Comparison of homogeneous and heterogeneous catalysts in dye-sensitised photoelectrochemical cells for alcohol oxidation coupled to dihydrogen formation†
Received
20th August 2021
, Accepted 30th September 2021
First published on 30th September 2021
Abstract
This study examines two strategies—homo- and heterogeneous approaches for the light-driven oxidation of benzyl alcohol in dye-sensitised photoelectrochemical cells (DSPECs). The DSPEC consists of a mesoporous anatase TiO2 film on FTO (fluorine-doped tin oxide), sensitised with the thienopyrroledione-based dye AP11 as the photoanode and an FTO–Pt cathode combined with a redox-mediating catalyst. The homogeneous catalyst approach entails the addition of the soluble 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) to the DSPEC anolyte, while the heterogeneous strategy employs immobilisation of a TEMPO analogue with a silatrane anchor (S-TEMPO) onto the photoanode. Irradiation of the photoanode oxidises the TEMPO-moiety to TEMPO+, both in the homogeneous and the heterogeneous system, which is a chemical oxidant for benzyl alcohol oxidation. Photoanodes containing the heterogeneous S-TEMPO+ demonstrate decreased photocurrent, attributed to introducing alternative pathways for electron recombination. Moreover, the immobilised S-TEMPO demonstrates an insufficient ability to mediate electron transfer from the organic substrate to the photooxidised dye, resulting in device instability. In contrast, the homogeneous approach with TEMPO as a redox-mediating catalyst in the anolyte is efficient in the light-driven oxidation of benzyl alcohol to benzaldehyde over 32 hours, promoted by the efficient electron mediation of TEMPO between AP11 and the organic substrate. Our work demonstrates that operational limitations in DSPECs can be solved by rational device design using diffusion-mediated electron transfer steps.
Introduction
Global warming urges countries to lower their CO2 emissions and reliance on ubiquitously used fossil fuels.1,2 Solar energy is highly advocated among CO2 neutral energy sources, with the potential to provide sufficient energy to sustain global energy consumption.3 Solar energy can be converted to electricity with photovoltaics (PVs). At the end of 2019, solar PV technology had achieved a global installed capacity of 600 GW with annual capacity additions amounting to ≥100 GW.4 However, the intermittent nature of solar energy supply requires solutions for (long-term) energy storage.5 The conversion of primary feedstocks, such as water and CO2, into fuels presents a potential solution.6 These “solar fuels” can be produced via indirect approaches (i.e., PV panels coupled to an electrolyser) to collect and store electrical energy into molecular bonds via electrochemical reactions.7 As a contrast, the largest natural solar-to-chemical energy converter—photosynthesis—uses a direct, molecular approach consisting of light-harvesting pigments, electron transport chains, redox complexes, and molecular catalysts.8 This inspired scientists to develop photosynthetic devices based on molecular components, in which light-harvesting processes are directly combined with oxidation and reduction reactions.9 Commonly used dye-sensitised photoelectrochemical cells (DSPECs) combine dye-sensitised solar cells (DSSCs) with additional catalytic conversion steps. DSSCs use molecular dyes, wide-bandgap semiconductors, and redox mediators to absorb light to facilitate a solar-to-electrical energy conversion.10–12 Redox mediators, responsible for dye regeneration in DSSCs, can be substituted for catalysts in DSPECs to realise oxidative and reductive transformations. Our focus lies on the oxidation reactions at the photoanode, as shown in Fig. 1.9,13–15
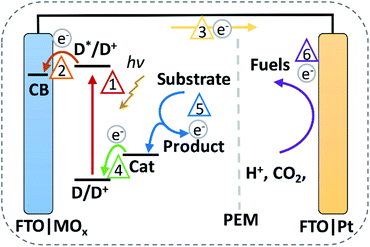 |
| | Fig. 1 Schematic representation of an operating DSPEC for substrate oxidation and reductive fuel production. The electron flow is as follows: (1) irradiation and excitation of a dye, (2) injection of e− in the MOx CB, (3) e− moves to the cathode, (4) Cat regenerates the dye and is oxidised, (5) Cat+ oxidises a substrate to generate e− and the product, (6) e− are reductively stored in fuels (D = dye, D+ = oxidised dye, D* = excited dye, e− = electrons, CB = conduction band, MOx = metal oxide, Cat = catalyst, Cat+ = oxidised catalyst). | |
In the past decades, the focus of solar fuel production (by both direct and indirect approaches) was on H2O oxidation to generate O2, electrons, and protons at the photoanode while simultaneously reducing protons with the generated electrons to H2 at the (photo)cathode.17,18 However, novel developments in DSPECs are moving towards alternative oxidative and reductive reactions to produce other types of chemicals, i.e., oxidation of alcohols at the photoanode.19–21 The ability to combine the generation of fuels and “solar products” increases the relevance of the technology. Interestingly, when using alternative oxidation reactions, lower cell potentials are required, which result in higher cell efficiencies.22–26 Improvement of cell performance is a motivation to study various DSPEC applications to give fundamental insights for the design of practical and real-world implementation of solar-to-product devices. Unfortunately, the DSPECs reported so far show short lifetimes (<1 week).27,28 Device stability is desired for long-term usage and pushes the need for intensive research and development to explore limiting processes.29 The design of DSPECs can be divided into two approaches. (1) A homogeneous approach in which the (molecular or inorganic) catalyst is dissolved in the electrolyte solution and thereby in contact with a dye-sensitised semiconductor.30 In this approach, inactive catalysts can be easily replaced by rejuvenating the bulk electrolyte, similar to batch reactors. However, one must consider that homogeneous catalysts are required in excess in the electrolyte solution to ensure sufficient catalyst concentration at the dye–semiconductor interface, where the catalyst can be activated. Additionally, post-reaction processing of the liquid-phase products is required.31(2) A heterogeneous approach where the catalyst and sensitiser independently or as an assembly are adsorbed on the surface of the semiconductor.17 In this method, the photoanode features a local high catalyst loading at the semiconductor surface and close proximity between the dye and the catalyst. By eliminating diffusion-dependent processes between the catalyst and the dye, electronic communication could be improved. In this setup, the strong binding of the catalyst at the surface is of great importance for device stability since catalyst replacement can be challenging.32 Undesirable surface electron recombination pathways can proliferate when the catalyst is attached at the surface, as observed from water splitting DSPECs, and may lower the efficiencies.33 With the pros and cons of both approaches in mind, we have constructed photoanodes that employ either a surface-bound (Fig. 2a) or solubilised molecular catalyst (Fig. 2b). As the photoanode, we use a highly oxidative thienopyrroledione-based dye (AP11)16 on a TiO2 semiconductor to ensure the catalyst's oxidation. The differences in the activity of both systems are monitored in a photoreactor with in situ monitoring of reaction progress and photoresponse. We employ a 6,6-tetramethylpiperidine-1-oxyl (TEMPO) catalyst and redox mediator for three reasons. Firstly in a catalytic sense, TEMPO+ is known as a sacrificial oxidant for many relevant oxidation reactions in solution organic syntheses.34–38 Secondly, TEMPO is active as a homogeneous and a heterogeneous electrocatalyst, i.e. both when in solution and when bound to the electrode surface.33,39 Furthermore, the TEMPO0/+ couple is a well-studied redox mediator in DSSCs.40–42 Changes in surface processes are studied using AP11–TEMPO+/0-DSSCs with and without surface-bound TEMPO (i.e.S-TEMPO). The addition of S-TEMPO to DSSC photoanodes reveals a decrease in photocurrent density compared to reference systems without an additional redox couple at the surface. The decrease in photocurrent density is probably due to unwanted electron recombination pathways similar to that observed in water splitting systems.33 Photosynthetic experiments indicate that the immobilised S-TEMPO is not capable of delivering sufficient electrons to the oxidised dye. This limitation results in low photocurrents and device degradation.
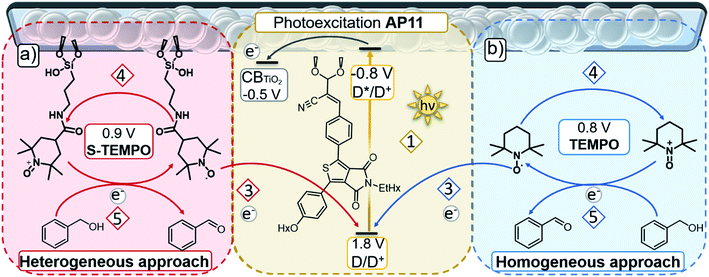 |
| | Fig. 2 Schematic representation of the two different approaches of an operating photoanode of a DSPEC: (a) heterogeneous, (b) homogeneous TEMPO mediated system for light-driven alcohol oxidation, with some of the potentials vs. NHE provided.16 The electron flow is as follows: (1) irradiation by light and excitation of AP11 (Hx = hexyl, EtHx = 2-ethylhexyl), (2) injection of e− in the CB of TiO2, (3) TEMPO regenerates AP11 and (4) is oxidised to TEMPO+ after which (5) TEMPO+ oxidises benzyl alcohol to generate benzaldehyde and obtain e− that are used to regenerate TEMPO acting as a redox mediator to repeat the cycle. | |
In contrast, experiments performed with unbound TEMPO show a ∼100 fold increase in photocurrent compared to the surface-bound species and around 10-fold higher than a previously reported system with a bound PhCH2OH oxidation catalyst.34,43 Near-quantitative faradaic yield is obtained, generating with the photocurrent only PhCHO and H2 in 32 hours.‡44 This paper provides new insights and design considerations for optimising DSPECs by focusing on the trade-off between the importance of preorganisation, electron harvesting properties of the photoanode, long-term device stability, and post-reaction product purification.
Results and discussion
Design and synthesis of building blocks
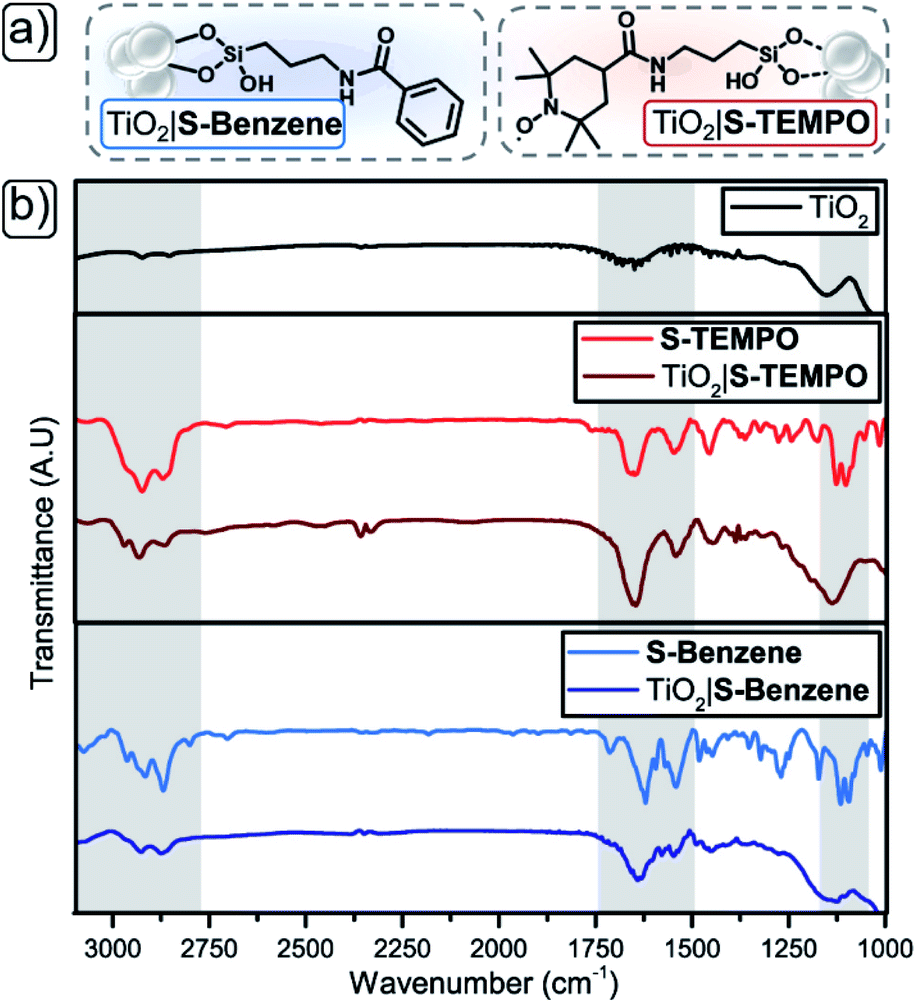
A strategy to enable a fair comparison between the solubilised and immobilised redox-mediating catalyst is of paramount importance to gain insight into the efficiency and stability improvements afforded by these respective approaches. As shown in Fig. 2b, we have chosen unfunctionalised TEMPO as the solution-based redox-mediating catalyst in the homogeneous catalytic oxidation of PhCH2OH. PhCH2OH acts as a model substrate for the operation of the device since the oxidation to PhCHO in combination with TEMPO is well studied.34 We employ S-TEMPO (Fig. 2a and 3a) for the heterogeneous approach. S-TEMPO features a silatrane linker known for creating (photo)electrochemically robust linkages with metal oxides.45–48 As a consequence, we eliminate the likelihood of cleavage of TEMPO from the surface. Silatrane-modified TEMPO (S-TEMPO) is synthesised, according to literature procedures (ESI†), by reacting the acyl chloride with 4-carboxy-TEMPO and subsequently with 3-aminopropyl silatrane.45 We use anatase TiO2 as the wide bandgap metal oxide semiconductor, as it is well studied and widely used in dye-sensitised photoelectrochemical devices.49 Surface modification of TiO2via silatrane anchoring could cause differentiation in semiconductor properties (e.g. chemical capacitance or valence band level).50 To account for differences in semiconductor properties by the silatrane linker, we synthesise an additional silatrane-modified benzene (S-Benzene) as a redox-innocent analogue of S-TEMPO, depicted in Fig. 3a. S-Benzene is prepared by reacting benzoyl chloride with 3-aminopropylsilatrane (ESI†).45
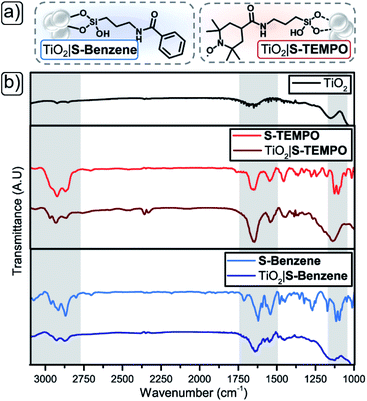 |
| | Fig. 3 (a) Schematic representation of immobilised S-Benzene (left) and S-TEMPO (right). (b) Stacked DRIFT spectrum of S-TEMPO (red) and S-Benzene (light blue) free and on TiO2 support (S-TEMPO = maroon, S-Benzene = marine) with unfunctionalised TiO2 black. | |
Infrared spectroscopic analysis of TiO2 particles functionalised with S-TEMPO or S-Benzene
The methods of functionalisation of TiO2 with S-TEMPO or S-Benzene (ESI†) were assessed by initial immobilisation of the silatranes on TiO2 particles (anatase, <5 μm), followed by characterisation with diffuse-reflectance infrared Fourier-transform (DRIFT) spectroscopy (Fig. 3b and Table 1; full DRIFT spectra in Fig. S1†). The TiO2 acts as a Lewis acid, aiding the hydrolysis of the silatrane moiety and concomitant formation of strong siloxane bonds at the metal oxide surface.51 First, the isolated molecules are discussed. S-Benzene shows a peak at 3060 cm−1 indicating aromatic protons (Table 1, entry 1). The pure compounds S-TEMPO and S-Benzene show aliphatic C–H stretches in the range of 2950–2850 cm−1 from the amide linker and in the case of S-TEMPO from the TEMPO moiety (Table 1, entries 2 and 3). In the fingerprint region, both compounds show two N–H stretches for the amide bond range of 1660–1620 cm−1 (Table 1, entries 4 and 5) and feature two Si–O stretches in the region of 1130–1090 cm−1 (Table 1, entries 6 and 7), originating from the Si–OR of the silatrane.52,53 When comparing the isolated and immobilised molecules on TiO2, similar stretches are visible for aliphatic and aromatic C–H in the range of 3060–2850 cm−1 (Table 1, entries 1–3) and two N–H stretches for the amide bond in the range of 1660–1620 cm−1 (Table 1, entries 4 and 5) indicating the presence of the molecules at the TiO2 particles. However, both TiO2|S-TEMPO and TiO2|S-Benzene spectra lack the two Si–OR stretches in the 1130–1090 cm−1 region (Table 1, entries 6 and 7). The disappearance of these stretches indicates hydrolysis of the silatrane moiety and formation of Si–O–Ti siloxane bonds and, therefore, the binding of the compounds.53
Table 1 Significant DRIFT peaks for isolated and immobilised S-Benzene and S-TEMPO on TiO2 particles
| Entry |
Bond |
S-TEMPO
ν (cm−1) |
TiO2|S-TEMPOν (cm−1) |
S-Benzene
ν (cm−1) |
TiO2|S-Benzeneν (cm−1) |
| 1 |
Ar–H |
— |
— |
3060 (w) |
3069 (w) |
| 2 |
C–H |
2923 (s) |
2926 (s) |
2920 (m) |
2933 (m) |
| 3 |
C–H |
2864 (s) |
2855 (s) |
2874 (m) |
2858 (m) |
| 4 |
N–H |
1657 (s) |
1646 (s) |
1624 (s) |
1634 (s) |
| 5 |
N–H |
1549 (m) |
1550 (m) |
1547 (m) |
1572 (m) |
| 6 |
Si–OR |
1133 (s) |
— |
1120 (s) |
— |
| 7 |
Si–OR |
1098 (s) |
— |
1099 (s) |
— |
Electrochemical characterisation of free compounds and those immobilised on FTO|TiO2
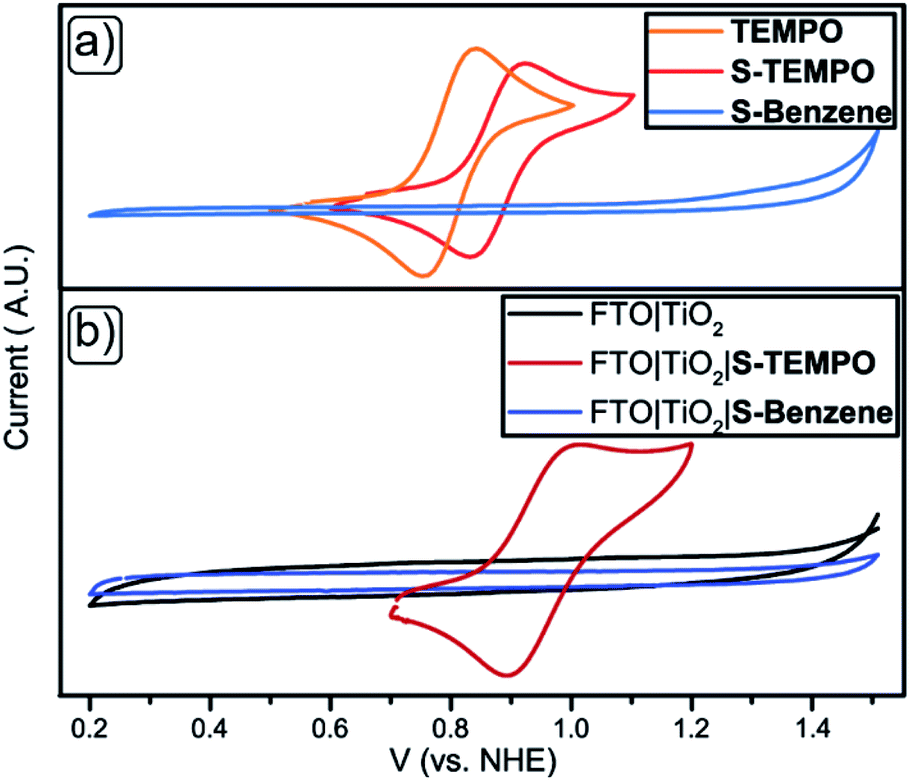
Redox properties of free TEMPO, S-TEMPO, and S-Benzene were compared with their TiO2-bound counterparts. FTO|TiO2 electrodes were prepared by screen printing a ∼4 μm active TiO2 layer onto the FTO glass. The electrodes were functionalised with S-Benzene and S-TEMPO akin to the TiO2 particles functionalisation (ESI†). Cyclic voltammetry (CV) scans of dissolved TEMPO, S-TEMPO, and S-Benzene are shown in Fig. 4a. TEMPO shows a reversible redox wave at E½ = 0.81 V vs. NHE, while S-TEMPO features a reversible redox event at a slightly higher potential of E½ = 0.89 V vs. NHE, corresponding to the nitroxide/oxoammonium species.54 CVs of FTO|TiO2, unfunctionalised and immobilised with S-TEMPO or S-Benzene, are shown in Fig. 4b. FTO|TiO2|S-TEMPO shows a reversible redox wave at E½ = 0.96 V vs. NHE, which is slightly more positive than the free S-TEMPO. Scan-rate (ν) dependent CV (Fig. S2†) was performed to determine the critical scan-rate (νc) and to confirm the surface-bound species. At ν < νc, a linear relationship between the peak current (ip) and the scan-rate appears, indicating a surface-bound species.55 (Fig. S3a and b†). The large peak-to-peak separation is strongly indicative of a slow, interfacial electron transfer due to the less conducting TiO2 (∼1.0 Ω−1 cm−1)56 compared to other electrode materials like for example indium-doped tin oxide (ITO, ∼104 Ω−1 cm−1).57,58 Multiple scans were performed to verify the stability of the electrode and are shown in Fig. S4.† No significant loss in current is observed after multiple scans (20 scans), and the electrodes showed no discolouration. Differential pulse voltammetry (DPV, Fig. S5†) was performed to determine the surface concentration of S-TEMPO, determined to be ∼18 nmol cm−1, comparable to ITO materials of similar thickness containing S-TEMPO.33S-Benzene shows no redox waves both in solution and immobilised state at the TiO2 surface. This observation confirms the redox innocence of S-Benzene at these potentials.
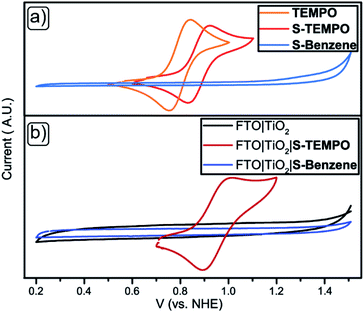 |
| | Fig. 4 CV scans for (a) TEMPO (orange), S-TEMPO (red) and S-Benzene (light blue) in solution (WE = glassy carbon, RE = Ag/AgCl + 0.21 V for NHE, CE = Pt-wire) and (b) functionalised to FTO|TiO2, WE = FTO|TiO2 (black), FTO|TiO2|S-TEMPO (maroon) or FTO|TiO2|S-Benzene (blue) (RE = Ag/AgCl + 0.21 V for NHE, CE = Pt-wire) in 100 mM LiTFSI in MeCN. | |
Design and preparation of the functionalised photoanodes
FTO|TiO2 electrodes were prepared by adding a TiO2 blocking layer onto the FTO glass, and screen printing an ∼4 μm active and a ∼6 μm reflection TiO2 layer, following sensitation with a thienopyrroledione-based (AP11) photosensitiser with a high oxidative power of 1.8 V vs. NHE upon illumination.16 A large Eox of the AP11 dye is needed to ensure the oxidation of all the TEMPO analogues used in this study. We opted for a sequential dipping process for photoanode production, with initial sensitisation using a solution of AP11 to a 50% loading level (as measured by UV-Vis, Fig. S6†). The 50% loading level was done to minimise any differences in photovoltaic performance arising from inconsistent dye loading. In this way, each plate is sensitised with an equal amount of photosensitiser (FTO|TiO2|AP110.5 contains ∼200 nmol cm−2 of AP11), and sufficient space remains on the semiconductor for subsequent silatrane anchoring. Finally, the sensitised electrodes were subjected to immobilisation with S-TEMPO or S-Benzene in a55 similar manner to previous experiments to yield the final photoanode. As a control, a set of unfunctionalised FTO|TiO2|AP110.5 electrodes was added to a blank second solution (i.e., no silatrane) to investigate any dye loss during the functionalisation process.
Photovoltaic performance of DSSCs with S-TEMPO
Sandwich DSSCs were assembled to investigate the influence of anchored S-TEMPO on photocurrent generation along with control solar cells using the redox innocent S-Benzene and unfunctionalised photoanodes. The photoanodes were combined with an electrodeposited Pt on an FTO counter electrode (CE) and a TEMPO+/0 couple solely as a redox mediator to close the electric circuit. Using the TEMPO0/+ couple instead of the regular iodide/triiodide (I−/I3−) couple, any unusual interaction of S-TEMPO and the alien molecules in the electrolyte is excluded. The photocurrent density–voltage (J–V) characteristics and IPCE of the resulting DSSCs were directly measured under a 100 mW cm−2 LED lamp (Fig. S7 and S17†), and the results are shown in Table 2.
Table 2 Data of TEMPO-based FTO|TiO2|AP110.5 sandwich DSSC devices with and without functionalisation with S-TEMPO or S-Benzene with a standard deviation of N = 3 in brackets. (WE = FTO|TiO2|AP110.5 with S-TEMPO or S-Benzene or without, CE = FTO–Pt electrodeposited, electrolyte: 1.2 M LiTFSI, 1.0 M TEMPO and 0.1 M TEMPO(BF4) in MeCN, 0.07 cm2 mask)
| Entry |
Functionalisation |
J
sc (mA cm−2) |
V
OC (V) |
FF |
η (%) |
| 1 |
None |
1.74 (±0.006) |
0.45 (±0.066) |
0.46 (±0.004) |
3.43 (±0.576) |
| 2 |
S-Benzene
|
2.12 (±0.030) |
0.38 (±0.050) |
0.46 (±0.004) |
3.62 (±0.496) |
| 3 |
S-TEMPO
|
0.47 (±0.016) |
0.42 (±0.028) |
0.45 (±0.001) |
0.88 (±0.087) |
The DSSCs prepared with unfunctionalised photoanodes (Table 2, entry 1) show stable short-circuit photocurrent density (Jsc) of 1.74 mA cm−2 and open-circuit voltage (VOC) of 0.45 V over various dipping times (Table S2, entries 1–3†). The invariability indicates the absence of dye leaching or decomposition by the S-TEMPO or S-Benzene functionalisation method over time. For S-Benzene-functionalised DSSCs, the Jsc of 2.12 mA cm−2 is slightly higher, and VOC of 0.38 V is slightly lower than for the unfunctionalised DSSCs, but are still in the same range (Table 2, entry 2). No exchange of AP11 for S-Benzene was observed as there was no discernible drop in Jsc. The slight increase in Jsc and decrease in VOC over dipping time (Table S1, entries 4–6†) can be an effect of surface protection of the benzene moiety, similar to the role of additives like 4-tert-butylpyridine and N-methyl benzimidazole in DSSCs. Here, the molecules adsorb to the TiO2 surface and passivate the surface-active recombination sites.59 In contrast to the S-Benzene-functionalised DSSCs, the DSSCs with an S-TEMPO-functionalised photoanode (Table 2, entry 3) show a lower Jsc of 0.47 mA cm−2 compared to the previous systems. Moreover, a decrease in Jsc is seen over S-TEMPO solution sensitisation time starting from 1.33 mA cm−2 at 1 hour to 0.47 mA cm−2 at 21 hours (Table S2, entries 7–9†). The decrease in Jsc upon anchoring S-TEMPO is likely caused by electron back transfer from the TiO2 to an oxidised S-TEMPO species at the surface. We surmise that additional redox-active TEMPO residues at the surface can scavenge injected electrons and provide an extra recombination pathway. This scavenging effect prevents injected electrons from moving towards the electric circuit and recombining with the oxidised S-TEMPO at the surface.60 In DSPECs, S-TEMPO+ is hypothesised to oxidise alcohols. Thus the back electron transfer from the TiO2 should be less apparent. S-TEMPO-based DSSCs show a VOC of 0.42 V comparable to the reference DSSCs. Also, the VOC appears to stay stable over dipping time, indicating that the injection dynamics of the excited AP11 dye into the conduction band of the TiO2 stay constant. We infer that the TEMPO+/0 couple in the electrolyte ensures sufficient electron injection leading to efficient charge collection. The indication of the generation of TEMPO+ at the surface and the unchanged dye injection dynamics imply a promising photoanode for alcohol oxidation.
Performance of DSPECs with immobilised S-TEMPO
Next, the capability of immobilised S-TEMPO to perform light-driven organic oxidations were examined using a DSPEC photoreactor (Fig. S8†), composed of two Teflon compartments separated by a Nafion-177 membrane. The working electrodes (WE) were prepared as per the photoanodes in the preceding DSSC experiments. Both S-TEMPO- and S-Benzene-functionalised photoanodes were fabricated to account for differences in photocurrent induced by the addition of substrates or by background electron generation processes. The anodic compartment contains the WE (either FTO|TiO2|AP110.5|S-TEMPO or FTO|TiO2|AP110.5|S-Benzene), an Ag/AgCl reference electrode (RE, near the WE), and an anolyte (1.2 M LiTFSI in MeCN). The counter electrode (CE) compartment contained an FTO electrode with an electrodeposited Pt layer as the CE and AcOH was added to the catholyte as an initial proton source. Benzyl alcohol (PhCH2OH) is chosen as a substrate, which will be oxidised to benzaldehyde (PhCHO) and provide protons and electrons for H2 generation. The photocurrent generated in the system originates from the absorption of light by the photosensitiser yielding a dye cation radical. We interpret the photocurrent density enhancement upon adding PhCH2OH at 10 minutes as an indication of effective electron generation as a consequence of substrate oxidation in the S-TEMPO system. The redox mediating properties of the S-TEMPO moiety should ensure dye generation in multiple cycles. In these cycles, S-TEMPO should be photooxidised by the AP11 photosensitiser to S-TEMPO+, after which S-TEMPO+ can oxidise the substrate. The oxidation reaction yields electrons and PhCHO while simultaneously converting S-TEMPO+ back to S-TEMPO needed for dye regeneration. Thus, the PhCH2OH acts as a chemical equivalent of the counter electrode used in a traditional DSSC, regenerating S-TEMPO to enable the system to perform further photocycles. Since redox innocent S-Benzene is incapable of oxidising PhCH2OH, photocurrent density should remain unaffected. Chopped-light measurements are shown in Fig. 5a (full spectrum Fig. S10†). The addition of PhCH2OH (at 10 minutes) did not increase the photocurrent significantly for the S-TEMPO-based system. However, slight variations in photocurrent density are observed upon the addition of PhCH2OH in both S-Benzene and S-TEMPO-based experiments, akin to S-Benzene-based DSSC experiments (vide supra). This invariable electron injection into the electric circuit upon PhCH2OH addition is ascribed to the incapability of S-TEMPO to mediate between the photooxidised photosensitiser and the substrate. S-TEMPO+ seems unable to oxidise PhCH2OH to PHCHO to generate S-TEMPO needed to shuttle electrons into the oxidised photosensitiser. The lack of fresh S-TEMPO results in AP11 being unable to inject electrons into the electric circuit and therefore failing to close the photocycle and increase photocurrent. Notably, the lack of substantial increase in photocurrent density by adding the substrate shows that the S-TEMPO system is incompetent for solar fuel production. We surmise that the biggest problem in the system is the inability of the surface-bound S-TEMPO to diffuse, which prevents S-TEMPO+ from being generated as a bulk chemical. A stable oxidised species is also needed to close the cycle in DSSCs and chemical oxidation to occur. S-TEMPO+ is short-lived due to the occurrence of the recombination processes (vide supra). Therefore a freely diffusing redox mediating catalyst is preferred (vide infra) to prevent these recombination processes.
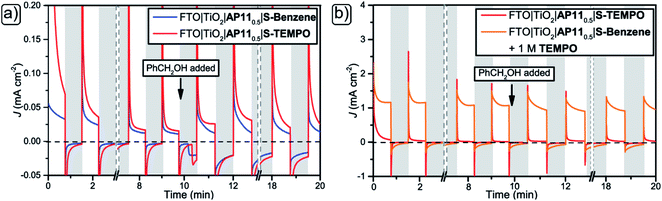 |
| | Fig. 5 Chopped light experiments at 100 mW cm−2 with 45 seconds light on (white background) 45 seconds off (grey background). (a) WE: FTO|TiO2|AP110.5|S-TEMPO (red) or FTO|TiO2|AP110.5|S-Benzene (blue) and (b) WE: FTO|TiO2|AP110.5|S-TEMPO (red) or FTO|TiO2|AP110.5|S-Benzene (orange) + 1 M TEMPO in the electrolyte. General conditions: 1.2 M LiTFSI in MeCN (3 mL), 30 μL PhCH2OH at 10 minutes, CE: FTO|Pt, 1.2 M LiTFSI, 1.0 M AcOH in MeCN (3 mL), separated by Nafion-117 proton exchange membrane, RE: 0 V vs. Ag/AgCl, masked area = 0.64 cm2. | |
Investigation in DSPEC decomposition
A clear observation was the profound disappearance of the typical colour of the AP11 sensitiser observed for DSPEC electrodes used in the heterogeneous approach upon a relatively short duration of the light illumination (30 minutes, Table 3, entries 1 and 2). This behaviour was not observed in the DSSC experiments where identical light intensity was used (Fig. S16†). It is known that UV light irradiation can induce direct bandgap excitation of TiO2, which is capable of oxidative photocatalytic activity. The excitation of TiO2 can produce free radical species, i.e. hydroxyl radicals, which can readily degrade organic matter, or in this case, the adsorbed AP11.61 However, since the LED white light source used contains barely any UV-properties (Fig. S7†), we surmise that this is not the case. Nevertheless, we still wanted to study the effect of over-illumination by reducing light intensity from 100 to 50 mW cm−2 (Fig. S9† and Table 3, entry 3). Here, a notable decrease in dye decomposition over 30 minutes of light exposure is still observed, although not significant enough to illustrate that excessive illumination is the only major cause for DSPEC instability. In contrast, DSSCs contain sufficient redox mediator concentrations for efficient dye regeneration.
Table 3 Images of used photoanodes after 30 minutes chopped-light photosynthetic experiments. WE: FTO|TiO2|AP110.5|S-TEMPO in 3 mL MeCN, 1.2 M LiTFSI, 30 μL PhCH2OH at 10 and 20 minutes, CE: FTO|Pt in 3 mL MeCN, 1.2 M LiTFSI, 1.0 M AcOH, separated by a Nafion-117 proton exchange membrane, RE: 0 V vs. Ag/AgCl
| Entry |
Photoanode |
Irradiation (30 min) |
Additions |
| 1 |

|
Unused |
— |
| 2 |

|
100 mW cm−2 |
— |
| 3 |

|
50 mW cm−2 |
— |
| 4 |

|
100 mW cm−2 |
1 M TEMPO in solution |
The redox mediator prevents the dye from remaining in its oxidised form. Our observations are in line with findings from electrochemical experiments that demonstrate irreversible oxidation at 1.8 V vs. NHE, suggesting decomposition of the oxidised AP11 when electrochemical dye oxidation is performed at the millisecond timescale.16 In photoexcited processes, faster (i.e. picosecond) timescales are preferred for dye regeneration to prevent stability problems in the dye molecule.62 To suppress this photodecomposition phenomenon, we added 1 M of TEMPO to the anolyte for sufficient dye regeneration, akin to the DSSCs. Chopped-light experiments comparing photocurrent density produced by free TEMPO to surface-bound S-TEMPO were performed and are shown in Fig. 5b (full spectrum Fig. S10†). By the addition of TEMPO to the solution, a 100 times increase in photocurrent density was achieved. The increase in photocurrent density suggests that efficient regeneration of the photooxidised dye by TEMPO occurs and that TEMPO+ is generated. It is crucial to notice that an increase in photocurrent density by adding PhCH2OH is not seen in this system and is irrelevant to validating the homogeneous system. The photocurrent density solely relies on the sufficient TEMPO at the surface. To ensure that the redox cycling of TEMPO in the device is not the rate-limiting step, TEMPO is added in a large amount (1 M) and therefore not depleted in a 30 minutes experiment. However, the stability of the photocurrent density is of great importance (vide infra). TEMPO+ is now generated in bulk where the chemical oxidation reaction takes place. As a result of facile electron transfer, the photoanode experienced little decomposition (Table 3, entry 4) after 30 minutes of light exposure. The increased stability of the photoanode, thanks to the presence of the homogeneous redox mediator, demonstrates the importance of fast regeneration of the oxidised photosensitiser in DSPEC design.
Performance of DSPECs with homogeneous TEMPO
Having established the requirement of unimpeded dye regeneration, we utilised homogeneous TEMPO in photosynthetic experiments to oxidise PhCH2OH to PhCHO.44 Here, TEMPO acts as a redox mediator akin to the oxidised dye and the CE in DSSCs. TEMPO is oxidised to TEMPO+ by the photooxidised AP11, after which TEMPO+ is converted back to TEMPO by oxidation of the substrate. Therefore, TEMPO is recycled within the device and only has to be added at the start of the light-driven experiment. Experiments were performed in which the electrodes were sensitised with full dye loading (400 nmol dye cm−2) to improve the light-harvesting properties.
Furthermore, the light intensity was lowered to 50 mW cm−2 to ensure excessive illumination was prevented. A chronoamperometric experiment started by direct illumination of the device and immediate addition of PhCH2OH. The photocurrents and PhCHO production were monitored over 32 hours and are shown in Fig. 6. The generated photocurrent overlapped with PhCHO production and showed a faradaic efficiency of nearly 100%. The CE compartment headspace was sampled by online gas-GC and H2 was confirmed to be the only reduction product (Fig. S11†). In contrast to the heterogeneous system, the homogeneous system shows an average photocurrent of 0.4 mA cm−2 with small fluctuations (Fig. S12†) over a 32 hour illumination experiment.
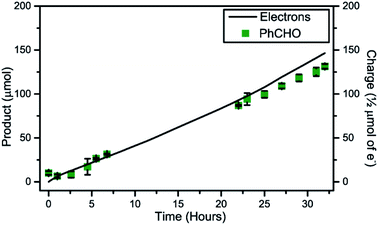 |
| | Fig. 6 GC quantification of light-driven PhCHO production (blue) with the anticipated amount of PhCHO produced based on photocurrent (black line) (WE: FTO|TiO2|AP11 in 3 mL MeCN, 1.2 M LiTFSI, 1.0 M TEMPO, 0.1 M PhCH2OH, 0.2 M C6H5Cl, CE: FTO|Pt in 3 mL MeCN, 1.2 M LiTFSI, 1.0 M AcOH, 0.2 M C6H5Cl separated by a Nafion-117 proton exchange membrane, RE: 0 V vs. Ag/AgCl masked area = 0.64 cm2) illuminated with a 50 mW cm−2 LED lamp for 32 hours. | |
Conclusions
In conclusion, a significant difference has been observed between a homogeneous approach and a heterogeneous one in TEMPO-based DSPEC in the light-driven chemical oxidation of organic alcohols into aldehydes coupled with reductive H2 production. The immobilisation of S-TEMPO results in photocurrent density losses in the DSSCs hypothesised to originate from electron scavenging pathways introduced by a surface-charge build-up upon S-TEMPO+ generation. S-TEMPO+ is unable to diffuse from the surface and, therefore, unable to participate in catalytic dye regeneration. In DSPECs, this inability to diffuse hampers the rapid injection of electrons from the substrate viaTEMPO into the photooxidised dye. Experiments containing surface-bound S-TEMPO showed no increase in photocurrent density after adding a substrate indicating no electron harvesting from substrate oxidation. Increased photocurrent density, leading to aldehyde production, is only achieved using a solubilised TEMPO redox-mediating catalyst. The DSPEC containing the homogeneous TEMPO generates a photocurrent density that is 100 times higher than the surface-bound S-TEMPO setup. Photosynthetic experiments are performed over 32 hours using the homogeneous TEMPO and show quantitative faradaic efficiency if the dye, light, and TEMPO are all present in the system. While the use of heterogeneous semiconductor catalyst assemblies seems appealing for facile product collection, this example demonstrates that these systems are also hampered by electron recombination pathways and dye instability issues. The homogeneous approach using a redox-mediating TEMPO catalyst appears to prevent both of these issues and generates a stable photocurrent of 0.4 mA cm−2 over a 32 hour illumination experiment. This system identifies that unimpeded dye regeneration is an essential element to keep in mind when designing DSPECs. The regeneration of the dye by the TEMPO redox-meditating catalyst in solution opens the way to many different substrates for the oxidation reactions and valuable products, i.e. the oxidation of bio-based feedstock. We are currently exploring biphasic DSPEC systems, featuring a catalytic phase (proximal to the photoanode, containing a freely diffusing redox-mediating catalyst, substrate, and product) and a substrate phase (preferential solubilisation of the substrate/product, present in bulk quantities while disfavouring the redox-mediating catalyst solubilisation). In such a manner, an efficient system that can meet the need for dye regeneration and catalytic properties is made while facilitating the requirement for easy product retrieval.
Experimental
Materials and methods
All reagents and solvents were obtained from Sigma-Aldrich, Fluorochem, or VWR and used without purification unless described otherwise. DCM and toluene were dried in a solvent purification system. AP11, S-TEMPO, S-Benzene, and TEMPO(BF4) are synthesised according to literature procedures with equal 1H-NMR and/or FD-MS as in literature values and are elaborated in the ESI.†16,45,63 Experiments were performed under atmospheric conditions unless otherwise noted. Column chromatography was performed using silica gel (SiliCycle, SiliaFlash P60, 40–63 μm, 230–400 mesh) while fractions were analysed using thin-layer chromatography (TLC silica gel 60 F254, Merck KGaA) visualised with 254/350 nm light. 1H-NMR spectra were recorded on a Bruker AV400 spectrometer, with chemical shifts reported in ppm relative to tetramethylsilane by referencing the residual solvent signal. High-resolution mass spectra (HR-MS) were recorded on an AccuTOF GC v 4g, JMS-T100GCV mass spectrometer (JEOL, Japan) equipped with a field desorption (FD) emitter, Carbotec (Germany), FD 13 μm. The current rate was 51.2 mA min−1, over 1.2 min period, using FD as the ionisation method. Diffuse Reflectance Infrared Fourier-Transform Spectroscopy (DRIFTS) measurements were performed at room temperature on a Nicolet iS50 FTIR (Thermo Fisher, United States) spectrometer equipped with a fast-recovery deuterated triglycine sulfate (DTGS) detector. Cyclic voltammograms were recorded on a PGSTAT10 potentiostat (Autolab) with either a WE = glassy carbon (MetrOhm, diameter 3 mm) or FTO|TiO2(|S-TEMPO or |S-Benzene) electrodes (0.19 cm2) clipped to a holder, RE = a leakless Ag/AgCl (ferrocene0/+ at 0.37 V vs. Ag/AgCl, eDAQ, ET069) and CE = Pt wire in 0.1 M LiTFSI in MeCN solution. Quantification and qualification of the conversion of PhCH2OH to PhCHO by Gas Chromatography (GC) was performed on a Trace GC Ultra machine (Interscience) with an RTX-1 column (30 m, 0.25 mm internal diameter, 0.25 μm film thickness), inlet temperature 70 °C, hold 2.00 minutes, ramp 10 °C min−1 to 340 °C and chlorobenzene as an internal standard (calibration curve, Fig. S13†). Quantification of hydrogen gas was performed on a CompactGC (Interscience) equipped with a 5 Å molecular sieve column (20 m length, 3.2 mm internal diameter), oven temperature of 34 °C, and an argon flow rate of 2 mL min−1.
Preparation of the (functionalised) photoanodes and counter electrodes
A complete elaboration on the (photo)electrode preparation is given in the ESI.† FTO electrodes (Solaronix, 2.2 mm, 15 Ω sq−1) (10 × 5 cm) were cleaned, and a TiCl4 blocking layer was added. One mesoporous active layer of TiO2 anatase nanoparticles (particle size 22–25 nm, Dyenamo) was screen printed (0.79 cm2 circles for DSPECs, 0.19 cm2 circles for DSSCs) and the thickness of the layer after annealing was 3.8 μm. (Fig. S14†);64 after that, a scattering layer of TiO2 (particle size > 100 nm, Solaronix Ti-Nanoxide R/SP) was added onto the plate giving the thickness of the layer after annealing (6.4 μm, Fig. S14†). The FTO|TiO2 electrodes were sensitised with 0.5 mM AP11 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 t-BuOH/MeCN solution in the dark for 45 minutes for silatrane surface-functionalised DSSC and DSPEC experiments and overnight for 1 M TEMPO DSPEC experiments, rinsed afterwards with a 1:1 t-BuOH/MeCN solution and kept in the dark (Fig. S15†). Electrodes used for silatrane functionalised DSSC and DSPEC with S-TEMPO or S-Benzene were prepared by adding the FTO|TiO2 electrodes to a blank, 2 mM S-TEMPO or 2 mM S-Benzene 1% H2O in MeCN solution. The mixture was heated to 70 °C for 1, 2, 5, or 21 hours in the dark under N2. The mixture was allowed to cool down, after which the electrodes were rinsed with MeCN. Modified TiO2 particles were prepared similarly. Counter electrodes were prepared with clean FTO electrodes (Solaronix, 2.2 mm, 15 Ω sq−1), after which a platinum layer was added by electrochemical deposition of platinum.
:1 t-BuOH/MeCN solution in the dark for 45 minutes for silatrane surface-functionalised DSSC and DSPEC experiments and overnight for 1 M TEMPO DSPEC experiments, rinsed afterwards with a 1:1 t-BuOH/MeCN solution and kept in the dark (Fig. S15†). Electrodes used for silatrane functionalised DSSC and DSPEC with S-TEMPO or S-Benzene were prepared by adding the FTO|TiO2 electrodes to a blank, 2 mM S-TEMPO or 2 mM S-Benzene 1% H2O in MeCN solution. The mixture was heated to 70 °C for 1, 2, 5, or 21 hours in the dark under N2. The mixture was allowed to cool down, after which the electrodes were rinsed with MeCN. Modified TiO2 particles were prepared similarly. Counter electrodes were prepared with clean FTO electrodes (Solaronix, 2.2 mm, 15 Ω sq−1), after which a platinum layer was added by electrochemical deposition of platinum.
Construction of dye-sensitised solar cells (DSSCs)
A complete elaboration on the DSSC preparation is given in the ESI.† Sensitised TiO2 photoanodes and Pt counter electrodes were sandwiched together using a hotmelt ionomer, after which the electrolyte (1.2 M LiTFSI, 1.0 M TEMPO, 0.1 M TEMPO (BF4) in MeCN) was introduced into the cells by vacuum backfilling, and the cells were sealed. The DSSCs were masked at 0.07 cm2 and measured using a P211 potentiostat (Zahner TLS3, 100 mW cm−1 LED lamp, Fig. S7†).
Illumination setup of the DSPEC
A complete elaboration on the DSPEC preparation is given in the ESI.† The photoreactor (Fig. S8†) is composed of 2 Teflon compartments, separated by a Nafion-177 membrane (FuelCellStore). The working compartment contained the photoanode (masked size 0.64 cm2) as a WE and Ag/AgCl (leakless, eDAQ, ET069) as a RE and was filled with a solution of 1.2 M LiTFSI in MeCN (3 mL). The counter electrode compartment contained an electrode with Pt electrodeposited on FTO and was filled with a solution of MeCN with 1.2 M LiTFSI and 1.0 M AcOH (3 mL). Chronoamperometric photocurrent measurements at 0 V vs. Ag/AgCl were carried out using a P211 potentiostat (Zahner). Chopped-light experiments were performed through 45 seconds illumination and 45 seconds darkness while using an LED light source (Zahner, TLS3, 100 mW cm−2, Fig. S7†) for a period of 30 minutes. Benzyl alcohol (30 μL, 290 μmol) was added to the working compartment after 10 and 20 minutes. For long-term photosynthesis experiments, the working electrode compartment was filled with a solution of 1.2 M LiTFSI, 1.0 M TEMPO, 0.1 M PhCH2OH, and 19.65 mM chlorobenzene as an internal standard in MeCN (3 mL), and the counter electrode compartment was filled with a solution of MeCN with 1.2 M LiTFSI, 1.0 M AcOH and 19.65 mM chlorobenzene (3 mL). The photoanode was illuminated with an LED light source (Zahner, TLS3, 50 mW cm−2, Fig. S7†) for a period of 32 hours. To monitor the reaction, GC samples were taken during the measurement. The counter electrode compartment was attached to a gas GC to measure H2 evolution. An initial spike of benzaldehyde was seen in all measurements and was subtracted for clarity (original data, Table S2†). The integration of half the photocurrent determines the number of electrons to account for two electrons needed per oxidation reaction.
Other experimental details can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We kindly acknowledge Merck and Netherlands Organization for Scientific Research (NWO) for financial support. Dr Eitan Oksenberg of AMOLF is gratefully thanked for the SEM measurements of the TiO2. Ed Zuidinga is kindly thanked for the FD-MS measurements. Hans Ellermeijer, Daan van Giesen and Tijmen Bakker are kindly thanked for their contribution to the development of the cell.
Notes and references
- X. D. Wu, J. L. Guo, X. Ji and G. Q. Chen, Energy Policy, 2019, 127, 287–298 CrossRef.
-
M. D. Kärkäs, T. M. Laine, E. V. Johnston and B. Akermark, in Applied Photosynthesis - New Progress, InTech, 2016 Search PubMed.
-
F. M. Guangul and G. T. Chala, in 2019 4th MEC International Conference on Big Data and Smart City, ICBDSC 2019, Institute of Electrical and Electronics Engineers Inc., 2019 Search PubMed.
-
IEA, Snapshot of Global PV Markets, 2014, http://www.iea-pvps.org/fileadmin/dam/public/report/technical/PVPS_report_-_A_Snapshot_of_Global_PV_-_1992-2014.pdf Search PubMed.
- J.-W. Schüttauf, M. A. Modestino, E. Chinello, D. Lambelet, A. Delfino, D. Dominé, A. Faes, M. Despeisse, J. Bailat, D. Psaltis, C. Moser and C. Ballif, J. Electrochem. Soc., 2016, 163, F1177–F1181 CrossRef.
- R. J. Detz, J. N. H. Reek and B. C. C. Van Der Zwaan, Energy Environ. Sci., 2018, 11, 1653–1669 RSC.
-
K. Scott, in RSC Energy and Environment Series, Royal Society of Chemistry, 2020, pp. 1–27 Search PubMed.
-
R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley, Oxford, UK, 2008 Search PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- S. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- R. R. Rodrigues, A. Peddapuram, A. L. Dorris, N. I. Hammer and J. H. Delcamp, ACS Appl. Energy Mater., 2019, 2, 5547–5556 CrossRef CAS.
- S. Zhang, H. Ye, J. Hua and H. Tian, EnergyChem, 2019, 1, 100015 CrossRef.
- C. Decavoli, C. L. Boldrini, N. Manfredi and A. Abbotto, Eur. J. Inorg. Chem., 2020, 2020, 978–999 CrossRef CAS.
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS.
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 1–5 CrossRef PubMed.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- D. Badgurjar, B. Shan, A. Nayak, L. Wu, R. Chitta and T. J. Meyer, ACS Appl. Mater. Interfaces, 2020, 12, 7768–7776 CrossRef CAS PubMed.
- J. Warnan and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 17344–17354 CrossRef CAS PubMed.
- C. Bozal-Ginesta, C. A. Mesa, A. Eisenschmidt, L. Francàs, R. B. Shankar, D. Antón-García, J. Warnan, J. Willkomm, A. Reynal, E. Reisner and J. R. Durrant, Chem. Sci., 2021, 12, 946–959 RSC.
- V. Balzani, G. Pacchioni, M. Prato and A. Zecchina, Rend. Lincei Sci. Fis. Nat., 2019, 30, 443–452 CrossRef.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- D. Wang, J. Hu, B. D. Sherman, M. V. Sheridan, L. Yan, C. J. Dares, Y. Zhu, F. Li, Q. Huang, W. You and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 13256–13260 CrossRef CAS PubMed.
- H. L. Tuller, Mater. Renew. Sustain. Energy, 2017, 6, 3 CrossRef PubMed.
- M. V. Sheridan, Y. Wang, D. Wang, L. Troian-Gautier, C. J. Dares, B. D. Sherman and T. J. Meyer, Angew. Chem., Int. Ed., 2018, 57, 3449–3453 CrossRef CAS PubMed.
- T. Li, Y. Cao, J. He and C. P. Berlinguette, ACS Cent. Sci., 2017, 3, 778–783 CrossRef CAS PubMed.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS PubMed.
- M. A. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. M. Roessler and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 15633–15641 CrossRef CAS PubMed.
- J. M. Bobbitt, C. BrüCkner and N. Merbouh, Org. React., 2009, 103–424 CrossRef CAS.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- M. F. Semmelhack, C. S. Chou and D. A. Cortes, J. Am. Chem. Soc., 1983, 105, 4492–4494 CrossRef CAS.
- Y. Zhang, Z. Wang and X. Lang, Catal.: Sci. Technol., 2017, 7, 4955–4963 RSC.
- B. J. Taitt, M. T. Bender and K. S. Choi, ACS Catal., 2020, 10, 265–275 CrossRef CAS.
- B. Karimi, M. Rafiee, S. Alizadeh and H. Vali, Green Chem., 2015, 17, 991–1000 RSC.
- R. Y. Y. Lin, T. C. Chu, P. W. Chen, J. S. Ni, P. C. Shih, Y. C. Chen, K. C. Ho and J. T. Lin, ChemSusChem, 2014, 7, 2221–2229 CrossRef CAS PubMed.
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341–346 CrossRef CAS.
- W. Yang, M. Söderberg, A. I. K. Eriksson and G. Boschloo, RSC Adv., 2015, 5, 26706–26709 RSC.
- D. Badgurjar, B. Shan, A. Nayak, L. Wu, R. Chitta and T. J. Meyer, ACS Appl. Mater. Interfaces, 2020, 12, 7768–7776 CrossRef CAS PubMed.
- D. F. Bruggeman, T. M. A. Bakker, S. Mathew and J. N. H. Reek, Chem.–Eur. J., 2021, 27, 218–221 CrossRef CAS PubMed.
- M. A. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. M. Roessler and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 15633–15641 CrossRef CAS PubMed.
- B. J. Brennan, M. J. Llansola Portolés, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605–16614 RSC.
- K. L. Materna, J. Jiang, R. H. Crabtree and G. W. Brudvig, ACS Appl. Mater. Interfaces, 2019, 11, 5602–5609 CrossRef CAS PubMed.
- K. L. Materna, B. J. Brennan and G. W. Brudvig, Dalton Trans., 2015, 44, 20312–20315 RSC.
- M. Shakeel Ahmad, A. K. Pandey and N. Abd Rahim, Renewable Sustainable Energy Rev., 2017, 77, 89–108 CrossRef CAS.
- K. Sharma, V. Sharma and S. S. Sharma, Nanoscale Res. Lett., 2018, 13, 381 CrossRef PubMed.
- N. Nakayama and T. Hayashi, Colloids Surf., A, 2008, 317, 543–550 CrossRef CAS.
-
I. Fleming and D. Williams, Spectroscopic Methods in Organic Chemistry, Springer International Publishing, Cham, 2019 Search PubMed.
- V. A. Zeitler and C. A. Brown, J. Phys. Chem., 1957, 61, 1174–1177 CrossRef CAS.
- Y. Kusumawati, Z. R. Puteri, A. L. Ivansyah, H. Fansuri and M. A. Martoprawiro, Theor. Chem. Acc., 2019, 138, 63 Search PubMed.
-
R. Bard, J. Allen and L. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., 2001 Search PubMed.
- A. Yildiz, S. B. Lisesivdin, M. Kasap and D. Mardare, J. Non-Cryst. Solids, 2008, 354, 4944–4947 CrossRef CAS.
- M. J. Alam and D. C. Cameron, Thin Solid Films, 2000, 377–378, 455–459 CrossRef CAS.
- A. Fattori, L. M. Peter, K. L. McCall, N. Robertson and F. Marken, J. Solid State Electrochem., 2010, 14, 1929–1936 CrossRef CAS.
- G. Boschloo, L. Häggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS PubMed.
- J. R. Swierk, N. S. McCool and T. E. Mallouk, J. Phys. Chem. C, 2015, 119, 13858–13867 CrossRef CAS.
- N. Prabavathy, S. Shalini, R. Balasundaraprabhu, D. Velauthapillai, S. Prasanna and N. Muthukumarasamy, Int. J. Energy Res., 2017, 41, 1372–1396 CrossRef CAS.
- J. Jeon, Y. C. Park, S. S. Han, W. A. Goddard, Y. S. Lee and H. Kim, J. Phys. Chem. Lett., 2014, 5, 4285–4290 CrossRef CAS PubMed.
- M. A. Mercadante, C. B. Kelly, J. M. Bobbitt, L. J. Tilley and N. E. Leadbeater, Nat. Protoc., 2013, 8, 666–676 CrossRef CAS PubMed.
-
H. Cheema and K. S. Joya, in Titanium Dioxide - Material for a Sustainable Environment, InTech, 2018 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01275d |
| ‡ As published in our previous communication. |
|
| This journal is © The Royal Society of Chemistry 2021 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
R. J.
Detz
a,
R. J.
Detz