary structure transition towards a β-sheet.
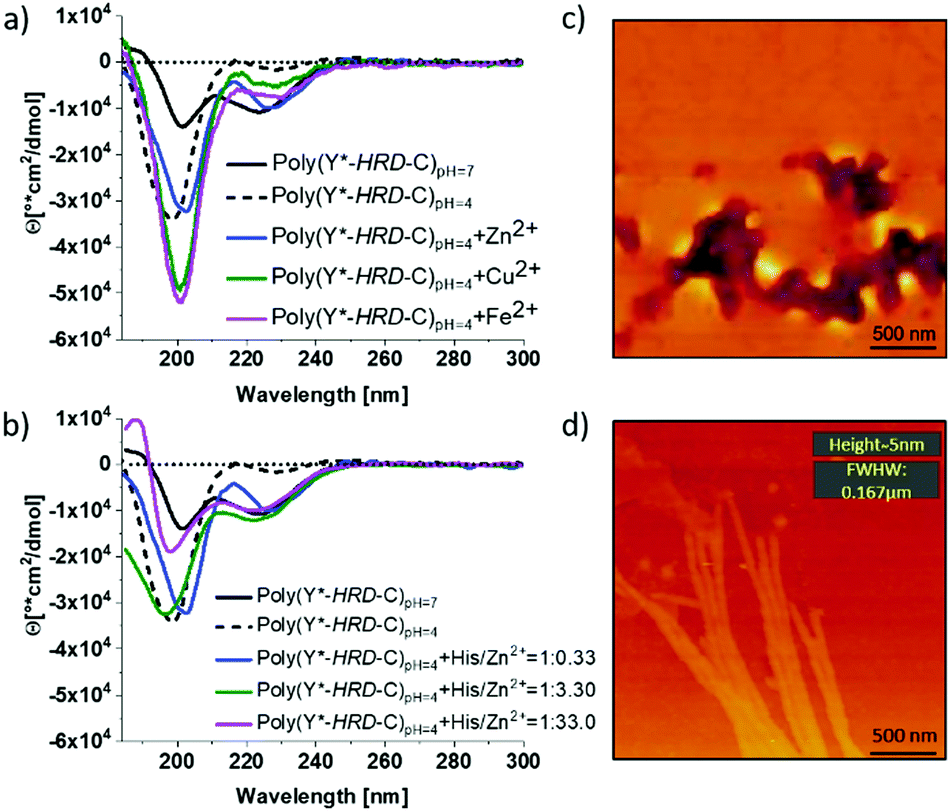 | ||
| Fig. 2 Triggering of β-sheet formation in poly(Y*-HRD-C) by pH changes or the addition of metal ions. CD spectra of polymer solutions at different pH and different metal ions (His/M2+ = 1:0.33) (a) and increase of the His/Zn2+ratio (b). AFM micrographs of poly(Y*-HRD-C) prior (c, pH 4) and after switching (d, pH7) revealing the formation β-sheet fibrils. | ||
Besides the pH responsiveness, the tendency of His residues to coordinate with ions is of high interest as these can stabilize a His rich face of β-sheets as well.3841 To investigate these effects, CD spectra of poly(Y*-HRD-C) solutions at pH 4 were recorded both in absence and presence of Zn2+, Cu2+ or Fe2+ as ions known to interact with imidazole ligands of the HRD. The addition of metal ions in a ratio of His/M2+ 1:0.33 leads only to marginal shifts towards β-sheet structures (Fig. 2a). The effect is most evident for Zn2+, where β-sheet cotton effects appeared, while the random coil fraction decreased with respect to the poly(Y*-HRD-C) at pH 4 without metal ions (ESI,† Fig. S10–S12 and Table S1). Taking into account, that both Cu2+ and Fe2+ induced β-sheets in the non-polymerized HRD domain,38 the marginal effect on the secondary structure of the poly(Y*-HRD-C) construct was unexpected. Considering the capability of Dopa residues to complex with metal ions45 an interaction with cysteinyldopa might be anticipated. These complexes could most likely not contribute to β-sheet stabilization as the geometry and orientation would not match structural requirements of the β-sheet.
In contrast to Cu2+ and Fe2+, Zn2+ ions trigger coil to β-sheet transitions and proved the interaction with the His-rich face of the poly(Y*-HRD-C) construct to stabilize β-sheets. This, however, does not exclude the occurrence of Zn2+/cysteinyldopa complexes, which apparently do not disturb the β-sheet structures. To evaluate this effect, the concentration of Zn2+ was systematically increased from 1:0.33 to 1:3.3 to 1:33 (molar ratio His/Zn2+). The higher the Zn2+ excess, the more pronounced are the β-sheet characteristic cotton effects at (−)223 nm. At 3.3 equiv. Zn2+ a similar band intensity at (−)223 nm was reached as in the salt free poly(Y*-HRD-C) solution at pH 7. Increasing the concentration of Zn2+ further did not increase the β-sheet band intensity at (−)223 nm, but decreased the band at (−)190 nm and thus suggest the reduction of the random coil structure fraction (Fig. 2b and ESI,† Table S1).
Nonetheless, the presence of Zn2+ or pH 7 proved to be suitable conditions to stabilize β-sheet structures in the poly(Y*-HRD-C) and both can act as sufficient triggers for secondary structure transitions. Moreover, it is noteworthy that the poly(Y*-HRD-C) at pH 7 adapts already a well expressed β-sheet, which in the presence of metal ions like Zn2+ could not be dramatically improved. Despite the polymeric character of poly(Y*-HRD-C), AFM micrographs confirmed the presence of distinct fibrillar aggregates at pH 7.0. Probably, those objects are formed via self-assembly processes of HRD-domains, folding the segmented poly(Y*-HRD-C) constructs into defined fibrils. Those appear to be rather persistent, reaching micrometres in length, exhibit heights of about 5 ± 0.5 nm and non-tip corrected full widths at half maximum (FWFM) of 170 ± 15 nm (Fig. 2d). Apparently, the height dimensions meet well that of β-sheet fibrils.46 The anisometric cross section probably reflects the packing constraints, which result from the segmented polymer chain, where β-sheet domains are disrupted by cysteinyldopa structure defects. This agrees well with previous studies on HRD-based peptides, describing the formation of hierarchical β-sheets organized across multiple length scales at neutral conditions.38 In control solutions of poly(Y*-HRD-C) at pH 4.0 no fibrils were found by AFM, which confirmed the CD results (Fig. 2c and ESI,† Fig. S13). Fibril visualization of Zn2+ ion stabilized poly(Y*-HRD-C) at pH 4.0 was difficult due to an increased salt load that interferes with the AFM imaging.
Besides the internal β-sheet organization that will dominantly modulate the cohesive properties, the polymerization process generates cysteinyldopa linkages at each repeat unit, which promise surface binding capabilities. A quartz crystal microbalance (QCM) with aluminium oxide coated sensors was used to gain insights into the adsorption behaviour and coating stability of poly(Y*-HRD-C). The QCM experiments show the adsorption of poly(Y*-HRD-C) from pH 4.0 solutions, to rapidly reach an equilibrium at frequency shifts of Δf = 6 Hz. According to the Voight model this corresponds to mass deposition of about 1.1 g m−2 (Fig. 3a).47 The low mass deposition was expected as the polymer repeat sequence exhibits a net charge of +8 at pH 4, which results in strong Coulomb repulsion. Moreover, taking CD analysis into account, the polymer adopts a statistical coil, making β-sheet assisted stabilization of the coating unlikely. The coating properties of poly(Y*-HRD-C) change dramatically at pH 7 (Fig. 3a). QCM revealed rapid adsorption with multilayer formation that prevents reaching an equilibrium as it was often observed for protein deposition.48 Interestingly, the adsorption isotherm suggests two distinct processes that probably correspond to mono- and multilayer formation. While the first gives highly rapid mass deposition to reach Δf ≈ −6 Hz, the second proceeds more slowly and reaches a frequency shift of Δf = −30 Hz after e.g. 4 h, corresponding to mass deposition of about 5.3 g m−2. CD spectroscopy suggested under these conditions the partial formation of β-sheets, that might improve cohesion in the coatings and contribute to multilayer formation. Moreover, at pH 7 the net charge of the poly(Y*-HRD-C) repeats drops to +4 and Coulomb repulsion is of less relevance as driving force to prevent multilayer deposition.
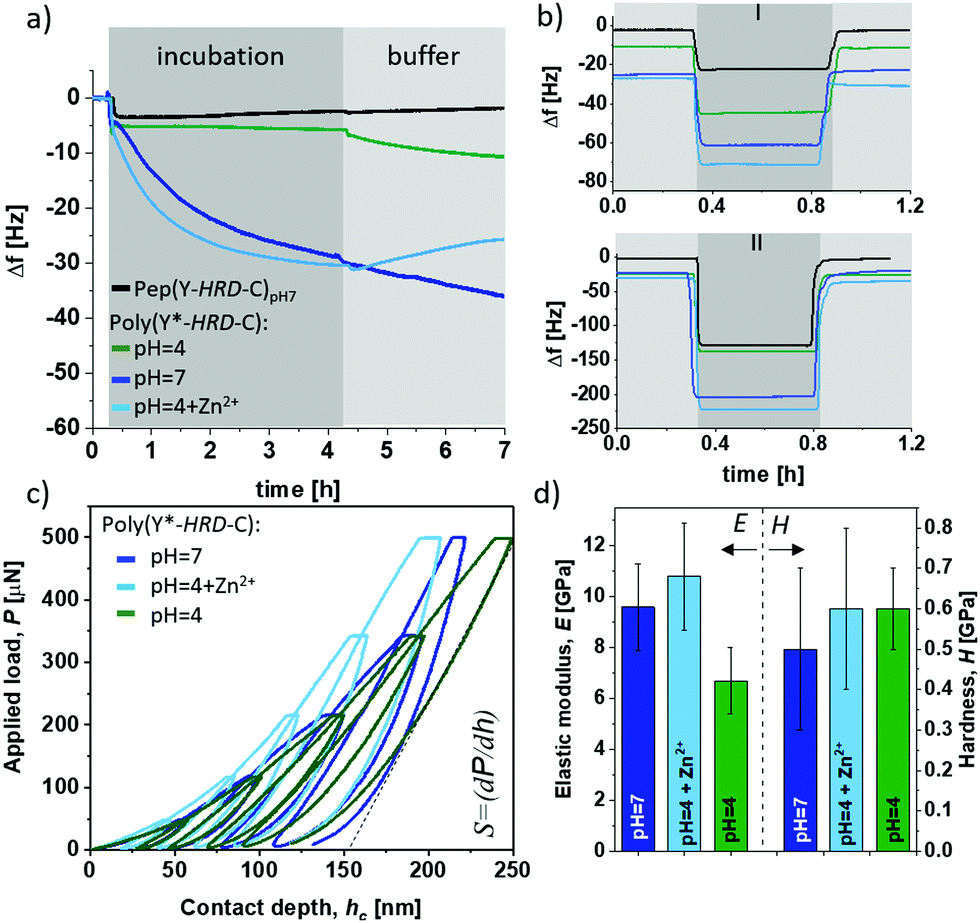 | ||
| Fig. 3 Coating behaviour and thick film mechanics of poly(Y*-HRD-C). Top: QCM adsorption isotherms of poly(Y*-HRD-C) on Al2O3 coated sensors (incubation step and subsequent buffer rinsing (a)) and stability tests of the coatings (b) by washing with 599 mM NaCl (I) and 4.2 M hypersaline solution (II)). Bottom: Mechanical properties obtained from depth-sensing nanoindentation studies (Depth profiling cyclic load function (c) and extracted mechanical responses at different pH (d)). | ||
The poly(Y*-HRD-C) coatings tolerate rinsing steps with buffer and irrespectively of the pH 7 or 4, a slight swelling was found indicating that the coatings accommodate more buffer (Fig. 3a). This effect was not expected, but it is not uncommon for cysteinyldopa presenting polymers.49 The mechanism at the molecular level requires further investigation, but apparently the deposition is fast and leads to kinetically controlled coatings, where the network structure can equilibrate under buffer conditions. The effect is most obvious for the coating, deposited at pH 7. A significant change in viscoelasticity of the coating was evident in the dissipation channels of QCM-D analysis that shows a strong splitting during buffer rinsing (ESI,† Fig. S14a).
As anticipated, the adsorption of poly(Y*-HRD-C) at pH 4 profits from the presence of Zn2+ due to β-sheet stabilization. The adsorption of poly(Y*-HRD-C) with 1:3.3 molar ratio His/Zn2+ proceeds already similarly to that at pH 7.0 without salt, both reaching Δf about −31 Hz after 4 h (Fig. 3a). However, the metal assisted deposition proceeds slightly faster, but the coating was more sensitive against rinsing. Probably due to Zn2+ wash-off from polymer-Zn2+ complexes the buffer wash leads to −23% mass loss and results in stable coating with 4.4 g m−2.
All coatings proved notable adhesion and robust stability by withstanding rinsing steps of harsh sea water model solutions and even hypersaline conditions as present in water of the Dead Sea (Fig. 3b). Most impressively the Zn2+ stabilized poly(Y*-HRD-C) coating, which is supposed to be most sensitive, shows a mass loss of only 5%, while the coating deposited at pH 7 has 9% mass loss during the hypersaline rinsing step (ESI,† Table S2).
As β-sheet structures are bridging multimer assemblies, a significant effect on mechanical bulk properties can be expected, depending on secondary structure in the poly(Y*-HRD-C). The elastic-inelastic response was characterized on thicker films casted from poly(Y*-HRD-C) at pH 7, pH 4 and pH 4 with Zn2+ (1:3.3 His/Zn2+) using depth-sensing nanoindentation (Fig. 3c and d). A change of pH from 4 → 7 induces β-sheet formation and increases the elastic (E) modulus from EpH4 = 6.7 ± 1.3 GPa to EpH7 = 9.6 ± 1.7 GPa. Interestingly, the Zn2+ stabilized poly(Y*-HRD-C) at pH 4 reached EpH4+Zn2+ = 10.8 ± 2.1 GPa. This might be rationalized by the fact, that metal ions not only contribute to stabilization of β-sheet structures, but also lead to interactions between β-sheet-tapes to generate further net-point contacts that influence the elastic responses of the polymer bulk.
It should be noted, that the poly(Y*-HRD-C) construct contains His and cysteinyldopa units, exhibiting imidazole and catechol functionalities, respectively. These enable two different types of metal ion complexations and provide two modes of crosslinking. Zn2+ has a strong preference to imidazole ligands of His residues and is found e.g. in protein structure motifs of the zinc finger.50 While a Zn2+ complexation to the catechol sites cannot be excluded, the metal induced β-sheet formation seems to be highly relevant to modulate the internal material mechanics. In contrast to the bridging interaction in a Zn2+/catechol complex, the β-sheet-fold multiplies the number of interactions between the poly(Y*-HRD-C) chains as several H-bonds and hydrophobic/entropic stabilizations get feasible between extended β-strands as well as between β-sheet tapes.
According to the Oliver–Pharr method the contact stiffness (S) directly correlates to the calculated elastic modulus (Fig. 3c). Hardness (H), which refers to the inelastic response of the materials, was not affected giving similar values of H(pH7−pH4+Zn2+−pH4) = 0.6 ± 0.1 GPa for all scenarios (Fig. 3d). Such differentiated behaviour in elastic response suggests that triggered formation of β-sheets promotes the resistance of the bulk adhesive particularly in presence of Zn2+ to cope with reversible/elastic deformations, indicating improved cohesion. With these mechanic properties, the peptide-based adhesive materials are promising as they reach the upper regime of common commodity polymers.51
In conclusion, a peptide originating from the histidine-rich domain (HRD) of a mussel byssus protein (preCollagen-D) was derivatized by Dopa and Cys flanks (Y*-HRD-C) to enable a chemically activated mussel-inspired polymerization. This yielded an artificial mussel glue protein (poly(Y*-HRD-C)), presenting adhesive cysteinyldopa connectivities in each repeat unit. IR, CD and AFM confirmed the formation of β-sheets by moderate pH changes from 4 → 7 or by the addition of Zn2+ at pH 4. The activation of this cohesion mechanism results in strong adsorption onto Al2O3 and the resulting coatings tolerate washing with 4.2 M hypersaline solutions. Nanoindentation evidenced an E-modulus of up to 10.8 GPa and hardness of 0.6 GPa. These artificial mussel glue proteins advent a versatile material platform that bridges the gap between mussel-glue inspired polymers and mussel foot proteins by realizing more complex functions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge M. Gräwert (MPIKG Golm) for GPC. Financial support is recognized by S. A. from Alexander von Humboldt Foundation (AvH postdoctoral fellowship) and by H. G. B. from German Research Council DFG/BO1762/9-2.Notes and references
- J. H. Waite and M. L. Tanzer, Science, 1981, 212, 1038–1040 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS.
- P. Wilke, N. Helfricht, A. Mark, G. Papastavrou, D. Faivre and H. G. Börner, J. Am. Chem. Soc., 2014, 136, 12667–12674 CrossRef CAS.
- B. K. Ahn, J. Am. Chem. Soc., 2017, 139, 10166–10171 CrossRef CAS.
- A. Lampel, S. A. McPhee, H. A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T. D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS.
- S. M. M. Reddy, E. Rasslenberg, S. Sloan-Dennison, T. Hesketh, O. Silberbush, T. Tuttle, E. Smith, D. Graham, K. Faulds, R. V. Ulijn, N. Ashkenasy and A. Lampel, Adv. Mater., 2020, 32, e2003511 CrossRef.
- L. Petrone, A. Kumar, C. N. Sutanto, N. J. Patil, S. Kannan, A. Palaniappan, S. Amini, B. Zappone, C. Verma and A. Miserez, Nat. Commun., 2015, 28, 8737 CrossRef.
- F. Jehle, E. Macias-Sanchez, P. Fratzl, L. Bertinetti and M. J. Harrington, Nat. Commun., 2020, 11, 862–870 CrossRef CAS.
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CrossRef CAS.
- J. H. Waite, J. Exp. Biol., 2017, 220, 517–530 CrossRef.
- C. R. Matos-Perez, J. D. White and J. J. Wilker, J. Am. Chem. Soc., 2012, 134, 9498–9505 CrossRef CAS.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216–220 CrossRef CAS.
- Q. Xu, M. Xu, C. Y. Lin, Q. Zhao, R. Zhang, X. Dong, Y. Zhang, S. Tian, Y. Tian and Z. Xia, Adv. Sci., 2019, 6, 1902043 CrossRef CAS.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS.
- J. L. Dalsin, B.-H. Hu, B. P. Lee and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 4253–4258 CrossRef CAS.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P. L. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS.
- P. Wilke and H. G. Börner, ACS Macro Lett., 2012, 1, 871–875 CrossRef CAS.
- N. L. Venkatareddy, P. Wilke, N. Ernst, J. Horsch, M. Weber, A. Dallmann and H. G. Börner, Adv. Mater. Interfaces, 2019, 6, 1900501 CrossRef.
- J. Horsch, P. Wilke, H. Stephanowitz, E. Krause and H. G. Börner, ACS Macro Lett., 2019, 8, 724–729 CrossRef.
- M. Meißler, A. Taden and H. G. Börner, ACS Macro Lett., 2016, 5, 583–587 CrossRef.
- B. D. B. Tiu, P. Delparastan, M. R. Ney, M. Gerst and P. B. Messersmith, Angew. Chem., Int. Ed., 2020, 59, 16616–16624 CrossRef CAS.
- A. Li, Y. Mu, W. Jiang and X. Wan, Chem. Commun., 2015, 51, 9117–9120 RSC.
- C. Juds, J. Schmidt, M. G. Weller, T. Lange, U. Beck, T. Conrad and H. G. Börner, J. Am. Chem. Soc., 2020, 142, 10624–10628 CrossRef CAS.
- S. Große, P. Wilke and H. G. Börner, Angew. Chem., Int. Ed., 2016, 55, 11266–11270 CrossRef.
- L. A. Burzio, Biochemistry, 2000, 39, 11147–11153 CrossRef CAS.
- S. Arias, S. Amini, J. Horsch, M. Pretzler, A. Rompel, I. Melnyk, D. Sychev, A. Fery and H. G. Börner, Angew. Chem., Int. Ed., 2020, 59, 18495–18499 CrossRef CAS.
- C. Zhong, T. Gurry, A. A. Cheng, J. Downey, Z. Deng, C. M. Stultz and T. K. Lu, Nat. Nanotechnol., 2014, 9, 858–866 CrossRef CAS.
- J. Wang and T. Scheibel, Biotechnol. J., 2018, 13, 1800146 CrossRef.
- J. Horsch, P. Wilke, M. Pretzler, M. Seuss, I. Melnyk, D. Remmler, A. Fery, A. Rompel and H. G. Börner, Angew. Chem., Int. Ed., 2018, 57, 15728–15732 CrossRef CAS.
- M. Pretzler, A. Bijelic and A. Rompel, Sci. Rep., 2017, 7, 1810–1819 CrossRef.
- J. M. Kohn, J. Riedel, J. Horsch, H. Stephanowitz and H. G. Börner, Macromol. Rapid Commun., 2020, 41, 1900431 CrossRef CAS.
- J. M. Krüger and H. G. Börner, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202015833.
- J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp and K. Savage, Philos. Trans. R. Soc., B, 2002, 357, 121–132 CrossRef CAS.
- J. H. Waite, E. Vaccaro, C. Sun and J. M. Lucas, Philos. Trans. R. Soc., B, 2002, 357, 143–153 CrossRef CAS.
- K. Kamino, Mar. Biotechnol., 2008, 10, 111–121 CrossRef CAS.
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS.
- F. Jehle, P. Fratzl and M. J. Harrington, ACS Nano, 2018, 12, 2160–2168 CrossRef CAS.
- J. M. Lucas, E. Vaccaro and J. H. Waite, J. Exp. Biol., 2002, 205, 1807–1817 CAS.
- M. J. Harrington and J. H. Waite, Biomacromolecules, 2008, 9, 1480–1486 CrossRef CAS.
- R. I. Kühnle and H. G. Börner, Angew. Chem., Int. Ed., 2011, 50, 4499–4502 CrossRef.
- H. Xiong, B. L. Buckwalter, H. Shieh and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 6349–6353 CrossRef CAS.
- A. Reinecke, G. Brezesinski and M. J. Harrington, Adv. Mater. Interfaces, 2017, 4, 1600416 CrossRef.
- A. Barth, Biochim. Biophys. Acta, 2007, 1767, 1073–1101 CrossRef CAS.
- K. S. Rajan, S. Mainer and J. M. Davis, Bioinorg. Chem., 1978, 9, 187–203 CrossRef CAS.
- R. P. W. Davies, A. Aggeli, A. J. Beevers, N. Boden, L. M. Carrick, C. W. G. Fishwick, T. C. B. McLeish, I. Nyrkova and A. N. Semenov, Supramol. Chem., 2006, 18, 435–443 CrossRef CAS.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391–396 CrossRef CAS.
- S. M. Notley, M. Eriksson and L. Wagberg, J. Colloid Interface Sci., 2005, 292, 29–37 CrossRef CAS.
- Y. Akdogan, W. Wei, K. Y. Huang, Y. Kageyama, E. W. Danner, D. R. Miller, N. R. Martinez Rodriguez, J. H. Waite and S. Han, Angew. Chem., Int. Ed., 2014, 53, 11253–11256 CrossRef CAS.
- A. Klug and D. Rhodes, Cold Spring Harbor Symp. Quant. Biol., 1987, 52, 473–482 CrossRef CAS.
- P.-E. Mazerana, M. Beyaouib, M. Bigerellea and M. Guigon, Int. J. Mater. Res., 2012, 103, 715–722 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, material synthesis, characterization and property analysis. See DOI: 10.1039/d0sm02118k |
| This journal is © The Royal Society of Chemistry 2021 |