
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operation of calcium-birnessite water-oxidation anodes: interactions of the catalyst with phosphate buffer anions†
Emanuel
Ronge‡
 a,
Jonas
Ohms
b,
Vladimir
Roddatis
a,
Travis
Jones
e,
Frederic
Sulzmann
e,
Axel
Knop-Gericke
de,
Robert
Schlögl
de,
Philipp
Kurz
*b,
Christian
Jooss
*ac and
Katarzyna
Skorupska
*de
a,
Jonas
Ohms
b,
Vladimir
Roddatis
a,
Travis
Jones
e,
Frederic
Sulzmann
e,
Axel
Knop-Gericke
de,
Robert
Schlögl
de,
Philipp
Kurz
*b,
Christian
Jooss
*ac and
Katarzyna
Skorupska
*de
aInstitut für Materialphysik, Georg-August-Universität Göttingen, Friedrich-Hund-Platz 1, Göttingen 37077, Germany. E-mail: cjooss@gwdg.de
bInstitut für Anorganische und Analytische Chemie, Freiburger Material forschungszentrum (FMF), Albert-Ludwigs-Universität Freiburg, Albertstraße 21, Freiburg 79104, Germany. E-mail: philipp.kurz@ac.uni-freiburg.de
cInternational Center for Advanced Energy Studies, Georg-August-Universität Göttingen, Tammannstr. 4, Göttingen 37077, Germany. E-mail: cjooss@gwdg.de
dMax-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, Mülheim an der Ruhr 45470, Germany
eFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, Berlin 14195, Germany. E-mail: skorupska@fhi-berlin.mpg.de
First published on 27th September 2021
Abstract
Investigating the interfaces between electrolytes and electrocatalysts during electrochemical water oxidation is of great importance for an understanding of the factors influencing catalytic activity and stability. Here, the interaction of a well-established, nanocrystalline and mesoporous Ca-birnessite catalyst material (initial composition K0.2Ca0.21MnO2.21·1.4H2O, initial Mn-oxidation state ∼+3.8) with an aqueous potassium phosphate buffer electrolyte at pH 7 was studied mainly by using various electron microscopy and X-ray spectroscopy techniques. In comparison to electrolyte solutions not containing phosphate, the investigated Ca-birnessite electrodes show especially high and stable oxygen evolution activity in phosphate buffer. During electrolysis, partial ion substitutions of Ca2+ by K+ and OH−/O2− by HnPO4(3−n)− were observed, leading to the formation of a stable, partially disordered Ca–K–Mn–HnPO4–H2O layer on the outer and the pore surfaces of the active electrocatalyst material. In this surface layer, Mn3+ ions are stabilized, which are often assumed to be of key importance for oxygen evolution catalysis. Furthermore, evidence for the formation of [Ca/PO4/H2O]− complexes located between the [MnO6] layers of the birnessite was found using the soft Ca 2p and Ca L-edge X-ray spectroscopy. A possible way to interpret the observed, obviously very favorable “special relationship” between (hydrogen)phosphates and Ca-birnessites in electrocatalytic water oxidation would be that HnPO4(3−n)− anions are incorporated into the catalyst material where they act as stabilizing units for Mn3+ highly active centers and also as “internal bases” for the protons released during the water-oxidation reaction.
Introduction
One major bottleneck for the production of hydrogen by electrochemical water splitting is the sluggish kinetics of the oxygen evolution reaction (OER).1,2 Here, a rational design of improved electrocatalysts requires a better understanding of the mechanisms controlling OER activity and stability.Inspired by the oxygen-evolving complex (OEC) of photosystem II – a Mn4CaO5 cluster which constitutes the OER active site in biological photosynthesis – a large variety of manganese oxide (MnOx) catalysts has been studied as potential synthetic OER catalyst materials.3–11 Beside some crystalline binary or ternary manganese oxides, disordered MnOx materials with a low degree of long-range order often show a high intrinsic catalytic activity.11–19 For some crystalline systems, the formation of catalytically much more active nanocrystals or disordered surface layers during the OER process has been observed.5,6,20–22
Among the different less-ordered manganese oxides, birnessites have been identified as especially promising OER catalysts showing both high activity and stability.13,16,23 The birnessite structure is built up from edge-sharing [MnO6]-octahedra forming extended layers. The stacking of the layers in c-direction depends on the amount of incorporated water and the type of additional cations and typically features interlayer distances of about 7 Å.13,16,24 Many different cations can be present in birnessite-type materials (s-block metal cations are most common) and these lead to different degrees of crystalline order as well as different ion exchange properties in contact with aqueous solutions.25,26
Previous studies have shown that the OER activity of birnessites is influenced by various parameters such as their crystallinity, type and concentration of additional cations and synthesis parameters (especially sintering temperatures).13,27 Here, especially Ca-birnessites show good activities.13 For cation incorporation into the birnessite structure, an upper concentration limit is usually observed, which is e.g. Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ∼ 0.6 for Ca2+.27
:Mn ∼ 0.6 for Ca2+.27
In previous investigations on birnessite-catalyzed OER by some of us, a synthetic birnessite with an approximate composition of K0.20Ca0.21MnO2.21·1.4H2O emerged as especially active catalyst material in both chemical (Ce4+ oxidation) and electrochemical OER screenings.23,27 When immobilized onto FTO substrates, stable current densities of ∼1 mA cm−2 could be reached in neutral, phosphate buffered electrolytes at overpotentials of η ∼ 500 mV.28
Detailed X-ray diffraction and X-ray absorption spectroscopy investigations of this and closely related synthetic Ca-birnessites indicated little long-range, in-plane order and also irregular stackings of the [MnO6]-layers.13,14,16 N2 sorption experiments revealed a mesoporous structure with quite high surface areas of SBET ∼ 50–250 m2 g−1 (mainly depending on the reaction temperatures used during post-synthetic sintering) and average Mn oxidation states were found to be in the range between +3.3 and +4.0.13,14,16,27 The latter observation indicates, that the Mn valence state within the birnessite structure can be reversibly changed – a material property which might be of general, crucial importance for OER catalysis by transition metal oxides.11,14,29–33
Additionally, manganese oxides with oxidation states close to +3.0 exhibit labile Mn–O bonds due to the typical Jahn–Teller distortions of the octahedral coordination spheres of Mn3+ cations (hs-d4), which could also promote OER catalysis.3 Indeed, oxides rich in Mn3+ often show much higher OER activities than Mn(IV) materials.15 On the other hand, the higher charge of Mn4+ seems to inhibit the decomposition of Mn(IV) compounds, as e.g. exemplified by the OEC of photosystem II.33
Concerning MnOx corrosion in aqueous media, the Pourbaix diagram for Mn suggests dissolution via solvation of Mn2+ for acidic pH, whereas dissolution of Mn via the formation of permanganate MnO4− at high anodic potentials is expected for neutral to alkaline pH.34 Both corrosion routes would result in Mn depleted surface layers, and these have indeed been found experimentally.35 On the other hand, such corrosion processes might be compensated by repair steps, as e.g. found by Najafpour et al. for reactions of a K0.25-birnessite with Ce4+. Here, the corrosion products MnO4− and Mn2+ were found to react back to a stable, layered (K,Ce)MnOx compound, which catalyzed the OER for 15 days without degradation or leaching of manganese.35,36 Finally, it has been proposed that redox inert ions like Ca2+ can stabilize MnOx and/or facilitate the formation of modified and open structures for water oxidation.35,37
Overall the literature data discussed so far hints at a general importance of additional cations for the OER performances of birnessites, not only by influencing the Mn valence state but also by increasing the stability of these MnOx-based (electro)catalysts. Consequently, the composition of the electrolyte has to be carefully chosen in order to control possible cation exchange processes.
In this study, Ca-birnessite coated electrodes were prepared by screen printing the previously mentioned OER catalyst material K0.20Ca0.21MnO2.21·1.4H2O on fluorine-doped tin oxide (FTO) as conductive support material following an established and already optimized method.28 In order to study the influence of ion exchange reactions on the electrochemical OER activity and stability as well as the material's microstructure, the Ca-birnessite anodes were mainly operated in three different, near neutral aqueous electrolyte solutions: a potassium phosphate buffer (pH 7), solutions of imidazolium sulphate (pH 7.4) and potassium sulphate (pH 7). Structural analyses of the electrodes' surfaces and bulk material were performed using transmission and scanning electron microscopy (TEM/SEM). To gain further insights into the interaction of the electrolyte with the MnOx catalyst, we investigated the chemical compositions, Mn valence states and coordination environments by electron energy-loss spectroscopy (EELS) element mapping, energy-dispersive X-ray spectroscopy (EDX), X-ray photoelectron spectroscopy (XPS) and near edge X-ray absorption fine structure (NEXAFS) at the Ca and Mn L-edges. The results hint at a special, favorable interaction between birnessites and phosphate-containing electrolytes, explaining why phosphate buffers might be the medium of choice for birnessite-catalyzed OER at near-neutral pH.
Experimental
Please consult previous publications and the ESI† for details of the synthesis of the Ca-birnessite catalyst material, the preparation of MnOx-coated electrodes by screen-printing, the exact compositions of the electrolytes as well as the electrochemical measurements.23,28SEM, EDX and cross-section imaging
SEM analysis was carried out with a Nova NanoSem 650 in situ SEM from FEI. A “through the lens” (TTL) detector was used to take images at an acceleration voltage of 10 kV. For energy-dispersive X-ray spectroscopy (EDX), an Oxford Instruments X-Max detector was employed. The quantification of the Ca/Mn and P/Mn ratios was performed with line-scans of a minimum of 10 points and 100 s of acquisition time per point over a range of ∼60 μm. The EDX acceleration voltage was 20 kV. Cross-section samples used for SEM imaging were cut using a Nova Nanolab 600 focused ion beam (FIB) system from FEI (ion acceleration voltage 30 kV). For the pristine sample, a protective layer of platinum was first deposited via electron beam induced deposition, followed by a thicker Pt layer fabricated by ion beam induced deposition. After rough cuts with 3 nA currents, cleaning cuts of the cross sections with 0.3 nA were carried out. The images of the cross sections were taken at an acceleration voltage of 5 kV and an angle of 52° using the TTL detector.TEM lamellae preparation and (S)TEM analysis
Electron-transparent cross-section lamellae of ∼4 μm thick electrodes before and after electrolysis were prepared by means of the FIB system described above. In order to avoid an overlap of the P and Pt signals, no protective Pt layer was applied in this case. For the 200 nm thin electrodes, the lamellae were prepared by conventional cutting, dragging and ion milling using a Gatan Model 691 precision ion polishing system (PIPS). The TEM investigations including imaging, electron energy loss spectroscopy (EELS) and EDX-measurements were performed using an aberration corrected FEI Titan electron microscope with 300 keV electrons. For this microscope, the information limit in high vacuum is about 0.08 nm. High angle annular dark field (HAADF) imaging was performed in scanning transmission electron microscopy mode (STEM). The STEM capability combined with EELS gives access to the local chemical composition with a spatial resolution of ∼0.2 nm and an energy resolution of ∼1.3 eV. The following energy edges were used for the EELS-mapping: Ca-L-edge, Mn-L-edge, O-K-edge, P-K-edge. The TEM EDX analyses were carried out with an Oxford Instruments X-Max detector and electron diffraction patterns were collected with a Phillips CM12 at 120 kV.X-ray spectroscopy
Near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) measurements were carried out using a setup located at the Innovative Station for In Situ Spectroscopy (ISISS) beamline at the BESSY II synchrotron of the Helmholtz-Zentrum Berlin.38 For the as-prepared oxide, ∼20 mg of Ca-birnessite powder was pressed into a pellet (Ø ∼ 2 mm) by applying a force of 1 ton for 1 min. Ca-birnessite-coated FTO slides were directly used for measurements as prepared or after electrochemical operation. In each case, the samples were mounted on a sapphire sample holder and positioned ∼1 mm in front of the first aperture of the differentially pumped electrostatic lens system. Measurements were conducted using an incident X-ray beam spot of roughly 100 × 300 μm at a pressure of 10−4 mbar. A pass energy of 20 eV was used for XPS measurements and the inelastic mean free path (IMFP) of the photoelectrons was calculated using the model of Tanuma et al.39 The binding energies of measured XPS spectra were calibrated using an O 1s second order peak with a theoretical photon energy of 730 eV. The XPS spectra were recorded at different excitation energies in order to obtain IMFPs of 0.7 nm (for Ek = 200 eV) or 1.7 nm (Ek = 830 eV), respectively. The deconvolution of the XP spectra was performed using the software CasaXPS after subtraction of the Shirley background.To perform NEXAFS measurements, the photon energy was scanned while the total electron (TEY) and Auger electron (AEY) yields were counted. Ca L-edges spectra were recorded for photon energies between 342 and 362 eV, while the photon energy was scanned between 625 and 660 eV to cover the Mn L3- and L2-edges. For clarity, only the data for the Mn L3-edge is presented. To assess possible beam damage, which is a well-known problem for NEXAFS of manganese oxides,40 stability tests were performed. Three Mn L-edge scans recorded successively on the same spot of three studied samples are shown in Fig. S6 of the ESI.† The deviations between subsequent scans indicate that Mn ions in the samples are reduced by the beam, but also that the observed spectral changes are small enough so that a meaningful analysis of the spectra is well possible.
Results and discussion
Electrochemistry
Among others, the study presented here tries to provide answers to the following two questions concerning OER electrocatalysis by MnOx in near-neutral conditions: (1) why is the use of a buffering electrolyte generally beneficial and (2) why are phosphate containing media apparently especially well-suited?11 To investigate this, it has to be noted that the number of possible aqueous electrolytes for OER experiments is generally limited as it is of particular importance in OER electrocatalysis that the species making up the electrolyte are themselves redox-inert at strongly oxidizing conditions in water.In order to select electrolytes for our comparative study, we therefore first carried out screening experiments in which we measured cyclic voltammetry (CV) and chronoamperometry (CA) traces for batches of identically prepared Ca-birnessite/FTO electrodes in five different electrolytes: borate (pH 9.2), phosphate (pH 7.0), methylphosphonate (pH 7.0), imidazolium sulphate (pH 7.4), and potassium sulphate (pH 7). CVs in these media indicated an onset of the OER reactions for η > ∼350 mV in all electrolytes and showed very similar traces for phosphate, borate and methylphosphonate, when plotted on an ERHE-scale (see ESI, Fig. S1†). On the other hand, the CV trace for imidazolium sulphate shows a much larger difference between forward and reverse scans than the one for the first three buffers, and for potassium sulphate, only a very small catalytic current could be detected. With the two questions formulated above in mind, we thus decided to limit our study to the following three (near-)neutral electrolytes: (1) potassium (di)hydrogen phosphate as a typical neutral phosphate-buffered electrolyte; (2) imidazolium sulphate as a buffering, but phosphate-free medium and (3) potassium sulphate as a solution which is both phosphate-free and not possessing any buffer qualities at pH 7.
In the CVs, two distinct features are noticeable for all three electrolytes (ESI, Fig. S1,† scan rate 20 mV s−1): (a) a “pre-wave” in the region of +1.0 to +1.4 V with current densities <0.2 mA cm−2 and (b) a much larger irreversible oxidative wave at potentials above +1.4 V, caused by the onset of water oxidation. Both features can show a splitting of the current-density traces between the forward and the return scans. Interestingly, these splits are very different between the three solvent systems: in phosphate, forward and return scans mainly differ in the pre-wave region, in imidazolium the splits are substantial for both parts, while in sulphate, the splits are clearly visible over the entire scanned potential range (but currents are – as already mentioned – overall very low).
In Fig. 1, CA traces for the three different electrolyte systems are presented. In each case, the Ca-birnessite/FTO-anodes were immersed in the respective media and an overpotential of η ∼ 500 mV was applied for a time of 1 h, while the OER current density was recorded. For details concerning these measurements, please consult ref. 27. Interestingly, we find very different j vs. t-traces for the three investigated solutions, despite their virtually identical pH-values and the fact that the applied oxidation potentials were iR-compensated to correct for conductivity differences.
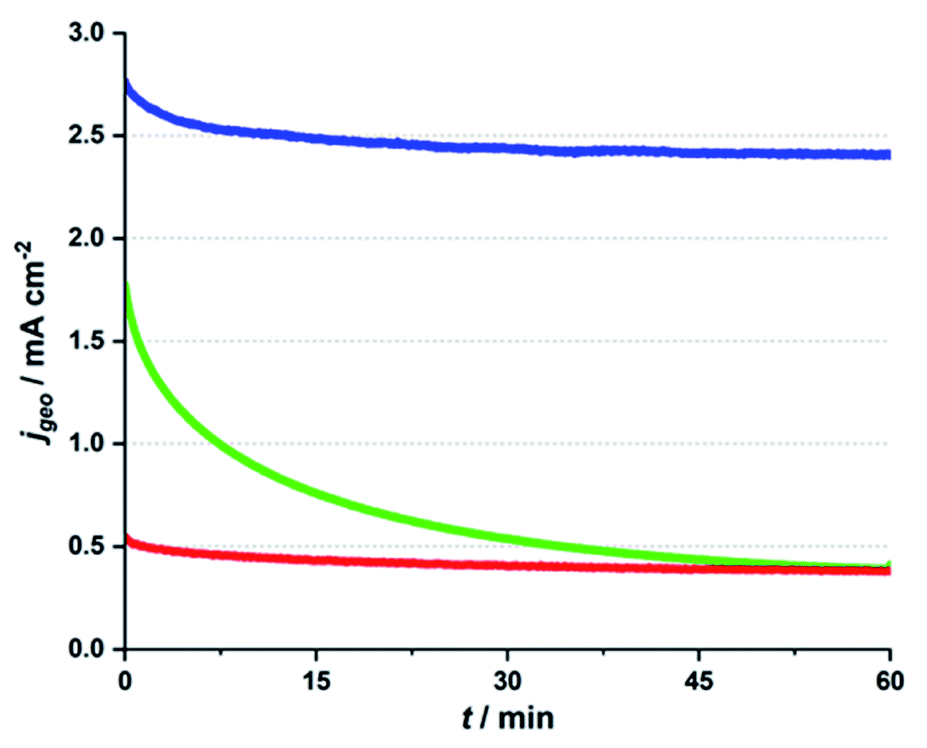 | ||
| Fig. 1 Chronoamperometry traces for Ca-birnessite/FTO electrodes at a potential of +1.71 V vs. RHE (η = 490 mV) in three different, near neutral electrolytes (all 70 mM): phosphate buffer (pH 7, blue trace), imidazolium sulphate buffer (pH 7.4, green trace) and potassium sulphate solution (pH 7, red trace). | ||
In all three cases, we find that current densities are significantly higher for the first ∼2 min of the experiment, but already here the j observed during the very first phase for phosphate (∼2.7 mA cm−2) is about 50% higher than the value for imidazolium (∼1.8 mA cm−2), while the current for potassium sulphate is very low (∼0.2 mA cm−2). As already observed in other studies, we find that the current density in phosphate reaches a phase of steady, but rather slow decline after ∼1 h of operation (here with a current density after 60 min of ∼2.4 mA cm−2).27 However, it should be noted that Ca-birnessite/FTO electrodes also lose most of their OER activity if electrolysis is continued over much longer time periods (see ESI, Fig. S1d† for a CA trace recorded over 16 h). The main reason for this loss in activity is most likely a gradual deterioration of the contact between the manganese oxide and the FTO back contact caused by the mechanical stress of the evolution of oxygen gas. This results in a depletion of the electrical contact as well as in particle loss. In a related study on MnOx/FTO-electrodes prepared by the same method, we found that this mechanical instability can generally be observed for different MnOx. However, it varies greatly depending on the choice of birnessite-type powder (and thus most likely the strength of the MnOx–FTO interaction).41 At the same time, corrosion and/or significant phase changes of the manganese oxides could be excluded as main reasons for the decrease of the catalytic current: both IR spectroscopy and SEM measurements revealed rather unchanged oxide materials after 20 h of OER electrolysis in neutral phosphate buffer.41 Furthermore, using carbon fiber paper as an improved support for a very similar birnessite OER electrocatalyst material showed a much improved long term stability of the catalytic current while again the MnOx material remained virtually unchanged by the electrolysis process.42
Both the “pre-waves” in the CV traces and the higher currents found during the first phase of the CA experiments have previously been explained by Mn-centered oxidation events (mainly Mn3+ → Mn4+) occurring before (CV) or “on top” of (CA) the OER currents.11 This assignment is e.g. supported by XAS data for the Mn K-edge (increase of the average Mn oxidation state of “our” Ca-birnessite from ∼+3.3 to ∼+3.7 under electrocatalytic conditions28) and also matches our own X-ray spectroscopy data (see below).
For imidazolium, a rather stable CA trace is reached after ∼1 h as well, but here the initial decrease in current density is dramatic, leaving a remaining current density of only ∼0.5 mA cm−2 after 1 h – and thus less than a third of the starting value. This low current density is the same as observed for the non-buffering potassium sulphate electrolyte during the entire chronoamperometry experiment. However, it is important to note that at ∼+1.7 V vs. RHE, bare FTO supports show current densities below 0.06 mA cm−2 in all three electrolytes (see orange traces in ESI, Fig. S1†), so that the currents of 0.4 mA cm−2 measured for imidazolium and sulphate electrolytes indicate that OER electrocatalysis by the Ca-birnessite still takes place to some degree in these solutions after 1 h.
The OER electrocatalysis traces shown in Fig. 1 thus clearly demonstrate that (1) a buffering electrolyte solution seems to be generally necessary if significant OER currents are to be reached over significant electrolysis times (phosphate/imidazolium buffers vs. potassium sulphate); (2) a phosphate buffer is apparently an exceptionally good choice for OER electrocatalysis by MnOx at pH 7. The also buffering, but phosphate-free imidazolium system fails to reach the initial current densities found for phosphate by far and also lacks long-term stability.
We surely realized that numerous further electrochemical investigations such as detailed Tafel analyses, buffer concentration variations, scan rate studies etc. might be of interest at this point. However, for this study we decided to focus on the identification of possible reasons for the observed profound, buffer-type dependent differences in the electrolysis experiments presented above. For this, we first turned to electron microscopy and X-ray spectroscopy in order to identify differences in morphology and/or elemental composition of the catalytic Ca-birnessite layer after continuous electrocatalysis in the three different solutions.
Investigations of morphology changes and ion-exchange processes of the catalyst layer by SEM, TEM and EDX
Four different types of samples were investigated by electron microscopy: (1) reference samples of pristine Ca-birnessite electrodes; (2) electrodes after use as water oxidation anodes for different electrolysis times in 70 mM phosphate or (3) 70 mM imidazolium sulphate buffer and (4) samples which had been immersed in a 70 mM phosphate buffer without applying an electrochemical potential. To facilitate the electron microscopy measurements, we prepared Ca-birnessite electrodes with a reduced catalyst layer thickness of ∼0.2 μm for some of the measurements.The scanning electron microscopy (SEM) images of Fig. 2A–D show representative parts of the surface as well as cross-sections of the electrode layer. Clearly visible are agglomerations of particles with a size of a few hundred nanometers forming a rough surface structure. In addition, there are surface cracks which are most likely a result of gaseous H2O and CO2 (the latter from carbon-oxidations, see XPS section below) leaving the electrode during the annealing procedure. These observations confirm that the already known mesoporosity and large surface area of the Ca-birnessite powder27 are not greatly affected by the fabrication process. The pore morphology is e.g. well illustrated by the SEM cross-sections, where small pores of diameters between 10 and 300 nm as well as large crack-like openings with a width of several micrometers are visible (Fig. 2C and D), supporting a previous characterization of the Ca-birnessite as a mesoporous material with a large surface area.23
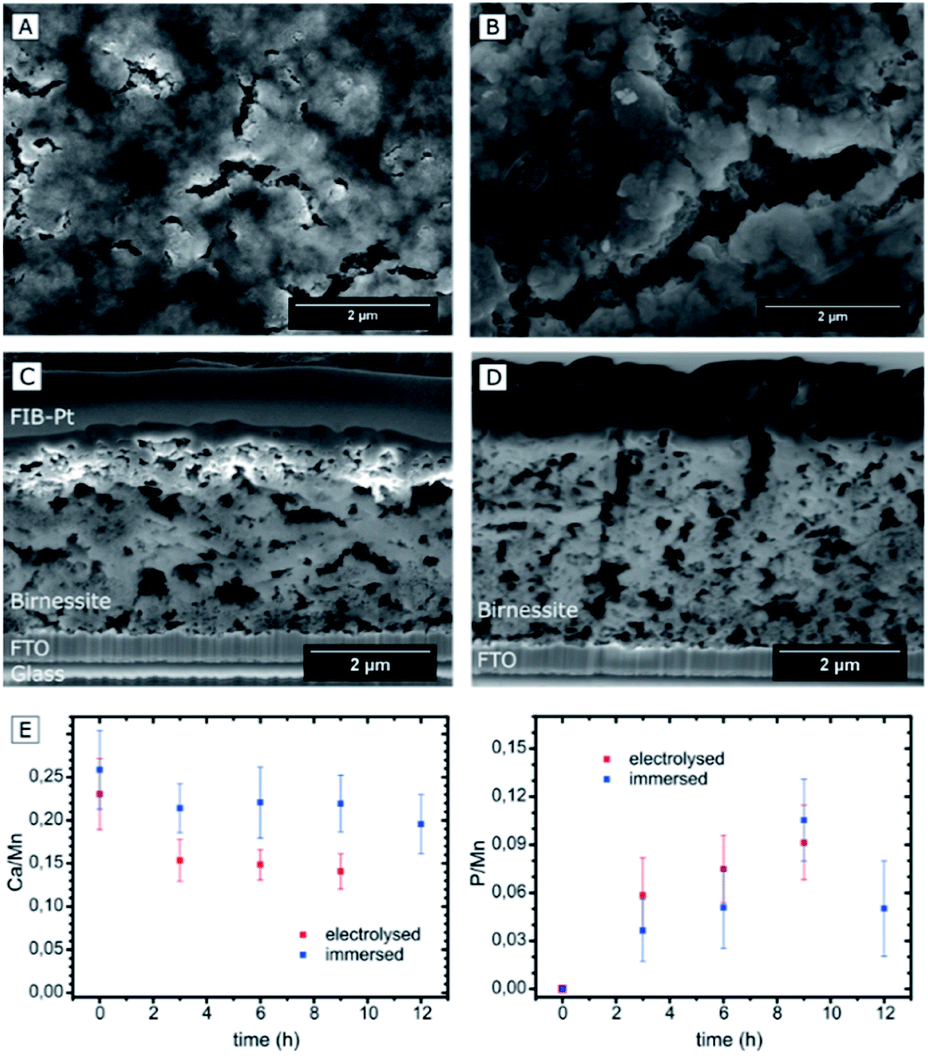 | ||
| Fig. 2 SEM images of Ca-birnessite electrodes. Top views of (A) a pristine electrode; (B) an electrode after 9 h of electrolysis in 0.07 M phosphate buffer (pH 7, η = 530 mV); cross-sections of a pristine sample with a protective layer of platinum (C) and a sample after electrolysis (D) without protection layer; (E) EDX-SEM measurements of changes of the Ca/Mn and P/Mn ratios over time for electrodes subjected to electrolysis in phosphate buffer (red data points) in comparison to samples immersed in phosphate buffer without applying an electrochemical potential (blue data points). | ||
High-resolution transmission electron microscopy (HRTEM) images show the presence of nanocrystals for both the freshly prepared electrodes and also the electrodes after electrolysis for which a birnessite phase could be assigned by the analysis of the electron diffraction data (see ESI, Fig. S2 and S3†).
Post-operando measurements using electron diffraction, XRD and SEM show that neither 9 h of electrochemical water-oxidation electrolysis nor extended immersion of the electrode into a phosphate-containing electrolyte lead to significant changes of the Ca-birnessite layer, therefore confirming previous Raman and XAS results that the birnessite oxide phase and the overall morphology of the catalyst layer is retained during OER.28 An important exception to this is the morphology of the electrode surface, where a thin new surface layer formed during electrolysis as visible in the TEM images (Fig. 4 and ESI, Fig. S2†).
 | ||
| Fig. 3 (A) EDX element mapping for a ∼4 μm thick birnessite electrode after 9 h of electrolysis (η = 530 mV). Left: HAADF STEM image of the mesoporous oxide structure; right: EDX map of phosphorus for the same part of the oxide layer EDX sum-spectra in EDX line scan from a pore surface into a nano-crystalline dense area of the same lamella as shown in (B). Foreground: EDX line scan of the P:Mn ratio; background: (HAADF) STEM image of the area of the line scan. The dashed line shows the location of the scan. | ||
Despite the apparently unchanged MnOx-structure, SEM-EDX indicates that the electrocatalytic process induced significant changes of the average elemental composition for the entire volume of the oxide layer. The calcium concentration decreased markedly, especially during the first 2 h of electrolysis from an initial Ca:Mn ratio of 0.24:1 to ∼0.14:1 (Fig. 2E). This value seems to represent a new equilibrium concentration as it then remains constant after ∼6 h for the following hours of the electrolysis. In contrast, the Ca:Mn ratio of samples which are just immersed in phosphate buffer does not change significantly within the error margin of the detection method. From this, we conclude that calcium is not simply dissolved out of the material but rather actively exchanged during the electrocatalytic process.
Given the importance of the electrolyte composition to reach high catalytic current densities (see Fig. 1), a closer look at the distribution of the elements within the material is required. Thus, high-angle annular dark field (HAADF) STEM images and EELS mappings of a 200 nm thick electrode after 12 h of electrolysis at η = 530 mV were recorded (Fig. 4). EELS confirmed the formation of a ∼25 nm thick, phosphorous-rich surface layer (far right part of the images in Fig. 4). This surface layer also contains manganese, oxygen and (very little) calcium. Based on the EELS mapping, we however cannot make a statement about the presence of phosphorous in the entire volume of the catalyst layer, because the phosphorous K edge at 2146 eV is too weak to detect phosphorous concentrations below ∼5 atom%. On the other hand, we were able to detect P in the entire birnessite-layer using TEM-EDX, as EDX is much more sensitive for P but unfortunately does not offer space resolution. For a sample with a thickness of ∼4 μm that had been operated as OER anode for 9 h, the average P:Mn ratio for bulk birnessite was determined by TEM-EDX as ∼0.15:1 (Fig. 3) and thus as very similar to the ratio reported above as the final state of the exchange equilibrium after 9 h of operation (Fig. 2E). However, despite of the good agreement of the average values, the fluctuations of the local P:Mn ratios are in the order of ∼65%. As can be seen in Fig. 3A, there is a correlation between the phosphorous signal in the EDX map and the contrast of the STEM-image (which is a measure of the local lamella thickness).
 | ||
| Fig. 4 HAADF STEM image (left) and EELS element mapping (right) of a 200 nm thick birnessite layer used during 12 h for electrolysis. The FTO substrate is on the far left, the exposed anode surface on the far right part of the images. | ||
Altogether, the element mapping data offers strong evidence for the presence of phosphorous in the entire birnessite volume. The detected variations of the phosphorous concentration can partially be explained by variations of the sample thickness, but also reflect real fluctuations within the nanocrystalline material. A further explanation can be found by a closer look at Fig. 3B. Here, an EDX line scan over a distance of only 100 nm indicates a much higher P:Mn ratio close to the surface of an inner pore of the birnessite film in comparison to the denser bulk of the material. This observation might also represent an early stage of the formation of a phosphorous-rich surface layer similar to the feature at the outer electrode surface shown in Fig. 4. Additional EDX line scans at high spatial resolution were recorded and these are also in agreement with the general conclusion that phosphorous is mainly located on the surfaces of the birnessite nanocrystals (see ESI, Fig. S5†).
In order to investigate whether the anion-exchange-processes during electrolysis are unique for the P/Mn couple, we compared the observations for the phosphate buffer to the buffering, but phosphate free imidazolium electrolyte. Here, the ion exchange processes were analyzed by transmission electron microscopy for 200 nm thick Ca-birnessite layers after 12 h of electrolysis. In Fig. 5A, C and E, EDX line scans of the electrode after an electrolysis in the phosphate buffer are shown. Similar to Fig. 3, a significantly higher P:Mn ratio can be observed. Despite the overlap off the Pt and P peaks in den EDX spectra, the data indicates a significant P concentration in the subsurface region down to 10 nm. In contrast, Fig. 5B, D and F show the same dataset for a sample after electrolysis in the imidazolium sulphate electrolyte. Interestingly, here the sulfur signals (which would indicate sulphate binding) are barely above the noise level. We conclude that, unlike for phosphate, no sulphate containing surface layer is apparently formed during OER electrocatalysis in the imidazolium sulphate electrolyte. This interpretation is also supported by SEM-EDX measurements for a 4 μm thick sample after 9 h of electrolysis in imidazolium sulphate, where no sulfur signal was detected (see ESI, Fig. S4B†). For the Ca/Mn ratio, a value of 0.11 ± 0.06 could be calculated from the SEM-EDX data, indicating partial removal of calcium from the material to a very similar degree as found before for phosphate.
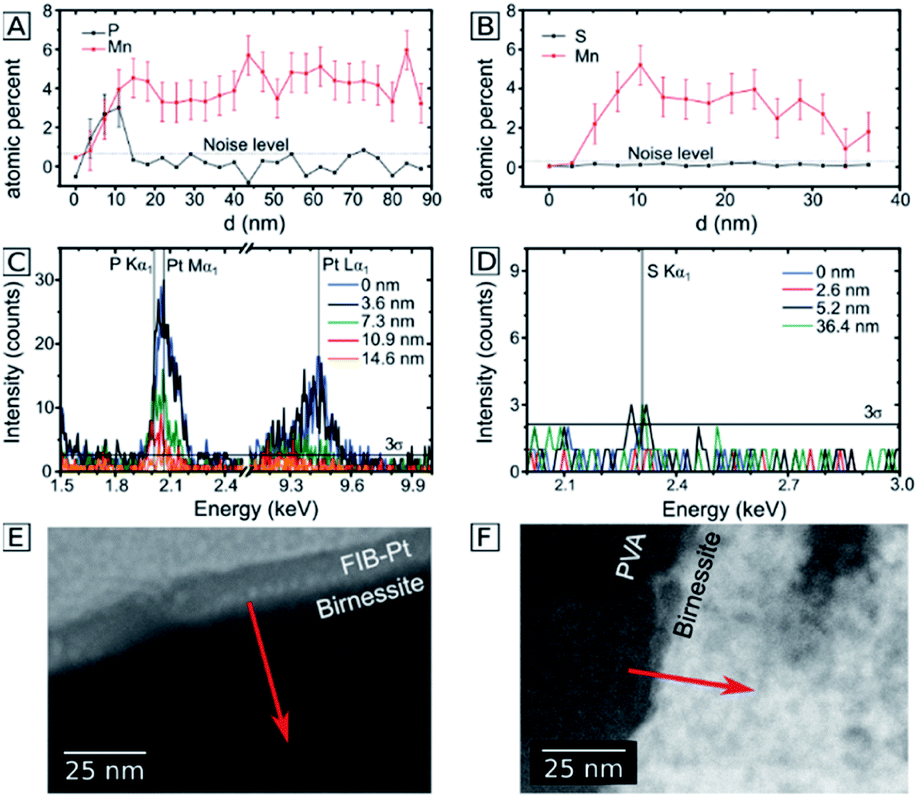 | ||
| Fig. 5 Comparison of 200 nm thick birnessite electrodes after 12 h of electrolysis in 70 mM phosphate buffer (left column) and imidazolium sulphate (right column). (A and B) EDX line scans taken at cross sections of the electrode surface, where the surface position is set to 0 nm, the dotted line indicates the noise level of the EDX measurement; (C and D) corresponding EDX spectra taken at different positions, the horizontal line represents the 3σ level of the noise; (E and F) HAADF TEM images of the position of the line scans. | ||
In summary, the presented electron microscopy results confirm that the mesoporous calcium–manganese oxide used here as electrocatalyst retains its nanocrystalline, birnessite-like structure during the electrolysis process, even as at least one third of the initially present Ca2+ ions are lost and/or replaced during electrocatalysis. For the phosphate buffered electrolyte, P enters the entire volume of the birnessite material. In addition, a P-rich oxide layer with a thickness of 15–20 nm forms at the outer surface of the 200 nm thick electrode. After extended electrolysis, phosphorous can finally be found all over the 4 μm thick sample, but its distribution appears to be inhomogeneous as the inner surfaces of the pores and the surfaces of the nanocrystals show much higher P-concentrations than the dense, nanocrystalline parts of the Ca-birnessite. The observed high and stable catalytic OER activity might thus be correlated to the formation of a P-containing, calcium–manganese oxide surface layer acting as “true” OER electrocatalyst. This is supported by recent in situ ETEM studies which revealed high atomic surface dynamics of birnessite-type MnOx under electrocatalytic OER conditions.43
Ca-birnessite/electrolyte interactions probed by XPS and NEXAFS
The results from the previous sections indicate that different ion exchange processes take place when the Ca-birnessite material is used as OER electrocatalyst in phosphate buffer: Ca2+ ions are partially exchanged and phosphate ions enter the material. In contrast, no sulfur incorporation was detectable after electrolysis in imidazolium sulphate. These different ion interactions, the accompanying changes of the Mn oxidation states and the role of calcium were now studied in more detail using X-ray spectroscopy methods.X-ray photoelectron spectroscopy (XPS) and near edge X-ray absorption fine structure spectroscopy (NEXAFS) were performed with the aim to gain insights into both the elemental composition and the chemical bonding situation. Such measurements typically probe the material close to the electrode surface (the penetration depth of a few nanometers) and thus have to be seen as complimentary e.g. to the previously carried out XAS measurements at the Mn K-edge,30 as well as the EDX and SEM experiments described above, all of which provide information about the entire μm-thick oxide layer. Similar to the previous section, XPS and NEXAFS spectra were recorded for four different types of samples: (1) the synthetic Ca-birnessite powder, (2) pristine Ca-birnessite electrodes, (3) electrodes after 16 h of operation at +1.77 V vs. RHE (η = 540 mV) in 70 mM phosphate buffer and for comparison (4) in 70 mM imidazolium sulphate buffer.
XPS survey spectra for samples 1 to 3 between 0 and 1200 eV are shown in Fig. 6. As expected for a calcium containing manganese oxide, the most intense signals in all spectra are observed for the O 1s and the Mn 2p electrons and calcium lines can also be found in all cases. The presence of weak tin signals from the underlying FTO substrate confirms the already detected cracks and/or the porosity of the catalyst layers, which allows some photons to reach the underlying SnO2:F support material. In addition, freshly prepared electrodes contain a significant amount of carbon, which must originate from the binder substances used during the printing process and wasn't completely removed during the calcination. Carbon visible in the spectrum of the synthetic oxide must originate from carbonates formed by interaction of atmospheric CO2 with alkaline solution used during the synthesis.
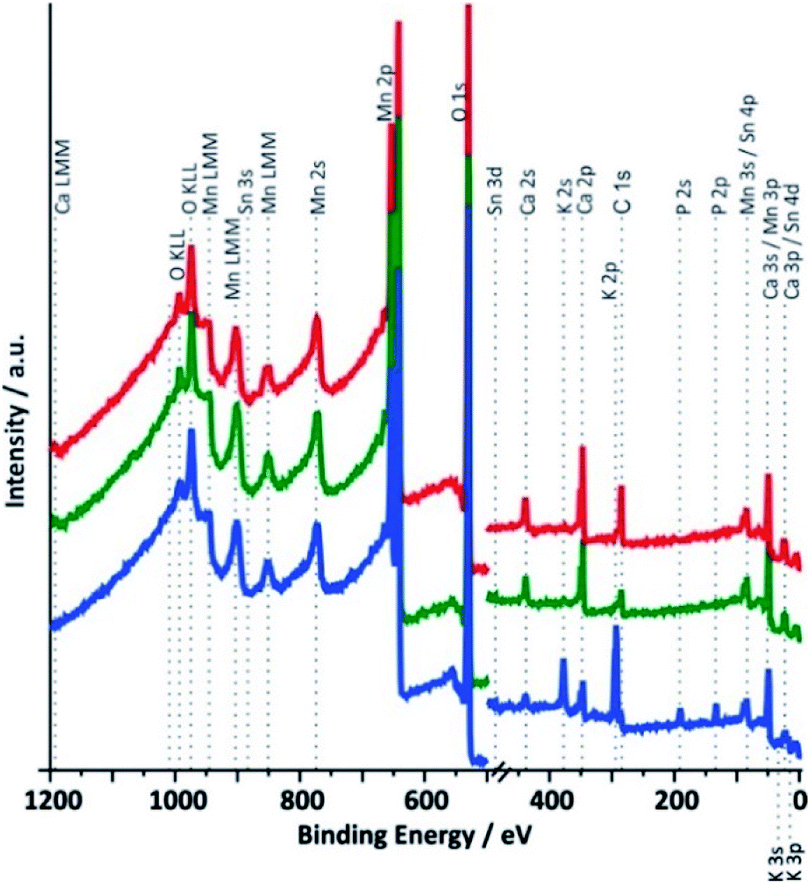 | ||
| Fig. 6 X-ray photoelectron survey spectra (XPS, hν = 1486.7 eV) of different Ca-birnessite samples. Red: synthetic Ca-birnessite powder, green: screen printed, 10 μm thick Ca-birnessite layer on FTO substrate after annealing at 450 °C and blue: XPS of an electrode after 16 h of OER electrocatalysis in 0.1 M phosphate buffer (η = 540 mV). | ||
The most obvious differences between the spectra recorded for Ca-birnessite electrodes before and after 16 h of OER electro-catalysis in phosphate buffer are the appearances of additional signals for potassium and phosphorous in the spectral region between 100 and 400 eV (Fig. 6, blue trace), which coincide with a marked decrease of the Ca peak intensity. On the other hand, the fact that the XPS spectra of Fig. 6 are overall very similar again confirms that the electrocatalyst layer retains the characteristics of a layered calcium manganese oxide even after extended electrolysis time. However – and in full agreement with the results from the previous section – we also observe significant changes of the chemical composition caused by the electrocatalytic process (this time for the zone close to the surface accessible to XPS): most of the carbon and some calcium leave the MnOx-layer, while potassium and phosphate from the electrolyte enter the material. In case of the imidazolium sulphate electrolyte, no S 1s line was found and additionally the Ca signals in the survey spectrum are not detectable any more after electrochemistry (see ESI, Fig. S7†). Thus, XPS confirms that very different ion exchange processes are at play when these two buffer systems interact with the Ca-birnessite catalyst during OER.
In order to obtain more detailed information about the chemical environment of the different elements, we also analyzed the Mn and Ca L-edge spectra in combination with the XPS data for Mn 2p, O 1s, P 2p and Ca 2p.28,44–47 The spectra were measured at kinetic electron energies of 200 eV or 830 eV which relate to inelastic mean free paths (IMFP) for the electrons of ∼1 or ∼2 nm, respectively. The NEXAFS spectra were taken both in the detection modes total electron yield (TEY, probing depth 3–10 nm) as well as Auger electron yield (AEY, probing depth < 1 nm). The same spectral features were found in both modes and thus only the TEY spectra are shown in Fig. 7 and 8 (see also ESI, Fig. S6†).
 | ||
| Fig. 7 Mn L3-edge NEXAFS (TEY) spectra of the three different Ca-birnessite samples studied by XAS. Color code and sample descriptions are identical to Fig. 6. | ||
The O 1s XPS spectra show contributions from water, Mn-bound hydroxide and bridging μ-oxido ligands between two Mn centers (see ESI, Fig S8†). A comparison of the data for the different photon energies indicates (as expected) a higher concentration of hydroxide groups and water at the surface of the samples compared to the underlying oxide volume. Furthermore, the spectral differences between the three samples show that the hydroxide concentration of the precursor powder is higher than that of the annealed electrode, but smaller than in the used electrodes. This is also expected, as the calcination step at 450 °C used for the electrode preparation should result in the elimination of OH− and/or H2O from the material, while the exposure to the aqueous medium during electrolysis will result in a renewed binding of these species. The shoulder appearing at 530.8 eV for the sample after electrochemistry can be connected to phosphates originating from the electrolyte.48,49 As can be seen in Fig. S8,† the surface of the electrode (Ek = 200 eV) shows a more pronounced signal related to the phosphate electrolyte residuals.
To gain insights into the oxidation states of the manganese species located on the surface of the electrode, NEXAFS spectra were taken at the Mn L-edges and the recorded Mn L3-edge spectra are shown in Fig. 7. The Mn 2p spectra for all the samples were also measured (ESI, Fig. S15†). Due to significant multiplet splitting of the three oxidation states of manganese(II, III, IV), and the overlapping binding energy for these multiplet splitting structures we decided to focus on the Mn L-edge (NEXAFS) analysis. From comparisons of reference data50 with the spectra measured for reference oxides and used for the difference spectra analysis (Fig. S9†) and also for linear combination analysis (LCA) (Fig. S9a†), we conclude that the as-prepared Ca-birnessite powder mainly contains Mn in the form of Mn4+ ions, giving rise to two pronounced NEXAFS peaks at 641.3 and 643.5 eV, respectively (59% Mn4+vs. 41% Mn3+ from LCA). During electrode preparation, manganese is partially reduced (most likely by the organic components of the ink, see XPS results for carbon) to an oxide material containing Mn predominantly in its Mn3+ oxidation state (LCA here: 7% Mn2+, 6% Mn2+/Mn3+, 65% Mn3+ and 22% Mn4+). This is manifested by two additional NEXAFS signals at 641.8 and 642.4 eV, while the feature at 643.5 eV almost disappears from the spectrum. Electrolysis at OER conditions results in a re-oxidation of manganese and the Mn4+-peak at 643.5 eV reappears. However, the peaks assigned to Mn4+ do not reach the same intensities after electrolysis as initially found for the powder sample, which suggests that a certain concentration of Mn3+ is still present on the material surface even after extended electrolysis at OER potentials (LCA after electrolysis: 2% Mn2+, 5% Mn2+/Mn3+, 45% Mn3+ and 48% Mn4+). This observation differs from the behavior of birnessites containing only potassium as additional cation51,52 and it is thus feasible that the Ca2+ ions have a stabilizing effect on the Mn2+ and Mn3+ oxidation states.
When an oxidizing potential was applied to the electrode in buffered electrolyte solutions, we detected electrochemical “pre-waves” in the CVs (see ESI, Fig. S1†) and also some reorganization of the material at its surface by electron microscopy. Both processes most likely involve complex Mn-centered redox reactions.28 In the light of the changes observed in the Mn NEXAFS spectra, we can now identify electrochemical oxidations of Mn3+ and possibly also Mn2+, (see Fig. S6 in the ESI†) centers to Mn4+ occurring for E > 1 V to be responsible for these events.
Next, the XPS and NEXAFS data for calcium were analyzed (Fig. 8 and ESI, Fig. S10†). The Ca 2p spectra were measured at kinetic energies of Ek = 200 eV and Ek = 830 eV, respectively. A quantitative analysis of the Ca 2p spectra shows that the calcium ions are eliminated from the structure resulting in a decrease of the Ca:Mn ratio from 0.3:1 for the powder sample and 0.18:1 for the freshly prepared electrode to 0.11:1 for the electrode after electrochemical performance in phosphate buffer. These numbers are in very good agreement with EDX-SEM measurements described above. Charge neutrality of the system is preserved by equilibrium between amount of Ca2+ ions that left the system and the oxidation state of manganese as well as incorporation of K+ ions (ESI, Table S4†).
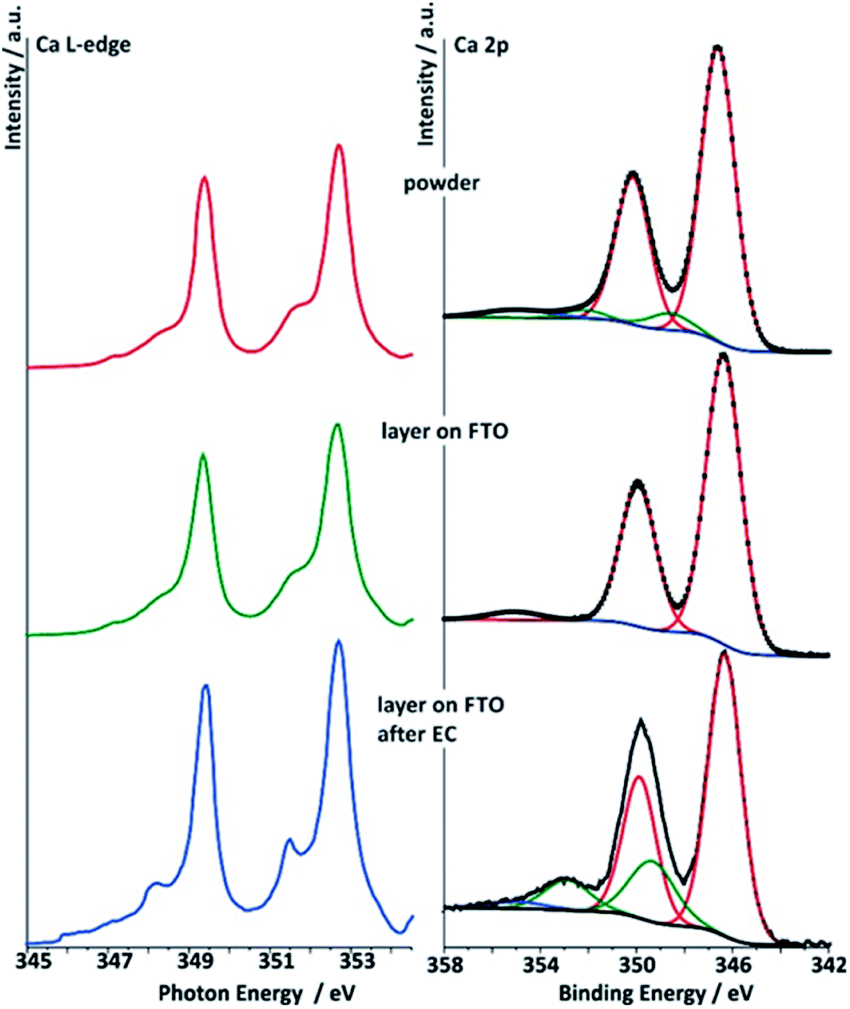 | ||
| Fig. 8 Right: deconvoluted Ca 2p spectra (data for Ek = 830 eV) of the three different Ca-birnessite samples studied by XAS: powder (top), printed Ca-birnessite layer on FTO before (middle) and after electrolysis in phosphate buffer (bottom). Left: Ca L-edge NEXAFS spectra of Ca-birnessite samples in total electron yield (TEY) mode. Color code and sample descriptions are identical to Fig. 6. | ||
Deconvolutions of the Ca 2p lines yielded a main doublet at 346.4 eV for all samples (Fig. 8). From the literature, this is typical for calcium manganese oxides like CaMnO3 or CaMn2O4, where Ca2+ interacts with neighboring Mn3+/4+ cations via μ-oxido bridges.53 After electrolysis in phosphate buffer, both the Ca 2p (XPS) and the Ca L-edges (NEXAFS) spectra show additional contributions when compared to those of the powder sample or the pristine electrode, indicating that some of the Ca2+ ions undergo modification to their ligand shell during electrochemical OER in phosphate buffer. In the XPS, after electrolysis an additional peak is found in the Ca 2p spectrum at a higher binding energy of 353.5 eV. Furthermore, NEXAFS shows an additional resonance at a lower excitation energy. This effect was observed only when phosphate buffer was used as electrolyte, while in the case of imidazolium sulphate no additional Ca L-edge feature was observed (see ESI, Fig. S11†).
The calcium ions in the interlayer space of birnessite-type oxides are expected to show strong Coulomb interactions with the oxido anions of the [MnO6] layers and neighboring water molecules. On the other hand, we found that most of the Ca2+ is removed from the birnessite material during OER in imidazolium sulphate (see ESI, Fig. S7†). In this case, the Mn L3-edge spectrum is also dominated by Mn4+ (Fig. S12†) and an analysis of the data shows that there is ca. 20% less Mn3+ present in comparison to the sample obtained after electrolysis in phosphate buffer (LCA here: 6% Mn2+, 7% Mn2+/Mn3+, 21% Mn3+ and 66% Mn4+; see ESI, Fig. S9a†). To us, it seems that all these observations might be explainable by the following interwoven effects: (1) Ca2+–phosphate interactions obviously stabilize Ca2+ within the birnessite, (2) it is known that secondary cations like Ca2+ influence the energetics of the conversion of Mn4+ to Mn3+,54 so that in consequence (3) the presence of phosphate, mediated via the Ca2+ ions in the material, stabilize lower oxidation states of manganese in the Ca-birnessite catalyst and thus make it more active in OER catalysis.
To test this assertion, we noticed that all XPS spectra of Fig. 8 feature a weak Ca 2p satellite peak at about 355 eV. At ∼10 eV of energy above the main line, these signals could well be exciton or charge transfer satellites associated with Ca2+.55,56 However, the spectra for the Ca-birnessite post-operando spectrum from the phosphate electrolyte additionally contain an intense satellite just 3 eV above the main line. It is accepted that such additional high energy features in XPS are charge transfer satellites resulting from the hybridization between valence orbitals of cations and anions, in our case Ca2+ and O2− (for more details see ESI, chapter 4 and Fig. S13b, d†).55–57 This hybridization can effectively be described by charge transfer from the ligand to the metal, resulting in a d1![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) configuration, where denotes a ligand hole and d1 indicates an additional d electron on the metal. The energy separation between these satellites and the main Ca 2p line is indicative of the degree of hybridization.52 Modeling the XPS spectra within a charge transfer multiplet approach, using the Quanty package,67 leads to the conclusion that a slight increase in the covalency of the Ca–ligand interaction through a drop in the ligand to Ca charge transfer energy can account for the changes observed after electrochemistry (Fig. S13d†).
configuration, where denotes a ligand hole and d1 indicates an additional d electron on the metal. The energy separation between these satellites and the main Ca 2p line is indicative of the degree of hybridization.52 Modeling the XPS spectra within a charge transfer multiplet approach, using the Quanty package,67 leads to the conclusion that a slight increase in the covalency of the Ca–ligand interaction through a drop in the ligand to Ca charge transfer energy can account for the changes observed after electrochemistry (Fig. S13d†).
For material obtained after printing onto the FTO substrate, we see that the charge transfer satellite seen in the after electrochemistry sample is lost (Fig. 8). Unlike the partially covalent ground state found after electrochemistry, the ground state after printing on FTO is dominated by the ionic d0 configuration. The origin of this change in covalency can be seen on the basis of hybrid density functional theory calculations which show an increase in both band gap and Ca–O distance in the presence of a high concentration of Mn3+ ions due to the Jahn–Teller distortion of these centres.58 Thus, the appearance of a high concentration of Mn3+ may lead to a loss of the charge transfer satellite in the Ca 2p XPS and a decrease in covalency of the Ca oxygen bonds. To test if this is the case, we turned to the Mn L-edge measurements (Fig. 7), from these spectra it seems that the existence of the Ca 2p charge transfer satellite may be connected with the presence of Mn4+. After printing, the sample is composed primarily of Mn3+ and there is no charge transfer satellite in the Ca 2p XPS spectrum. However, when Mn4+ dominates the Mn L-edge spectra as in the sample after electrochemistry, the charge transfer satellite is seen in the Ca 2p XPS spectrum.
The analysis of the Ca L-edge spectra shows that the spectrum recorded for the electrode after OER electrocatalysis in phosphate buffer shows an additional feature similar to what was found for the XPS Ca 2p lines (Fig. 8). The main spin–orbit related peaks L3 (2p3/2) and L2 (2p1/2) at 349.4 eV and 252.7 eV, respectively, are present in all three spectra. The position of these main absorptions is nearly the same for all three samples. For the sample treated electrochemically in phosphate buffer, peaks appear at 348.2 eV and 351.47 eV, while spectra of the two others samples show only small shoulders at these photon energies.
The Ca L-edge spectra is known to be dominated by transitions into localized Ca 3d states meaning that p → s transitions can be ignored to first approximation.59 Thus, for Ca2+ without hybridization, the final state in NEXAFS is 2p5d1. In spherical symmetry this would lead to two primary lines associated with the p3/2 and p1/2 initial states, while in an octahedral field the d orbitals are no longer degenerate and two final states become available for each initial state, and the ionic contribution to the crystal field splitting dominates changes to the spectrum.60
We calculated the X-ray absorption spectra using a multiplet Hamiltonian that includes hybridization with the same parameters as for the XPS calculations described above. A comparison of calculated L-edge spectra with the experimental results shows that Ca2+ interacts weakly with its surroundings in the as prepared birnessite powder, with a value of only 10Dq = 0.6 eV required to model the shoulders at 348.2 eV and 351.5 eV. For comparison, 10Dq is 1.2 eV in CaO.60 After electrochemistry 10Dq increases to 1 eV, consistent with a change in the ligand shell expected from the XPS results (Fig. S13c†).
The agreement between the theoretical description of the NEXAFS and XPS spectra allows us to conclude that the additional peaks in the Ca L-edge spectra recorded for the electrode sample after OER electrocatalysis in phosphate buffer can be assigned to a partial change in environment of the calcium ions. This indicates the formation of a new Ca2+-containing phase, in which the Ca–ligand interaction becomes slightly more covalent, and perhaps therefore resistant to dissolution into the electrolyte. Note, however, from the small orbital overlap the Ca2+ is likely intercalated in all cases, which can also be seen by comparison of the Ca 2p XPS spectra shown here and those for Ca oxides, hydroxides, carbonates, and phosphates,61–63 where the latter have satellites at much higher energies dues to larger Ca–ligand hopping. For the sample obtained from the imidazolium sulphate electrolyte, such additional features at 348.2 eV and 351.47 eV are not observed, suggesting that hybridization between Ca2+ and O2− is not as strong in this case and might thus be phosphate-dependent (see ESI, Fig. S11†).
The survey XPS spectrum of the sample after electrochemistry in phosphate buffer also revealed the presence of a P 2p signal, which must originate from phosphate ions incorporated into the oxide (Fig. 6). At this stage it is unclear whether this signal might also originate from calcium phosphates (Eb = 347–348 eV) as possible products from the reaction between Ca2+ from the Ca-birnessite and (H)PO42/3− anions from the electrolyte.64 Taking into account the well-studied chemistry of calcium phosphates and the TEM-EDX data presented above (where phosphorous was found close to the surfaces of the Ca-birnessite nanocrystals), the formation of some kind of calcium phosphate moieties seems plausible. Unfortunately, in our case calcium phosphate species cannot be identified by their Ca 2p XP spectra as the binding energies for calcium manganese oxides and calcium phosphates are separated by less than 1 eV.54
Nevertheless, in order to be able to detect possible contribution from calcium phosphates next to calcium manganese oxides, a P 2p XPS for the electrode after electrolysis was measured using two kinetic energies (Ek = 200 eV and 830 eV, see ESI, Fig. S14†). The two spectra differ by a greater bulk contribution (Ek = 820 eV vs. Ek = 200 eV) of a phase appearing at higher binding energy (∼136 eV). After deconvolution of the 830 eV spectrum, three doublets were found (Fig. 9). A comparison with literature data combined with theoretical calculations allows an analysis of the contributions to this P 2p peak: the first doublet located at 132.8 eV (red line) can be assigned to HPO42− and H2PO4−, the buffer anions present in the electrolyte.64 The second (133.7 eV) (blue line) and third (135.75 eV) (green line) doublet are related to each other. The energetic position of the second doublet is in very good agreement with the literature data for calcium phosphates and the formation of such compounds from the reaction between phosphate from the electrolyte and calcium ions located within the birnessite structure appears to us very plausible. The spectral contribution located at the highest binding energy was assigned to calcium phosphates containing H2O and/or OH− and its observation is in agreement with our findings for the Ca 2p line and the Ca L-edge spectra described above. Here, the formation of clusters like CaH(PO4) associated with water molecules is possible which are known from the minerals brushite (CaH(PO4)·2H2O) or octacalcium phosphate (Ca8H2(PO4)6·5H2O). Another possible reaction at pH 7 is the formation of hydroxyapatite (Ca5(PO4)3(OH)).
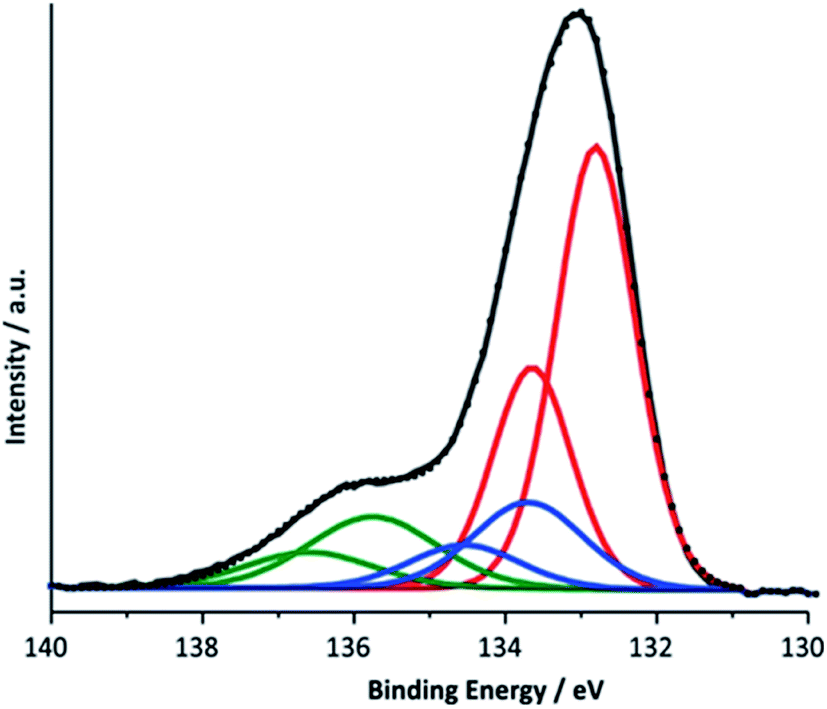 | ||
| Fig. 9 Deconvoluted P 2p XPS spectrum for a Ca-birnessite electrode after electrolysis under OER conditions (16 h in 0.1 M phosphate buffer at η = 540 mV). | ||
The most obvious explanation for the appearance of the contributions of the P 2p line is the reaction of phosphate ions with the surface of the manganese oxide layer, where oxygen point-defects could react especially well with phosphate units.65 For this, different phosphate binding motifs are possible: direct binding of phosphate to the [MnO6]n-layer could occur via a single oxygen vacancy or a phosphate bonded via two oxygen atoms to two neighboring O-defects. Another possibility is the reaction of phosphate with calcium ions located between the [MnO6]-layers, which are separated by a rather large 7 Å interlayer space for birnessites. This is supported by the reduced Ca2+ concentration in phosphate enriched areas.
In conclusion, the XPS results can be interpreted by equilibria involving dynamic exchanges of Ca2+, K+, PO43− and HPO42− ions bound to the surfaces, within the pores and probably even between the [MnO6]n layers of the birnessite material as depicted in Fig. 10. Among the anions, phosphate species seem to have an especially high affinity for birnessites, leading to a substantial incorporation of phosphate into the birnessites' surface layers where catalytic water oxidation most likely occurs. The combination of XPS, NEXAFS and theoretical approaches indicates that the binding of phosphate can either occur directly via O-vacancies in the [MnO6]-layers or also by the formation of [Ca/PO4/H2O]− complexes within the layers. The latter appear to be of significant importance because close calcium–manganese and calcium–phosphate interactions are indicated by the detailed analyses not only of the P 2p but also the Ca 2p and Ca, Mn L-edges signals. Finally, we would like to add that the described incorporation of phosphate is to a large extend reversible: if an anode “loaded” with phosphate by OER electrocatalysis is placed in deionised water for 12 h, no XPS peaks for P can be detected any more.
Conclusions
While few studies on water-oxidation catalysis by manganese oxides emphasize the critical role of the electrolyte for effective water oxidation catalysis, our results clearly show the potential benefits of a rational, concerted development of catalyst and electrolyte in parallel. For Ca-birnessite, we find that the use of buffering electrolytes is generally beneficial and that phosphate-based systems seem to be particularly advantageous for the operation of birnessite-anodes at near-neutral conditions.Our detailed investigation of the interactions between the Ca-birnessite material and (hydrogen) phosphate electrolytes during electrocatalysis provided proof for very dynamic, complicated ion exchange equilibria between the solution and the heterogeneous water-oxidation catalyst. Using a broad set of investigation methods (electron microscopy, EDX, XPS and NEXAFS), we could show that the elements carbon and calcium, which are part of the catalyst layer as a result of the preparation method, are fully (C) or to a large degree (Ca) eliminated from the material after some hours of operation as OER electrocatalyst. Further analysis of the XPS and NEXAFS spectra acquired after OER reveals that a large fraction of the remaining calcium–oxygen interactions is likely of covalent nature, preventing the complete dissolution of calcium from the material. It seems that phosphate plays a role in accruing this phenomenon as no hybridization was observed in the case of the imidazolium sulphate electrolyte, and much more Ca2+ ions left the structure. This in turn leads to a destabilization of the Mn3+ state resulting in a decrease of OER activity.
In contrast, for the neutral potassium phosphate electrolyte, our data suggests that both K+ and HnPO4(3−n)−, enter the mesoporous birnessite layer. After some time, these ions can be found in the entire volume of the catalyst, with especially high concentrations at the top of the catalyst layer (where a new, probably re-precipitated material is formed) and on the inner surfaces of the pores. In future experiments, it would be interesting to investigate whether such ion-exchange processes only occur between different s-block cations like K+, Na+, Ca2+etc. or also for (most likely much stronger bound) d-block cations like Ni2+ or Con+. The latter have already been successfully incorporated into birnessites resulting in improved electrochemical OER activity and stability.41
The interpretation of XPS and NEXAFS data for a nanocrystalline, partially disordered manganese oxide like birnessite is complex and far from routine. However, our analysis is in agreement with previous investigations which indicated that the redox equilibrium Mn3+ ⇆ Mn4+ is of central importance for OER catalysis by birnessites. A significant concentration of Mn3+ ions at the surface of the oxide might be key for the material to be catalytically active. Mediated by bridging oxygen atoms, Mn cations interact with both calcium (which seems to influence its oxidation state) and phosphorous during electrocatalysis and furthermore Ca2+ and HnPO4(3−n)− additionally seem to form some kind of aggregates within the material as detected by XPS.
Overall, the operation of Ca-birnessite as a water-oxidation catalyst in phosphate electrolytes significantly alters the manganese oxide material. Especially striking is the incorporation of (hydrogen)phosphate anions into the catalyst, where they stabilize Ca2+ in the structure and might additionally act as “internal bases” to shuttle protons released from the OER reaction at catalytic sites into the bulk of the solution as illustrated in Fig. 10. These special effects of the interaction of HnPO4(3−n)− with the catalyst might explain the markedly better OER-performance of the Ca-birnessite catalyst material in phosphate solutions in comparison to other buffering electrolytes. Finally, we would like to add that the processes described here might be closely related to the incorporation of phosphate ions into the much-studied “CoPi”-catalyst films generated by the electrodeposition of amorphous cobalt oxides from phosphate-containing solutions.66 Thus CoOx- and nanocrystalline, layered MnOx-electrocatalysts for water-oxidation show an important similarity: in addition to the atomic structure and morphology of the catalyst material, the interaction of the oxides with the ionic components of the electrolyte is of central importance for the catalytic process as well as for the formation and stabilization of the catalytically active surface.
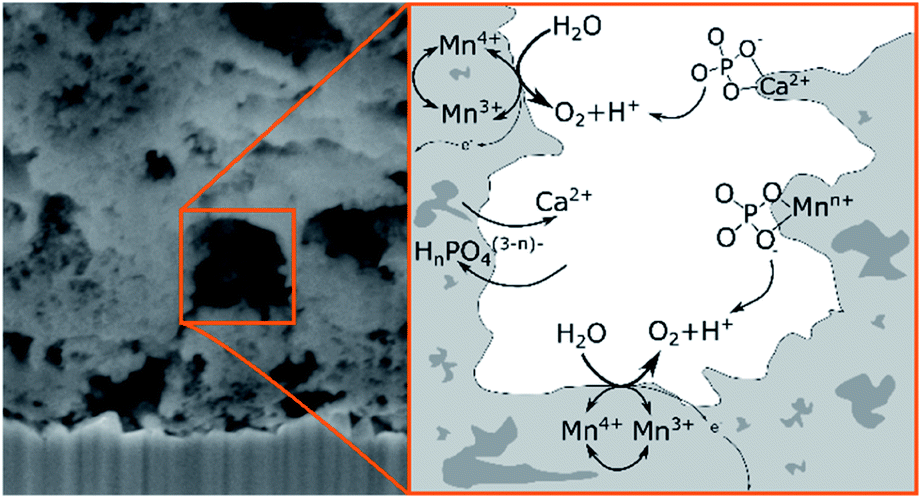 | ||
| Fig. 10 Schematic illustration of the suggested ion exchanges and proton shuttling processes occurring on the surface of the porous Ca-birnessite during OER electrocatalysis. | ||
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was made possible by the generous financial support by the Federal Ministry of Education and Research (BMBF cluster project MANGAN), the German Research Foundation (DFG) priority program SPP1613, grants KU2885/2-2 and JO348/11-1 and SFB 1073, project C02, funded by DFG. T. E. J. acknowledges the Alexander-von-Humboldt Foundation for financial support. Open Access funding provided by the Max Planck Society.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018 RSC.
- P. W. Menezes, A. Indra, P. Littlewood, M. Schwarze, C. Göbel, R. Schomäcker and M. Driess, ChemSusChem, 2014, 7, 2202–2211 CrossRef CAS PubMed.
- A. Ramírez, P. Hillebrand, D. Stellmach, M. M. May, P. Bogdanoff and S. Fiechter, J. Phys. Chem. C, 2014, 118, 14073–14081 CrossRef.
- P. F. Smith, B. J. Deibert, S. Kaushik, G. Gardner, S. Hwang, H. Wang, J. F. Al-Sharab, E. Garfunkel, L. Fabris, J. Li and G. C. Dismukes, ACS Catal., 2016, 6, 2089–2099 CrossRef CAS.
- S. Raabe, D. Mierwaldt, J. Ciston, M. Uijttewaal, H. Stein, J. Hoffmann, Y. Zhu, P. Blöchl and C. Jooss, Adv. Funct. Mater., 2012, 22, 3378–3388 CrossRef CAS.
- J. Scholz, M. Risch, K. A. Stoerzinger, G. Wartner, Y. Shao-Horn and C. Jooss, J. Phys. Chem. C, 2016, 120, 27746–27756 CrossRef CAS.
- J. Scholz, M. Risch, G. Wartner, C. Luderer, V. Roddatis and C. Jooss, Catalysts, 2017, 7, 139 CrossRef.
- J. Melder, P. Bogdanoff, I. Zaharieva, S. Fiechter, H. Dau and P. Kurz, Z. Phys. Chem., 2020, 234, 925–978 CrossRef CAS.
- G. Mattioli, I. Zaharieva, H. Dau and L. Guidoni, J. Am. Chem. Soc., 2015, 137, 10254–10267 CrossRef CAS PubMed.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330–2339 RSC.
- I. Zaharieva, D. González-Flores, B. Asfari, C. Pasquini, M. R. Mohammadi, K. Klingan, I. Zizak, S. Loos, P. Chernev and H. Dau, Energy Environ. Sci., 2016, 9, 2433–2443 RSC.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- I. Zaharieva, M. M. Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Energy Environ. Sci., 2011, 4, 2400–2408 RSC.
- D. Shevchenko, M. F. Anderlund, S. Styring, H. Dau, I. Zaharieva and A. Thapper, Phys. Chem. Chem. Phys., 2014, 16, 11965 RSC.
- D. González-Flores, I. Zaharieva, J. Heidkamp, P. Chernev, E. Martínez-Moreno, C. Pasquini, M. R. Mohammadi, K. Klingan, U. Gernet, A. Fischer and H. Dau, ChemSusChem, 2016, 9, 379–387 CrossRef PubMed.
- F. Jiao and H. Frei, Chem. Commun., 2010, 46, 2920 RSC.
- A. Indra, P. W. Menezes, I. Zaharieva, E. Baktash, J. Pfrommer, M. Schwarze, H. Dau and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 13206–13210 CrossRef CAS PubMed.
- M. Fekete, R. K. Hocking, S. L. Y. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222–2232 RSC.
- M. M. Najafpour, A. N. Moghaddam, H. Dau and I. Zaharieva, J. Am. Chem. Soc., 2014, 136, 7245–7248 CrossRef CAS PubMed.
- C. E. Frey and P. Kurz, Chem.–Eur. J., 2015, 21, 14958–14968 CrossRef CAS PubMed.
- M. M. Najafpour, F. Ebrahimi, M. Abasi and S. M. Hosseini, Int. J. Hydrogen Energy, 2016, 41, 18472–18477 CrossRef CAS.
- D. C. Golden, J. B. Dixon and C. C. Chen, Clays Clay Miner., 1986, 34, 511–520 CrossRef CAS.
- E. A. Johnson and J. E. Post, Am. Mineral., 2006, 91, 609–618 CrossRef CAS.
- C. E. Frey, M. Wiechen and P. Kurz, Dalton Trans., 2014, 43, 4370–4379 RSC.
- S. Y. Lee, D. González-Flores, J. Ohms, T. Trost, H. Dau, I. Zaharieva and P. Kurz, ChemSusChem, 2014, 7, 3442–3451 CrossRef CAS PubMed.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5, 542–549 CrossRef CAS PubMed.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, ChemSusChem, 2014, 7, 1301–1310 CrossRef CAS PubMed.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC.
- L. C. Seitz, T. J. P. Hersbach, D. Nordlund and T. F. Jaramillo, J. Phys. Chem. Lett., 2015, 6, 4178–4183 CrossRef CAS PubMed.
- M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- A. Izgorodin, O. Winther-Jensen and D. R. MacFarlane, Aust. J. Chem., 2012, 65, 638–642 CrossRef CAS.
- M. M. Najafpour, D. J. Sedigh, B. Pashaei and S. Nayeri, New J. Chem., 2013, 37, 2448–2459 RSC.
- M. M. Najafpour and D. J. Sedigh, Dalton Trans., 2013, 42, 12173–12178 RSC.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, Adv. Catal., 2009, 213–272 CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1993, 20, 77–89 CrossRef CAS.
- M. M. van Schooneveld and S. DeBeer, J. Electron Spectrosc. Relat. Phenom., 2015, 198, 31–56 CrossRef CAS.
- G. Elmacı, G. Özgenç, P. Kurz and B. Zumreoglu-Karan, Sustainable Energy Fuels, 2020, 4, 3157–3166 RSC.
- J. Melder, W. L. Kwong, D. Shevela, J. Messinger and P. Kurz, ChemSusChem, 2017, 10, 4491–4502 CrossRef CAS PubMed.
- E. Ronge, J. Lindner, U. Ross, J. Melder, J. Ohms, V. Roddatis, P. Kurz and C. Jooss, J. Phys. Chem. C, 2021, 125, 5037–5047 CrossRef CAS.
- B. Gilbert, B. H. Frazer, A. Belz, P. G. Conrad, K. H. Nealson, D. Haskel, J. C. Lang, G. Srajer and G. De Stasio, J. Phys. Chem. A, 2003, 107, 2839–2847 CrossRef CAS.
- R. Qiao, T. Chin, S. J. Harris, S. Yan and W. Yang, Curr. Appl. Phys., 2013, 13, 544–548 CrossRef.
- S. Voss, M. Fonin, U. Rüdiger, M. Burgert, U. Groth and Y. S. Dedkov, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 045102 CrossRef.
- M. Khan, E. Suljoti, A. Singh, S. A. Bonke, T. Brandenburg, K. Atak, R. Golnak, L. Spiccia and E. F. Aziz, J. Mater. Chem. A, 2014, 2, 18199–18203 RSC.
- W. E. Morgan, J. R. Van Wazer and W. J. Stec, J. Am. Chem. Soc., 1973, 95, 751–755 CrossRef CAS.
- P.-H. Lo, J. Electrochem. Soc., 1995, 142, 91 CrossRef CAS.
- J. Fernández-Rodríguez, B. Toby and M. Van Veenendaal, J. Electron Spectrosc. Relat. Phenom., 2015, 202, 81–88 CrossRef.
- F. Sulzmann, W. Jang, J. Melder, C. Rohner, F. Girgsdies, A. Knop-Gerickec, R. Schlögl, T. Jones, P. Kurz and K. Skorupska, manuscr. to be submitt.
- M. F. Tesch, S. A. Bonke, T. E. Jones, M. N. Shaker, J. Xiao, K. Skorupska, R. Mom, J. Melder, P. Kurz, A. Knop-Gericke, R. Schlögl, R. K. Hocking and A. N. Simonov, Angew. Chem., 2019, 131, 3464–3470 CrossRef.
- D. K. G. de Boer, C. Haas and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 4401–4419 CrossRef CAS.
- S. Kumar, V. S. Raju, S. Bera, K. Vijaynandhini and T. R. N. Kutty, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 237, 623–630 CrossRef CAS.
- A. E. Bocquet, T. Mizokawa, K. Morikawa, A. Fujimori, S. R. Barman, K. Maiti, D. D. Sarma, Y. Tokura and M. Onoda, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 1161–1170 CrossRef CAS PubMed.
- G. van der Laan, C. Westra, C. Haas and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 4369–4380 CrossRef CAS.
- B. A. Pinaud, Z. Chen, D. N. Abram and T. F. Jaramillo, J. Phys. Chem. C, 2011, 115, 11830–11838 CrossRef CAS.
- K. P. Lucht and J. L. Mendoza-Cortes, J. Phys. Chem. C, 2015, 119, 22838–22846 CrossRef CAS.
- M. W. D. Mansfield, Proc. R. Soc. A, 1976, 348, 143–151 CAS.
- F. J. Himpsel, U. O. Karlsson, A. B. McLean, L. J. Terminello, F. M. F. De Groot, M. Abbate, J. C. Fuggle, J. A. Yarmoff, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 6899–6907 CrossRef CAS PubMed.
- M. A. Stranick, Surf. Sci. Spectra, 1999, 6, 39–46 CrossRef CAS.
- D. R. Baer and J. F. Moulder, Surf. Sci. Spectra, 1993, 2, 1–7 CrossRef CAS.
- J. C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- W. J. Landis and J. R. Martin, J. Vac. Sci. Technol., A, 1984, 2, 1108–1111 CrossRef CAS.
- C. V. K. Sharma, C. C. Chusuei, R. Clérac, T. Möller, K. R. Dunbar, A. Clearfield, C. V. K. Sharma, C. C. Chusuei, R. Clérac, T. Möller, K. R. Dunbar and A. Clearfield, Inorg. Chem., 2003, 42, 8300–8308 CrossRef CAS PubMed.
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed.
- M. W. Haverkort, M. Zwierzycki and O. K. Andersen, Phys. Rev. B, 2012, 85(16), 165113–165133 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Material details and remarks on the methods, cyclic voltammograms of birnessite, complementary TEM analysis including HRTEM, electron diffraction and additional EDX linescans, additional spectroscopic information including reference spectra, linear combination analysis and hybridization and calculation details. See DOI: 10.1039/d1se01076j |
| ‡ Present address: Fraunhofer Institute for Silicon Technology ISIT, Fraunhoferstrasse 1, 25524 Itzehoe, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |