
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Near-zero surface pressure assembly of rectangular lattices of microgels at fluid interfaces for colloidal lithography†
Miguel Angel
Fernandez-Rodriguez
 *ab,
Maria-Nefeli
Antonopoulou‡
b and
Lucio
Isa
b
*ab,
Maria-Nefeli
Antonopoulou‡
b and
Lucio
Isa
b
aLaboratory of Surface and Interface Physics, Biocolloids and Fluid Physics group, Faculty of Sciences, University of Granada, Campus de Fuentenueva s/n, ES 18071 Granada, Spain. E-mail: mafernandez@ugr.es
bLaboratory for Soft Materials and Interfaces, Swiss Federal Institute of Technology Zürich, Vladimir-Prelog-Weg 1-5/10, 8093 Zürich, Switzerland
First published on 9th December 2020
Abstract
Understanding and engineering the self-assembly of soft colloidal particles (microgels) at liquid–liquid interfaces is broadening their use in colloidal lithography. Here, we present a new route to assemble rectangular lattices of microgels at near zero surface pressure relying on the balance between attractive quadrupolar capillary interactions and steric repulsion among the particles at water/oil interfaces. These self-assembled rectangular lattices are obtained for a broad range of particles and, after deposition, can be used as lithography masks to obtain regular arrays of vertically aligned nanowires via wet and dry etching processes.
Microgels are versatile soft colloids1 that self-assemble at water/air and water/oil interfaces2,3 enabling their controlled deposition on a solid substrate and their use for soft colloidal lithography.4,5 Although microgels made of poly(N-isopropylacrylamide) (PNIPAM) are hydrophilic and easily dispersed in water, they can be readily adsorbed at liquid–liquid interfaces using a spreading agent. Upon adsorption at the interface, each microgel spreads to reduce the area of the bare fluid–fluid interface until the deformation is counterbalanced by the microgel internal elasticity.6,7 The spreading can be tuned by adjusting the particle crosslinking density, leading to effective diameters at the interface greater than twice the diameter in bulk dispersions.8 Once at the interface, the structural and dynamical behaviour of assemblies of microgels is mainly dictated by capillary and steric interactions.9 Upon compression, microgel monolayers can reach a point at which all microgels enter into contact to form hexagonal lattices whose lattice spacing can be reduced by further compression.5,10 In order to expand the applications of soft colloidal lithography, new routes need to be developed to produce patterns with symmetries beyond the hexagonal one. A broad range of Bravais lattices can be obtained for particles interacting via soft electrostatic repulsion via the stretching of hexagonal lattices while they are being deposited on the substrate.11 However, this process relies on a delicate interplay between the wettability of the substrates, the angle of deposition and a thermal fixation of the colloids to circumvent the collapse of the structures due to capillary forces during drying. Additional structures, including binary colloidal alloys5 and Moiré and complex tessellations,12,13 can also be obtained by sequential depositions of microgels, respectively of two different sizes or of the same size. Nevertheless, they require multiple steps and a careful control of the surface pressure Π to achieve the target structure. In order to circumvent sensitive processes, it would be desirable to obtain directly non-hexagonal structures via robust self-assembly at the fluid interface. The existence of complex two-dimensional structures beyond the triangular symmetry was numerically predicted by Jagla twenty years ago14 for colloids with a repulsion potential exhibiting two distinct length scales (e.g. a hard core and a soft shell) and recently experimentally realised by Rey et al. for polystyrene colloids with a microgel soft shell confined at a water/oil interface.15 Non-triangular assemblies, including strings and rectangular lattices, occur at high values of Π, beyond compact hexagonal monolayers, where the microgel shells are under compression. However, the parameter space in the phase diagram where such structures are obtained is fairly small, and their kinetic access is delicate, making their experimental realization a complex task with limited tuneability.
We report here a novel, robust route to obtain self-assembled rectangular microgels lattices at fluid interfaces for a broad range of different particles. In contrast to the lattices predicted by Jagla,14 we find rectangular lattices at Π ≈ 0 mN m−1, arising from the combination of steric repulsion and leading-order quadrupolar capillary attraction between microgels at the interface, which are the result of the nanometre-scale roughness induced by the microgels adsorbed at the interface.16,17
The microgels used here were synthesized via precipitation polimerization of N-isopropylacrylamide (NIPAM) in water.8 The NIPAM monomer and crosslinker (BIS) were added in one or two steps to grow larger shells as reported in a previous work.13 Their labeling is CXSY, where X = 3,5,7% is the mass ratio between crosslinker and total monomer, whereas the total amount of monomer is constant, and Y = 0,1 is the number of steps for additional shell growth. This procedure leads to the synthesis of soft colloidal particles with consistent chemical composition and core–shell architecture, but varying degrees of crosslinking densities and diameters in bulk aqueous suspensions and at the interface. Our microgels have bulk hydrodynamic diameters ranging from (574 ± 73) nm of C7S0 to (879 ± 121) nm of C3S1, as measured by DLS (Malvern Zetasizer, z-averaged diameter) and interfacial sizes ranging from (786 ± 30) nm of C7S0 to (1578 ± 46) nm of C3S1 (measured ex situ after deposition on a silicon substrate via AFM, see ESI† for full synthesis details and dimensions). In Fig. 1, we show rectangular lattices obtained after the deposition of monolayers formed at water/hexane interfaces on silicon substrates using a Langmuir–Blodgett (LB) trough at Π ≈ 0 mN m−1 for all types of microgel studied.
 | ||
| Fig. 1 Lattices with four-fold symmetries and corresponding FFT, obtained by depositing the different microgels (a) C7S0, (b) C7S1, (c) C5S0, (d) C5S1, (e) C3S0, and (f) C3S1, at Π ≈ 0 mN m−1. Scale bar is 5 μm and the FFTs are enlarged 3 times. | ||
In order to understand in which region of a 2D phase diagram the structures are obtained, in Fig. 2 we show the compression curve of the C7S1 microgels and the corresponding monolayer microstructure after deposition. These experiments are performed on a LB trough, where we deposit the particles on a substrate, which crosses the water/hexane interface at the same time as the monolayer is gradually compressed by the moving barriers of the trough. This procedure allows transferring monolayers subjected to continuously varying surface pressure onto a single sample, so that different locations on the sample correspond to different values of pressure and surface coverage (see ESI† for full details). From the reading of Π using a Wilhelmy plate and the direct measurement of the area per particle Ap (extracted by image analysis after deposition), we observe that lattices with a four-fold coordination (e.g. rectangular and rhomboidal) are found for values of Π ≈ 0 mN m−1 in the compression curve. As Π grows, the structures transition to the well-known hexagonally packed monolayers. The transition between the two types of coordination is clearly displayed in the inset to Fig. 2, which shows the average order parameters 〈Ψ4〉 and 〈Ψ6〉 calculated as in eqn (1), where j = 4,6 represents the symmetry of the coordination described by 〈Ψj〉, N is the total number of particles, the index k refers to a given particle with Nl neighbours and θlk is the angle formed between the particle k and its neighbour l respect to a fixed axis.
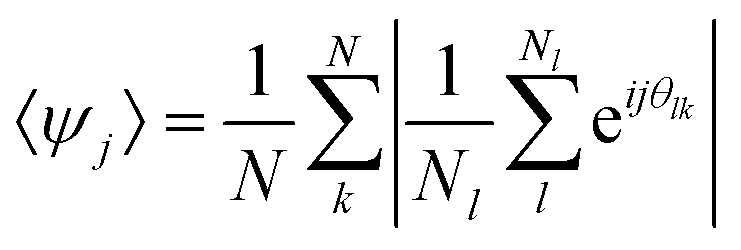 | (1) |
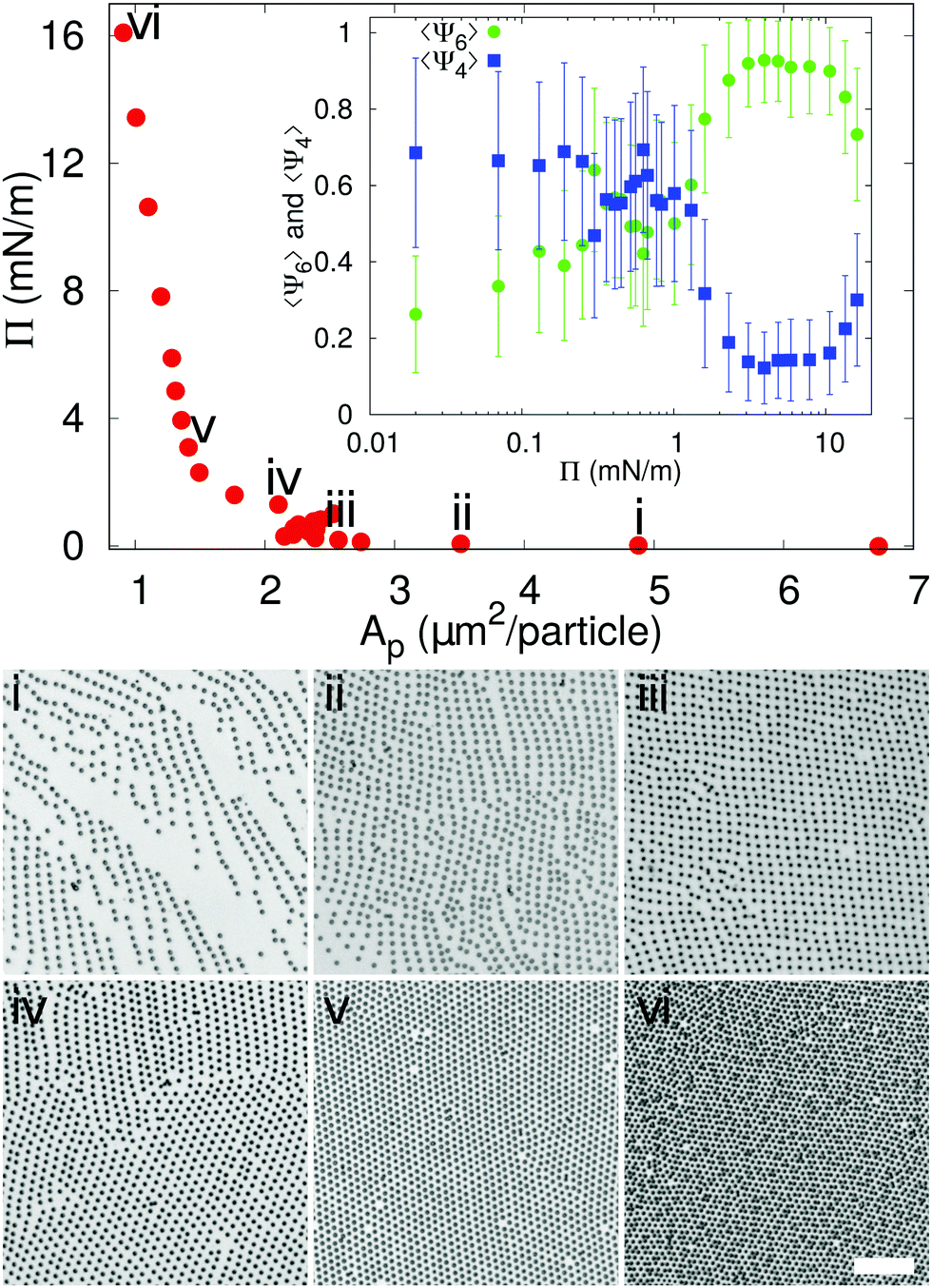 | ||
| Fig. 2 Compression curve for C7S1 microgels as they are deposited on a silicon substrate in a LB trough. Microscope images are provided for different points in the compression curve on the substrate (scale bar is 10 μm). The inset shows the corresponding order parameters 〈Ψ6〉 and 〈Ψ4〉 vs. Π. Error bars correspond to standard deviations. | ||
The average 〈Ψ4〉 and 〈Ψ6〉 parameters are closer to 1 when there is a mainly rectangular or hexagonal lattice, respectively.13 Therefore, from the data it is clear that for Π ≈ 0 mN m−1 〈Ψ4〉 > 〈Ψ6〉, while the point at which Π starts rising corresponds to a transition to a hexagonal layer, where 〈Ψ4〉 < 〈Ψ6〉. Above 10 mN m−1 an isostructural phase transition occurs, which results in a decrease of 〈Ψ6〉, as previously reported.10
In order to eliminate the possibility that the rectangular lattices are an artifact induced by the deposition process, e.g. as reported for drying droplets,18 we imaged these microgels directly at the water/hexadecane interface, as well as at an water/air interface, in conditions of Π ≈ 0 mN m−1 using several complementary approaches. The first approach is direct optical microscopy at the fluid interface using a 40× dipping objective (Nikon) to image the interface through the oil. To reduce the large convection flows present in the system, we corralled the particles using a polymeric ring of 800 μm-diameter fabricated with a Nanoscribe (fabrication described in ESI†) and placed at the microgel-laden water/hexadecane interface with a micropositioner. The field of view is 85 × 85 μm2 and we imaged close to the center of the ring, avoiding possible confinement effects that are known to provide capillary forces inducing square lattice formation at interfaces19,20 (see Fig. 3a). Within the ring, microgels exhibit Brownian motion at the interface (see Movie S1 in ESI†) and the overall structure of the interface is globally disordered, as reflected by the FFT in the inset to Fig. 3a. However, local, clearly four-fold coordinated particle clusters are present at the interface (Fig. 3a). These clusters are weakly bound, and continuously form and disappear under thermal fluctuations and the residual small drift at the interface (see Fig. 3b and Movies S1–S3, ESI†). These observations already demonstrate that short-ranged four-fold coordinated assemblies exist at an unperturbed interface. However, the deposition process in an LB trough promotes the creation of larger-area structures. In particular, the presence of the macroscopically straight three-phase (water–oil–silicon wafer) contact line aligns the formed clusters with respect to the deposition direction and the continuous compression of the barriers produces extended regions with the right surface coverage to obtain longer-ranged order. This alignment at the meniscus also explains the formation of lines in Fig. 2i for lower surface coverages, with insufficient particle numbers to create a continuous monolayer. As a complementary set of measurements, we employed two different ways of immobilizing and imaging the particles at a fluid interface. Rectangular lattices were first observed in freeze-fracture shadow-cast (FreSCa) cryo-SEM images at the water/decane interface (see Fig. 3c and ESI†), a technique that exploits shock-freezing to immobilize and characterize the microgels in situ at the water/oil interface.8,21 In a last set of measurements, we placed a silicon wafer in a Teflon beaker filled with water, forming a ≈30° angle with the interface and partially crossing it. We then deposited C7S1 microgels at the water/air interface ensuring that we are close to Π ≈ 0 mN m−1. Then, the Teflon beaker was enclosed in a methacrylate box next to a droplet of cyanoacrylate. The evaporating cyanoacrylate reacts at the water/air interface, and after an overnight wait, it created an acrylate film that fixed the particles at the interface.22 Next, the substrate was extracted from water, collecting the film, which was characterized by SEM and AFM and showed the presence of rectangular lattices (Fig. 3d and ESI†). This experiment indicates that the self-assembly of rectangular lattices is not specific to water/alkane oil interfaces, but it is also found at water/air interfaces, expanding its generality and applicability.
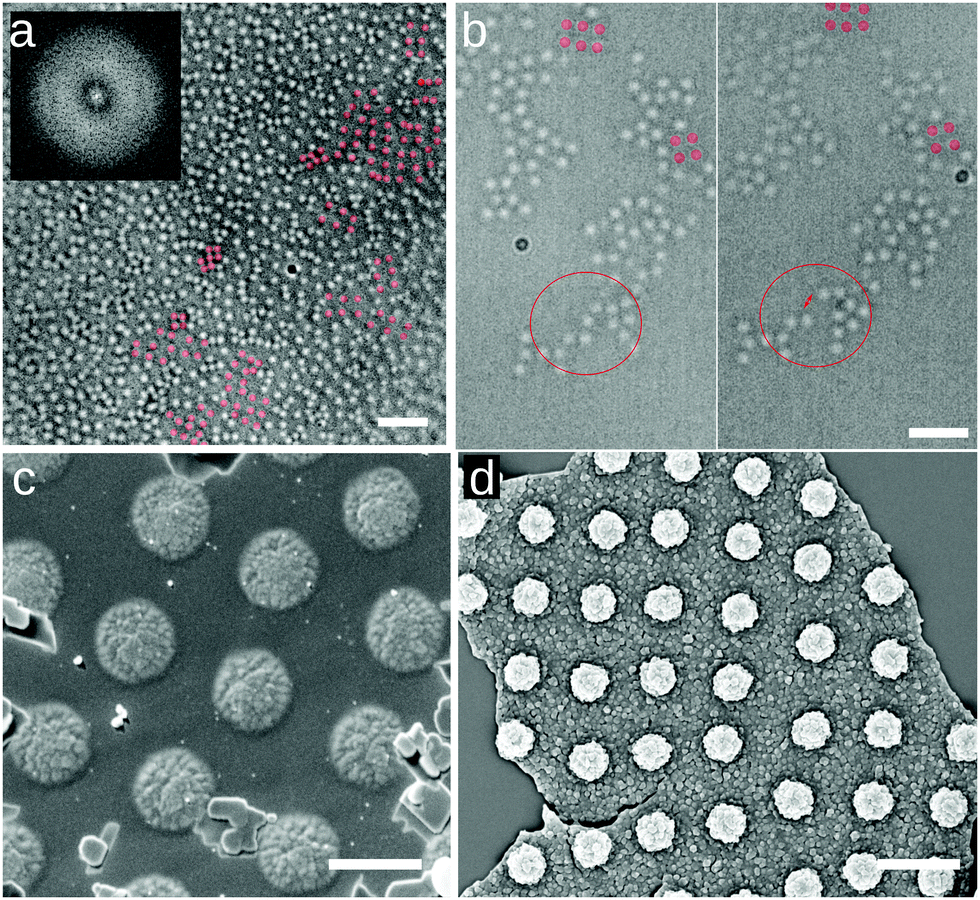 | ||
| Fig. 3 Spontaneous assembly of square lattices of C7S1 microgels at the interface. (a) Frame of Movie S1 (ESI†) at the water/hexadecane interface at the center of a 800 μm-diameter ring (scale bar: 10 μm). The inset shows the FFT, with the diffuse ring characteristic of 2D disordered liquids. (b) Two frames of Movie S2 (ESI†) of a water/hexadecane interface showing the spontaneous detachment of a microgel from a cluster, marked for clarity (scale bar: 5 μm). Representative four-fold coordinated clusters are highlighted in red in (a) and (b). (c) Cryo-SEM image of a water/decane interface where the interface has been fractured and the oil phase removed, showing the microgels protruding from the vitrified water phase (scale bar: 1 μm). (d) SEM image of an upside-down fragment of the microgel-cyanoacrylate film, showing the portion immersed in water, with the microgels in a square lattice configuration (scale bar: 2 μm). | ||
After reporting these observations, we show that the formation of the four-fold coordinated structures can be rationalized by balance between capillary interactions and steric repulsion among the microgels. Although capillary forces are typically associated with interfacial deformations imparted by hard colloids, they have also been reported for soft particles.9,23 In general, the capillary forces between colloidal particles trapped at a fluid interface and displaying an arbitrarily undulated contact line can be described as a multipole expansion, where, in the absence of gravity, the leading-order term has a quadrupolar symmetry.24 Even if higher-order terms may not be neglected for a full quantification of capillary interactions at close separations,17,25,26 the clear presence of four-fold symmetries in our experiments confirms that the aggregation behavior can be captured by the leading quadrupolar term.
The steric repulsion between microgels at interfaces has been described via Hertzian or generalized Hertzian potentials.13,27 As we are in the regime where the microgels are not externally compressed, the specific form of the steric potential is not relevant and its contribution is effectively null until the particles overlap. However, we can consider the steric repulsion, once the particles enter into contact, to be much steeper than the quadrupolar attractive interaction. This implies that, essentially, capillary forces bring the particles at a center-to-center distance approximately equal to their single-particle diameter at the interface, and not closer. In order to reduce this separation further, external compression, i.e. with the barriers of a LB trough, is required and leads to the formation of 2D hexagonal crystals with lattice parameters smaller than the single-particle diameter. The fact that the microgels can rearrange at the interface, i.e. that the clusters spontaneously form and disappear under thermal fluctuations, furthermore indicates that the attractive capillary potential at contact is of the order of a few KBT. With these assumptions, we can estimate the capillary quadrupolar potential between two microgels as a function of their center-to-center distance d as
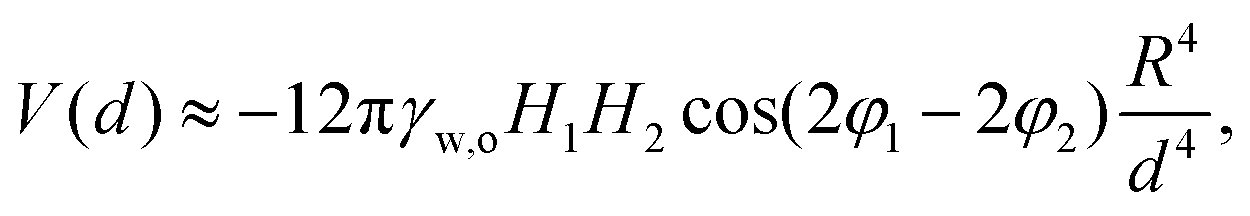 | (2) |
Taking the example of 7CS1 microgels, we measure an experimental value of d ≈ 1.8 μm between particles in rectangular configurations, while 2R = 1.2 μm (see ESI†). Assuming T = 25 °C, and the orientation of the phase angles that gives the maximum attraction, we find that H1,2 = [0.4,1.3] nm for V(1.8 μm) = [1,10]KBT. These interface roughness values are compatible with the ones we measure from the outer part of the microgels via AFM, after deposition on a substrate, as reported here (see ESI†) and in previous works.8 These considerations indicate that the formation of four-fold coordinated structures can emerge as a consequence of attractive capillary interactions with a quadrupolar leading order balanced by steric repulsion upon contact in the absence of external compression, i.e. for Π ≈ 0 mN m−1.
Exploiting the spontaneous formation of non-hexagonal structures opens up new opportunities for soft colloidal lithography. As a demonstration, Fig. 4a shows a square array of C7S0 microgels, which is subsequently used as a mask for the metal-assisted chemical “wet” etching, of vertically aligned silicon nanowires (VA-NWs) in a square lattice configuration (Fig. 4b). To achieve this, a silicon substrate similar to the ones in Fig. 1 was prepared. In a next step, the microgels were swollen in photoresist, effectively turning into lithography masks4 (see ESI†). After the swelling, the substrate was sputter-coated by a 10 nm-thick gold layer and immersed in an etching solution containing HF and H2O2 for 4 min. The photoresist-swollen microgels protect the underlying silicon wafer from the metal-assisted etching, leading to the formation of nanowires in correspondence of each microgel. The final structure was imaged via SEM, tilting the substrate by 30°. The VA-NWs are ≈2.6 μm-long and the partially removed microgels in the washing process are still visible, and can be removed via further O2 plasma treatment.5 Furthermore, we tested the capability of such soft colloidal masks to be used in conventional deep reactive ion exchange, “dry” etching. To this end, we deposited C3S0 microgels in a square lattice configuration and swelled them in photoresist. Next, we performed the dry etching for 60 s, obtaining the VA-NWs shown in the SEM image in Fig. 4c. The pillars were ≈640 nm long and the microgels were etched away in the process. Although the dry etching could not produce VA-NWs as long as in the equivalent wet etching process, it leads to a very homogeneous patterning over the whole substrate, while wet etching is more heterogeneous and leads to a wider distribution of VA-NWs lengths. Ideally, one could improve the performance of soft colloidal masks for dry etching by improving the photoresist swelling process and combining it with a Bosch etching process where etching steps are alternated with the deposition of protective passivizing layers.28 The use of microgels with different core-to-shell size ratios allows for the independent tuning of diameter and spacing of the VA-NWs.
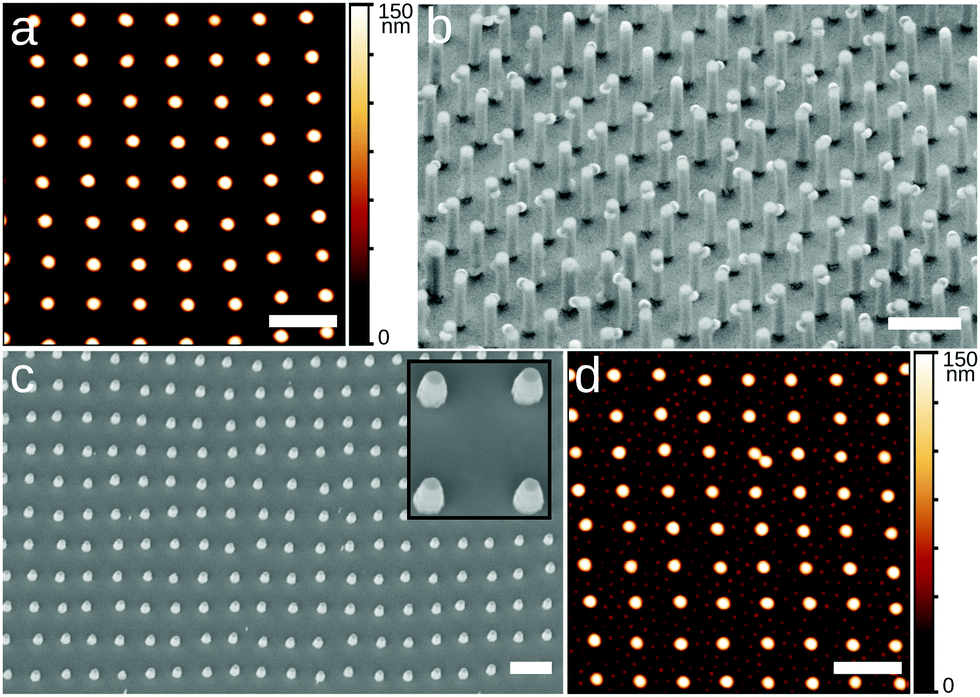 | ||
| Fig. 4 Square lattices for colloidal soft lithography applications. (a) AFM height image of a square lattice of C7S0 microgels deposited on a silicon substrate at Π ≈ 0 mN m−1. (b) Substrate prepared similar to the one in (a) and subjected to metal assisted chemical wet etching. (c) Square lattice of C3S0 microgels deposited on a silicon wafer and dry etched with deep reactive ion exchange. (d) Same substrate as in (a) subjected to a second deposition of smaller microgels with Π ≈ 25 mN m−1. All scale bars are 2 μm. The inset in (c) is 2 μm-wide. | ||
Finally, Fig. 4d, shows the possibility of realizing sequential depositions of microgels where the first layer has a square symmetry instead of a hexagonal one, as previously shown.5,13 First, a square lattice of C7S0 is produced at Π ≈ 0 mN m−1 (Fig. 4a). Next, the same substrate is immersed in water and a second deposition of smaller P(NIPAM-co-MAA) microgels at Π ≈ 25 mN m−1 is performed to produce a 2D binary colloidal alloy (see ESI† for further details). The combination of different symmetries for sequentially deposited microgel layers offers exciting opportunities for the future realization of complex structures.
Concluding, we show that interesting possibilities emerge from the interfacial self-assembly of soft colloids at close to zero surface pressures. This region of the compression curves is seemingly overlooked, as usually one finds 2D random gas and liquid phases for repulsion-dominated systems, or isolated aggregates if there are strong attractive forces. Here, by carefully exploring this region for soft microgels, we find that, at the right values of surface coverage, large-scale assemblies with four-fold symmetry emerge, which we hypothesize are caused by weak capillary attraction counterbalanced by stiff steric repulsion. These assemblies are found for a broad range of core–shell PNIPAM microgels, with different sizes and crosslinking densities, and for both water/oil and water/air interfaces. Their formation thus seems general and future work to prove if the same structures are found for microgels with different chemistries and architectures will be very interesting. We expect that these lattices with four-fold symmetries can not only be added to the extensive library of self-assembled 2D structures for colloidal lithography, but that they can also inspire further engineering of particle pair potentials at interfaces, e.g. through targeted particle-induced deformation of the interface as realized for larger objects.29 Detailed numerical studies of the interplay between particle architecture and interactions at the interface, paying close attention to near-field capillary forces, may finally direct the design of tailored soft colloids for the fabrication of new functional materials.
Author contributions
Author contributions are defined based on the CRediT (Contributor Roles Taxonomy) and listed alphabetically. Conceptualization: MNA, MAFR, LI. Formal analysis: MNA, MAFR. Funding acquisition: LI. Investigation: MNA, MAFR. Methodology: MNA, MAFR, LI. Project administration: LI. Software: MAFR. Supervision: MAFR, LI. Validation: MNA, MAFR. Visualization: MNA, MAFR, LI. Writing original draft: MAFR, LI. Writing review and editing: MNA, MAFR, LI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors acknowledge Prof. Walter Richtering for providing the P(NIPAM-co-MAA) microgels. M. A. F. R. and L. I. acknowledge financial support from the Swiss National Science Foundation Grant PP00P2-172913/1. M. A. F. R. has received funding from the postdoctoral fellowships programme Beatriu de Pinós, funded by the Secretary of Universities and Research (Government of Catalonia) and by the Horizon 2020 programme of research and innovation of the European Union under the Marie Skłodowska-Curie grant agreement No 801370 (Grant 2018 BP 00029) and the Spanish Juan de la Cierva Programme 2018 – Incorporation Grants (IJC2018-035946-I).Notes and references
- L. A. Lyon and A. Fernandez-Nieves, Annu. Rev. Phys. Chem., 2012, 63, 25–43 CrossRef CAS.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. Lyon, J. Crassous, D. Suzuki, R. Gumerov, S. Schneider, I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS.
- M. Rey, M. A. Fernandez Rodriguez, M. Karg, L. Isa and N. Vogel, Acc. Chem. Res., 2020, 53, 414–424 CrossRef CAS.
- M. Rey, R. Elnathan, R. Ditcovski, K. Geisel, M. Zanini, M. A. Fernandez-Rodriguez, V. V. Naik, A. Frutiger, W. Richtering, T. Ellenbogen, N. H. Voelcker and L. Isa, Nano Lett., 2016, 16, 157–163 CrossRef.
- M. Á. Fernández-Rodrguez, R. Elnathan, R. Ditcovski, F. Grillo, G. M. Conley, F. Timpu, A. Rauh, K. Geisel, T. Ellenbogen, R. Grange, F. Scheffold, M. Karg, W. Richtering, N. H. Voelcker and L. Isa, Nanoscale, 2018, 10, 22189–22195 RSC.
- R. W. Style, L. Isa and E. R. Dufresne, Soft Matter, 2015, 11, 7412–7419 RSC.
- H. Mehrabian, J. Harting and J. H. Snoeijer, Soft Matter, 2016, 12, 1062–1073 RSC.
- F. Camerin, M. A. Fernández-Rodríguez, L. Rovigatti, M.-N. Antonopoulou, N. Gnan, A. Ninarello, L. Isa and E. Zaccarelli, ACS Nano, 2019, 13, 4548–4559 CrossRef CAS.
- S. Huang, K. Gawlitza, R. von Klitzing, L. Gilson, J. Nowak, S. Odenbach, W. Steffen and G. K. Auernhammer, Langmuir, 2016, 32, 712–722 CrossRef CAS.
- M. Rey, M. A. Fernandez-Rodriguez, M. Steinacher, L. Scheidegger, K. Geisel, W. Richtering, T. M. Squires and L. Isa, Soft Matter, 2016, 12, 3545–3557 RSC.
- M. E. J. Hummel, C. Stelling, B. A. F. Kopera, F. A. Nutz, M. Karg, M. Retsch and S. Förster, Langmuir, 2019, 35, 973–979 CrossRef CAS.
- K. Volk, F. Deißenbeck, S. Mandal, H. Löwen and M. Karg, Phys. Chem. Chem. Phys., 2019, 21, 19153–19162 RSC.
- F. Grillo, M. A. Fernandez-Rodriguez, M.-N. Antonopoulou, D. Gerber and L. Isa, Nature, 2020, 582, 219–224 CrossRef CAS.
- E. A. Jagla, J. Chem. Phys., 1999, 110, 451–456 CrossRef CAS.
- M. Rey, A. D. Law, D. M. A. Buzza and N. Vogel, J. Am. Chem. Soc., 2018, 139, 17464–17473 CrossRef.
- K. D. Danov, P. A. Kralchevsky, B. N. Naydenov and G. Brenn, J. Colloid Interface Sci., 2005, 287, 121–134 CrossRef CAS.
- I. B. Liu, N. Sharifi-Mood and K. J. Stebe, Annu. Rev. Condens. Matter Phys., 2018, 9, 283–305 CrossRef CAS.
- A. G. Marín, H. Gelderblom, D. Lohse and J. H. Snoeijer, Phys. Rev. Lett., 2011, 107, 085502 CrossRef.
- A. Würger, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 74, 041402 CrossRef.
- D. Ershov, J. Sprakel, J. Appel, M. A. Cohen Stuart and J. van der Gucht, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9220–9224 CrossRef CAS.
- K. Geisel, L. Isa and W. Richtering, Langmuir, 2012, 28, 15770–15776 CrossRef CAS.
- N. Vogel, J. Ally, K. Bley, M. Kappl, K. Landfester and C. K. Weiss, Nanoscale, 2014, 6, 6879–6885 RSC.
- A. Rauh, M. Rey, L. Barbera, M. Zanini, M. Karg and L. Isa, Soft Matter, 2017, 13, 158–169 RSC.
- D. Stamou, C. Duschl and D. Johannsmann, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 5263–5272 CrossRef CAS.
- L. Yao, L. Botto, M. Cavallaro, Jr, B. J. Bleier, V. Garbin and K. J. Stebe, Soft Matter, 2013, 9, 779–786 RSC.
- N. Bowden, A. Terfort, J. Carbeck and G. M. Whitesides, Science, 1997, 276, 233–235 CrossRef CAS.
- F. Camerin, N. Gnan, J. Ruiz-Franco, A. Ninarello, L. Rovigatti and E. Zaccarelli, Phys. Rev. X, 2020, 10, 031012 CAS.
- F. Laermer and A. Schilp, Method of anisotropically etching silicon, US Pat., 5501893, 1996 Search PubMed.
- J. Bae, N. P. Bende, A. A. Evans, J.-H. Na, C. D. Santangelo and R. C. Hayward, Mater. Horiz., 2017, 4, 228–235 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01823f |
| ‡ Current address: Polymeric Materials, Department of Materials, Swiss Federal Institute of Technology Zürich, Vladimir-Prelog-Weg 1-5/10, 8093 Zürich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2021 |