
The effect of amide bond orientation and symmetry on the self-assembly and gelation of discotic tripeptides†
Santosh
Kumar
,
Santu
Bera
 ,
Sujay Kumar
Nandi
and
Debasish
Haldar
*
,
Sujay Kumar
Nandi
and
Debasish
Haldar
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur-741246, West Bengal, India. E-mail: deba_h76@yahoo.com; deba_h76@iiserkol.ac.in
First published on 20th October 2020
Abstract
A series of discotic tripeptides containing a rigid aromatic core and L-phenylalanine have been developed. The orientation of the amide bonds yielded variations of the structure and self-assembly properties of the compounds. The aggregation behavior of the discotic tripeptides was studied by various spectroscopic techniques. The morphology of the resulting aggregates was studied by field emission electron microscopy and atomic force microscopy. These studies showed that the orientation of the amide bonds has a strong influence on the intermolecular interactions, resulting in huge differences in the aggregation properties, and morphology of the discotic tripeptides. Only the C3-symmetric discotic tripeptides formed organogels. The supramolecular aggregation mechanism of N-centered and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-centered discotic tripeptides for forming 3-fold intermolecular H-bonded helical column were the same, there was only a smaller enthalpy change due to the occurrence of longer distances for the N–H⋯OC bonds of the N-centered discotic tripeptide. Whereas, the C2-symmetric discotic tripeptides 2 and 3 adopted a 6-fold intermolecular H-bonded dimer structure. Thus, this report presents a valuable approach for the fine-tuning of the discotic tripeptide based functional material.
O-centered discotic tripeptides for forming 3-fold intermolecular H-bonded helical column were the same, there was only a smaller enthalpy change due to the occurrence of longer distances for the N–H⋯OC bonds of the N-centered discotic tripeptide. Whereas, the C2-symmetric discotic tripeptides 2 and 3 adopted a 6-fold intermolecular H-bonded dimer structure. Thus, this report presents a valuable approach for the fine-tuning of the discotic tripeptide based functional material.
Introduction
Exploring the mechanisms that produce the supramolecular order structure from simple molecular building blocks is highly important to understand the fundamental processes in material science and biology.1–4 The designer building blocks can self-assemble and this leads to supramolecular polymers with predictable shapes and controlled properties.5–7 The self-assembly mainly happens through molecular recognition and non-covalent interactions such as hydrogen bonding, π–π stacking and hydrophobic interactions, between the building blocks.8 A discotic building block, namely, 1,3,5-benzene tricarboxylic acid, was synthesized by Curtius in 1915.9 However, Matsunaga et al. developed the 1,3,5-benzene tricarboxamides (BTAs) and reported the formation of hexagonal columnar liquid crystals in 1986.10 Since then, 1,3,5-benzene tricarboxamide derivatives have been extensively studied because of their advanced technological applications as discotic liquid crystals, organogels and hydrogels, conducting materials and MRI contrast agents.11 The discotic compound contains a C3-symmetric aromatic core functionalized with three amide groups. Depending on the orientation of the amide groups, there are two distinct types of discotic tricarboxamides, namely, N-centered and CO-centered. Both types of discotic compounds self-aggregate resulting in well-defined supramolecular architectures.12,13 The aggregation process has been extensively studied in the solid state by FT-IR,14 powder X-ray diffraction (PXRD)15 and single crystal X-ray diffraction,16 and in the solution state using CD,17 NMR,18 UV-vis spectrometry19 and fluorescence spectroscopic techniques.20
The simple structure, easy synthesis, wide variation of functionalization and detailed understanding of their supramolecular self-assembly process allows full utilization of this versatile, supramolecular building block in nanotechnology and polymer science.21 In most of this report, the discotic tricarboxamides self-assemble into one-dimensional, nanometer-sized rod-like structures stabilized by threefold H-bonding.22 Previously, it has been reported that the side chain-core interactions in the discotic tricarboxyamide leads to porous organic material.23 The influence of side-chain interactions on the self-assembly of discotic tricarboxyamides has also been discussed.24
With reference to the previously mentioned reports, in the research reported here, the effect of amide bond orientation on the assembly of discotic tripeptides containing a rigid aromatic core and L-phenylalanine was investigated. Previously, Srinivasulu et al. have reported the crystal structure of benzene-1,3,5-tricarbonyl phenylalanine (discotic tripeptide 1) with an edge to edge supramolecular helical arrangement.25 Ishioka et al. have developed a supramolecular gel of benzene-1,3,5-tricarbonyl phenylalanine (discotic tripeptide 1) in ionic liquids.26 Also, the sonication responsive gelation of discotic tripeptide 4 has been reported.27 The orientation of amide bonds changes from discotic tripeptide 1 to 2 to 3 and then completely opposite in 4 (Fig. 1), i.e., discotic tripeptide 1 is CO-centered and discotic tripeptide 4 is N-centered. Compounds 1 and 4 have C3 symmetry, whereas compounds 2 and 3 have C2 symmetry (Fig. 1). Previous reports claimed that the C3 symmetric discotic tripeptide 1 adopt a 3-fold intermolecular H-bonded helical columnar structure and form a gel. But in the current research we were curious about the self-assembly of other compounds. From UV-vis, FT-IR and CD spectroscopic studies, the discotic tripeptides 1 and 4 have a similar self-assembly pattern. Interestingly the C3-symmetric discotic tripeptide 4 also formed an organogel in aromatic solvents. But compounds 2 and 3 failed to form a gel under the same conditions. The field emission – scanning electron microscopy (FE-SEM) images show that the discotic tripeptide 1 has unbranched, rod-like morphology which further entangled to form flower-like structures. The discotic tripeptides 2 and 3 are self-assembled to form a polydisperse disk-like morphology. But, the discotic tripeptide 4 self-assembled to form a fiber network structure. These studies showed that the orientation of the amide bonds has a strong influence on the intermolecular interactions, resulting in huge differences in aggregation properties, and morphology of the discotic tripeptides.
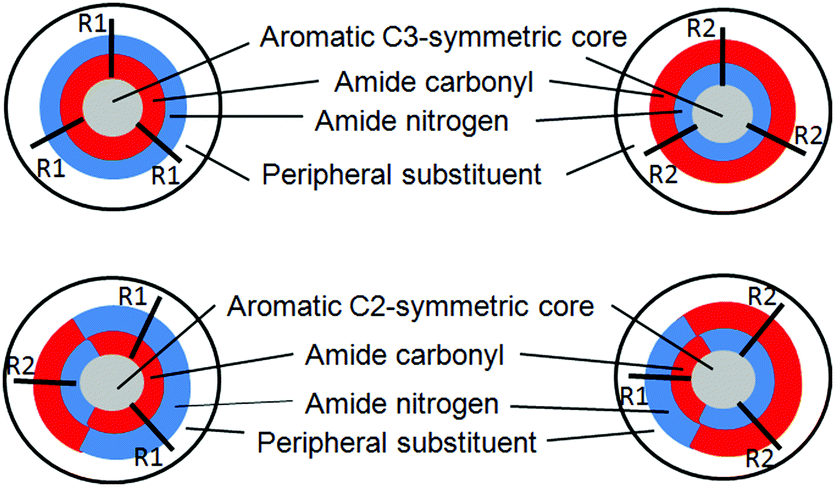 | ||
| Fig. 1 The schematic presentation of diverse discotic tripeptides with rigid aromatic core (gray color), surrounded by amide bonds (red: oxygen, blue: nitrogen) and a peripheral substituent (white). | ||
Experimental
General
L-Phenylalanine and 1,3,5-benzenetricarboxylic acid were purchased from Sigma Chemicals.Synthesis
Compounds 1–4 were synthesized using conventional solution phase methods using a racemization free fragment condensation strategy. The Boc group was used for N-terminal protection and the C-terminus was protected by a methyl ester. Couplings were mediated by dicyclohexylcarbodiimide-1-hydroxybenzotriazole (DCC/HOBt). The final compounds were fully characterized using 400 MHz 1H-NMR spectroscopy, 100 MHz 13C-NMR spectroscopy and FT-IR spectroscopy.NMR studies
The 1H-NMR and 13C-NMR spectrometry of the compound (1–10 mM) in DMSO-d6 solution was perfomed using a Bruker 400 MHz spectrometer. Tetramethylsilane (d = 0.0 ppm) was used as the internal standard.Mass spectrometry
The compound was characterized by mass spectrometry on a Waters Corporation Q-Tof Micro YA263 high-resolution mass spectrometer with electrospray ionization (positive-mode).FT-IR spectroscopy
All the reported solution-state FT-IR spectra were obtained with a Bruker spectrophotometer in DCM solution.UV-vis spectroscopy
UV-vis absorption spectra were recorded on a Hitachi UV-vis spectrophotometer.Fluorescence spectroscopy study
Fluorescence spectra were recorded on a Cary Eclipse fluorescence spectrophotometer. In a typical experiment, compounds were dissolved in methanol. The spectra were recorded by exciting the system at 274 nm, 300 nm and 258 nm.Circular dichroism (CD) spectroscopy
A solution state CD study of peptide 1 in benzene (1 mg mL−1) was carried out before and after sonication on a Jasco J-815-150S spectrometer at a temperature of 25 °C.Gelation study
The gel forming ability of the synthetic compounds was studied in different organic solvents. A portion (5–20 mg) of the corresponding compound was added to 0.5 mL of the solvent under investigation and a clear solution was obtained by heating. The gel appeared after sonication for 30 or 40 s. The gel melting temperature of the resultant organogel was determined by the inverted test tube method.Dynamic light scattering
The particle size of discotic tripeptides 2 and 3 were determined by using a Malvern Instruments Ltd (UK) Zetasizer Nano ZS dynamic light scattering (DLS) instrument. Samples were filtered through a 0.45 μm syringe filter prior to analysis and each measurement was repeated three times to obtain the average value. All the concentrations were kept in the μM range.Polarized optical microscopy (POM)
The morphologies of the discotic tripeptides were determined by polarized optical microscopy. A small amount of the sample was drop-cast on a clean glass cover slip and then dried under vacuum at 30 °C for two days and was then visualized at 40× or 100× magnification on an Olympus optical microscope equipped with a polarizer and CCD camera.Field emission scanning electron microscopy
The FE-SEM samples were prepared following a similar protocol as that used for gelation. Briefly, a slice of gel of discotic peptides 1 and 4 were placed on a glass cover slip and dried under vacuum at 30 °C for two days before proceeding with the imaging. For discotic peptides 2 and 3, a solution of the peptide in toluene was drop-casted on a glass cover slip and dried at room temperature, and then dried under vacuum at 30 °C for two days before proceeding with the imaging. The images of the by gold coated samples were captured with an FE-SEM apparatus in a Zeiss DSM 950 scanning electron microscope.Cryogenic transmission electron microscopy
The microstructure of the reported discotic tripeptides were investigated using cryogenic transmission electron microscopy (Cryo-TEM). The micrographs were captured in Jeol JEM 2010 electron microscope.Results and discussion
The discotic tripeptides were designed with an assumption that the rigid aromatic core would help with self-assemble through hydrogen bonds and π–π stacking interactions (Fig. 2). The orientation of the amide bonds changed from discotic tripeptide 1 to 2 to 3 and then was completely opposite in 4 (Fig. 2). In this context the folding and interactions of hydrophobic L-phenylalanine groups was interesting. The target discotic tripeptides 1–4 were synthesized by conventional solution phase synthesis methodology. The resulting discotic tripeptides were purified by column chromatography and characterized by 1H-NMR, 13C-NMR, FT-IR spectroscopy and mass spectrometry. Previously, the supramolecular aggregation mechanism of N-centered BTA derivatives had been carried out by Stals et al. using temperature-dependent CD and UV-vis analysis and it was concluded that the same cooperative supramolecular aggregation mechanism occurred but with a smaller enthalpy change associated with the elongation step when compared with the analog CO-centered compound.28 From the reported X-ray structures, the H-bond distances (N—H….O=C) for the CO-centered discotic tripeptides were between 1.58 to 2.05 Å.16 However, the N-centered discotic tripeptides showed longer N—H….O=C distances for H-bonds 2.11 to 2.14 Å long, a characteristic for weaker interactions.29 Stals et al. have reported that the rotation along the CAr–N bond (N-centered) was less favourable than the rotation of the CAr–C bond (CO-centered) and a lower cohesive energy for the aggregation process of the N-centered BTA compared with the CO-centered (ΔE of 10 kJ mol−1) due to the occurrence of longer distances for the N–H⋯OC bonds of the N-centered BTA.28 This increased distance was interpreted as a sign of the decrease of the intermolecular H-bond strength.30
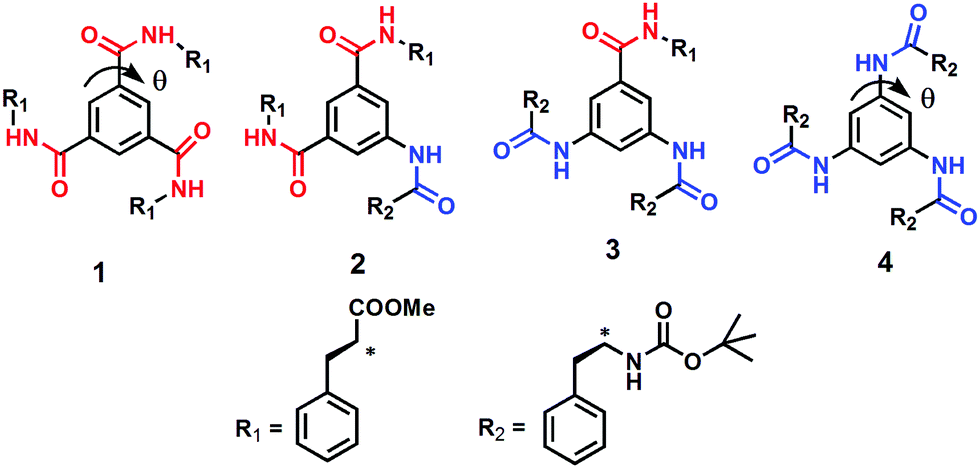 | ||
| Fig. 2 The discotic tripeptides 1–4. | ||
To study the self-assembly properties of discotic tripeptides 1–4, a wide variety of different spectroscopic techniques were used. The typical UV-vis absorption spectra of discotic tripeptide 1 in methanol (2.0 × 10−4 M) exhibited an absorption band at 207 nm for π to π* transition (Fig. S1a, ESI†). However, increasing the concentration of peptide 1 induced stacking interactions between the molecules. The bathochromic shift of 6 nm of the absorption band with increasing concentrations indicated J-type aggregation of discotic tripeptide 1.29 The absorption spectra of discotic tripeptide 2 in methanol also showed absorption band at 300nm (Fig. S1b, ESI†). The absorption spectra of discotic tripeptide 3 in methanol (2.0 × 10−4 M) also showed two band at 302nm (Fig. S1c, ESI†). However, with increasing concentration of 3, the intensity of the bands increased gradually. Hence, the discotic tripeptides 2 and 3 have a close aggregation pattern. The absorption spectra of discotic tripeptide 4 in methanol exhibited an absorption band at 208 nm and a bathochromic shift of 4 nm which indicated J-type aggregation with increasing concentration (Fig. S1d, ESI†). These results showed that the discotic tripeptides 1 and 4 have a similar aggregation pattern. In addition the discoid tripeptides were studied by emission spectroscopy. The emission spectra of discotic tripeptide 1 in methanol (2.0 × 10−4 M, excitation at 274 nm) exhibited bands at 294 nm and 395 nm (Fig. 3a). However, when increasing the concentration of 1 only the intensity of the bands increased. The emission spectra of discotic tripeptide 2 in methanol (2.0 × 10−4 M, excitation at 300 nm) showed a band at 345 nm (Fig. 3b). The hypsochromic shift of the emission band (10 nm) was observed with an increasing concentration of discotic tripeptide 2. The emission spectra of discotic tripeptide 3 in methanol (2.0 × 10−4 M, excitation at 300 nm) showed a band at 350 nm (Fig. 3c) which exhibited a hypsochromic shift 7 nm with increasing concentration. Hence, the discotic tripeptides 2 and 3 have the same cheracteristics. The DLS studies also showed that discotic tripeptides 2 and 3 had the same aggregation pattern (Fig. S2, ESI†). The emission spectra of discotic tripeptide 4 in methanol (2.0 × 10−4 M, excitation at 258 nm) exhibited absorption bands at 286 nm and 375 nm. However, by increasing the concentration of 4 only the intensity of the bands increased (Fig. 3d). Hence, the discotic tripeptides 1 and 4 show the same aggregation pattern.
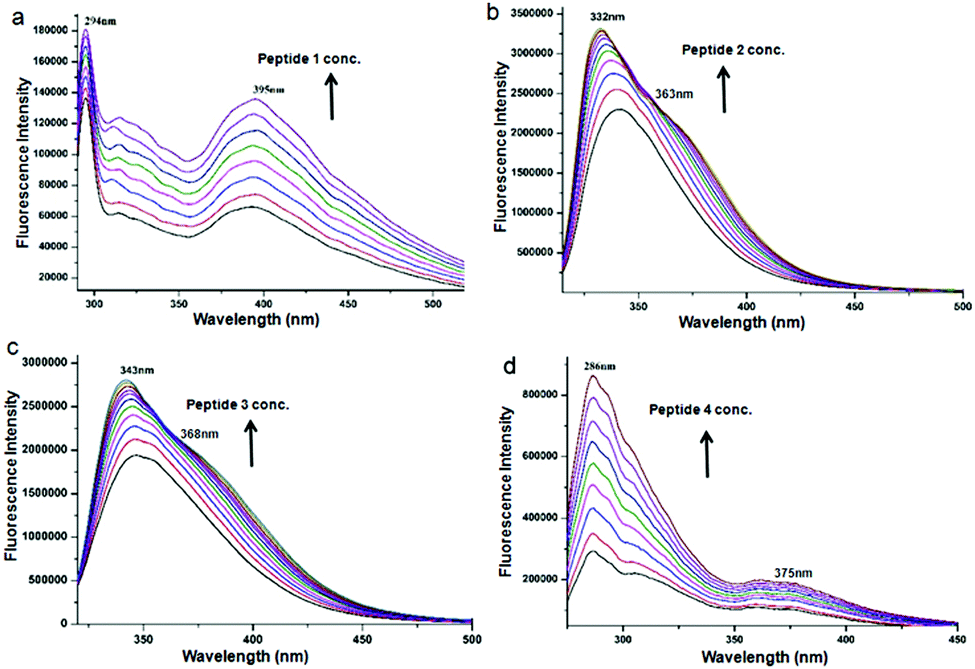 | ||
| Fig. 3 The concentration dependent emission spectra of (a) discotic tripeptide 1, (b) discotic tripeptide 2, (c) discotic tripeptide 3 and (d) discotic tripeptide 4 in methanol. | ||
To study the aggregation behavior of the discotic tripeptides 1–4 further, CD spectroscopy was performed. The shape and intensity of the CD spectra significantly differ for discotic tripeptides 1–4. Discotic tripeptide 1 shows a positive Cotton effect at 198 nm and negative Cotton effects around 208 nm and 228 nm (Fig. 4). It was concluded that such a strong CD signal originated from a rigid self-assembly of the compound. The discotic tripeptide 2 had a positive band at 200 nm and negative bands at 211 nm and 226 nm and the shape was close to that of 1 (Fig. 4). However, discotic tripeptide 3 showed a positive band at 199 nm and a negative band at 207 nm (Fig. 4). The discotic tripeptide 4 exhibited positive bands at 199 nm and 223 nm and a negative band at 210 nm (Fig. 4). Although the orientation of amide bonds in discotic tripeptides 1 and 4 were completely opposite, the shapes of the CD spectra were very similar. These results showed that the discotic tripeptides 1 and 4 have a similar aggregation pattern.
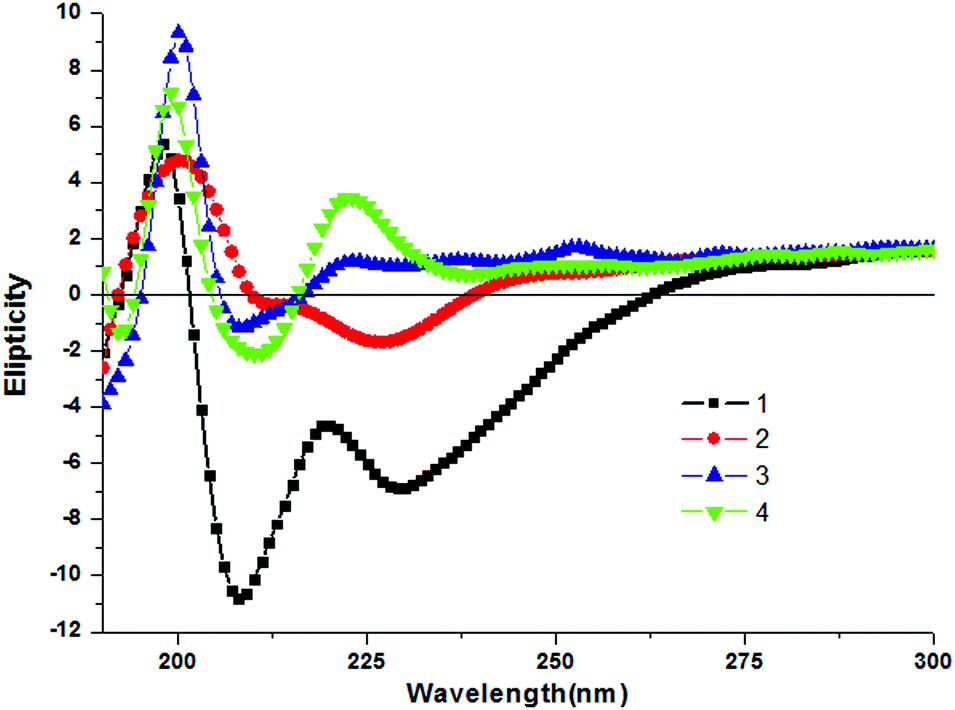 | ||
| Fig. 4 The CD spectra of discotic tripeptides 1 (black), 2 (red), 3 (blue) and 4 (green). | ||
The solution state FT-IR spectroscopy was an excellent method to examine the self-assembly property of the discotic tripeptides. In FT-IR, the region of 3500–3200 cm−1 is important for the N–H stretching vibrations, however the range 1800–1500 cm−1 is assigned to the stretching band of amide I and the bending peak of amide II and ester groups.31 The FT-IR spectra of discotic tripeptide 1 (Fig. 5a) exhibited an N–H stretching frequency at 3188 cm−1 for hydrogen bonded N–H and amide peaks at 1619 and 1535 cm−1 which indicated the presence of threefold intermolecular H-bonding between neighboring molecules within the columnar structures.31 The peak around 1725 cm−1 was responsible for the non hydrogen bonded ester carbonyls. The discotic tripeptide 2 exhibited a peak at 3260 cm−1 for the N–H stretching frequency. The amide and methyl ester appeared at 1631, 1513 and 1717 cm−1 (Fig. 5b). Discotic tripeptide 3 (Fig. 5c) exhibited a N–H stretching frequency at 3265 cm−1 for hydrogen bonded N–H and amide peaks at 1656 and 1505 cm−1. The compound 4 exhibited a peak at 3304 cm−1 for N–H stretching frequency. The amide peaks appeared at 1637 and 1499 cm−1 (Fig. 5d).
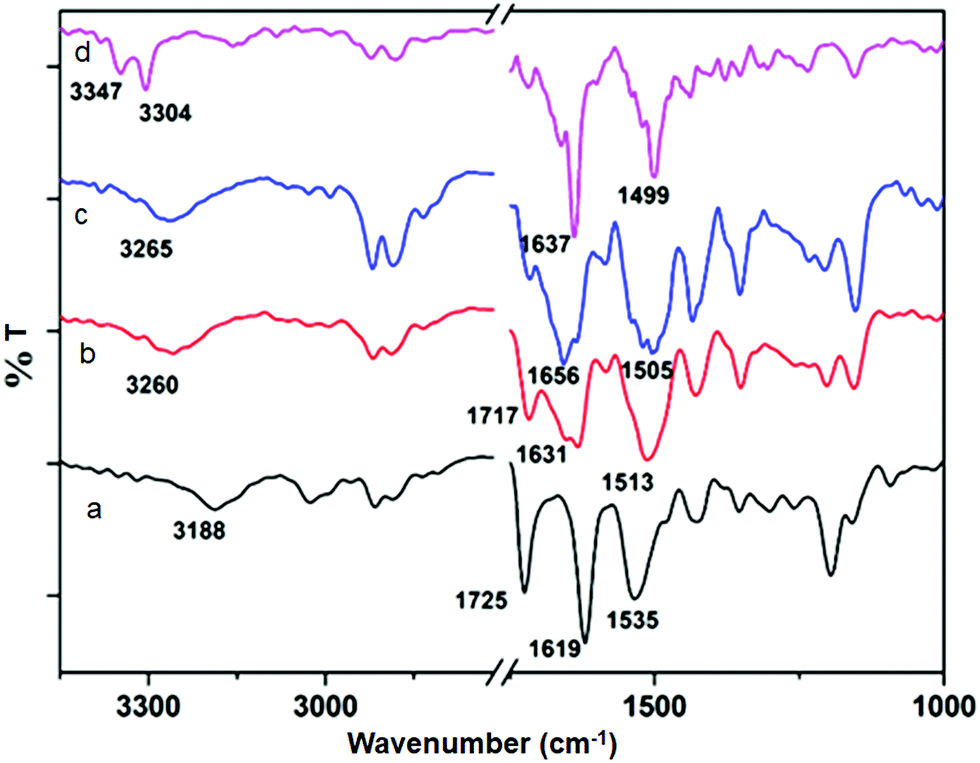 | ||
| Fig. 5 The FT-IR spectra of discotic tripeptides: (a) 1, (b) 2, (c) 3, and (d) 4 in DCM solution. | ||
The aggregation ability of discotic tripeptides 1–4 containing a rigid aromatic core was investigated using the gelation studies. The gelation properties of discotic tripeptides 1–4 were studied in aromatic organic solvents using conventional heating–cooling and sonication techniques.32 The tripodal peptide 1 forms a weak gel in aromatic hydrocarbons such as toluene, o-xylene, m-xylene and p-xylene (6 mg mL−1) (Fig. 6a). With increasing peptide concentration, the gel became strong. In contrast, it was found that discotic tripeptides 2 and 3 did not form a gel in toluene or xylene (Fig. 6b and c). However, a viscous solution was formed at a high concentration, which indicated the formation of higher aggregates. But when solutions of the discotic tripeptide 4 in various aromatic hydrocarbons such as toluene, o-xylene, m-xylene, p-xylene (Table S1, ESI†) were subjected to sonication and cooling, homogeneous gelation was observed (Fig. 6d). The gelation was confirmed by the inverted vial method.32 The gels were thermoreversible. The opaque gels of discotic tripeptides 1 and 4 were stable for 1–2 months at room temperature. The discotic tripeptides 1 and 4 formed a 1-D columnar structure because of the 3-fold intermolecular H-bonding interactions between the amide functionalities, which further self-assembled to form an entangled fiber-like a network and immobilized solvent molecules inside to form a gel. But, the C2-symmetric discotic tripeptides 2 and 3 adopted a 6-fold intermolecular H-bonded dimer structure, which failed to form a fiber network or a gel.
 | ||
| Fig. 6 (a) Weak gel of discotic tripeptide 1 in toluene (6 mg mL−1), (b) solution of discotic tripeptide 2 (20 mg mL−1), (c) solution of discotic tripeptide 3 (20 mg mL−1) and (d) gel of discotic tripeptide 4 in toluene (6 mg mL−1). | ||
The organogel formed by discotic tripeptide 4 in toluene was so stable that the gel could be shaped into any self-supporting geometric shape.33 For discotic tripeptide 1 and 4 gel in toluene, the Tgels (°C) were 101 ± 0.5 °C and 93 ± 0.5 °C, respectively. The gel could be suspended by holding one side (Fig. 7a). Also a big gel block could be sliced into various small pieces with a razor (Fig. 7b).34 Moreover, the organogel formed by discotic tripeptide 4 showed remarkable self-healing properties. When a block of gel was cut into two pieces and then joined together, the pieces merged into a continuous block (Fig. 7c).34 Also, several small blocks of gels could be joined together to form a stable self-supporting bar (Fig. 7d). The fusion of a rhodamine 6G-doped gel with an undoped gel showed the diffusion of rhodamine 6G to the undoped piece, thus suggesting the dynamic exchange of dissolved molecules across the fusion interface (Fig. 7b–d). This self-healing process showed that the gel was living and dynamic in nature.35,36 The dynamic equilibrium between the formation of new fibers and dissociation of old fibers helped the fusion process. The POM images show that the discotic tripeptides do not have liquid crystal property (Fig. S3, ESI†).
 | ||
| Fig. 7 (a) A rhodamine 6G-doped gel cylinder held in the hand. (b) Alternate arrangement of rhodamine 6G-doped and undoped gel cylinders. (c) Self-healing of the gel and the diffusion of rhodamine 6G from the doped to the undoped piece. (d) A cylinder made from four small gel discs suspended on two glass stoppers. The gel shown in these images was prepared in toluene (10 mg mL−1). | ||
Furthermore, FE-SEM was used to study the morphology of the discotic tripeptides. For FE-SEM, the xerogels of discotic tripeptides 1 and 4 from toluene were studied, and for discotic tripeptides 2 and 3, the toluene solutions were placed on microscopic glass slides and then dried under vacuum for two days. The FE-SEM images of the discotic tripeptide 1 xerogel exhibited an entangled fiber morphology (Fig. 8a and Fig. S4, ESI†). The twisted fibers had diameters of approximately 400 nm and were several micrometers long (Fig. 8b). The FE-SEM images of the discotic tripeptide 4 xerogel showed the fibrillar entangled morphology (Fig. 8c). The diameters of the peptide 4 fibers were in the range 80–100 nm (Fig. 8d and Fig. S4, ESI†) and several micrometers in length. These fiber networks immobilized the solvents and helped to form the gel.
 | ||
| Fig. 8 (a) The FE-SEM images of the discotic tripeptide 1 xerogel show the morphology of the entangled fibers. (b) The FE-SEM image of the twisted fibers of the discotic tripeptide 1 in xerogel. (c) FE-SEM image of the long twisted fibers of discotic tripeptide 4 in xerogel prepared from toluene. (d) The fibers of discotic tripeptide 4 form a highly entangled network. | ||
The FE-SEM images of the discotic tripeptide 2 showed that the peptide forms a polydisperse disk-like structure with a diameter in the range of 0.5–2 μm (Fig. 9a). The disks are made of multiple layers and a pore (Fig. 9b). However, the FE-SEM images show that discotic tripeptide 3 also forms disk-like structures (Fig. 9c). The discotic tripeptide 3 disks are polydisperse in nature (Fig. 9d). The average diameter of the disks was 500 nm.
 | ||
| Fig. 9 (a) The FE-SEM image of the discotic tripeptide 2 show disk-like morphology. (b) The disk-like structures of the discotic tripeptide 2 are polydisperse in nature. (c) The FE-SEM image of the disk-like structure of the discotic tripeptide 3. (d) The FE-SEM image showing the disk of the discotic tripeptide 3 are monodisperse in nature. | ||
The morphology of the reported discotic tripeptides was also studied by cryo-TEM. For the cryo-TEM experiments, the solutions of discotic tripeptides 2 and 3 were placed on a grid-mesh and plunge-frozen immediately. The Cryo-TEM images of the discotic tripeptide 2 exhibit a polydisperse disk-like structure where the surface of the disk is very sharp (Fig. 10a). The discotic tripeptide 3 also shows a polydisperse disk-like morphology, but the surface of the disk is very rough (Fig. 10b).
 | ||
| Fig. 10 (a) The cryo-TEM image of discotic tripeptide 2 showing a polydisperse disk-like structure. Inset shows that the surface of the disk is very sharp. (b) The cryo-TEM image of discotic tripeptide 3 showing a polydisperse disk-like structure. Inset shows that the surface of the disk is very rough. | ||
Moreover, to gain a better insight about the self-assembly of the discotic tripeptides, X-ray diffraction experiments were performed.37,38 The PXRD pattern of the xerogels of discotic tripeptides 1 and 4 shows that the materials are crystalline in the xerogel and sharp reflections appeared in the 2θ range of 5–40°. For compounds 1 and 4, the sharp peaks with long d spacing values indicated the presence of a sheet-like packing arrangement (Fig. S5, ESI†).39,40 However, from the PXRD pattern, discotic tripeptides 2 and 3 were non crystalline in nature (Fig. S5, ESI†).
To account for all these data, it was proposed that, irrespective of amide bond orientation, the discotic tripeptides with the C3-symmetry (discotic tripeptides 1 and 4) adopted a 1-D columnar structure (Fig. 11) by 3-fold intermolecular H-bonding interactions between the amide functionalities.41 Because the 1-D column can represent thermodynamic equilibrium, they may be a thermodynamically favorable assembly structure for C3-symmetric discotic tripeptides. These helical columns further self-assembled to form an entangled fiber-like network which immobilized solvent molecules and helped to form the gel (Fig. 11).42–44 Apart from hydrogen-bonding, π–π and hydrophobic interactions also play an important role in driving the self-assembly of the C3-symmetric discotic tripeptides. But, the C2-symmetric discotic tripeptides 2 and 3 adopted a 6-fold intermolecular H-bonded dimer structure (Fig. 11), which prevented further unidirectional fibrillar growth and gelation. The kinetic factors actually influenced the thermodynamic interactions between discotic tripeptide molecules, as well as between discotic tripeptide molecules and the solvents.
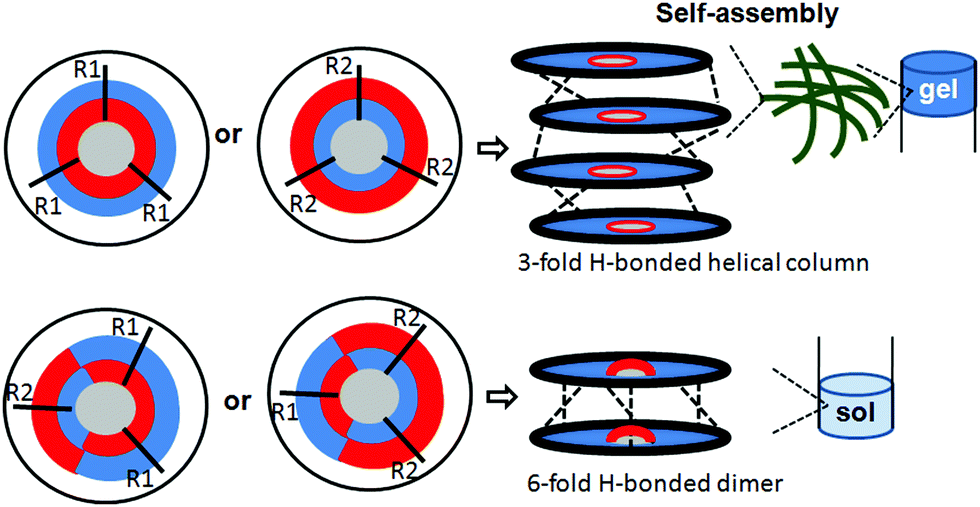 | ||
| Fig. 11 Proposed molecular assembly pattern of the discotic tripeptides. | ||
Conclusion
In conclusion, a series of discotic tripeptides containing a rigid aromatic core and L-phenylalanine were designed and successfully synthesized. The orientation of the amide bonds changed from discotic tripeptide 1 to 2 to 3 and was completely opposite in 4. These structural alterations have a strong influence on the intermolecular interactions, resulting in significant variation of the aggregation properties, gelation abilities and morphology of the compounds. The UV-vis spectroscopy, FT-IR spectroscopy, and CD spectroscopy show that the amide bond orientations have a strong influence on the aggregation ability. Irrespective of the amide bond orientation, the C3-symmetric discotic tripeptides 1 and 4 adopt a 3-fold intermolecular H-bonded helical columnar structure and form an organogel in aromatic solvents. Field emission scanning electron microscopy and atomic force microscopy show the fiber morphology for discotic tripeptides 1 and 4. However, in the absence of C3-symmetry, the discotic tripeptides 2 and 3 have a polydisperse microsphere morphology. These studies show that the orientation of the amide bonds and symmetry has a strong influence on the intermolecular interactions, resulting in huge differences in aggregation properties, and morphology of the discotic tripeptides. The findings provide a simple but valuable approach for the fine-tuning of next-generation functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK and SKN thank CSIR, India for their research fellowships. SB thanks UGC, India for his research fellowship. We thank IISER, Kolkata for use of their analytical facilities.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 1329, 1304–1319 CrossRef.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 144, 1973 CrossRef.
- A. Ciferri, Chem. Rev., 2016, 116, 1353–1374 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS.
- S. Hecht and I. Huc, Foldamers: structure, properties and applications, Wiley-VCH Verlag Gmbh, Weinheim, 2007 Search PubMed.
- T. Curtius, J. Prakt. Chem., 1915, 91, 39–102 CrossRef CAS.
- Y. Matsunaga, Y. Nakayasu, S. Sakai and M. Yonenaga, Mol. Cryst. Liq. Cryst., 1986, 141, 327–333 CrossRef CAS.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolfs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116(3), 1139–1241 CrossRef.
- M. de Loos, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron, 2007, 63, 7285–7301 CrossRef CAS.
- K. P. van den Hout, R. M. Rapffln, J. A. J. M. Vekemans and E. W. Meijer, Chem. – Eur. J., 2007, 13, 8111–8123 CrossRef CAS.
- M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. E. Warren, Chem. Commun., 1999, 1945–1946 RSC.
- A. Desmarchelier, M. Raynal, P. Brocorens, N. Vanthuyne and L. Bouteiller, Chem. Commun., 2015, 51, 7397–7400 RSC.
- A. Paikar and D. Haldar, RSC Adv., 2017, 7, 47170–47176 RSC.
- A. Paikar, A. Pramanik and D. Haldar, RSC Adv., 2015, 5, 31845–31851 RSC.
- S. Dinda, M. Ghosh and P. K. Das, Langmuir, 2016, 32, 6701–6712 CrossRef CAS.
- E. Banach, C. Invernizzi, M. Baudin, R. Neier and D. Carnevale, Phys. Chem. Chem. Phys., 2017, 19, 5525–5539 RSC.
- P. P. Bose, M. G. B. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196–3198 RSC.
- P. Jana, A. Paikar, S. Bera, S. K. Maity and D. Haldar, Org. Lett., 2014, 16, 3841 CrossRef.
- A. Paikar, A. Pramanik and D. Haldar, RSC Adv., 2015, 5, 31845–31851 RSC.
- G. Srinivasulu, B. Sridhar, K. R. Kumar, B. Sreedhar, V. Ramesh, R. Srinivas and A. C. Kunwar, J. Mol. Struct., 2011, 1006, 180–184 CrossRef CAS.
- Y. Ishioka, N. Minakuchi, M. Mizuhata and T. Maruyama, Soft Matter, 2014, 10, 965–971 RSC.
- S. Maity, S. Sarkar, P. Jana, S. K. Maity, S. Bera, V. Mahalingam and D. Haldar, Soft Matter, 2012, 8, 7960–7966 RSC.
- P. J. M. Stals, J. C. Everts, R. de Bruijn, I. A. W. Filot, M. M. J. Smulders, R. Martin-Rapun, E. A. Pidko, T. F. A. de Greef, A. R. A. Palmans and E. W. Meijer, Chem. – Eur. J., 2010, 16, 810–821 CrossRef CAS.
- M. Schmidt, J. J. Wittmann, R. Kress, D. Schneider, S. Steuernagel, H.-W. Schmidt and J. Senker, Cryst. Growth Des., 2012, 12, 2543–2551 CrossRef CAS.
- M. Rozenberg, A. Loewenschus and Y. Marcus, Phys. Chem. Chem. Phys., 2000, 2, 2699–2702 RSC.
- N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K. C. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS.
- S. Maity, P. Kumar and D. Haldar, Soft Matter, 2011, 7, 5239–5245 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS.
- A. Vidyasagar, K. Handore and K. M. Sureshan, Angew. Chem., Int. Ed., 2011, 50, 8021–8024 CrossRef CAS.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- Z. Xu, J. Peng, N. Yan, H. Yu, S. Zhang, K. Liu and Y. Fang, Soft Matter, 2013, 9, 1091–1099 RSC.
- Q. Jin, J. Li, L. Zhang, S. Fanga and M. Liu, CrystEngComm, 2015, 17, 8058–8063 RSC.
- M. Dubey, A. Kumar, R. K. Gupta and D. S. Pandey, Chem. Commun., 2014, 50, 8144–8147 RSC.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- S. Basak, N. Nandi and A. Banerjee, Chem. Commun., 2014, 50, 6917–6919 RSC.
- X. Lou, R. P. M. Lafleur, C. M. A. Leenders, S. M. C. Schoenmakers, N. M. Matsumoto, M. B. Baker, J. L. J. Van Dongen, A. R. A. Palmans and E. W. Meijer, Nat. Commun., 2017, 8, 15420 CrossRef.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- J. Li, R. Xing, S. Bai and X. Yan, Soft Matter, 2019, 15, 1704–1715 RSC.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S17, synthesis, and characterizations. See DOI: 10.1039/d0sm01804j |
| This journal is © The Royal Society of Chemistry 2021 |