
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncreasing complexity in small-angle X-ray and neutron scattering experiments: from biological membrane mimics to live cells
Enrico F.
Semeraro
 ab,
Lisa
Marx
ab,
Moritz P. K.
Frewein
abc and
Georg
Pabst
*ab
ab,
Lisa
Marx
ab,
Moritz P. K.
Frewein
abc and
Georg
Pabst
*ab
aUniversity of Graz, Institute of Molecular Biosciences, Biophysics Division, NAWI Graz, 8010 Graz, Austria. E-mail: georg.pabst@uni-graz.at; Tel: +43 316 380 4989
bBioTechMed Graz, 8010 Graz, Austria
cInstitut Laue-Langevin, 38000 Grenoble, France
First published on 17th February 2020
Abstract
Small-angle X-ray and neutron scattering are well-established, non-invasive experimental techniques to interrogate global structural properties of biological membrane mimicking systems under physiologically relevant conditions. Recent developments, both in bottom-up sample preparation techniques for increasingly complex model systems, and in data analysis techniques have opened the path toward addressing long standing issues of biological membrane remodelling processes. These efforts also include emerging quantitative scattering studies on live cells, thus enabling a bridging of molecular to cellular length scales. Here, we review recent progress in devising compositional models for joint small-angle X-ray and neutron scattering studies on diverse membrane mimics – with a specific focus on membrane structural coupling to amphiphatic peptides and integral proteins – and live Escherichia coli. In particular, we outline the present state-of-the-art in small-angle scattering methods applied to complex membrane systems, highlighting how increasing system complexity must be followed by an advance in compositional modelling and data-analysis tools.
1 Introduction
X-ray scattering experiments on membranes date back to the 1930–1940ies, when Gundo Boehm, Francis O. Schmitt and coworkers analyzed the fine structure of nerve myelin.1,2 Yet, as a first step, it was necessary to explore the physicochemical properties of membrane lipids, which form the structural matrix of cellular envelopes. Vittorio Luzzati and coworkers, using small-angle X-ray scattering (SAXS), described in seminal studies performed in the 1960s3 most of the diverse mesophases of membrane lipids, well before the fluid mosaic model for the structure of biological membranes was conceived by Singer and Nicholson.4The following steadily growing awareness for lipid-only membranes as valuable tools for studying diverse aspects of membrane physiology under chemically well-defined conditions, created the need to resolve details of internal membrane structure. Note that scattering techniques, unlike most other structure-sensitive techniques, do not require the use of bulky labels, which potentially influence the structural equilibrium. Instead, selective deuterium labelling combined with small-angle neutron scattering (SANS), first demonstrated in groundbreaking studies by Büldt and Zaccai,5,6 is often used to enhance structural resolution.
Another way to enhance structural resolution is to work at reduced levels of sample hydration. Wiener and White used this approach, when introducing a joint analysis of SAXS/SANS data of highly aligned lipid multibilayers,7,8 and likewise John Nagle and coworkers, when evolving an earlier reported theory to accurately describe the line-shape of the Bragg peaks of multilamellar vesicles (MLVs).9 But partial dehydration of samples affects the chemical equilibrium of lipid bilayers, which in turn influences interactions with membrane active compounds such as peptides or drugs. For such issues, it is highly desirable to work at full hydration. This was resolved a few years later by developing an analysis, which also took diffuse scattering contributions into account,10 enabling a detailed structural analysis of fully hydrated MLVs made up of either single lipid species or homogeneous lipid mixtures. Working at full levels of hydration may, however, also lead to the formation of unilamellar vesicles, e.g., due to membrane surface charges. Also sample preparation techniques, such as extrusion or sonication are often used to prepare single-shelled vesicles with a specific size. For the analysis of SAXS/SANS data from such samples, different techniques were developed.11,12 In particular, the achievements of Kučerka and coworkers, who advanced the joint SAXS/SANS analysis technique by Wiener and White to unilamellar vesicles by coupling the analysis to molecular dynamics simulations, need to be mentioned in this context.13
The observation of fluid domains in cholesterol-containing ternary lipid mixtures at the turn of the century14 changed gears in biomembrane research. Besides providing the ability to study the physicochemical properties of the previously proposed membrane rafts,15 these systems also created the need to resolve structural details of the coexisting domains to understand e.g. protein partitioning into specific domains based on hydrophobic matching.16 While SANS and SAXS are in principle sensitive to this information, further developments in data analysis were required. Pencer et al., were the first to demonstrate the detection of submicron sized domains by SANS and subsequently developed tools to analyze their shape and size (see, ref. 17, for review). Later, our laboratory developed a method to analyze SAXS data for coexisting domains in MLVs,18,19 gaining access to their specific transbilayer structural features. Finally, the advent of protocols to fabricate lipid vesicles with transbilayer asymmetry20 not only made it possible to study more realistic plasma membrane mimics, but again demanded an evolution of SAXS/SANS data analysis to detail their leaflet-specific structure, which was achieved recently by Eicher et al.21 Thus, increasing the complexity of membrane mimics always caused an adaptation of the scattering analysis.
In addition to all these bottom-up approaches of increasing system complexity, several groups have started to work out the scattering contributions of natural systems, starting from the early work on nerve myelin1,2 and red blood cells22 (which were still studied under quite dehydrated conditions) to live cells such as bacteria.23,24 This opens up new avenues to bridge the gap between studies on model membranes and cells using the same techniques, i.e. with comparable experimental windows. Research along these directions is spurred by the ease of accessing large-scale facilities hosting either a neutron or synchrotron source. This allows researchers to combine the power of H/D contrast variation of neutron scattering with an array of scattering experiments enabled by the high photon flux available at synchrotron sources,25 culminating at single particle/cell experiments using X-ray free electron lasers.
Focusing mainly on the remodelling of membrane systems mediated by peptides and proteins, the present review provides a timeline from past toward future scattering experiments on membrane mimics and live cells biased by the authors' contributions and knowledge of the field. Besides providing a general overview of structural details, we will also discuss the ability of scattering techniques to provide insight on intrinsically stored elastic properties of membranes which are of relevance for the function of integral membrane proteins.
2 General aspects of contrast in small-angle scattering experiments
SAXS and SANS data can be analysed by using the same formalism. In both cases, the scattering intensity, I(q), is proportional to the square of the scattering density contrast, which is the difference between the scattering length densities (SLDs), ρ, of the molecule of interest and the suspension medium, I(q) ∝ (ρ − ρmedium)2. The quantity ρ is expressed as: | (1) |
| Element/isotope | b X-ray (fm) | b neutron (fm) |
|---|---|---|
| Hydrogen (1H) | 2.82 | −3.74 |
| Deuterium (2D) | 2.82 | 6.67 |
| Carbon (12C) | 16.9 | 6.65 |
| Nitrogen (14N) | 19.7 | 9.36 |
| Oxygen (16O) | 22.5 | 5.80 |
| Phosphorous (31P) | 42.3 | 5.13 |
The difference in contrast between hydrogen and deuterium or light and heavy water (Table 2) allows for the powerful tool of H/D contrast variation.28 Contrast-variation SANS measurements are usually performed by varying the percentage of heavy water in the suspension medium and/or by selectively substituting hydrogen with deuterium atoms in the macromolecules within the samples.29 With this approach, specific regions of the system can be contrast matched regarding their SLD values and hence their contributions to the scattering intensity can be tuned.
| Molecule | (×10−4 nm−2) | |
|---|---|---|
| ρ X-ray | ρ neutron | |
| a Effective SLD in the case of complete H/D exchange of weakly bound hydrogens in 100 wt% D2O (two for PG and one for PE). | ||
| H2O | 9.39 | −0.56 |
| D2O | 9.38 | 6.73 |
| Phosphatidylglycerol (PG) (head-group) | 15.1 | 2.47/3.19a |
| Phosphatidylethanolamine (PE) (head-group) | 16.1 | 2.55/2.98a |
| Palmitic acid | 8.12 | −0.03 |
| Deuterated palmitic acid | 8.12 | 6.64 |
Even though contrast-matching is also possible for SAXS, SANS offers a much wider range of contrast variation, without the need to alter the chemical composition of the suspension medium. Nonetheless, it is good practice to always verify if heavy water or deuterated macromolecules are affecting the native microstructure, kinetics and dynamics of the system. For example, the exchange of H2O by D2O may lead to a clear shift of the melting transition of single component lipid bilayers.30
3 Compositional modelling of membranes
Reconstruction of structural information from scattering intensities in general involves either the application of Fourier transformation techniques or real-space modelling.31,32 Using the latter approach enables detailed structural investigations of complex lipid systems considering their molecular composition. This was particularly demonstrated for membranes with heterogeneities, such as domains or transbilayer asymmetry. Below, we briefly summarize recent developments in the field. For more detailed reviews on these aspects, see, e.g.ref. 33 and 34.3.1 Homogenous membranes
Over the years, diverse models for the transbilayer structure of lipid membranes have been reported.35–39 Currently, the most refined way of describing a lipid bilayer are the scattering length density profile (SDP)13 or the continuous distribution models.37 In both models the scattering contrast is characterized in terms of a composition-dependent lipid parsing using volume-filling probability distribution functions. Specifically, the lipid bilayer is parsed into several quasimolecular fragments and their spatial distribution along the bilayer normal is described with either Gaussian13 or error functions,37 respectively see (Fig. 1A). This procedure is aided by comparison to molecular dynamics (MD) simulations. This yields composition-specific structural models that require supplementary information, such as the molecular lipid volumes as input parameters.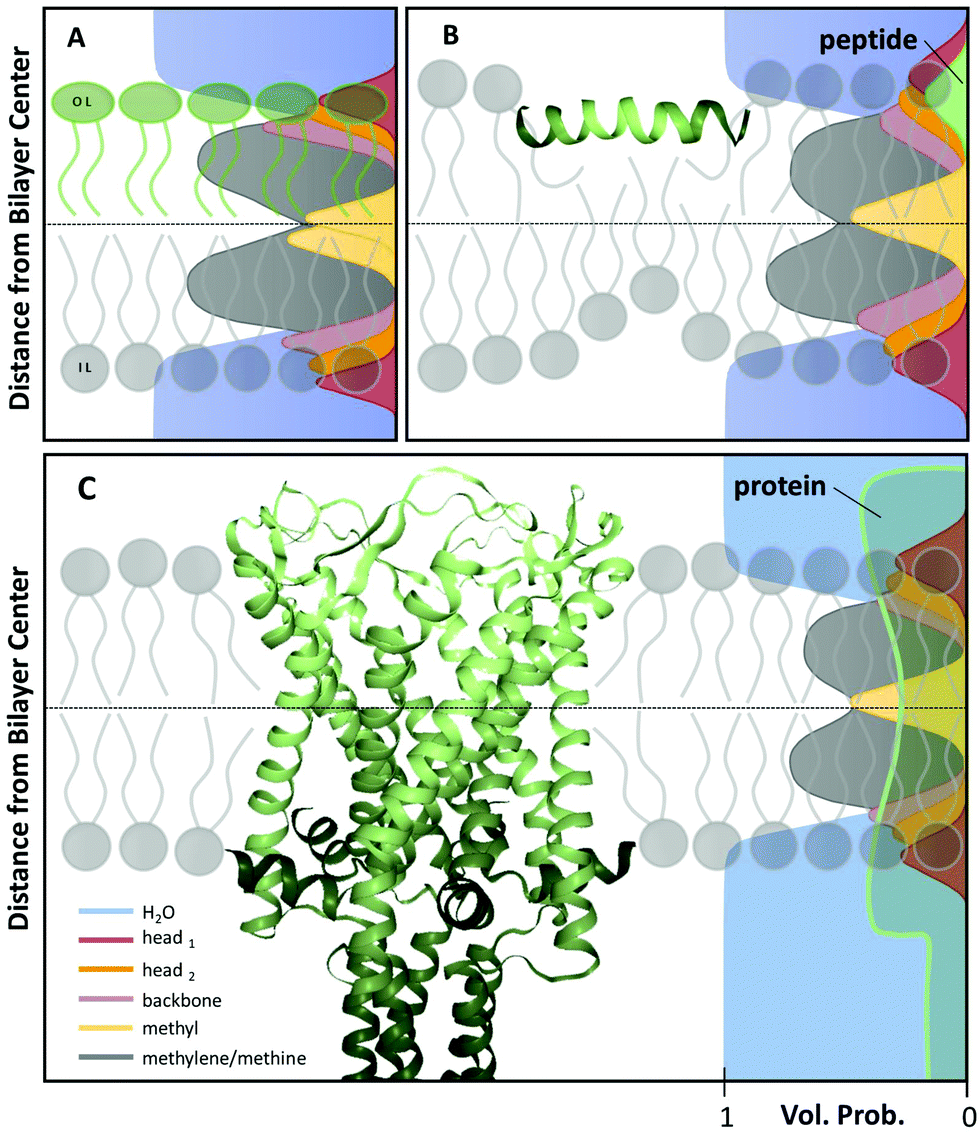 | ||
| Fig. 1 Scattering length density models with distribution functions of Gaussian type for quasimolecular fragments of lipids in a bilayer (A) with asymmetric lipid composition, (B) including a surface-aligned amphipathic peptide (magainin 2; PDB code: 2MAG), or (C), an integral membrane protein (mechanosensitive channel of large conductance; PDB code: 2OAR). | ||
The benefit of using volume distribution functions is the ability to jointly analyze SAXS and differently-contrasted SANS data, which increases structural fidelity. Typically reported parameters are the lateral area per lipid, the bilayer thickness and the length of the hydrocarbon chains. Moreover, this method can be readily extended to multilamellar vesicles,40 which enables the analysis of bilayer interactions using the osmotic stress technique.41 For a recent overview of published high-resolution structures of lipid membranes, see ref. 34.
3.2 Heterogeneous membranes
Heterogeneity in lipid-only membranes arises either from domains in symmetric bilayers, from leaflet-compositional asymmetry, or from a combination of both. The first type of lipid systems is a frequently applied model for gaining insight on membrane-rafts, whose proof of existence is still highly controversial in live cells.42–44Lipid membranes composed of low-melting and high-melting lipids, as well as cholesterol, display coexisting liquid-ordered (Lo)/liquid-disorderd (Ld) domains, over a broad range of compositions and temperatures,45 although it is important to discriminate between microscopic (i.e. micron-sized) and nanoscopic (few nanometers) domains. In the first case, scattering data can be modeled by a linear combination of homogeneous membranes (one representing Lo and the other Ld domains), while cross-correlations between Lo and Ld domains need to be considered for nanoscopic domains.19 In both cases, modelling each lipid species individually would lead to an inordinate number of adjustable parameters. Therefore molecular averages of the individual lipid properties are utilized by defining a virtual hybrid molecule.18 Using this approach, coexisting Lo/Ld domains can be investigated in situ, gaining insight into structural details like cholesterol content.19 In addition, information on domain size can be obtained. This is particularly important for nanoscopic domains that “escape” observation by light microscopy due to diffraction limitations. In-plane contrast is commonly much smaller than transverse contrast, but can be enhanced using SANS in combination with appropriate H/D variation schemes (Fig. 2). To investigate in-plane scattering contributions, transverse contrast has to be suppressed experimentally while enhancing lateral contrast.12 This can be achieved by appropriate mixing of protiated and deuterated lipids, and adjusting the H2O/D2O ratio of the surrounding aqueous solution. This way, nanoscopic domains with sizes as small as 13.6 nm were determined46 Notably, highly ordered nanoscopic lipid domains enriched in cholesterol were also reported in binary mixtures with phosphatidylcholines using neutron diffraction on highly aligned multibilayers.47,48 This supported the idea that Lo and Ld phases consist of inhomogenities due to fluctuations in cholesterol concentration within each domain (see, e.g., ref. 33, for a recent review of this topic).
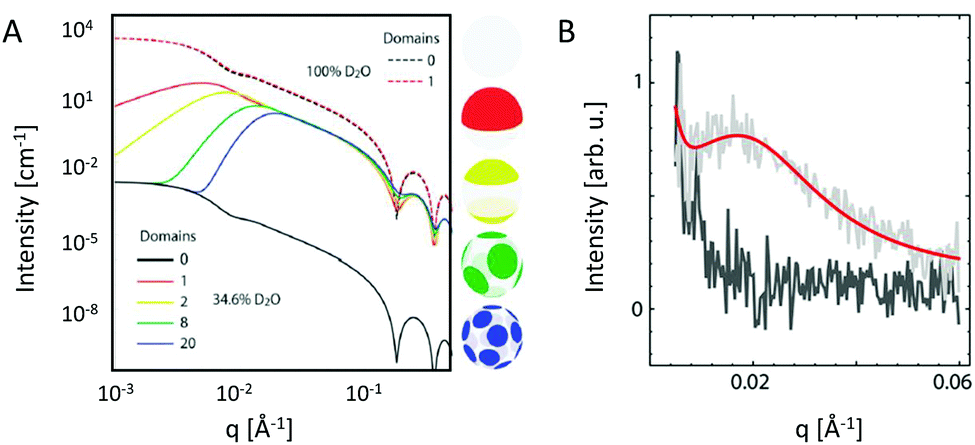 | ||
| Fig. 2 Nanoscopic lipid domains investigated with SANS in combination with contrast variation. Panel A shows theoretical scattering curves for multidomain vesicles in either 100% D2O (dashed-lines) or 34.6% D2O (solid lines). Only the latter condition allows to differentiate between the different domains. Note that the transbilayer contrast is identical for all systems. Panel B shows the analysis of domain exhibiting vesicles (red line), or in a non phase separated (homogeneous) state (dark grey). Panel A: reproduced from ref. 33 with permission from The Royal Society of Chemistry, copyright 2015. Panel B: reproduced (adapted) from ref. 49 with permission from The American Chemical Society, copyright 2013. | ||
The second case of bilayer compositional complexity originates from an asymmetric distribution of lipids across the membrane leaflets. Interest in studying such systems arises from ubiquity of membrane asymmetry in biological cells.50 Recent advances in developing protocols for fabrication of asymmetric large unilamellar vesicles (aLUVs)51–53 have paved the way for systematic studies of asymmetric membranes using an array of biophysical techniques, including SAXS and SANS. The unique advantage of neutron scattering in this regard is the ability to distinguish between the two leaflets, by placing chain deuterated lipids in one of the two monolayers. In this case, the compositional asymmetry is directly linked to a lift-off of the first minimum in the SANS pattern (Fig. 3). Further, it is possible to match the contrast of the aqueous solution with either leaflet to isolate the scattering of a single monolayer. The joint analysis of SAXS and SANS data then enables in-depth interrogation of leaflet-specific structural features, addressing e.g. transbilayer coupling in asymmetric bilayers.21,51,54
 | ||
| Fig. 3 SANS patterns of symmetric and asymmetric LUVs composed of chain-deuterated 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC-d62) and 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) in 100% D2O (unpublished data recorded at the Institute Laue-Langevin, Grenoble, France, beamline D22). Symmetric LUVs show a distinct minimum around q = 10−1 Å−1, which is absent for aLUVs. The inset shows the corresponding trans-bilayer neutron-SLD profiles. | ||
3.3 Inverted hexagonal phases – intrinsic lipid curvatures
Inverted hexagonal phases (HII) are frequently formed by lipids of cone-like shape, such as e.g. phosphatidylethanolamines or glycolipids. Although inverted hexagonal structures have not been observed in vivo so far, membranes usually contain significant fractions of HII-forming lipids. Thylakoid membranes, for example, were demonstrated to exhibit HII phases, if deprived of their protein content.55 It has been hypothesized already in the 1980ies that the high content of non-bilayer forming lipids in membranes indicates mechanical coupling to protein function.56 This can be rationalized on theoretical grounds in terms of the lateral pressure profile57 or a line tension58 that membranes exert on proteins.A fundamental parameter, involved in both considerations is the intrinsic (or spontaneous) lipid curvature C0, which describes the tendency of a lipid monolayer to curl away from a flat surface.59 The lipid intrinsic curvature gives rise, e.g. to a stored elastic energy E0 = kCC02/2, where kC is the monolayer bending rigidity. Importantly, C0 must not be confused with the spontaneous bilayer curvature, which can be determined experimentally from tether-pulling experiments.60
The HII phase offers a valuable template for determining C0 using SAXS.61,62 Essentially, the problem condenses into finding the radius of the neutral plane R0, since C0 = −R0−1. It is reasonable to assume that R0, which describes the plane where lipid bending and stretching modes are decoupled, occurs at the glycerol backbone.63 One way to evaluate SAXS data of HII phases is reconstructing the electron density profile from the peak intensities via Fourier synthesis.63 This readily yields the position of the lipid headgroups, from which R0 can be estimated by adding a constant for the distance to the backbone determined from other experiments.
Compositional modelling of HII phases has the advantage of defining the position of R0 within the model, thus increasing the reliability of C0 determination. In this case, the lipid unit cell has a pie shape (Fig. 4A). For computational reasons it has been proven useful to parse the lipid molecule into different slabs, where R0 is located in the center of the lipid backbone slab.65
 | ||
| Fig. 4 Global analysis of HII phases. Panel A shows a cartoon of an inverted hexagonal phase and the compositional model of the lipid unit cell divided into H2O, H (head), BB (backbone) and HC (hydrocarbon) slabs. Panel B shows successful fits to SAXS data of DMPE (dimyristoyl-phosphatidylethanolamine), di16:1PE (dipalmitoleoyl-phosphatidylethanolamine), and POPE (palmitoyl-oleoyl phosphatidylethanolamine) in the HII phase. Figures and data reproduced (adapted) from ref. 64 with permission of Springer, copyright 2009, and ref. 65 with permission from IUCr Journals, copyright 2019. | ||
Beside the headgroup size, C0 is closely connected to the hydrocarbon chain composition (length and number of double bonds), which causes some disaturated phosphatidylethanolamines such as DMPE (dimyristoyl-phospatidylethanolamine) to have their Lα-HII phase transition, TH, at physiologially unrealistic temperatures. Experiments with HII phases are frequently performed with additional hydrophobic agents such as alkanes61,66 or alkenes.67 These agents do not only significantly lower TH, but also reduce deformations of the cylinders by filling their interstices, thus yielding better estimates of the stress-free monolayer curvature. Interestingly, also ions significantly affect C0. This can be understood by the reorientation of the lipid headgroups toward the aqueous phase,68 which leads to a shift of C0 to more negative values as observed for palmitoyl-oleoyl-phosphatidylethanolamine (POPE) (Table 3).
| Lipid | C 0 (nm−1) | Lipid | C 0 (nm−1) |
|---|---|---|---|
| a Global analysis. b Fourier synthesis from peak intensities. c Linear extrapolation. d T = 80 °C. e Prepared in NaPi-buffer (20 mM Na-phosphate, 130 mM NaCl, pH 7.4). | |||
| DMPEa,d | −0.314 ± 0.00665 | Cholesterolc | −0.494 ± 0.01363 |
di16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1PEa :1PEa |
−0.382 ± 0.00965 | DPPCc | +0.068 ± 0.03263 |
| POPEa | −0.317 ± 0.00765 | POPCc | −0.022 ± 0.01063 |
| DOPEa | −0.409 ± 0.01065 | DOPCc | −0.091 ± 0.00863 |
| POPEb,e | −0.350 ± 0.00769 | DLPEc+ | −0.22 ± 0.0270 |
| DPPEc,e | −0.38 ± 0.1970 | Lyso-PEc,e | +0.18 ± 0.1269 |
| POPGc,e | −0.02 ± 0.0369 | DOPGc,e | +0.03 ± 0.0670 |
Lipids with approximately zero or positive C0 do not form HII phases by themselves and have to be measured indirectly by mixing them with templates of HII-forming (host-) lipids. Here, it is crucial to find the right way of accounting for the contributions of the guest lipids to the mixtures. As a zero order approximation, one can measure the total curvature from samples with varying host/guest lipid ratios and extrapolate linearly to 100% guest lipid content.63 However, even approximating the lipids as rigid cones or cylinders can lead to a non-linear relationship for the total curvature, as for instance, if the headgroup sizes differ. In these cases, a more complex model is more appropriate to account also for lipid-specific interactions. We are currently exploring such approaches.
4 Membrane remodelling by proteins and peptides and vice versa
4.1 Amphipathic peptides
Interactions of amphiphatic peptides, such as e.g. antimicrobial peptides, with lipid model membranes have been widely studied with scattering techniques (see, e.g.ref. 71–73). Depending on the physicochemical properties of the peptides (size/length, polarity, charge, etc.) and the lipid membrane (thickness, charge, intrinsic curvature, elasticity, etc.), the peptides may either remain in a surface-aligned topology or insert at a given angle into the membrane.69,74,75 In addition, aggregation of the peptides atop of the bilayer, or in the membrane-inserted topology in the case of pore formation, may lead to distinct membrane remodelling effects (see, e.g.ref. 76). Often these states are only transient or strongly fluctuate.Peptides in the surface-aligned state typically cause significant membrane perturbation due to the need to bring the lipids of the opposing leaflet close to the hydrophobic surface of the peptide. This leads to the well-described effect of membrane thinning.72 Fully inserted peptides, in turn, may not cause this effect, especially if the peptide length matches the hydrophobic thickness of the bilayer, although significant membrane softening may be induced.77
When peptides do not lead to an aggregation of LUVs (e.g. in the case of low peptide/lipid ratios) SAXS/SANS data can be analyzed in terms of transbilayer scattering length density models, similar to pure lipid membranes as discussed in Section 3. Specifically, peptides can be added to the volume probability distributions using either Gaussian or error functions (Fig. 1B). In the case of a transmembrane topology, only the latter one applies. However, adding the peptides requires appropriate removal of lipid contributions, hence, again, modelling largely benefits from coupling to MD simulations.77,78 With this analysis, the peptide position in the bilayer can be determined with high accuracy by simultaneous statistical analysis of differently contrasted SAXS/SANS experiments.78
Using this approach we have most recently demonstrated distinct membrane effects for the surface-aligned antimicrobial peptides Magainin 2 (MG2a), PGLa and their equimolar mixture.78 Specifically, by combining the scattering data analysis with MD simulations we found that PGLa, because of its lower mean hydrophobic moment, adsorbs slightly deeper into the headgroup region of POPE/palmitoyl-oleoyl-phosphatidylglycerol (POPG) mixtures than MG2a (Fig. 5). Moreover, we observed pronounced membrane thinning for the peptide mixtures at peptide/lipid ratios as low as 1/200, indicating an early onset of peptide heterodimer formation, consistent with MD simulations. The peptide heterodimers, which were found to be stabilized by salt bridges and hydrophobic forces, provide the nucleus of the well-known synergistic activity of MG2a and PGLa at elevated peptide concentrations.69
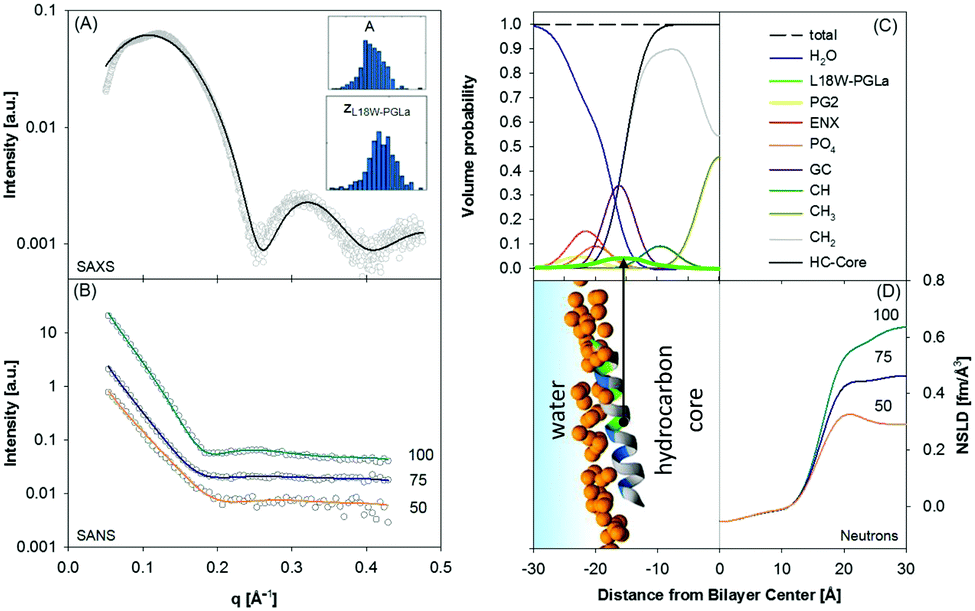 | ||
| Fig. 5 Compositional modelling of surface-aligned PGLa in POPE/POPG (3:1 mol/mol) vesicles (peptide/lipid = 1/200). Panels (A) and (B) present SAXS and contrast variation SANS data, respectively. The inserts show histograms of the area per unit cell, AU, and the position of the peptide in the bilayer, zP, from 400 independent optimization runs. Panel (C) shows the volume probability distributions of the bilayer, and panel (D) displays a snapshot of an all-atom MD simulation (left) and neutron SLD profiles (right) for the different D2O concentrations. Figure reproduced from data reported in ref. 70. | ||
4.2 Integral proteins
Introducing bitopic or polytopic proteins adds even more degrees of freedom to the available parameter space for compositional models. These include vertical translation or tilting of the protein, but also its structural conformation and perturbation of the bilayer in the vicinity of the protein inclusion. Often the latter component is avoided by performing scattering experiments on membrane proteins reconstituted into detergent micelles (see, e.g.ref. 79–81). Naturally, the focus then is on protein structure and not on membrane remodeling by proteins. For a general review on the study of protein/lipid complexes using SANS, see ref. 82.For proteins reconstituted into lipid membranes, essentially two approaches have been reported. None of them rely on proteoliposomes (i.e. proteins reconstituted into lipid vesicles), which suffer from ambiguities in transmembrane protein directionality (inside-out vs. outside-in). That is, detailed information on protein structure is challenged by its variable transmembrane orientations in proteoliposomes (see, e.g.ref. 83). The first strategy involves lipid nanodiscs.84 In this case the orientations of the protein and the lipid disc are coupled (i.e. they diffuse/rotate in the aqueous phase as one entity). Scattering data has been described by a hybrid of a compositional model for the protein surrounding lipid bilayer and a bead-based model, frequently used for interpreting protein solution scattering.85 The second approach for studying membrane inserted proteins has been developed for neutron reflectometry using bilayers sparsely tethered to a solid support (for review, see ref. 86). Briefly, the protein contribution to the scattering unit cell is considered by envelope functions determined by the proteins' cross-sectional areas along the transbilayer coordinate. When crystallographic information on the protein is available, its contribution can be considered as a rigid body. For proteins of unknown structure or highly flexible domains in the aqueous phase, model-independent parameterizations are also feasible. In particular, Hermite splines have proven to be a tractable route for representing the protein envelope (Fig. 1C). Upon combining the protein envelope with the lipid bilayer, it has been shown to be sufficient to adjust the positions of the lipid quasimolecular groups, while leaving the shape of their distribution unaltered.
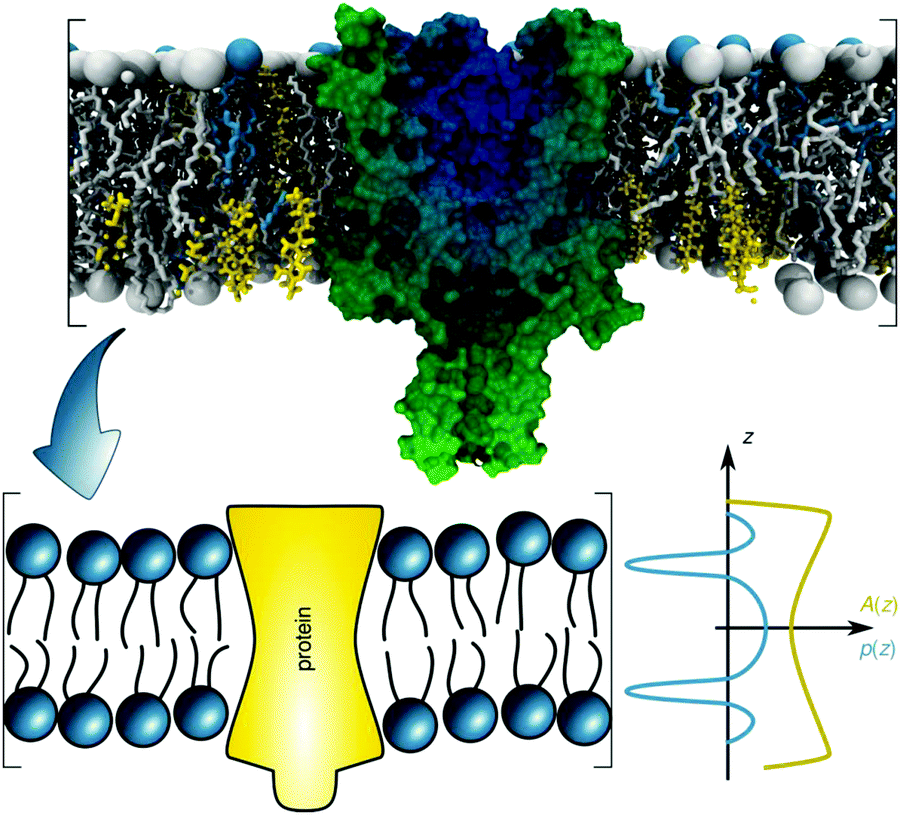 | ||
| Fig. 6 Schematic overview of the lateral pressure profile p(z) and its coupling to a membrane protein, where z is the coordinate normal to the bilayer surface. For calculations, the complex shape of a membrane protein is transferred into a simple rotationally symmetric body with cross sectional area A(z). Reproduced from ref. 92 with permission of The Royal Society of Chemistry, copyright 2016. | ||
The underlying theoretical framework enables calculating the contributions of lateral pressures to conformational changes of integral proteins (e.g. ion channels)93 or preferential protein partitioning in a given lipid environment.92 For example, we have derived the corresponding parameters from a ternary lipid mixture displaying a coexistence of liquid-ordered (Lo) and liquid-disordered (Ld) domains (Fig. 7A–C). Lo-domains exhibit, due to their increased concentration of cholesterol, a significantly more negative C0 (see also Table 3). Further, due to the well-known membrane condensing effect of cholesterol, also h and kC values are higher for Lo (Fig. 7C). Using these parameters we calculated the work for moving a protein of specific shape into different lipid domains. Since parameters were derived for lipid monolayers, we were able to discriminate leaflet registered and antiregistered Lo/Ld domains (i.e. upper/lower leaflet: Ld/Ld, Lo/Lo, Ld/Lo, Lo/Ld). Fig. 7D shows the results for five different protein shapes, where the preferred state is the one with the lowest strain energy. Clearly, the LPP of this specific lipid mixture drives proteins with concave geometries into Lo-domains, convex proteins into Ld-domains and cone-shaped ones into asymmetric membrane compartments (Fig. 7D). The effect is proportional to the protein radius, which means that the propensity for a certain state is increased for larger proteins.92 Interestingly, asymmetric proteins prefer to be located within antiregistered (or asymmetric) domains. This suggests that integral proteins, most of which do have significant degrees of transmembrane asymmetry, energetically prefer an asymmetric lipid environment – alluding to a potential link to the abundance of asymmetry in natural membranes.
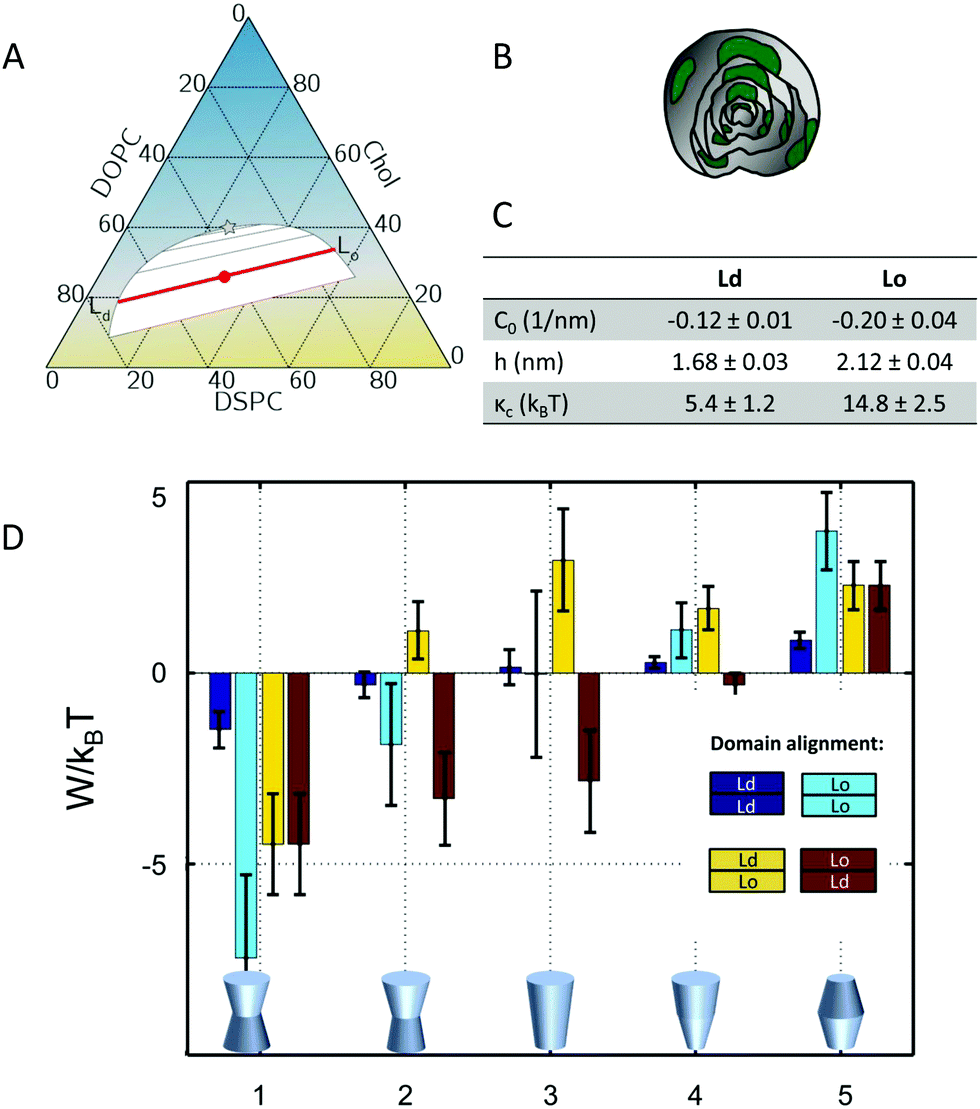 | ||
| Fig. 7 Protein partitioning in a ternary mixture of dioleoyl-phosphatidylcholine (DOPC)/distearoyl-phosphatidylcholine (DSPC)/cholesterol (Chol) (0.42:0.37:0.21 mol/mol/mol). Panel A shows the compositional phase diagram (adapted from ref. 94 with permission of Elsevier, copyright 2007) and indicates the studied mixture as well its separation along the tie-line (red circle and red line). Panels B and C give a cartoon of the studied multilamellar vesicles containing domains and the corresponding structural/elastic parameters determined by SAXS, respectively. Panel D: stored lateral strain energy W for proteins of different shapes in symmetric or asymmetric Lo and Ld domains. The overall preferred lipid environment for a given protein shape is given by the lowest W/kBT-value. Reproduced from ref. 92 with permission of The Royal Society of Chemistry, copyright 2016. | ||
5 Structural insights into live cells
Modelling scattering from live cells seems a taunting task in view of the vast complexity at various levels of structural hierarchy. Indeed, compared to lipid or lipid/protein systems, the degree of retrievable structural details decreases further with the increasing complexity of microorganisms. For example, SANS has been employed to investigate the periodicity of intracellular thylakoid membranes and its variations in connection to the photosynthetic processes in Gram-negative cyanobacteria.95,96 More recently Nickels and coworkers23 performed a nanoscale characterization of membrane heterogeneities of the Gram-positive bacterium Bacillus subtilis. The authors used chemical and genetical manipulations to contrast match all cellular components except for the membrane. This approach allowed them to demonstrate the existence of nanoscopic lipid domains in the bacteria's cell envelope.Here, we focus on a multiscale (nm to μm) scattering model of Escherichia coli24 – one of the most widely studied bacterial strains. The ultrastructure of bacteria is typically resolved by transmission electron microscopy (TEM).97–101 However, TEM imaging is a massively invasive technique and requires fixation in combination with either staining,97 or sample sectioning,100 which might produce artifacts. The advantages of using high-resolution SAS are (i) the possibility to study live bacteria at variable sample conditions (pH, temperature, cosolutes, etc.); (ii) the exclusion of any artifacts originating from staining or labeling and (iii) the determination of structural features from ensemble averages from over 104 to 1010 cell units from one single measurement. Nevertheless, TEM has been instrumental for constraining the multiscale scattering model of E. coli.24 The model accounts for all cellular contributions by combining ultra-SAXS (USAXS)/SAXS and contrast-variation SANS. Note that USAXS extends the range of studied length scales to a few μm and a recent upgrade in synchrotron X-ray instrumentation for USAXS102,103 allowed to fully exploit this technique for bacteria. To achieve this goal, several aspects need to be taken into account, which are summarized below.
E. coli are rod-shaped (length: 1.5–3 μm; diameter: 0.8–1 μm)104 and have a cell wall composed of two membranes,105 which envelope the cytoplasmic content. Specifically, the cytoplasm contains a long, folded DNA ring with associated proteins, which defines the nucleoid region; while the non-nucleoid volume includes ribosomes, proteins, RNA, plasmids, etc. In both regions, the macromolecules are diffusing in an aqueous suspension of up to 1200 low-molecular-weight molecules (including nucleotides, aminoacids, alcohols, etc.) and ions.106 Notably, DNA, tRNA and mRNA fill only 2% of the available volume, while ribosomes and proteins take up around 6–7 vol% and 13–16 vol%, respectively (values adapted from ref. 107). The average radius of gyration, Rg, from these cytoplasmic proteins is ∼2 nm,108 whereas ribosomes of E. coli are much bigger (Rg = 8.8 nm) and have been well characterized in terms of their scattering form factor.109
The bacterial cell envelope of E. coli consists of two membranes enclosing the so-called periplasmic space. The periplasm has a thickness of about 20–30 nm100 and hosts the peptidoglycan, or murein, layer, which is a net-like, stiff structure made up of long disaccharides chains that are covalently bridged by 4–5 amino acid sequences.110 The lipid composition of the cytoplasmic membrane is dominated by phosphatidylethanolamine, phosphatidylglycerol and cardiolipin (about 85:5:10 molar ratio)111 where the predominant hydrocarbon chains are C16:0 and C18:1 fatty acids.112 The outer membrane is highly asymmetric, where the outer leaflet is almost exclusively composed of lipopolysaccharides and the inner leaflet consists of the same phospholipids found in the cytoplasmic membrane. The final structural components of E. coli to consider are flagella, which are protein-based helical tubes (length: 10–15 μm; diameter: ∼20 nm)113 that allow bacteria to perform their typical run-and-tumble dynamics.114
SANS is an indispensable tool to dissect the contributions of all these components making use of the so called scattering invariant, which is a measure of the overall sample contrast.32 Briefly, the square-root of the scattering invariant as a function of D2O wt% shows the contribution of a given entity to the overall scattering signal. In the case of E. coli, the average contrast for phospholipids is “nullified” at about 11 wt% D2O, while proteins and DNA are contrast-matched at 42 wt% and 65 wt% D2O, respectively (Fig. 8A and B).
 | ||
| Fig. 8 Dissecting scattering from live E. coli. Panel A displays a schematic of contrast-matching cellular compartments at diverse D2O concentrations and panel B shows the corresponding scattering invariant estimated from the cellular composition. These include the main components of the cell-wall (left column) and cytoplasmic region (right column). Panel C shows the scattering intensities and fits of E. coli (SANS data have been re-scaled for visibility). The SLD profile of the cell-wall is composed of inner (IM) and outer membrane (OM), peptidoglycan layer (PG), cytoplasm (CP) and periplasm domain (PP). The arrows highlight the q-ranges where scattering features mostly refer to cell-body information (blue) or cell-wall characteristics (red). The inset shows the obtained X-ray SLD of the cell envelope. Reproduced (adapted) from ref. 24 with permission of IUCr Journals, copyright 2017. | ||
On the base of these estimations, the cytoplasm of E. coli was approximated by a prolate of homogeneous SLD, enveloped by several “shells” of different SLDs, describing the cell wall architecture including inner and outer membrane, the peptidoglycan layer and the periplasmic space (Fig. 8C). Note that scattering contributions from macromolecules (proteins, DNA, RNA, ribosomes, etc.) within the cell are negligible, i.e. they have poor contrast when compared to the average SLDs of their surroundings. In addition, the contribution from flagella was included as a smooth background via the asymptotic power law of the self-avoiding-walk polymer model,115i.e. a scattering intensity contribution proportional to q−1.7.
The application of this scattering model provided quantitative measures of the bacterial cell envelope. For example, the average width of the periplasmic space was found to be 23 nm, which is in perfect agreement with the range of values (11–25 nm) reported from TEM.98,100 Further, the periplasm was found to be much more dilute than observed from cryo-TEM using chemically fixed bacteria.99 Indeed, results support more recent TEM data, which minimized sample alterations during preparation,100 alluding again to potential artefacts introduced by labelling techniques. The distance between the peptidoglycan layer and the outer membrane is much more difficult to resolve with TEM. Semeraro et al.24 reported a value of 11 nm for the center-to-center distance between the two layers, consistent with the length of the cylindrical Braun's lipoprotein Lpp-56, which connects the peptidoglycan and the outer membrane.116 Finally, the inner membrane was observed to be slightly thinner (∼4 nm) than the outer membrane (∼6 nm),24 which is most likely due to the larger headgroups of the outer LPS layer, although fine details, such as details of membrane asymmetry or headgroup structure are beyond experimental resolution.
6 Conclusions and outlook
Elastic scattering techniques (SAXS, SANS) provide a wealth of information on membrane structures and have a history of almost one hundred years. The benefits of these techniques definitely originate from the non-invasive nature and the ability to study systems under physiologically relevant conditions, without the need to resort to bulky labels. While most of its applications have been focused on homogeneous lipid-only membranes, the level of complexity of interrogated systems is steadily increasing in order to match those studied by other, complementary techniques. These systems include domains, lipid asymmetry, as well as peptide–lipid or protein–lipid assemblies. Here, we have primarily focused on the latter systems. At the same time, possibly as a corollary to these efforts on bottom-up systems, researchers have commenced with quantitative scattering studies on live cells.Admittedly, a broad application of quantitative elastic scattering on complex mimics or even live cells is challenged by the need of extensive data modelling. Specifically, scattering contributions from all sample components (e.g., lipids, proteins, DNA) are convoluted within one signal. Hence, compositional modelling, i.e., supplying the models with complementary information, limits the physically available/realistic parameter space, but also has to be carefully balanced against overparameterization. Thus, the overall large number of parameters requires application of an advanced global search optimization, including genetic algorithms, statistical data evaluation schemes, or machine learning methods, see e.g.ref. 19, 24, 65, 78, 86 and 117. Moreover, these latter techniques allow also simultaneous analysis of scattering data with complementary experiments or even optimization of the experimental setup, as reported recently for neutron reflectometry.118
Of particular interest for future studies are time-resolved experiments in combination with compositional modelling, exploiting the high photon fluxes available at synchrotron sources. For example, we are currently exploring the effect of antimicrobial peptides on live bacteria, which will yield insight into peptide targets and the evolution of peptide impact on bacteria on the nanoscopic to macroscopic length scales. Current limitations, such as averaging over dead and live cells, could be overcome by single-particle scattering measurements at X-ray free electron laser facilities. Note that SAXS and USAXS have already been used on qualitative grounds for screening of antibiotic effects on E. coli.119,120
Finally, efforts in sample preparation of bottom-up systems will allow to fabricate membrane mimics with controlled membrane asymmetry and integral protein directionality. This will boost the structural insight on membrane remodelling processes that can be gained. Hence, the future is bright in many aspects for elastic scattering techniques.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Austrian Science Funds (FWF), grant no. P30921.References
- G. Boehm, Kolloid-Z., 1933, 62, 22–26 CrossRef CAS.
- F. O. Schmitt, R. S. Bear and K. J. Palmer, J. Cell. Comp. Physiol., 1941, 18, 31–42 CrossRef CAS.
- V. Luzzati, in Biological membranes, ed. D. Chapman, Academic Press, New York, 1968, pp. 71–123 Search PubMed.
- S. J. Singer and G. L. Nicholson, Science, 1972, 175, 720–731 CrossRef CAS PubMed.
- G. Büldt, H. U. Gally, A. Seelig, J. Seelig and G. Zaccai, Nature, 1978, 271, 182–184 CrossRef PubMed.
- G. Zaccai, G. Büldt, A. Seelig and J. Seelig, J. Mol. Biol., 1979, 134, 693–706 CrossRef CAS PubMed.
- M. C. Wiener and S. H. White, Biophys. J., 1991, 59, 174–185 CrossRef CAS PubMed.
- M. C. Wiener and S. H. White, Biophys. J., 1991, 59, 162–173 CrossRef CAS.
- R. Zhang, R. M. Suter and J. F. Nagle, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 5047–5059 CrossRef CAS PubMed.
- G. Pabst, M. Rappolt, H. Amenitsch and P. Laggner, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4000–4009 CrossRef CAS PubMed.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 CrossRef.
- J. Pencer, S. Krueger, C. P. Adams and J. Katsaras, J. Appl. Crystallogr., 2006, 39, 293–303 CrossRef CAS.
- N. Kučerka, J. F. Nagle, J. N. Sachs, S. E. Feller, J. Pencer, A. Jackson and J. Katsaras, Biophys. J., 2008, 95, 2356–2367 CrossRef PubMed.
- L. A. Bagatolli and E. Gratton, Biophys. J., 2000, 79, 434–447 CrossRef CAS PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- D. Marsh, Biophys. J., 2008, 94, 3996–4013 CrossRef CAS PubMed.
- J. Pencer, T. T. Mills, N. Kucerka, M. P. Nieh and J. Katsaras, Methods Mol. Biol., 2007, 398, 231–244 CrossRef CAS PubMed.
- P. Heftberger, B. Kollmitzer, A. A. Rieder, H. Amenitsch and G. Pabst, Biophys. J., 2015, 108, 854–862 CrossRef CAS PubMed.
- M. Belička, A. Weitzer and G. Pabst, Soft Matter, 2017, 13, 1823–1833 RSC.
- H. T. Cheng, Megha and E. London, J. Biol. Chem., 2009, 284, 6079–6092 CrossRef CAS PubMed.
- B. Eicher, F. A. Heberle, D. Marquardt, G. N. Rechberger, J. Katsaras and G. Pabst, J. Appl. Crystallogr., 2017, 50, 419–429 CrossRef CAS PubMed.
- L. McCaughan and S. Krimm, Science, 1980, 207, 1481–1483 CrossRef CAS PubMed.
- J. D. Nickels, S. Chatterjee, C. B. Stanley, S. Qian, X. Cheng, D. A. A. Myles, R. F. Standaert, J. G. Elkins and J. Katsaras, PLoS Biol., 2017, 15, e2002214 CrossRef PubMed.
- E. F. Semeraro, J. M. Devos, L. Porcar, V. T. Forsyth and T. Narayanan, IUCrJ, 2017, 4, 751–757 CrossRef CAS PubMed.
- T. Narayanan and O. Konovalov, Materials, 2020, 13, 752 CrossRef PubMed.
- X-ray Data Booklet, ed. A. C. Thompson, Lawrence Berkeley National Laboratory, Berkeley, 3rd edn, 2009 Search PubMed.
- Neutron Data Booklet, ed. A.-J. Dianoux and G. Lander, Institut Laue-Langevin, Grenoble, 2nd edn, 2003 Search PubMed.
- P. Schurtenberger, in Neutrons, X-rays and light: Scattering methods applied to soft condensed matter, ed. P. Lindner and T. Zemb, Norh-Holland Elsevier, Amsterdam, 2002, ch. 7 Search PubMed.
- E. Mahieu and F. Gabel, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74, 715–726 CrossRef CAS PubMed.
- M. A. Kiselev and D. Lombardo, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3700–3717 CrossRef CAS PubMed.
- B. E. Warren, X-ray diffraction, Dover Publications, New York, 1990 Search PubMed.
- O. Glatter, Scattering Methods and Their Application in Colloid and Interface Science, Elsevier, 1st edn, 2018 Search PubMed.
- D. Marquardt, F. A. Heberle, J. D. Nickels, G. Pabst and J. Katsaras, Soft Matter, 2015, 11, 9055–9072 RSC.
- F. A. Heberle and G. Pabst, Biophys. Rev., 2017, 9, 353–373 CrossRef CAS PubMed.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, 2000, 1469, 159–195 CrossRef CAS.
- G. Pabst, N. Kučerka, M.-P. Nieh, M. C. Rheinstädter and J. Katsaras, Chem. Phys. Lipids, 2010, 163, 460–479 CrossRef CAS PubMed.
- P. Shekhar, H. Nanda, M. Lösche and F. Heinrich, J. Appl. Phys., 2011, 110, 102216 CrossRef.
- F. A. Heberle, J. Pan, R. F. Standaert, P. Drazba, N. Kučerka and J. Katsaras, Eur. Biophys. J., 2012, 41, 875–890 CrossRef CAS PubMed.
- J. C. Fogarty, M. Arjunwadkar, S. A. Pandit and J. Pan, Biochim. Biophys. Acta, 2015, 1848, 662–672 CrossRef CAS PubMed.
- P. Heftberger, B. Kollmitzer, F. A. Heberle, J. Pan, M. Rappolt, H. Amenitsch, N. Kučerka, J. Katsaras and G. Pabst, J. Appl. Crystallogr., 2014, 47, 173–180 CrossRef CAS PubMed.
- B. Kollmitzer, P. Heftberger, R. Podgornik, J. F. Nagle and G. Pabst, Biophys. J., 2015, 108, 2833–2842 CrossRef CAS PubMed.
- C. Eggeling, C. Ringemann, R. Medda, G. Schwarzmann, K. Sandhoff, S. Polyakova, V. N. Belov, B. Hein, C. von Middendorff, A. Schonle and S. W. Hell, Nature, 2009, 457, 1159–1162 CrossRef CAS PubMed.
- M. L. Kraft, Mol. Biol. Cell, 2013, 24, 2765–2768 CrossRef CAS PubMed.
- E. Sevcsik, M. Brameshuber, M. Fölser, J. Weghuber, A. Honigmann and G. J. Schütz, Nat. Commun., 2015, 6, 6969 CrossRef CAS PubMed.
- D. G. Ackerman and G. W. Feigenson, Essays Biochem., 2015, 57, 33–42 CrossRef PubMed.
- F. A. Heberle, M. Doktorova, S. L. Goh, R. F. Standaert, J. Katsaras and G. W. Feigenson, J. Am. Chem. Soc., 2013, 135, 14932–14935 CrossRef CAS PubMed.
- C. L. Armstrong, D. Marquardt, H. Dies, N. Kučerka, Z. Yamani, T. A. Harroun, J. Katsaras, A.-C. Shi and M. C. Rheinstädter, PLoS One, 2013, 8, e66162 CrossRef CAS PubMed.
- L. Toppozini, S. Meinhardt, C. L. Armstrong, Z. Yamani, N. Kučerka, F. Schmid and M. C. Rheinstädter, Phys. Rev. Lett., 2014, 113, 228101 CrossRef PubMed.
- F. A. Heberle, R. S. Petruzielo, J. Pan, P. Drazba, N. KuKučerka, R. F. Standaert, G. W. Feigenson and J. Katsaras, J. Am. Chem. Soc., 2013, 135, 6853–6859 CrossRef CAS PubMed.
- J. M. Boon and B. D. Smith, Med. Res. Rev., 2002, 22, 251–281 CrossRef CAS PubMed.
- F. A. Heberle, D. Marquardt, M. Doktorova, B. Geier, R. F. Standaert, P. Heftberger, B. Kollmitzer, J. D. Nickels, R. A. Dick and G. W. Feigenson, et al. , Langmuir, 2016, 32, 5195–5200 CrossRef CAS PubMed.
- M. Doktorova, F. A. Heberle, B. Eicher, R. F. Standaert, J. Katsaras, E. London, G. Pabst and D. Marquardt, Nat. Protoc., 2018, 13, 2086–2101 CrossRef CAS PubMed.
- M. Markones, C. Drechsler, M. Kaiser, L. Kalie, H. Heerklotz and S. Fiedler, Langmuir, 2018, 34, 1999–2005 CrossRef CAS PubMed.
- B. Eicher, D. Marquardt, F. A. Heberle, I. Letofsky-Papst, G. N. Rechberger, M.-S. Appavou, J. Katsaras and G. Pabst, Biophys. J., 2018, 146–157 CrossRef CAS PubMed.
- I. Simidjiev, S. Stoylova, H. Amenitsch, T. Javorfi, L. Mustardy, P. Laggner, A. Holzenburg and G. Garab, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1473–1476 CrossRef CAS PubMed.
- S. M. Gruner, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3665–3669 CrossRef CAS PubMed.
- R. S. Cantor, J. Phys. Chem. B, 1997, 101, 1723–1725 CrossRef CAS.
- N. Dan and S. A. Safran, Biophys. J., 1998, 75, 1410–1414 CrossRef CAS PubMed.
- J. M. Seddon and R. H. Templer, in Structure and dynamics of membranes, ed. R. Lipowsky and E. Sackmann, North-Holland, Amsterdam, 1995, pp. 97–160 Search PubMed.
- A. Callan-Jones, B. Sorre and P. Bassereau, Cold Spring Harbor Perspect. Biol., 2011, 3, a004648 Search PubMed.
- R. P. Rand, N. L. Fuller, S. M. Gruner and V. A. Parsegian, Biochemistry, 1990, 29, 76–87 CrossRef CAS.
- S. H. Alley, O. Ces, M. Barahona and R. H. Templer, Chem. Phys. Lipids, 2008, 154, 64–67 CrossRef CAS PubMed.
- B. Kollmitzer, P. Heftberger, M. Rappolt and G. Pabst, Soft Matter, 2013, 9, 10877–10884 RSC.
- G. Tresset, PMC Biophys., 2009, 2, 3 CrossRef PubMed.
- M. P. K. Frewein, M. Rumetshofer and G. Pabst, J. Appl. Crystallogr., 2019, 52, 403–414 CrossRef CAS PubMed.
- M. W. Tate and S. M. Gruner, Biochemistry, 1989, 28, 4245–4253 CrossRef CAS PubMed.
- H. P. Vacklin, B. J. Khoo, K. H. Madan, J. M. Seddon and R. H. Templer, Langmuir, 2000, 16, 4741–4748 CrossRef CAS.
- J. Seelig, P. M. Macdonald and P. G. Scherer, Biochemistry, 1987, 26, 7535–7541 CrossRef CAS PubMed.
- R. Leber, M. Pachler, I. Kabelka, I. Svoboda, D. Enkoller, R. Vácha, K. Lohner and G. Pabst, Biophys. J., 2018, 114, 1945–1954 CrossRef CAS PubMed.
- M. Pachler, PhD thesis, TU Graz, Graz, 2019.
- K. Lohner and E. J. Prenner, Biochim. Biophys. Acta, 1999, 1462, 141–156 CrossRef CAS.
- H. W. Huang, Biochim. Biophys. Acta, 2006, 1758, 1292–1302 CrossRef CAS PubMed.
- A. Kumagai, F. G. Dupuy, Z. Arsov, Y. Elhady, D. Moody, R. K. Ernst, B. Deslouches, R. C. Montelaro, Y. Peter Di and S. Tristram-Nagle, Soft Matter, 2019, 15, 1860–1868 RSC.
- B. Bechinger, Curr. Opin. Colloid Interface Sci., 2009, 14, 349–355 CrossRef CAS.
- E. S. Salnikov and B. Bechinger, Biophys. J., 2011, 100, 1473–1480 CrossRef CAS PubMed.
- W. C. Wimley and K. Hristova, J. Membr. Biol., 2011, 239, 27–34 CrossRef CAS PubMed.
- J. Pan, D. P. Tieleman, J. F. Nagle, N. Kucerka and S. Tristram-Nagle, Biochim. Biophys. Acta, 2009, 1788, 1387–1397 CrossRef CAS PubMed.
- M. Pachler, I. Kabelka, M.-S. Appavou, K. Lohner, R. Vácha and G. Pabst, Biophys. J., 2019, 117, 1858–1869 CrossRef CAS PubMed.
- A. Berthaud, J. Manzi, J. Pérez and S. Mangenot, J. Am. Chem. Soc., 2012, 134, 10080–10088 CrossRef CAS PubMed.
- A. Calcutta, C. M. Jessen, M. A. Behrens, C. L. P. Oliveira, M. L. Renart, J. M. González-Ros, D. E. Otzen, J. S. Pedersen, A. Malmendal and N. C. Nielsen, Biochim. Biophys. Acta, 2012, 1818, 2290–2301 CrossRef CAS PubMed.
- C. Breyton, F. Gabel, M. Lethier, A. Flayhan, G. Durand, J.-M. Jault, C. Juillan-Binard, L. Imbert, M. Moulin, S. Ravaud, M. Härtlein and C. Ebel, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 71 CrossRef PubMed.
- L. A. Clifton, C. Neylon and J. H. Lakey, Methods Mol. Biol., 2013, 974, 119–150 CrossRef CAS.
- K. A. Rubinson, C. Pokalsky, S. Krueger and L. J. Prochaska, Protein J., 2013, 32, 27–38 CrossRef CAS PubMed.
- M. A. Schuler, I. G. Denisov and S. G. Sligar, in Lipid-protein Interactions, ed. J. H. Kleinschmidt, Humana Press and Springer, New York, 2013, pp. 415–433 Search PubMed.
- S. A. R. Kynde, N. Skar-Gislinge, M. C. Pedersen, S. R. Midtgaard, J. B. Simonsen, R. Schweins, K. Mortensen and L. Arleth, Acta Crystallogr., Sect. D: Struct. Biol., 2014, 70, 371–383 CrossRef CAS PubMed.
- F. Heinrich and M. Lösche, Biochim. Biophys. Acta, 2014, 1838, 2341–2349 CrossRef CAS PubMed.
- S. Gupta, J. A. Dura, J. A. Freites, D. J. Tobias and J. K. Blasie, Langmuir, 2012, 28, 10504–10520 CrossRef CAS PubMed.
- Y. D. Paila, S. Tiwari and A. Chattopadhyay, Biochim. Biophys. Acta, 2009, 1788, 295–302 CrossRef CAS PubMed.
- A. G. Lee, Biochim. Biophys. Acta, 2004, 1666, 62–87 CrossRef CAS PubMed.
- O. H. Ollila and I. Vattulainen, in Molecular Simulations and Biomembranes, ed. M. S. P. Sansom and P. C. Biggin, RSC Publishing, London, UK, 2010, pp. 26–55 Search PubMed.
- R. S. Cantor, Chem. Phys. Lipids, 1999, 101, 45–56 CrossRef CAS.
- M. Frewein, B. Kollmitzer, P. Heftberger and G. Pabst, Soft Matter, 2016, 12, 3189–3195 RSC.
- G. Pabst, S. L. Grage, S. Danner-Pongratz, W. Jing, A. S. Ulrich, A. Watts, K. Lohner and A. Hickel, Biophys. J., 2008, 95, 5779–5788 CrossRef CAS PubMed.
- J. Zhao, J. Wu, H. Shao, F. Kong, N. Jain, G. Hunt and G. Feigenson, Biochim. Biophys. Acta, 2007, 1768, 2777–2786 CrossRef CAS PubMed.
- M. Liberton, L. E. Page, W. B. O'Dell, H. O'Neill, E. Mamontov, V. S. Urban and H. B. Pakrasi, J. Biol. Chem., 2013, 288, 3632–3640 CrossRef CAS.
- R. Ünnep, G. Nagy, M. Markó and G. Garab, Plant Physiol. Biochem., 2014, 81, 197–207 CrossRef.
- J. A. Hobot, E. Carlemalm, W. Villiger and E. Kellenberger, J. Bacteriol., 1984, 160, 143–152 CrossRef CAS.
- L. L. Graham, T. J. Beveridge and N. Nanninga, Trends Biochem. Sci., 1991, 16, 328–329 CrossRef CAS.
- T. J. Beveridge, J. Bacteriol., 1999, 181, 4725–4733 CrossRef CAS.
- V. R. F. Matias, A. Al-amoudi, J. Dubochet and T. J. Beveridge, J. Bacteriol., 2003, 185, 6112–6118 CrossRef CAS.
- J. Milne and S. Subramaniam, Nat. Rev. Microbiol., 2009, 7, 666 CrossRef CAS.
- T. Narayanan, H. Wacklin, O. Konovalov and R. Lund, Crystallogr. Rev., 2017, 160–226 CrossRef CAS.
- T. Narayanan, M. Sztucki, P. van Vaerenbergh, J. Léonardon, J. Gorini, L. Claustre, F. Sever, J. Morse and P. Boesecke, J. Appl. Crystallogr., 2018, 51, 1–14 CrossRef PubMed.
- A.-C. Chien, N. S. Hill and P. A. Levin, Curr. Biol., 2012, 22, R340–R349 CrossRef CAS PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- R. P. Maharjan and T. Ferenci, Anal. Biochem., 2003, 313, 145–154 CrossRef.
- Escherichia coli and Salmonella typhimurium - Cellular and Molecular Biology, ed. F. C. Neidhardt, American Society for Microbiology, Washington, DC, 1987 Search PubMed.
- S. B. Zimmerman and S. O. Trach, J. Mol. Biol., 1991, 222, 599–620 CrossRef CAS.
- D. Lebedev, A. Paleskava, A. Shvetcov, M. Polyakova, V. Isaev-Ivanov and A. L. Konevega, Time-resolved structural rearrangements of translocating ribosome: Experimental report LS 2406 (ESRF).
- L. Gan, S. Chen and G. J. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18953–18957 CrossRef CAS.
- K. Lohner, E. Sevcsik and G. Pabst, Liposome-Based Biomembrane Mimetic Systems: Implications for Lipid-Peptide Interactions, 2008 Search PubMed.
- A. J. de Siervo, J. Bacteriol., 1969, 100, 1342–1349 CrossRef CAS.
- I. Yamashita, K. Hasegawa, H. Suzuki, F. Vonderviszt, Y. Mimori-Kiyosue and K. Namba, Nat. Struct. Biol., 1998, 5, 125–132 CrossRef CAS.
- L. Turner, R. Zhang, N. C. Darnton and H. C. Berg, J. Bacteriol., 2010, 192, 3259–3267 CrossRef CAS.
- P. Schurtenberger, in Neutrons, X-rays and light: Scattering methods applied to soft condensed matter, ed. P. Lindner and T. Zemb, Norh-Holland Elsevier, Amsterdam, 2002, ch. 11 Search PubMed.
- W. Shu, J. Liu, H. Ji and M. Lu, J. Mol. Biol., 2000, 299, 1101–1112 CrossRef CAS.
- D. Franke, C. M. Jeffries and D. I. Svergun, Biophys. J., 2018, 114, 2485–2492 CrossRef CAS PubMed.
- B. W. Treece, P. A. Kienzle, D. P. Hoogerheide, C. F. Majkrzak, M. Lösche and F. Heinrich, J. Appl. Crystallogr., 2019, 52, 47–59 CrossRef CAS PubMed.
- A. R. von Gundlach, V. M. Garamus, T. Gorniak, H. A. Davies, M. Reischl, R. Mikut, K. Hilpert and A. Rosenhahn, Biochim. Biophys. Acta, 2015, 1858, 918–925 CrossRef PubMed.
- A. R. von Gundlach, V. M. Garamus, T. M. Willey, J. Ilavsky, K. Hilpert and A. Rosenhahn, J. Appl. Crystallogr., 2016, 49, 2210–2216 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |