
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ formation of a molecular cobalt(III)/AgCl photocatalyst for visible-light water oxidation†
Hiroto
Mogi
a,
Megumi
Okazaki
 ab,
Shunta
Nishioka
a and
Kazuhiko
Maeda
*a
ab,
Shunta
Nishioka
a and
Kazuhiko
Maeda
*a
aDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: maedak@chem.titech.ac.jp
bJapan Society for the Promotion of Science, Kojimachi Business Center Building, 5-3-1 Kojimachi, Chiyoda-ku, Tokyo 102-0083, Japan
First published on 4th September 2021
Abstract
Hexaamminecobalt(III) chloride underwent reaction in an aqueous AgNO3 solution containing a buffer base (pH 7–9), producing insoluble AgCl that showed no visible light photoresponse. The resultant molecular Co(III)/AgCl system showed water oxidation activity under visible light (λ > 400 nm), approximately five times greater than that of a previously reported CoOx/anatase-TiO2 photocatalyst.
Photocatalytic water splitting under irradiation with sunlight is an attractive solution to various energy and environmental issues.1–7 Because O2 evolution requires the transfer of four electrons from two H2O molecules, it is considered a difficult process from a kinetics perspective, hence necessitating a catalyst. Therefore, the development of a water oxidation catalyst (or photocatalyst) is an important research target in both heterogeneous and homogeneous systems,8–10 even if a sacrificial electron acceptor is employed.11–16
In metal (or metal oxide)/semiconductor composites, one may expect electron transitions from the loaded metal species to the conduction band of the semiconductor, and the metal-to-semiconductor electron transitions can be utilized for water oxidation photocatalysis.17–19 Our group has reported that modifying a wide-bandgap semiconductor (e.g., TiO2) with cobalt oxide (CoOx) or hydroxide (Co(OH)2) leads to the appearance of a new visible-light absorption band attributable to electron transitions from the loaded Co species to the conduction band of the semiconductor.20–22 For example, Co/TiO2 hybrid materials are active as photocatalysts for water oxidation in the presence of AgNO3 as an electron acceptor under visible light, whereas unmodified TiO2 is inactive because of its large bandgap. We achieved O2 evolution even without physical contact between the Co species and the TiO2 support. Overall water splitting without a sacrificial reagent was also possible when Co/TiO2 was used as an anode in a photoelectrochemical cell.23 Certain Co compounds are known to be good catalysts (or cocatalysts) for water oxidation.8–12,24,25 Therefore, the Co species in these Co/semiconductor hybrid systems can be regarded as a “catalytic photosensitizer” for water oxidation (Scheme 1), which is unique in heterogeneous photocatalysis. However, the photocatalytic performance of the Co/semiconductor hybrid systems for water oxidation is still unsatisfactory, and the development of more active photocatalysts based on a catalytic Co photosensitizer is strongly desired.
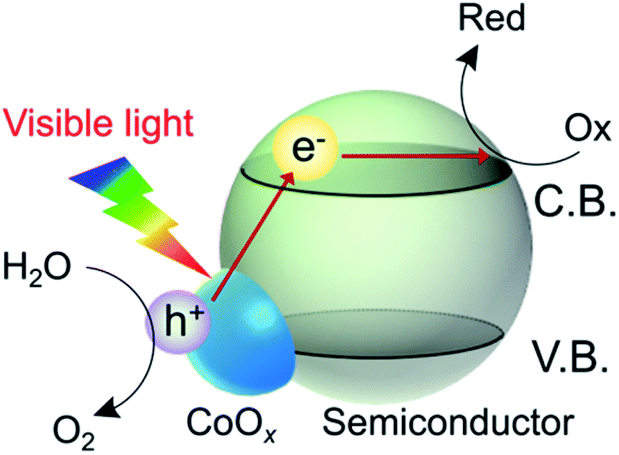 | ||
| Scheme 1 Water oxidation driven by charge transfer from a loaded Co species (e.g., CoOx) to a semiconductor support. Even though the bandgap photoexcitation of the semiconductor from the valence band (V.B.) to the conduction band (C.B.) under visible light is not possible because of the large bandgap of the semiconductor, interfacial charge transfer from Co to the semiconductor may occur under visible light. | ||
In the present work, we report a new visible-light-driven water oxidation system based on a molecular Co(III) complex and a wide-gap semiconductor. We found that hexaamminecobalt(III) chloride ([Co(NH3)6]Cl3) undergoes reaction in an aqueous AgNO3 solution containing a buffer base (pH 7–9), producing a mixture of insoluble AgCl and aqueous [Co(NH3)6]3+ species. Surprisingly, the Co(III)/AgCl system, even without any physical connection, exhibited approximately fivefold greater activity for visible-light water oxidation (λ > 400 nm) compared with the activity of a previously reported CoOx/anatase-TiO2 photocatalyst.
[Co(NH3)6]3+ has previously been reported to function as a catalyst for water oxidation in the presence of [Ru(bpy)3]3+ as an oxidant.26 The absorption spectrum of [Co(NH3)6]3+ shows a characteristic absorption band in the visible-light region (Fig. 1), which is assigned to d–d transitions (1A1g → 1T1g). AgCl has a cubic rocksalt structure and an indirect bandgap of 3.28 eV.27 The absorption spectrum of our AgCl prepared via a precipitation reaction between AgNO3 and HCl (see the ESI† for details) showed an absorption edge at ∼410 nm (Fig. 1 and S1†), with an estimated bandgap of 3.0 eV, which is similar to the reported value. The spectrum of the as-prepared AgCl also showed an increased background in the visible-light region, presumably because of self-sensitization caused by reduced Ag species.28 Although AgCl alone does not efficiently absorb visible light because of its large bandgap, AgCl modified with metals such as Au or Ag has been reported to act as a visible-light-driven “plasmonic” photocatalyst and/or photoanode.28–30 However, the combination of AgCl with Co species for visible-light water oxidation has not yet been reported.
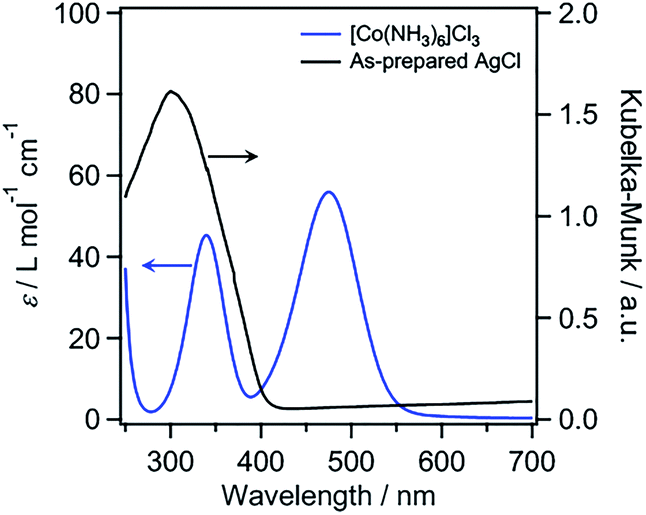 | ||
| Fig. 1 UV-visible absorption spectrum of [Co(NH3)6]Cl3 (5 mM) in H2O and the diffuse reflectance spectrum of the as-prepared AgCl. | ||
The water oxidation reaction was performed at room temperature using a top-irradiation-type reactor connected to a closed gas-circulation system.31 The reaction solutions were prepared by adding different Co sources (e.g., [Co(NH3)6]Cl3, CoCl2 and Co(NO3)2) as “precatalysts” to a 5 mM AgNO3 solution (140 mL) containing 100 mg of La2O3 as a pH buffering agent (pH ∼ 8).32 After the reaction solution was degassed, irradiation was carried out using a 300 W Xe lamp and a cutoff filter. The evolved gases were analysed using an online gas chromatograph.
As shown in Fig. 2a, the [Co(NH3)6]Cl3 and CoCl2 systems produced O2 when irradiated with visible light (λ > 400 nm), with initial rates of approximately 68 and 10 μmol h−1, respectively. By contrast, the use of Co(NO3)2 did not yield O2. Consequently, among the investigated precatalysts, [Co(NH3)6]Cl3 was determined to be the most appropriate for visible-light water oxidation. Under the optimal conditions, the activity of the [Co(NH3)6]Cl3 system was five times greater than that of the photocatalyst CoOx/TiO2, which was previously reported to exhibit the highest activity among Co/wide-bandgap-semiconductor combinations.22 The initial rate of O2 evolution over the [Co(NH3)6]Cl3 system was comparable to that recorded for representative visible-light-driven photocatalysts (e.g., TaON and LaTiO2N),16 which exhibited high water oxidation activities in aqueous AgNO3 solution with La2O3.
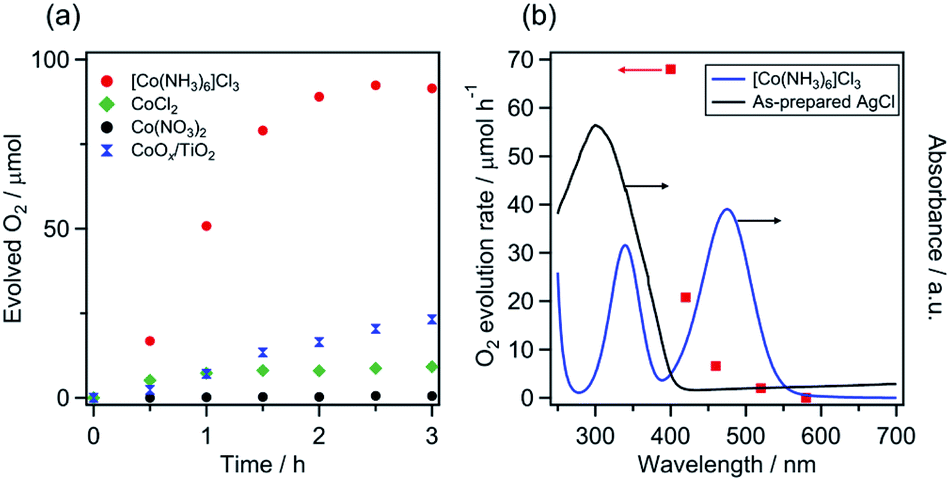 | ||
| Fig. 2 (a) Time courses of O2 evolution over various precatalyst systems under visible light (λ > 400 nm) and (b) the dependence of the rate of O2 evolution by the [Co(NH3)6]Cl3 system on the cutoff wavelength of incident light. Reaction conditions: precatalyst, 30 mg; 5 mM aqueous AgNO3 solution containing La2O3 (100 mg), 140 mL; light source, 300 W Xe lamp with a CM-1 mirror and cutoff filter (L42, Y44, Y48, O54 or R60). In the case of the CoOx/TiO2: catalyst, 100 mg; 10 mM aqueous AgNO3 solution containing La2O3 (200 mg). The actual time course data for the O2 evolution are shown in Fig. S6.† | ||
In the absence of La2O3, however, the O2 evolution activity of the [Co(NH3)6]Cl3 system decreased to almost 13% of its value when La2O3 was present (Fig. S2†), which suggests that a pH of ∼8 is desirable for the efficient production of O2. The effect of reaction pH on the O2 evolution activity was investigated in sodium tetraborate aqueous solutions without La2O3. As shown in Fig. S3,† relatively high O2 evolution rates were obtained at pH 7–9. However, O2 evolution was negligible above pH 10. At pH 10, it was found that the visible light absorption capability of [Co(NH3)6]Cl3 disappeared completely (Fig. S4†), which would be the main reason for the very low activity. These results again indicate the importance of reaction pH. Moreover, no O2 evolution was observed in the absence of either [Co(NH3)6]Cl3, AgNO3 or light irradiation. The fact that no O2 evolution was observed in the dark precludes a stoichiometric reaction between the high-valent [Co(NH3)6]Cl3 and H2O. The apparent quantum yield of the [Co(NH3)6]Cl3 system was 0.20% under irradiation with 460 nm light (Fig. S5†).
Fig. 2b shows the dependence of the rate of O2 evolution by the [Co(NH3)6]Cl3 system on the wavelength of the incident light. The O2 evolution rate decreased with increasing wavelength of the incident light. O2 evolution was observable under irradiation with >520 nm light but became almost zero under irradiation with >580 nm light. The trend of O2 evolution appears to be associated with light absorption by [Co(NH3)6]Cl3. However, the abrupt decrease observed when comparing the O2 evolution rate under λ > 400 nm irradiation with that under λ > 420 nm irradiation (Fig. 2b and S6†) strongly suggests that the activity is not simply correlated with the light-absorption behaviour of [Co(NH3)6]Cl3. These results will be discussed again in later sections.
Because the solubility product (Ksp) of AgCl is 1.8 × 10−10 mol2 L−2,33 AgCl should be generated under the present reaction conditions, where 0.8 mM [Co(NH3)6]Cl3 and 5 mM AgNO3 coexist. The products obtained after photocatalytic reaction for 1 h, at which point the photocatalyst suspension exhibited sufficient activity to generate O2 (Fig. 2), were characterized by X-ray diffraction (XRD) and UV-visible spectroscopy measurements. As displayed in Fig. 3a, the XRD patterns show that the obtained product was a mixture of La(OH)3, AgCl and Ag, with no peaks assignable to [Co(NH3)6]Cl3. The production of Ag0, as a result of the reduction of Ag+, was also confirmed (Fig. S7†). Energy-dispersive X-ray spectroscopy analysis was conducted for the solid product; however, Co species were not measurable presumably because of the low concentration. These results strongly suggest that Co species existed predominantly in the solution phase under the investigated reaction conditions, and also preclude the possibility of Co nanoparticle formation. Fig. 3b shows the UV-visible absorption spectra of the reaction solution after filtration of the solid products. The peak intensity at 473 nm was almost unchanged after 1 h of reaction; however, a decrease in the intensity of the absorption at 340 nm (1A1g → 1T2g) was observed, along with the appearance of a new absorption band at ∼300 nm. The 300 nm-band is assigned to the absorption of NO3− species.34 A substantial reduction of the intensity of the visible-light absorption band was observed after 3 h of reaction, indicating the decomposition of [Co(NH3)6]3+. This decomposition is likely one of the causes of photocatalyst deactivation, but the main cause of the deactivation is the consumption of Ag+ in the solution (see additional discussion in the ESI†).
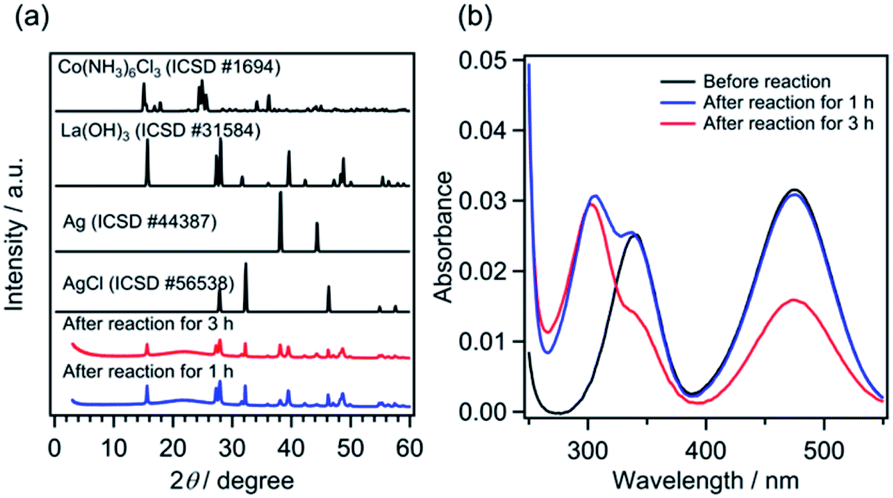 | ||
| Fig. 3 (a) XRD patterns of the obtained products and (b) UV-visible absorption spectra of reaction solutions after different reaction times. The solution collected after filtration was diluted with H2O to prepare a 200 mL solution for measurements. The sample “Before reaction” in panel (b) did not include AgNO3, but only contained 0.56 mM [Co(NH3)6]Cl3. | ||
On the basis of the aforementioned results, we concluded that the active photosystem consisted of aqueous [Co(NH3)6]3+ and AgCl as essential components. Note that approximately 0.36 mmol of AgNO3 still remain even if the formation of AgCl from [Co(NH3)6]Cl3 and AgNO3 in the initial reaction solution occurs stoichiometrically. Taken together, a possible reaction mechanism for O2 evolution by the [Co(NH3)6]Cl3 system is proposed (Scheme 2). Under dark conditions before irradiation, AgCl is formed by a reaction between [Co(NH3)6]Cl3 and AgNO3 in the reaction solution, as confirmed by XRD measurement (Fig. S1†). Visible-light irradiation of the suspension initiates electron transfer from water-soluble [Co(NH3)6]3+ species to the conduction band of AgCl, followed by the oxidation of water to O2 and the reduction of Ag+ to Ag, which occur on higher-valent (i.e., electron-deficient) Co species and AgCl, respectively. It has been reported that the ζ-potentials of a AgCl single crystal measured in aqueous KNO3 solution (1 and 10 mM) were negative at pH > 5.35 This implies that there is an electrostatic attraction between the in situ formed AgCl and [Co(NH3)6]3+ species in the reactant solution, which benefits electron transfer from Co to AgCl. It appears, however, that the electrostatic attraction is too weak to bind [Co(NH3)6]3+ on the AgCl surface. Another possibility that [Co(NH3)6]3+ species undergo photoexcitation and then inject an electron into AgCl is not realistic because [Co(NH3)6]3+ is a non-emissive complex (i.e., the lifetime of the photoexcited state is too short to induce a photoinduced electron transfer reaction from its excited state).
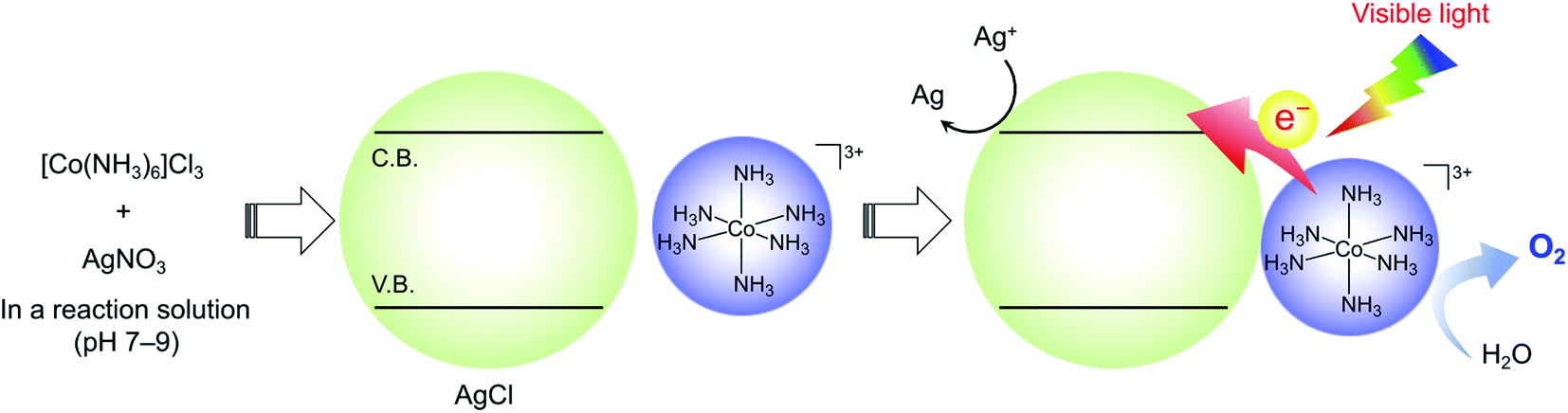 | ||
| Scheme 2 Proposed reaction mechanism for visible-light-driven water oxidation over the [Co(NH3)6]3+/AgCl system. | ||
The presence of AgCl formed in situ in the reaction solution was found to be indispensable for obtaining a measurable amount of O2. When AgCl particles were intentionally removed from the solution by filtration prior to photoreaction, the O2 evolution activity decreased substantially (black squares, Fig. S8†). This confirms that AgCl is essential for achieving visible-light water oxidation with [Co(NH3)6]3+. When the homemade AgCl was added to the filtered (i.e., AgCl-free) suspension and the photoreaction was conducted, a small amount of O2 evolution was observed (green diamonds). The addition of NaCl to the filtered suspension produced AgCl again, resulting in measurable O2 evolution (green squares). However, the thus-obtained activities were much lower than the activity of the standard [Co(NH3)6]3+/AgCl suspension. As shown in Fig. 2a, the Co(NO3)2 system produced negligible O2. However, the coexistence of NaCl in the Co(NO3)2 system led to an improvement of the O2 evolution rate to 14 μmol h−1 (blue diamonds, Fig. S8†). In this case, AgCl formation by the reaction between NaCl and AgNO3 was clearly indicated by naked-eye observation of a white precipitate in the solution. All these results indicate the importance of the in situ-generated AgCl, which plays a key role in oxidizing water to O2 in the presence of Co species. Interestingly, O2 evolution from a mixed system consisting of [Co(NH3)6]3+ and TiO2 was very slow, as compared to that recorded for the standard [Co(NH3)6]3+/AgCl system (Fig. S2†).
Notably, even in the presence of a Cl−-containing precatalyst that can form AgCl during the reaction, precatalysts with higher-valent Co (here [Co(NH3)6]Cl3) exhibited better performance (Fig. 2a). This result is likely attributable to the higher-valence metal species which are generally advantageous in water oxidation catalysis because of their strong oxidizing ability. Water oxidation in the presence of NaCl without a Co precatalyst hardly occurred as well (orange triangles, Fig. S8†). The AgCl formed by the reaction between NaCl and AgNO3 showed a small absorption band in the visible region (Fig. S1†). However, it is clear that the visible light absorption of AgCl does not contribute to O2 evolution. Therefore, if the direct photoexcitation of AgCl occurs at all, it is a minor reaction path during water oxidation by the [Co(NH3)6]Cl3 system.
Conclusions
In summary, we developed a new visible-light-driven water oxidation system that consists of in situ-formed AgCl and [Co(NH3)6]3+. Although AgCl and [Co(NH3)6]3+ were individually almost inactive toward visible-light water oxidation, the combination of the two, even without distinct chemical bond linkage, yielded clear photocatalytic activity for water oxidation at pH 7–9 under visible-light irradiation; the activity was five times greater than that of a previously reported CoOx/anatase TiO2 system. The results of the present study also suggest that the resultant photocatalytic activities were dependent on the valence state of the Co species initially employed, and that suitable combinations of a molecular catalyst and a semiconductor may give us more active photosystems.It has been shown that in metal-complex/semiconductor hybrid photocatalysts for CO2 reduction, electron transfer from suspended semiconductor particles to dissolved metal complexes can occur, even without any chemical bond linkage between the two components.36,37 Therefore, the present water oxidation system is another example of the electron transfer type reaction between a molecule and a semiconductor. As mentioned, [Co(NH3)6]3+ has been reported to act as a catalyst for water oxidation,26 but is generally considered to be inactive for ligand exchange. Further investigations of the underlying mechanism and the development of other photocatalytic reactions using this strategy are underway in our laboratory.
Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (Project JP19H02511). M. O. wishes to acknowledge support by a JSPS Fellowship for Young Scientists (JP19J21858).Notes and references
- R. Abe, Bull. Chem. Soc. Jpn., 2011, 84, 1000–1030 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- K. Maeda and K. Domen, Bull. Chem. Soc. Jpn., 2016, 89, 627–648 CrossRef CAS.
- K. Maeda and T. E. Mallouk, Bull. Chem. Soc. Jpn., 2019, 92, 38–54 CrossRef CAS.
- A. Miyoshi and K. Maeda, Sol. RRL, 2020, 5, 2000521 CrossRef.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018 RSC.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS PubMed.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- S. Chen, Y. Qi, G. Liu, J. Yang, F. Zhang and C. Li, Chem. Commun., 2014, 50, 14415–14417 RSC.
- S. Chen, S. Shen, G. Liu, Y. Qi, F. Zhang and C. Li, Angew. Chem., Int. Ed., 2015, 54, 3047–3051 CrossRef CAS PubMed.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS.
- T. Kanazawa, K. Kato, R. Yamaguchi, T. Uchiyama, D. L. Lu, S. Nozawa, A. Yamakata, Y. Uchimoto and K. Maeda, ACS Catal., 2020, 10, 4960–4966 CrossRef CAS.
- Y. Nishijima, K. Ueno, Y. Kotake, K. Murakoshi, H. Inoue and H. Misawa, J. Phys. Chem. Lett., 2012, 3, 1248–1252 CrossRef CAS PubMed.
- A. Tanaka, K. Nakanishi, R. Hamada, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 1886–1891 CrossRef CAS.
- M. Kajita, K. Saito, N. Abe, A. Shoji, K. Matsubara, T. Yui and M. Yagi, Chem. Commun., 2014, 50, 1241–1243 RSC.
- K. Maeda, K. Ishimaki, Y. Tokunaga, D. Lu and M. Eguchi, Angew. Chem., Int. Ed., 2016, 55, 8309–8313 CrossRef CAS PubMed.
- K. Maeda, K. Ishimaki, M. Okazaki, T. Kanazawa, D. Lu, S. Nozawa, H. Kato and M. Kakihana, ACS Appl. Mater. Interfaces, 2017, 9, 6114–6122 CrossRef CAS PubMed.
- M. Okazaki, Y. Wang, T. Yokoi and K. Maeda, J. Phys. Chem. C, 2019, 123, 10429–10434 CrossRef CAS.
- H. Tanaka, T. Uchiyama, N. Kawakami, M. Okazaki, Y. Uchimoto and K. Maeda, ACS Appl. Mater. Interfaces, 2020, 12, 9219–9225 CrossRef CAS PubMed.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795–2806 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- G. L. Elizarova, L. G. Matvienko, N. V. Lozhkina, V. N. Parmon and K. I. Zamaraev, React. Kinet. Catal. Lett., 1981, 16, 191–194 CrossRef CAS.
- P. M. Scop, Phys. Rev., 1965, 139, A934–A940 CrossRef.
- P. Wang, B. Huang, X. Qin, X. Zhang, Y. Dai, J. Wei and M. H. Whangbo, Angew. Chem., Int. Ed., 2008, 47, 7931–7933 CrossRef CAS PubMed.
- A. Currao, V. R. Reddy and G. Calzaferri, ChemPhysChem, 2004, 5, 720–724 CrossRef CAS PubMed.
- L. Han, P. Wang, C. Zhu, Y. Zhai and S. Dong, Nanoscale, 2011, 3, 2931–2935 RSC.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed.
- A. Kasahara, K. Nukumizu, G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. A, 2002, 106, 6750–6753 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 84th edn, 2003 Search PubMed.
- A. Ono, R. Matsumoto and N. Yamaguchi, Trans. Iron Steel Inst. Jpn., 1982, 22, 121–125 CrossRef.
- A. Selmani, J. Lutzenkirchen, N. Kallay and T. Preocanin, J. Phys.: Condens. Matter, 2014, 26, 244104 CrossRef PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- C. Cometto, R. Kuriki, L. Chen, K. Maeda, T. C. Lau, O. Ishitani and M. Robert, J. Am. Chem. Soc., 2018, 140, 7437–7440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization and photocatalytic reaction data. See DOI: 10.1039/d1se01075a |
| This journal is © The Royal Society of Chemistry 2021 |