
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How to go beyond C1 products with electrochemical reduction of CO2
Da
Li†
ab,
Hao
Zhang†
a,
Hang
Xiang
b,
Shahid
Rasul
c,
Jean-Marie
Fontmorin
bh,
Paniz
Izadi
bi,
Alberto
Roldan
 d,
Rebecca
Taylor
e,
Yujie
Feng
f,
Liam
Banerji
g,
Alexander
Cowan
*g,
Eileen Hao
Yu
*abf and
Jin
Xuan
*a
d,
Rebecca
Taylor
e,
Yujie
Feng
f,
Liam
Banerji
g,
Alexander
Cowan
*g,
Eileen Hao
Yu
*abf and
Jin
Xuan
*a
aDepartment of Chemical Engineering, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: e.yu@lboro.ac.uk; J.Xuan@lboro.ac.uk
bSchool of Engineering, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK
cFaculty of Engineering and Environment, Northumbria University, Newcastle Upon Tyne, NE1 8ST, UK
dCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF 10 3AT, UK
eDepartment of Chemical Engineering and Analysis Science, University of Manchester, Manchester, M13 9PL, UK
fSchool of Environment, Harbin Institute of Technology, Harbin, 150090, PR China
gDepartment of Chemistry, University of Liverpool, Liverpool, L69 7ZF, UK. E-mail: A.J.Cowan@liverpool.ac.uk
hUniv Rennes, CNRS, ISCR, UMR 6226, 35000 Rennes, France
iHelmholtz Centre for Environmental Research GmbH - UFZ, Department of Environmental Microbiology, Permoserstraße 15, 04318 Leipzig, Germany
First published on 20th October 2021
Abstract
The electrochemical reduction of CO2 to produce fuels and value-added organic chemicals is of great potential, providing a mechanism to convert and store renewable energy within a carbon-neutral energy circle. Currently the majority of studies report C1 products such as carbon monoxide and formate as the major CO2 reduction products. A particularly challenging goal within CO2 electrochemical reduction is the pursuit of multi-carbon (C2+) products which have been proposed to enable a more economically viable value chain. This review summaries recent development across electro-, photoelectro- and bioelectro-catalyst developments. It also explores the role of device design and operating conditions in enabling C–C bond generation.
1. Introduction
The concentration of CO2 in the atmosphere has hit its highest level in history, reaching over 420 ppm in 2021.1 It is widely accepted that the greenhouse effect caused by CO2 accumulation is the main reason for global warming and climate change, which is threatening sustainable life on earth. Carbon Capture and Utilization (CCU) is one way to deal with the ever-increasing CO2 concentration in the atmosphere. Electrochemical CO2 reduction (ECO2R), utilizing renewable energy resources is potentially an efficient process to convert the waste CO2, captured from the atmosphere and/or industrial processes. ECO2R has an added advantage in its use as “energy storage” technology, where intermittent renewable energy is utilized to store electrons in high energy density chemical bonds.2–4 At present, ECO2R conversion to C1 is well established, however, there are several challenges for the ECO2R process to scale-up at industrial scale i.e. process energy efficiency, product selectivity and inaccessibility to multi-carbon (C2+) products from the reaction. Indeed, multi-carbon (C2+) products from ECO2R are attractive from the industrial application point of view due to their higher energy density, well-established chemistry and available infrastructure for processing and transportation.2–4The reduction of CO2 to stable C2+ species is extremely challenging, for example, for C2 compounds such as C2H4 and C2H5OH, 12-electrons need to be transferred to the reaction centre, whilst for C3 compounds such as C3H7OH 18 electrons are transferred.5,6 To date two main strategies have been used to achieve C2+ products. Firstly, the discovery and design of a single electrocatalyst able to convert CO2 to C2+ at a low overpotential has been targeted. However, the large number of proton and electron transfers required leads to complex, multiple reaction pathways being accessible and the similarity of the equilibrium potentials for many products (e.g. C2H4, C2H5OH, and C3H7OH are ∼0.1 V vs. RHE, Fig. 1) makes the generation of a wide product distribution likely in the absence of a highly selective catalyst.7,8 The difficulty of generating a single catalytic centre, or several centres within a single material, which have optimized binding energies for the wide-range of intermediates to be formed is clearly extremely challenging and to date this, coupled to the low aqueous solubility of CO2, has typically led to low partial current densities and selectivities for C2+ production. An alternative strategy is to separate the overall reduction of CO2 to products into two separate steps: (i) initial reduction of CO2 electrochemically to C1 compounds (CO), and (ii) reduction of C1 products into C2, C3 and higher carbon-containing liquid fuels.9 By focusing on the further reduction of C1 compounds, the demands on the catalyst can be reduced with a lower number of intermediates as compared to the reduction of CO2 as starting reactant. Furthermore, CO is found to be an intermediate7,10 in the reduction of CO2 to fuels and other useful chemicals, which makes this method more attractive.
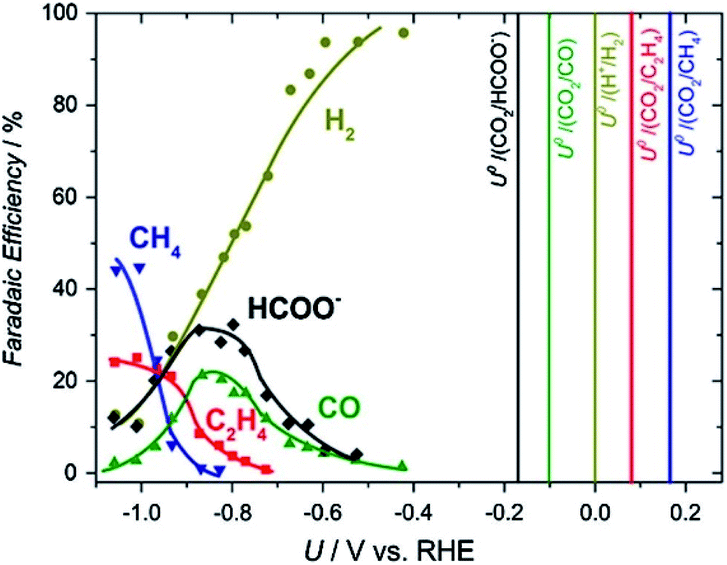 | ||
| Fig. 1 Product faradaic efficiency (FE) vs. potential at −5 mA cm−2 on a Cu electrode in 0.1 M KHCO3. The vertical lines show the standard thermodynamic potentials.11 Reproduced from ref. 11 with permission from Elsevier, copyright 2016. | ||
From a mechanistic point of view, it is well known that during the ECO2R, once CO2 is activated, CO is formed on the electrode surface and the rate-determining step for the ECO2R is a reaction involving adsorbed *CO.11,12 Also, adsorbed intermediates such as HCOO* (ref. 7 and 13) and COOH* (ref. 14 and 15) have been identified experimentally and validated from theoretical calculations. Although computational simulations of electrochemical systems are exceptionally challenging,16 various types of model approximations have been developed to theoretically investigate the electrode–electrolyte interface: (i) the hydrogen electrode model,17 and (ii) the water solvated and shuttling model.18
Among many of the metallic electrocatalysts tested, only Cu based electrocatalysts are proven to show electrocatalytic activity for either CO2 or CO reduction to produce hydrocarbons at reasonable FEs and oxygenates with a reasonable selectively however at the cost of a high overpotential.19,20 This is a developing field with promising strategies revolving around Cu based structures with low coordinated active sites generated by engineered defects, tuned oxidation states and modified electronic structures with the addition of binary/ternary metallic structures.
In this review, after only briefly summarizing the state-of-the-art in the development of Cu based electrodes for the generation of C2+ products by ECO2R, we focus instead on strategies beyond Cu electrode design. A summary of the current state of the art of alternative approaches to metal electrodes; such as the use molecular electrocatalysts and bio-electrochemical approaches for the synthesis of hydrocarbons from ECO2R are reported. We then present the recent developments, and critical role, that electrochemical reactor design and electrolyte engineering (e.g. through the use of novel ionic liquids) can have in controlling activity towards C2+ production formation. Finally, the challenges and strategies of developing advanced catalysts, mechanistic understanding and reactors design are proposed to guide the future R&D of ECO2R.
2. Electrocatalysts for C2+ products
2.1 Metals/metal oxide electrocatalysts
Cu with the specific electron configuration in d sub-shell is able to activate CO2 with more than two pairs of the proton-electron transfer. Of the transition metals Cu is the only elemental metal reported to catalyse ECO2R to multi-carbon products. Research from Kuhl et al.38 observed 16 types of carbonaceous products, i.e., 4 C1, 6 C2, and 5 C3, from ECO2R catalysed by polycrystalline Cu in CO2-saturated 0.1 M KHCO3. Among the C2+ products, only C2H4 and C2H5OH produced with higher than 10% FE. Table 1 summarizes works that produce C2+ products from ECO2R using heterogeneous electrocatalysts including metal (or metal-oxide) and carbon materials. Generally, C2H4 and C2H5OH have been achieved with higher selectivity than other C2+ products. The highest FEs reported so far are 87% (ref. 39) (C2H4) and 93.2% (ref. 40) (C2H5OH), indicating the greatest potential for further development and application. Thus, it appears promising to try to enhance the selectivity of these two valuable C2 products by modification of Cu catalyst. This has been approached by the following four ways.
| Catalyst/cathode material | Applied potential (V vs. RHE) | Current density (mA cm−2) | Electrolyte | C≥2 FEs (%) | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C2H4 | C2H5OH | C2H6 | CH3COOH | C3H7OH | C3H5OH | Other C2+ | ||||||
| a RHE, reversible hydrogen electrode; FEs, faradaic efficiencies; N.G., not given. | ||||||||||||
| Altering the lattice/crystal structure of Cu | Polycrystalline Cu | −1.0 | 5 | 0.1 M KHCO3 | 30.1 | 6.9 | N.G. | N.G. | 3.0 | N.G. | N.G. | 51 |
| Polycrystalline Cu | −1.05 | ∼6.2 | 0.1 M KHCO3 | 26.0 | 9.8 | N.G. | 0.3 | 2.3 | 1.1 | 0.28 (C2H4O2) | 38 | |
| 0.33 (C2H4O) | ||||||||||||
| 0.12 (C2H6O2) | ||||||||||||
| 0.08 (C3H6O) | ||||||||||||
| Cu(100) | N.G. | 5 | 0.1 M KHCO3 | 40.7 | 12.8 | N.G. | 2.5 | 1.6 | 0.7 | 1.0 (C2H4O) | 43 | |
| 1.2 (C3H6O) | ||||||||||||
| Cu(111) | N.G. | 5 | 0.1 M KHCO3 | 8.8 | 5.3 | N.G. | 1.1 | <0.05 | <0.05 | 2.6 (C2H4O) | 43 | |
| Cu(S)-[4(100) × (111)] | N.G. | 5 | 0.1 M KHCO3 | 50.0 | 7.4 | N.G. | 0.9 | 4.6 | 2.2 | 1.2 (C2H4O) | 47 | |
| 5.2 (C3H6O) | ||||||||||||
| Enhancing the surface roughness of Cu | Cu NPs surface | −1.1 | N.G. | 0.1 M KClO4 | 35.5 | N.G. | <0.5 | N.G. | N.G. | N.G. | N.G. | 52 |
| Electro-deposited Cu | −1.3 | N.G. | 0.1 M KHCO3 | 33.3 | N.G. | <0.5 | N.G. | N.G. | N.G. | N.G. | 53 | |
| Ex situ electro-deposited Cu | −1.3 | N.G. | 0.1 M KHCO3 | 11 | N.G. | 2.1 | N.G. | N.G. | N.G. | N.G. | 54 | |
| Cu nanofoam | −1.1 | N.G. | 0.1 M KHCO3 | 1.4 | N.G. | 1.2 | N.G. | N.G. | N.G. | 0.13 (C3H6) | 55 | |
| 15 nm Cu NPs | −1.1 | 22 | 0.1 M KHCO3 | 3 | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | 56 | |
| Cu nano-flower | −1.3 | ∼18 | 0.1 M KHCO3 | 19 | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | 57 | |
| 44 nm Cu nano-cube | −1.1 | N.G. | 0.1 M KHCO3 | 41 | 4 | N.G. | N.G. | 3 | <0.1 | 2 (C2H4O) | 58 | |
| Glycine treated Cu nano-wire | −1.3 | N.G. | 0.1 M KHCO3 | 12.7 | N.G. | 21.1 | N.G. | N.G. | N.G. | 0.3 (C3H6) | 59 | |
| Cu inverse opal-6 layers | −0.81 | ∼15 | 0.1 M KHCO3 | 29.2 | 9.32 | N.G. | 0.36 | 3.35 | N.G. | 1 (C2H2O2) | 60 | |
| 0.12 (C2H4O2) | ||||||||||||
| 0.13 (C2H6O2) | ||||||||||||
| O2 plasma-modified Cu dendrites | −0.9 | ∼30 | 0.1 M KHCO3 | 29 | 12 | N.G. | N.G. | N.G. | N.G. | N.G. | 61 | |
| Oxide-derived Cu catalysts | [100] Cu2O film | −1.1 | N.G. | 0.1 M KHCO3 | 21.5 | N.G. | 1.5 | N.G. | N.G. | N.G. | N.G. | 62 |
| Oxygen-evacuated Cu2O | −1.2 | N.G. | 0.5 M KHCO3 | 25 | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | 63 | |
| 0.9 μm Cu2O film | −0.99 | 25 | 0.1 M KHCO3 | 40.3 | 8.66 | 0.04 | N.G. | N.G. | N.G. | N.G. | 64 | |
| OH-mediated Cu NPs | −0.54 | 225 | 10 M KOH | 66 | 11 | N.G. | 7 | N.G. | N.G. | N.G. | 65 | |
| OH-mediated Cu NPs | −0.79 | ∼300 | 0.5 M KHCO3 | 45.6 | 17.4 | N.G. | 2.5 | 4.5 | N.G. | N.G. | 66 | |
| Alloying and compositing Cu with other components | Au NPs covered Cu2O | −1.05 | 28.08 | 0.1 M KHCO3 | 38.7 | 22.6 | N.G. | 0.85 | 6.83 | N.G. | 1.10 (C2H4O) | 67 |
| Phase-blended Ag–Cu2O | −1.20 | ∼2.5 | 0.1 M KHCO3 | 9.5 | 34.15 | 0.1 | N.G. | N.G. | N.G. | 5.65 (CH3COOH + C2H4O) | 68 | |
| Ag-saturated Cu(100) + Ag surface alloy | −1.0 | N.G. | 0.05 M Cs2CO3 | 42 | 12.5 | N.G. | 2 | 6 | 2.5 | 1 (C2H4O) | 69 | |
| 2.5 (C3H6O) | ||||||||||||
| CuSn alloy | −0.8 | 225 | 1 M KOH | 56.7 | 22.9 | N.G. | 4.7 | 0 | N.G. | N.G. | 70 | |
| Bimetallic Ag/Cu | −0.67 | 250 | 1 M KOH | 35 | 41 | N.G. | 6 | N.G. | N.G. | N.G. | 71 | |
| N-Doped graphene supported Cu NPs | −1.2 | N.G. | 0.1 M KHCO3 | N.G. | 63 | N.G. | N.G. | N.G. | N.G. | N.G. | 72 | |
| Organics-modified Cu | Pyridinic-N rich graphene supported Cu NPs (7 nm) | −0.9 | N.G. | 0.5 M KHCO3 | 19 | N.G. | <2 | N.G. | N.G. | N.G. | N.G. | 73 |
| Cu–N–C | −1.2 | 16.2 | 0.1 M CsHCO3 | 0 | 43 | N.G. | N.G. | N.G. | N.G. | N.G. | 74 | |
| Polycrystalline Cu with N-substituted pyridinium | −1.1 | 1.02 | 0.1 M KHCO3 | 40.5 | 30.6 | N.G. | N.G. | 7.1 | N.G. | N.G. | 75 | |
| Catalysts besides Cu | N-Doped nanodiamond/Si rod array | −1.0 | ∼6.5 | 0.5 M NaHCO3 | N.G. | N.G. | N.G. | 77.6 | N.G. | N.G. | N.G. | 76 |
| N-Doped mesoporous carbon | −0.56 | 0.5 | 0.1 M KHCO3 | N.G. | 77 | N.G. | N.G. | N.G. | N.G. | N.G. | 77 | |
| Pd-multiwalled carbon nanotubes | 4 V (cell voltage) | ∼5.3 | 0.8 M KHCO3 | N.G. | N.G. | N.G. | 38.7 | N.G. | N.G. | N.G. | 78 | |
Altering the lattice/crystal structure of Cu. A facet-dependent activity for CO2 electroreduction toward C2+ products has been demonstrated.41,42 Hori et al.43 in 2002 used a series of single-crystal Cu electrodes to investigate the effect of the crystal structure of the Cu electrode on ECO2R performance. The results demonstrated the dependence of product selectivity on the Cu crystal structure: Cu(111) typically generates CH4, while Cu(100) gives preference to C2+ products, often C2H4. These experimental results can be rationalised by the relative stability of CO on different Cu surfaces. CO dimerization is an important route to C–C bond formation44,45 and it has been found that the formation of absorbed CO dimer occurs more easily, and is most stable, on the square arrangement of 4 surface atoms, i.e., the (100) crystal plane.46 In 2003 Hori et al.47 tested a Cu electrode with a mixed crystal surface, which was Cu[4(100) × (111)] composed of 4 atomic rows of (100) terraces and one atomic height of the (111) step. It was shown to be an effective material for C2H4 formation (the FE reached up to 50% with a C2H4/CH4 ratio around 13.5), indicating that the stepped surface mixed with (100) and (111) is more favourable for ethylene formation. Therefore, synthesis of Cu nanoparticles with high Cu(100) exposure serves as a guideline for designing a catalyst towards C2+ products.48 Cu cubes dominated by Cu(100) catalysed CO2-to-C2H4 with a FE of 57% at a moderate potential of −0.65 V versus RHE. By contrast, Cu octahedrons with Cu(111) terraces reached a selectivity for CH4.41 Sargent et al. developed Cu(100)-rich Cu nanoparticles which showed a strong adsorption capability to intermediates, such an electrode achieved nearly 90% FE for C2 products.49 A recent calculation work demonstrated that increasing CO coverage on the Cu(111) surface decreased the C–C coupling energy, favouring to C2 products, especially ethanol.50
Enhancing the surface roughness of Cu. Roughened Cu electrodes have a high density of exposed surface grain boundaries, such as steps, edges, and defects. These grain boundaries are commonly considered as the active sites for ECO2R.79–82 Wang et al. utilized a facile molten salt decomposition method to prepare a nanostructured Cu@Cu2(OH)3NO3 electrode. Cu2(OH)3NO3 is converted into metallic Cu during CO2 electroreduction, leaving abundant defects on the dendritic rough surface. Benefiting from the highly rough surface, this electrode achieved a high selectivity for C2H4 production with a FE of 31.80%.81 The concave octahedral Cu2O catalyst with the abundant active sites derived from the in situ-generated crystal defects/grain boundaries were suggested to account for the improved C–C coupling during the ECO2R.82
Different shapes or textures of the micro- or nano-sized Cu catalysts present different types of grain boundaries. A leaf-like CuO nanosheet fabricated on nitrogen doped graphene displayed a high-curvature structure and multiple distinguished grain boundaries to enhance the selectivity to C2H4 with a high FE of 30%.83 Porous copper nanospheres with higher specific surface area and richer pores were designed. Such a porous structure greatly improved the C–C coupling process by enriching *CO intermediates in the pore structure. As a result, a high C2 selectivity with a FE of 57.22% was achieved, about 2.5-fold than the compact Cu.84
The plasma treated Cu foil was efficient to create a rough surface with numerous undercoordinated sites that bound CO preferentially.85 Roughened surfaces also contained square sites with neighbouring step sites, which adsorbed the precursor *OC–COH for C2+ products. As a result, the FE for C2 products (C2H4 and C2H5OH) increased from ∼30% to 55%. More importantly, the increased roughening of the Cu surface increased the ratio of current densities for C2+ to C1 products up to a maximum value of ∼9 for a surface roughness factor of ∼3.
Oxide-derived Cu catalysts. In reality, Cu electrodes prepared by electrodeposition in aqueous solution always have an oxidized outer layer which was reported to reduce the over-potential for the formation of carbonaceous products. Therefore, there has been intense interests in the role that oxide-derived (OD) Cu may have had in achieving enhanced ECO2R performance.86–92 Similar to the metallic Cu electrode, OD-Cu generally show a wide C1–C3 product distribution from ECO2R, but seem to be more preferable in the production of C2+ products.89,90 Cuprous oxide (Cu2O) is widely proposed to be a more efficient OD-Cu for CH3OH production, due to its moderate binding energy to methoxy (CH3O*) adsorbates (key intermediates for CH3OH formation).62 Theoretical studies showed that the intermediate CH3O* can transform to CH4 or CH3OH depending on whether the interacting surface is metallic Cu or oxidized Cu.93 Chang et al.94 proposed that the key factor was the availability of surface oxygen vacancies, particularly those formed by the reduction of Cu4O3 surface, which could stabilize the CO dimer. However it is important to note that Cu oxides can be fully reduced to metallic Cu at less-negative potential than ECO2R.64,88,95 This has led some to state that the active phase for ECO2R is metallic Cu.89,96,97 For instance, Cu oxides were fully reduced to metallic Cu during ECO2R, which showed even better C2+ selectivity compared to the untreated counterparts, suggesting the C2+ selectivities did not link to the specific oxidation states of Cu.98 However, this has been disputed as some studies demonstrated the existence of subsurface oxygen of OD-Cu during ECO2R, confirmed by 18O isotope labelling,99in situ surface-enhanced Raman spectroscopy,88 and in situ ambient pressure X-ray photoelectron spectroscopy.100 The residual subsurface oxygen was proposed to enhance the binding energy and coverage of *CO100 and thus raise the possibility of CO dimerization.101 Also, a higher local pH offering a substantial amount of OH groups was assumed to facilitate the stabilization of the residual oxygen.99,102
Alloying and compositing Cu with other components. Alloying Cu with other metals is a widely studied approach to reduce the reaction overpotential and adjust the product distribution. Alloying modifies the surface lattice strain that affects the intermediates adsorption energies.103,104 In addition, the second metal generates high abundance of CO which is subsequently spilled over to Cu sites that are active for the C–C coupling.105,106 To date, reports increasingly see alloy catalysts to show better selectivity of C2+ from ECO2R than Cu alone, and these all use Cu-rich alloys (or metallic composites) containing another CO-selective metal, such as Au,107–111 Ag,27,105,106,112–114 and Zn.115–119 Alloying Cu with a CO producing metal is considered to deliver synergetic effects derived by the availability of C–C coupling sites (Cu) to CO-producing atoms (e.g. Au, Ag, and Zn).120 Compared to the ∼55% FE with Cu2O electrode, C2–C3 with up to 65% FE were produced by using a Ag–Cu2O bimetallic electrode, with the greatest selectivity towards C2H4.113 The mechanism of improved C2 activity was proposed to be due to the expanded Cu–Ag distances compared to metallic Cu–Cu distances, which changed the binding energies of adsorbates and intermediates, thus favouring the formation of C2+ products. A controlled selectivity of C2H5OH production from ECO2R was achieved by varying the elemental arrangement of Ag and Cu in a Ag–Cu2O composite.68 More specifically, the FE of C2H5OH increased from 20% to 35% with the enhanced surface atomic composition of Ag from 0.12 to 0.22. A CO-insertion mechanism was hypothesised, which is briefly CO reacting with *CH2 species on the Ag–Cu biphasic boundary. Phase-segregated Ag/Cu composites obtained an enhancement in the FE for C2H5OH by 2.3-fold and in the partial current density for CO2 reduction by 5-fold compared with the pure Cu counterpart.106 Results also indicated C2H5OH production scaled with the amount of CO evolved from Ag sites and the abundance of Cu–Ag boundaries.
Zn is also known for its high selectivity to CO, therefore CO intermediates could first be generated at its Zn sites, which would then be transferred to adjacent Cu sites for further reduction into hydrocarbon products. B-doped Cu–Zn gas diffusion electrodes exhibited excellent selectivity for C2+ (C2H4, C2H5OH, C3H7OH) with a FE value of 70% at −0.54 V vs. RHE.118 The close proximity of Cu and Zn atoms on the surface of the catalyst was found to facilitate both stabilization of the CO* intermediate and its transfer from Zn atoms to their neighboured Cu for further dimerization and protonation.121 Similarly, hierarchically porous Cu/Zn alloy catalysts also showed a higher selectivity towards C2 products. The FE of C2H5OH reached 46.6% and the total FE of liquid C2 products closed to 60% at −0.8 V vs. RHE. The Cu/Zn catalysts promoted the adsorption of CO2 and simultaneously suppressed adsorption of protons. The tendency of C–C coupling could be controlled by the Zn content in Cu/Zn alloys.117
Non-metals as components in Cu composites also have shown an ability to promote C–C coupling from ECO2R. Sulphur, an alternative chalcogen to oxygen, has been reported to be an efficient promoter for C2H5OH formation by a Cu2S–Cu core–shell catalyst containing high levels of Cu vacancies.122 The synergy between subsurface sulphur atoms and copper vacancy defects shifts the product preference to C2H5OH with suppressed C2H4 production. Boron-doped CuO nanobundles designed for CO2 reduction have abundant oxygen vacancies, the increased exposure of accessible active sites and the CO2 adsorption capacity facilitated to the C2H4 production with the FE of 58.4%.123
2.2 Molecular catalysts
Small molecular electrocatalysts provide well defined active sites that can be synthetically tuned, offering a pathway to control the mechanisms of inner-sphere electron transfer between the catalyst and substrate that occur following initial reduction(s) of the catalyst at the electrode surface. Therefore, molecular electrocatalysts may provide access to lower overpotentials and improved selectivity towards the desired products.128 Indeed, turn-over frequencies in excess 106 per catalyst at overpotentials as low as 0.2 V have now been reported129 for the reduction of CO2 to CO in aprotic solvents and the field continues to develop rapidly. However, it is striking that the vast majority of studies using molecular electrocatalysts only report the formation of the simplest C1 products (CO, HCOOH), formed by the 2-electron, 2-proton-reduction of CO2 and progress towards these products is covered in multiple recent reviews.128,130,131 This can be readily understood as the majority of molecular electrocatalysts for CO2 reduction contain a single metal site, with limited coordination flexibility, preventing either accumulation or stabilization of initially reduced C1 species to enable subsequent C–C bond formation. Despite these challenges there are examples of catalysts that achieve C–C coupling. Two recent comprehensive reviews include molecular catalysis towards >2e− products and have extensive coverage on many of the advances to date.132,133 The highest performing of these are covered in this section. The relatively small number of C2 producing molecular catalysts can be broadly classed into 3 categories, each of which has a distinct approach to enabling C–C bond formation; (i) those that produce oxalate, typically via the use of catalysts with multiple CO2 binding sites, (ii) Ru based catalysts that stabilize reduced carbon intermediates and (iii) heterogenized molecular electrocatalysts.Oxalate (C2O42−) is arguably the simplest C2 product and its formation during the reduction of CO2 has been reported to occur with high FEs on relatively inert metal electrodes such as Hg and Pb in aprotic solvents at very negative potentials.128 There, the direct 1-electron reduction of CO2 leads to the formation of CO2˙− which dimerizes. Several molecular catalysts have now been reported to facilitate oxalate formation at greatly reduced overpotentials. In early works, Tanaka and colleagues described the production of oxalate using an Ir catalyst containing three metal centres in acetonitrile.134 Infrared spectroscopy was used to follow the mechanism allowing the authors to show that in contrast to the behaviour of CO2 on metal electrodes, free CO2˙− was not formed. Instead coupling of two reduced, bound CO2 molecules within the catalyst was proposed to occur. A common feature of many oxalate producing catalysts134–136 is that they consist of multiple metal centres which may be envisaged to provide an advantageous mechanism for C–C bond formation. However, intriguingly experiments on both bi- and monometallic Ru complexes demonstrated that in the absence of water both could produce C2O4.129–131,134–136 Despite only having a single metal centre, mechanistic studies clearly demonstrated again that free CO2˙− was not formed and instead it was proposed that reversible ligand loss could occur to enable binding of a second CO2 molecule to the same metal atom. Of particular note is a more recent report of binuclear Cu complex that was shown to produced oxalate at very low overpotentials with >96% selectivity, which remarkably could even reduce CO2 at the concentrations found in air, as shown in Fig. 2.135
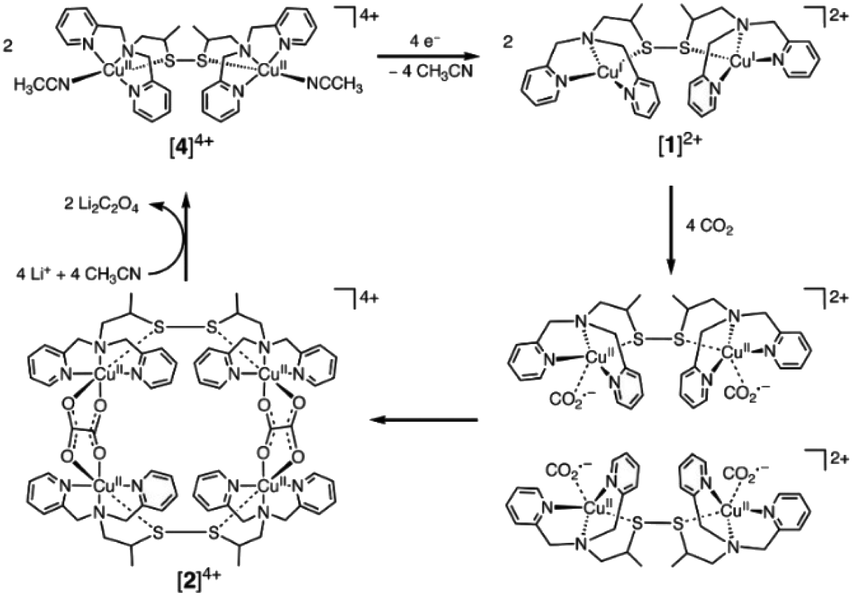 | ||
| Fig. 2 Multimetallic Cu complexes have been shown to enable C–C bond formation through the coupling of multiple bound CO2˙− species within the cluster giving rise to very low overpotentials and high selectivity to oxalate formation even with air as the CO2 source.135 Reproduced from ref. 135 with permission from American Association for the Advancement of Science, copyright 2010. | ||
A series of papers from Tanaka and colleagues used Ru based polypridyl electrocatalysts for the production of a wide range of complex multicarbon products.137–142 Early studies on the chemical, and electrochemical reduction of [Ru(bpy)(trpy)(CO)2]2+ in the presence of CO2 showed marked temperature dependencies. At low temperature (−20 °C) in C2H5OH/H2O mixes CO2 electrolysis led to the formation of a range of products including formate, glycolic acid (HOCH2CO2H) and 3-oxopropanoic acid with isotopic labelling demonstrating that both carbons were derived from CO2.137 In contrast, at room temperature CO and formate were the dominant products. The ability of the catalyst to generate C–C bonds at low temperature was proposed to be due to the stability of a [Ru(bpy)(trpy)CO]2+ intermediate, allowing for further reduction and the formation of [Ru(bpy)(trpy)(CHO)]+, which could then be reduced and interact with a CO2 molecule bound at the vacant site on the Ru centre. On the other hand, at room temperature CO loss could compete preventing the formation of the key formyl intermediate.68,142 However, it was noted that even at low temperature formate production dominated as the strong hydride donating ability of the formyl complex led to the direct reaction of CO2 and loss of formate. Further studies by the team using naphthyridine ligands that can stabilize Ru–CO intermediates to reductive CO loss have since achieved C–C formation with high yields and selectivity even at room temperature, however, in these cases only one carbon is shown to come from CO2 with the 2nd proposed to come from the supporting tetramethyl ammonium cation of the electrolyte via a methylation reaction.138–142 Nonetheless, the approach clearly demonstrates that through careful ligand design it is possible to stabilize the initial C1 products at a molecular catalyst to allow for C–C bond formation.
The studies of molecular catalysts described so far either use aprotic or organic/water mixes at low temperature to control the stability of key intermediates or, in the case of oxalate formation, dry solvents. However, it is widely recognised that for electrocatalytic CO2 reduction to become viable at the scale it will need to be coupled to water oxidation, making the discovery of catalysts that can operate in aqueous conditions highly desirable.143 Of particular interest is the reported electrocatalytic reduction of CO2 to form C2 and C3 products in aqueous electrolytes. Early studies using metal porphyrin and phthalocyanine (e.g. M-5,10,15,20-tetrakis-(4-methoxyphenyl)porphyrinato Co(II) and Cu(II))144 complexes immobilised in a polymer matrix on electrode surfaces primarily yielded C1 products but traces of C2 products144,145 including C2H6 and C2H4, however as will be discussed below in the following section more careful in situ analysis is required to confirm whether the transition metal complex is the true catalytic centre. Work by Ogura and colleagues also explored the use of polymer-coated electrodes with multiple potential catalytic centers and CO2 binding sites for C2–C3 formation in water. Initial experiments with films containing both iron cyanide complexes and a cobalt co-catalyst in polyaniline (PANI) demonstrated the formation of CO2 reduction products including C2H5OH and C3H6O3, although FEs were very low (<2% for each).146 Further studies showed that by replacing the cobalt catalyst with a 2nd iron(II) complex FEs in excess of 10% could be achieved for CO2 reduction to C3H6O3 at a pH of 3.147 Subsequent mechanistic studies highlighted the important role of the PANI in CO2 accumulation close to the catalyst centres thus enabling C–C bond formation.148,149 It is therefore interesting to note that the majority of studies now use PTFE based polymers, which cannot facilitate such an interaction with CO2 to aid catalyst adhesion to the electroactive support. We propose that a promising, simple avenue that should be explored by the community is the potentially beneficial role of the use of a “non-innocent” supporting polymer such as PANI.
Immobilisation of molecular catalysts onto electrodes continues to be a common factor in studies where the electrocatalytic reduction of CO2 to C2 occurs, however careful attention must be paid to the true nature of the active site. One study used a copper-porphyrin complex with pendant hydroxyl groups (PorCu) heterogenized on a carbon paper electrode, Fig. 3, to produce ethylene at a high partial current density (8.4 mA cm−2, ∼18% FE) in addition to CH4, CO and H2 at −0.976 V vs. RHE in KHCO3 electrolyte.150 The catalytic current and activity towards C2H4 production increased with experiment time, so a series of ex situ control experiments (absorption/fluorescence spectroscopy, mass spectroscopy, TEM, XPS) were performed to test the nature of the active catalyst. Whilst these found no evidence for degradation ex situ, a follow-up study by the same group151 utilised in situ and operando X-ray adsorption spectroscopy (XAS) to investigate three copper based molecular catalysts for ECO2R and noted that at −1.06 V vs. RHE the Cu(0) state was dominant. Extended X-ray adsorption fine structure (EXAFS) demonstrated the appearance of a bond peak at 2.2 Å, consistent with Cu–Cu bonds in metallic copper under reducing conditions. Interestingly the initial Cu(II) state was regenerated upon switching back to the open circuit potential, demonstrating reversible metal nanoparticle formation from the molecular Cu complex. As reversible nanoparticle formation has now been confirmed to occur under reducing conditions for a range of catalysts including PorCu, it seems likely that similar behavior was also occurring in the early studies where trace C2 products were formed using Cu based molecular catalysts in polymer supports144 and also these more recent studies. Indeed a dinuclear copper tris(2-benzimidazolylmethyl)amine (Cu2(NTB)) complex which shows amongst the highest FE for C2H4 (42% at −1.28 V vs. RHE) has also been recently reported whilst also showing similar reversible Cu nanoparticle formation, with the metallic nanoparticle the likely active catalyst.152
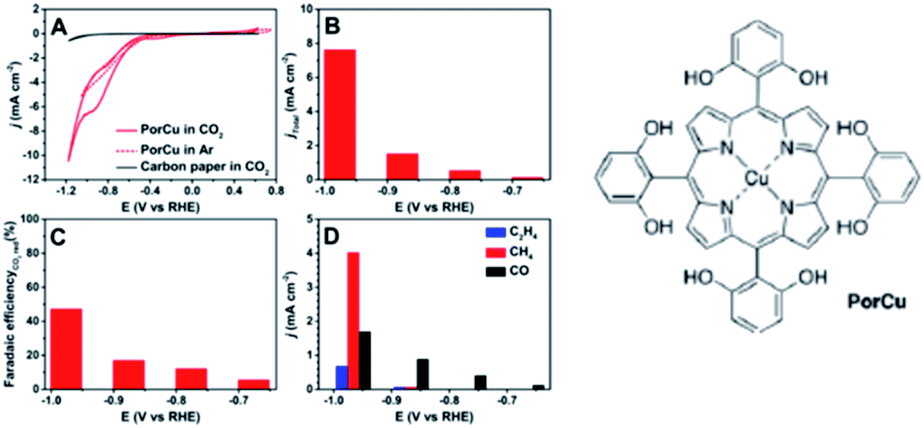 | ||
| Fig. 3 Electrocatalytic activity of a copper-porphyrin complex (PorCu) with pendant hydroxyl pendant hydroxyl groups heterogenized on a carbon paper electrode: (A) CV curves, (B) total curent densities, (C) FE, and (D) partial curent densities of gaseous products at various potentials.150 Reproduced from ref. 150 with permission from American Chemistry Society, copyright 2016. The catalyst was later found to reversibly reconstruct into Cu nanoparticles, which provide the active site for ethylene formation.151 | ||
There is evidence that both reversible and irreversible metal nanoparticle formation also occurs also with some Cu based covalent frameworks153 and metal–organic frameworks154,155 and even with Cu doped carbon structures. For example, one study investigated a copper/nitrogen doped carbon network (Cu0.5NC) and found it to be particularly active for C2H5OH formation, yielding 55% in 0.1 M CsHCO3 at −1.2 V vs. RHE.156 XANES and EXAFS were again employed to examine the catalytically active species throughout electrochemical reduction. The operando XANES analysis showed a change in oxidation state from Cu(II) to Cu(0) whilst the EXAFS data showed the appearance of Cu–Cu bonds, consistent with the formation of copper nanoparticles. The activity of this catalyst is highly comparable to an engineered copper nanoparticle/N-doped graphene catalyst, which also produced ethanol at 63% FE at −1.2 V vs. RHE,157 providing further evidence that the active species in the catalyst is metallic copper.
However some catalysts, such as a self-assembled cuprous coordination polymer (Cu-SCP), are reported to be stable towards the formation of metallic Cu(0) nanostructures.153 The persistence of the Cu(I) oxidation state throughout catalysis is proposed to be responsible for superior C2 production, which reached a maximum of 33.5% for C2H5OH and 19.7% for C2H4. The hydrophobicity of the ligand was increased by adding phenyl groups, which was proposed as a strategy for suppressing the HER whilst increasing the production of the crucial *CO intermediate. XAS measurements pre and post electrolysis show no change in state of the copper, however a lack of operando measurements makes it difficult to confirm that metal nanoparticle formation isn't occurring reversibly under catalytic conditions.
Overall, the number of studies of transition metal complexes that act either as catalysts or pre-catalysts for ECO2R to C2+ products is relatively low when compared to the level of research on metallic Cu electrodes. However, there is a large number of transition metal complexes known to be able to reduce CO2 to CO and a recently reported new approach to C2+ production is to immobilize these on Cu surface.50 The aim is to produce CO, a crucial intermediate for C2+ products, at high turnover frequency and selectivity using the molecular catalyst, prior to CO reduction on Cu. This tandem approach is analogous to that described in Section 2.1.1 that explores the combining of copper with CO producing metals. The only example of a tandem system using a molecular catalyst is reported by Sargent et al.,50 in which a copper surface is functionalized with 5,10,15,20-tetraphenyl-21H,23H-porphine iron(III) chloride (FeTPP[Cl]), a well-established CO2 to CO molecular catalyst.158 A peak FE for C2H5OH of 41% was reached at −0.82 V vs. RHE, significantly out-performing bare Cu at 29% FE. The partial current density was 124 mA cm−2. In situ XAS showed the FeTPP to be stable against metallic Fe nanocluster formation under electrochemical conditions.
More widely, surface modifications of Cu electrodes with molecular species have shown success in promoting C2+ selectivity. Glycine has demonstrated impressive enhancement of C2H4 and C2H6 production on copper nanowires (Cu NW) across the entire studied potential range (−0.65 to −1.25 V vs. RHE), doubling their FE at −1.25 V to 35%.59 A noteworthy limitation of the catalyst is the stability of the adsorbed glycine, which will detach at potentials more negative than the studied range. A range of morphologies was investigated with all showing an enhancement in the presence of the glycine adsorbates. DFT calculations reveal stabilization of the *CHO intermediate, key to the formation of hydrocarbons, by glycine's –NH3+. N,N′-Ethylene-phenanthrolinium dibromide has been utilized to synthesize and stabilize nanostructured copper from planar polycrystalline foil, shifting the selectivity to C2+ products including C2H4 (FE = 45%) and C2H5OH (FE = 15%) at moderate potential (−1.05 V vs. RHE).159 The additive first corrodes the surface of the foil to form the nanostructure and then dimerises under applied potential to form a film which was shown to stabilize the structure for over 40 hours. Altering the C2 pathway to preferentially form C2H5OH over C2H4 has been explored using a nitrogen-doped carbon layer coated onto copper.160 DFT studies give evidence towards to simultaneous improvement in C–C coupling and suppression of deoxygenation of the fork-in-the-road HOCCH* intermediate which is a necessary step for the formation of C2H4. C2H5OH is selectively produced at 52% FE at, crucially, an industrially relevant partial current density of 156 mA cm−2 at −0.68 V vs. RHE.
2.3 Photoelectrodes and photocatalysts
Indirect solar CO2 reduction, where a photovoltaic is used to enable dark electrocatalysis with metal electrodes, is increasingly reported, but is not discussed here as the achieved product distribution invariably aligns with that achieved with the electrode material used conventionally. Instead, we briefly examine the reduction of CO2 to C2+ products using either semiconductor photocatalysts or photoelectrodes that generate suitably reducing photoelectrons to enable CO2 reduction at a catalytic site.Perhaps surprisingly, given the need to accumulate very large numbers of photoelectrons at a catalytic site (e.g. the production of ethane from CO2 is a 14-electrons process), and the typically low quantum yields of most photocatalysts coupled to the available solar flux, examples of materials that convert CO2 to C2+ products do exist. These are included within a comprehensive recent review on CO2 photoelectrodes and photocatalysts,161–166 where it is noted that when metal co-catalysts are deposited on the light absorbing semiconductor the achieved selectivity often matches that expected for the material with Cu co-catalysts often giving rise to a distribution of products including ethane and other C2 molecules. Highlights of Cu catalyzed systems include reports of p-Si nanowires with Cu deposited on the surface for the production a range of C2–C4 products,167 PbS sensitized TiO2 particles with Cu co-catalysts168 that could produce C2H6 in the presence of CO2 and bimetallic alloys of Pt/Cu on TiO2 for the production of C2H6 and C2H4.169
Cu containing photoelectrodes have also been reported to produce complex carbon products without the need for an additional co-catalyst, likely due to the availability of active Cu sites on the electrode surface.170–173 Selected examples include one-dimensional Cu2O@Cu metal–semiconductor heterostructured nanorods that produced C2H4.172 The reduction of CO2 to acetic on CuO photoelectrodes where the formation of thin surface films of Cu–In that inhibited H2 evolution and promoted C2H5OH production.174 Also a study where very high selectivity's towards C2H5OH was reported using a CuO/Cu2O photocathode with a micro-flow reactor which appeared to have an essential role in controlling pH and local CO2 concentration.175
Several studies do exist where non-Cu based co-catalysts have been used.176–179 In two reports180,181 Pd–TiO2 was shown to produce ethane under CO2 and in the presence of water, particularly at low temperature.180 Plasmonic Au nanoparticles are also proposed to be able to act as both the light absorbing and catalyst centre for C2H6 production from CO2,182 and although not containing a C–C bond, methyl formate production via the photoelectrochemical reduction of CO2 at InP electrodes can occur.183
Although both electro- and photocatalytic CO2 reduction offer efficient techniques for C2 products conversion, the reduction of CO2 to C2+ products, such as C3 are rarely reported.184 Moreover, the long-term stability of the widely used transition metal-based catalysts is still under investigation.185 Biological CO2 conversion as a cost-effective approach brings new opportunity to generate more complex chemical compounds than purely electrochemical methods. The excellent stability of bio-catalysts and the high coulombic efficiency of CO2 conversion make it a potentially scalable technology for CO2 utilization in the circular bioeconomy and high-value biofuel generation (Table 2).185,186
| Product | Catalyst | Potential (V vs. Ag/AgCl) | Current density (mA cm−2) | Production rate (g m−2 h−1) | FE (%) | Operation time (h) | Ref. |
|---|---|---|---|---|---|---|---|
| a Coulombic efficiency. | |||||||
| Acetate | Mixed microbial consortium | −1.3 | 20 | 55.4 | 99 | 1680 | 187 |
| Mo8@Cu | ∼−1.85 | 110 | N.G. | 48.7 | 3 | 188 | |
| Ethanol | Clostridium ljungdahlii ERI-2 | −0.78 | 0.19 | 0.25 | 66.1 | 168 | 189 |
| B, N-codoped nano diamond | ∼−1.65 | N.G. | 7.2 × 10−5 | 93.2 | 3 | 40 | |
| Mixed alcohols | Mixed microbial consortium | −1.0 | ∼1.0 | 0.15 | 49.5: ethanol, butanol and organic acid | 2160 | 190 |
| Mesoporous metallic Cu | ∼−1.65 | 18.5 | N.G. | 35: ethanol and propanol | 1 | 21 | |
2.4 Biological catalysts
In order to develop a robust process at a larger scale, mixed microbial communities will be preferred as they are more robust and versatile, but also easier to handle. Besides, mixed microbial communities can be directly enriched from environmental samples such as activated or digested sludge, anaerobic digester effluents or river sediments.199–203 In a controlled environment such as in Bio-Electrochemical Systems (BES) and the presence of methane inhibitor, the enrichment of acetogenic bacteria from environmental inocula was very often reported, which is consistent with the production of Volatile Fatty Acids (VFAs) and alcohols. For example, it was reported in several studies that organisms belonging to the genus Clostridium and Acetobacter dominated both the biofilm and the planktonic microbial communities of a BES producing VFAs and alcohols from CO2 conversion.198,204,205 The presence of Desulfovibrio species was also reported.206–209 This could be explained by their implication in biological hydrogen production in biocathodes and would thus support the thesis of a hydrogen-mediated mechanism.209–211 In our latest study, the activated sludge from a wastewater treatment plant was used as inoculum for MES which was poised potential of −1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V vs. Ag/AgCl and provided only gaseous CO2. A dense biofilm dominated by Acetobacterium (ca. 50% of biofilm) was gradually formed to produce a higher yield of acetate (106.9 ± 10.5 mM). The sharp drop of charge transfer resistance within the biofilm highlighted the advantage of mixed communities. In addition, the biofilm maintained a highly active performance during long-term operation even with the stress associated with the low pH as a result of accumulation of acidic products.204 Changing the operation from batch to continuous mode (hydraulic retention time of 3 days) further promoted the acetate production as it provided better control of pH and constant medium refreshment.212
V vs. Ag/AgCl and provided only gaseous CO2. A dense biofilm dominated by Acetobacterium (ca. 50% of biofilm) was gradually formed to produce a higher yield of acetate (106.9 ± 10.5 mM). The sharp drop of charge transfer resistance within the biofilm highlighted the advantage of mixed communities. In addition, the biofilm maintained a highly active performance during long-term operation even with the stress associated with the low pH as a result of accumulation of acidic products.204 Changing the operation from batch to continuous mode (hydraulic retention time of 3 days) further promoted the acetate production as it provided better control of pH and constant medium refreshment.212
| CO2 + 7H+ + 8e− → CH3COO− + 2H2O; E0 = −280 mV vs. SHE | (1) |
It is well understood that potential losses occur in BES. These losses are related to mass transport and kinetics limitations between the medium, the electrode and within the biofilm (when applicable), but also to ohmic voltage losses associated with the electrolytes, the membrane, the electrodes and connections. Therefore, it has been postulated that in order to overcome potential losses due to the components of BES, microbial acetogenesis requires potentials of −400 mV or much more negative.204,235–237
In order for the technology to be viable, reaching high productivity and selectivity is crucial. Therefore, different strategies have been investigated to increase the acetate production rate, ultimately limited by the applied current. For example, the continuous production and extraction of acetate were achieved in a 3-compartment microbial electrolysis cell (applied current of −50 mA) reaching an accumulated acetate concentration of 13.5 g L−1 (225 mM).208 In this design, the middle chamber was used for the continuous extraction of acetate which led to the high titer achieved. Electrode materials have also been subject to investigation with the utilization of reticulated vitreous carbon (RVC) modified with multi-walled carbon nanotubes (MWCN) by electrophoretic deposition (EPD).238 In this reactor polarized at −0.85 V vs. SHE, a high acetate production rate of 685 ± 30 g m−2 day−1 was achieved (product titer of 11 g L−1), which is according to the authors comparable to productivity levels of industrial fermentation processes.239 However, this experiment was carried out at a very small scale (electrode projected surface area of 1.36 cm2), and extrapolating such results is not straightforward despite the high productivity obtained. In addition, the viability of such electrode materials at a larger/industrial scale should be evaluated by economic and life cycle assessment analysis.
Although, as an end-product, it does not have a high market value (ca. 500€ per t),239 acetate and more specifically acetyl-CoA are excellent building blocks for the production of more valuable chemicals through the W–L pathway.193,240–242 Therefore, under specific conditions of pH and hydraulic retention time and following mechanisms derived from the W–L pathway, butyrate, ethanol, or butanol can be all produced from acetyl-CoA as a precursor (see Fig. 4a). Ethanol is an important biofuel and can be used as an additive to diesel. Butanol is an important industrial bulk as well as a promising biofuel as it can be used without modifications of car engines.236,243,244 In the W–L pathway, acetate is the prerequisite for any other VFA or alcohol, following which butyrate can be produced either via a linear extension of the acetyl-CoA to butyril-CoA, or via reverse β-oxidation which is also called microbial chain elongation (Fig. 4b).234
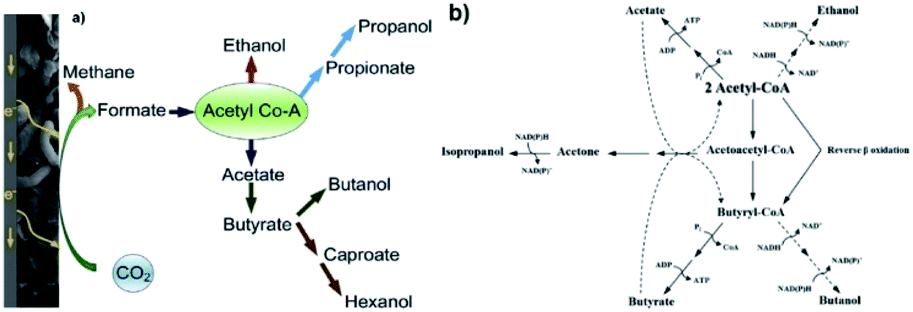 | ||
| Fig. 4 (a) Possible metabolic pathways for microbial electrochemical CO2 reduction that leads to the generation of a variety of high value organic compounds beyond acetic acid and CH4.224 Reproduced from ref. 224 with permission from Elsevier, copyright 2019. (b) Schematic diagram of the suggested metabolic pathways for the production of acetate, ethanol, acetone, isopropanol, butyrate and butanol from acetyl-CoA. In dashed lines are the pathways for solventogenesis after the production of acetate and butyrate.245 Reproduced from ref. 245 with permission from Elsevier, copyright 2012. | ||
As acetate and butyrate concentrations increase in the medium, pH will naturally drop creating harmful conditions for microorganisms. As a defensive mechanism, a switch of mechanism from acidogenesis (production of fatty acids) to solventogenesis (production of solvents, typically alcohols) can occur, leading to the production of butanol and ethanol.246 Concomitant of the production of butanol and ethanol, acetone can also be produced via acetone–butanol–ethanol (ABE) fermentation, which can be further reduced to iso-propanol via isopropanol–butanol–ethanol (IBE) fermentation.246 This mechanism, however, has rarely been reported in bioelectrochemical systems. During MES, operating parameters such as applied potential, pH or hydraulic retention time (HRT) have a crucial impact on the bioproduction. In a continuous reactor operated at −50 mA (cathode potential of −1.0 ± 0.6 V vs. SHE), it was shown that the bioproduction could be driven either towards acetate or towards product diversification by controlling the HRT and thus the pH.209 At HRT of 3.3 days, a production rate of 21 g mcathode−2 d−1 could be achieved for acetate with very low product diversification. Higher HRT of 5 days enhanced the production of butyrate and isopropanol as the pH would drop to 5, with maximum concentrations of 0.67 and 0.82 g L−1, respectively. In another semi-batch reactor operated for 450 days at an applied cathode potential of −0.80 V vs. SHE and with controlled pH ca. 5, the accumulation of acetate up to 4.9 g L−1 also led to the production of ethanol (up to 1.3 g L−1).209 As observed in other studies, the presence of both acetate and ethanol led to both solventogenesis and chain elongation. Therefore, high butyrate and isobutyrate titers of 3.1 and 1.6 g L−1 were measured. Caproic acid was also detected, reaching a maximum concentration of 1.2 g L−1 after 450 days of operation. Equivalent alcohols were produced: in addition to ethanol, butanol, isobutanol and hexanol were measured as high as 0.4, 0.2 and 0.2 g L−1, respectively.206 More recently, the system was adapted to a 3-chamber MES rector equipped with a dual biocathode arrangement for sequential acetogenesis/carbon chain elongation and solventogenesis, as presented in Fig. 5.206 In this study, the pH of both biocathode chambers were controlled without the addition of any chemicals but CO2 sparging and by controlling the applied potential and the abiotic reactions occurring at each cathode. In the first chamber (pH 6.9), acetate could be produced at a maximum concentration of 9.5 g L−1 (0.46 g L−1 day−1). As acetate would migrate to the second chamber (pH 4.9), solventogenesis would occur and ethanol would be produced. Very interestingly, after a connection was made between the headspaces of both cathodic chambers, a transfer of ethanol from the second chamber to the first chamber via gas stripping promoted chain elongation in the first chamber. C4+ products were then detected with levels up to 1.33 g L−1 isobutyrate, 1.49 g L−1 butyrate and 0.27 g L−1 caproate. Acidic conditions in the second chamber resulted in the biosynthesis of C4–C6 alcohols, with levels up to 0.33 g L−1 isobutanol, 0.82 g L−1 butanol and 0.11 g L−1 hexanol.247
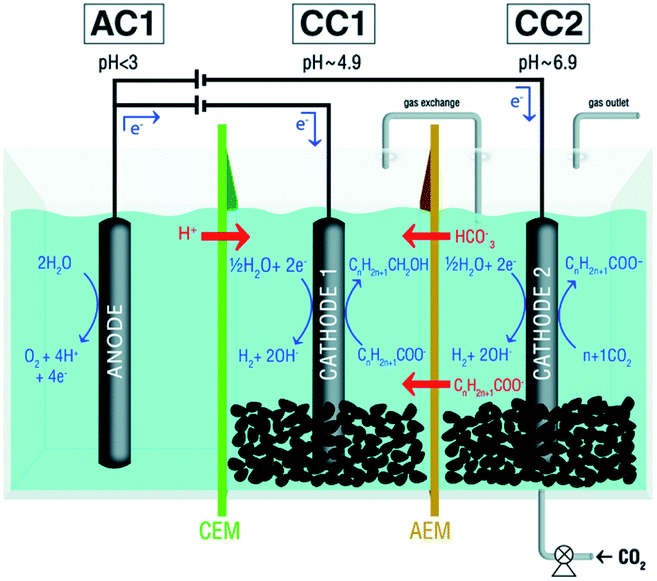 | ||
| Fig. 5 Schematic representation of a microbial electrosynthesis reactor with dual biocathode for the production of organic acids at neutral pH and simultaneous reduction of the produced organic acids to the corresponding alcohols at mildly acidic pH. CEM: cation-exchange membrane; AEM: anion exchange membrane.247 Reproduced from ref. 247 with permission from the Royal Society of Chemistry, copyright 2019. | ||
It is assumed that additional electron donors than a polarized cathode was required to steer the production towards longer chain of carboxylates than acetate.248 Based on our latest results, formate as additional electron donors were supplied into MES in which cathode was poised at −1.0 V vs. Ag/AgCl. Although acetate was still the major product, significant increase in the production of butyrate (3.8 times higher in maximum concentration) and butanol (maximum of 6.8 ± 0.3 mmol C L−1) was observed after supplying formate. However, the production of acetate and longer chain carboxylic acids ethanol stopped at open circuit condition. By comparison, ethanol as another one electron donor could trigger the C4 compounds production without polarized cathode.237
3. Reactor design
3.1 Reactor configurations and process integration
The design of a CO2 electrochemical reduction device requires multi-disciplinary efforts. Among the three sources of inefficiencies in ECO2R, which are kinetics, mass and charge transfer, potential losses caused by mass and charge transfer are closely determined by reactor configuration. The priority of device design is minimizing those energy losses as much as possible. To date, the most studied devices for ECO2R are membrane-based architectures. These are used in order to avoid cross over of products and the undesired consequent parasitic effects, e.g. ion-exchange membranes are employed to separate the catholyte and anolyte while maintaining the transfer of ion transfer across the membrane at the same time. According to the pH environment of electrolyte, cation-exchange or anion-exchange membranes can be used.In the studies of ECO2R at lab-scale, batch type reactors, shown in Fig. 6, are widely used in favor of precise control of reaction conditions and product characterization. In order to upscale the process, continuous flow configurations are designed to meet the demands of industrial applications as shown in Fig. 6. It is suggested by Delacourt et al. that a buffer layer is crucial to stabilize the pH of catholyte, and thus enhance the selective reduction of CO2 and prevent the evolution of hydrogen.249 However, due to the limited solubility of CO2 (1.5 g L−1 (ref. 250)) in aqueous solutions, flow cells fed with saturated CO2 electrolyte suffer from depletion of reagent showing a low mass transfer limiting current density at around 20 mA cm−2.251 Inspired by fuel cell configuration, the gas diffusion electrode (GDE) is integrated into the electrolyzer to enable direct gas CO2 feeding to overcome the mass transfer limitation of the reactant.252 By doing so, the operating current density reaches 102 to 103 mA cm−2.253–255 One can find a more detailed development of GDE for ECO2R as reported in a recent review.256 Besides, the rapid removal of reductive products along with CO2 stream lowers the issues arising due to ohmic loss caused by the bubble formation in the channel.
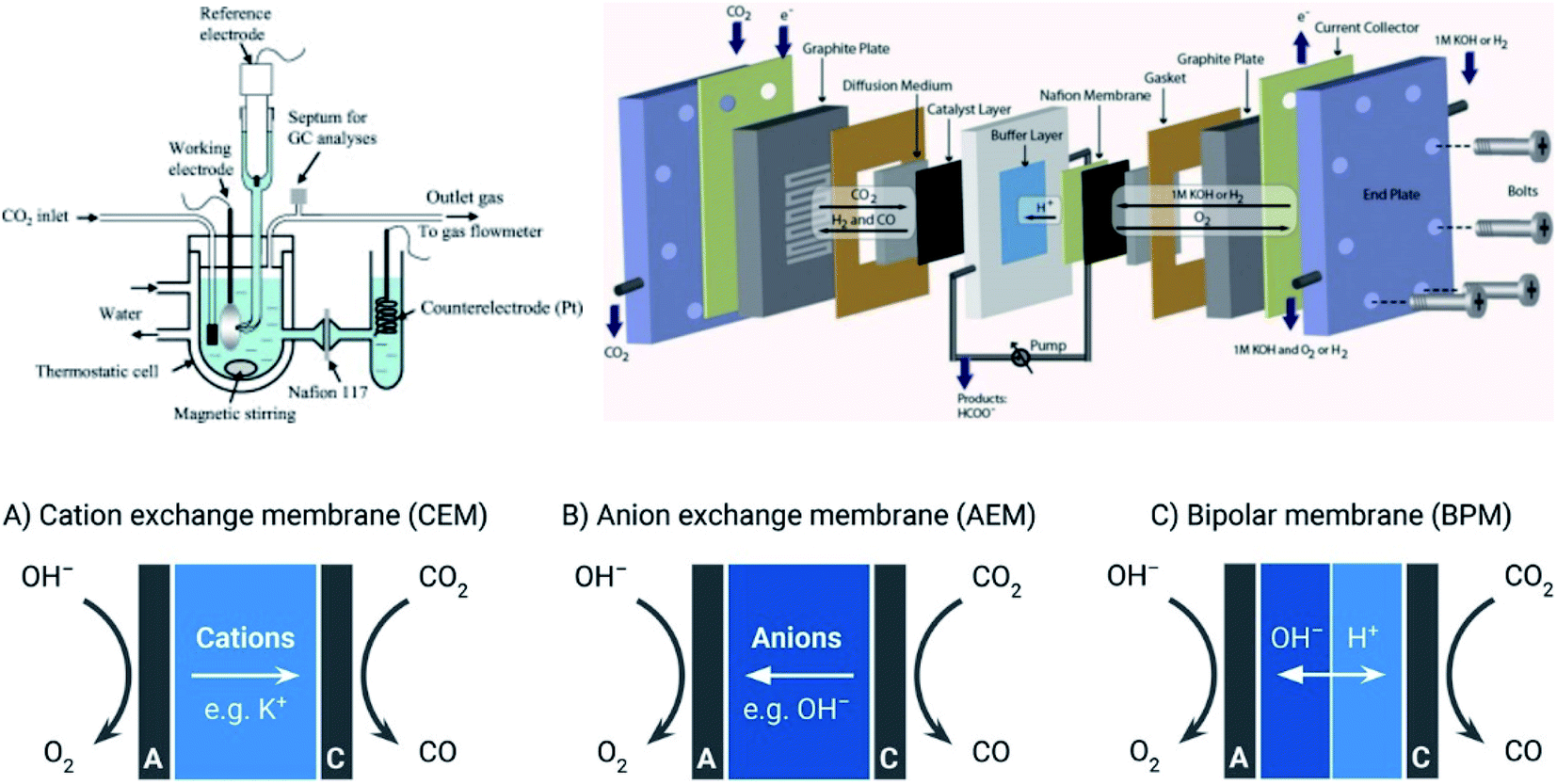 | ||
| Fig. 6 (Up) The schematic of batch and continuous flow reactor for CRR.273,274 Reproduced from ref. 273 and 274 with permission from IOP Publishing, copyright 2010 and 2013; (down) ion transport pathways in CEM (A), AEM (B), and BPM (C).261 Reproduced from ref. 261 with permission from American Chemical Society, copyright 2018. | ||
However, the use of mono cation or anion exchange membrane requires the consistency of the pH environment across the membrane. This brings a performance trade-off on anode and cathode because of the conflicting electrolyte requirements for desirable half-reactions.257,258 For example, the alkaline electrolyte is preferred for anodic oxygen evolution reaction (OER) to eliminate the use of catalysts based on precious metal, while there are still plenty of ECO2R attempted at a near-neutral catholyte. The bipolar membrane configuration enables a differential pH environment for each half-reaction, as shown in Fig. 6.259 The development of bipolar membranes with controlled local pH has been recently reported as one way to use the devices with the CEM in contact with the cathode for CO2 reduction260 although this configuration requires large membrane potential loss sometimes to drive ion transfer at the membrane, which can lead to the decline of energy efficiency.261 In addition, the loss of CO2 due to the reaction with OH− to form CO32− at the cathode usually needed a large excess of CO2 to support high current densities, which led to a lower carbon efficiency. BPM provided the possibility for CO32− protonation at the membrane–electrolyte interface to release CO2, reducing the losses imposed by CO32− formation.262 As an alternative to a physical membrane a microfluidic approach was implemented to introduce a thin laminar flow electrolyte layer served as a ‘virtual membrane’ to separate anode and cathode.263 High efficiency and precise control of operating conditions make microfluidics a powerful tool to boost the performance of ECO2R.264 By employing two GDEs, one as an anode and one a cathode, this configuration is able to utilize gaseous CO2 and transport byproduct O2 away. Performing ECO2R in a solid oxide electrolyzer cell (SOEC) under high temperature is more kinetically desired and energy efficient than room temperature membrane-based ECO2R configuration.265 Approximately a 73% energy efficiency and 95% FE was reported by Kaur et al.266 In addition, SOEC is capable of operating at high current density (up to 2 A cm−2), which is crucial for industrial scale application.267 More information about SOEC can be found in a recent review.268
One of the key concepts of carbon-neutral fuel generation by ECO2R is that electrical energy comes from renewable sources, like solar and wind energy.269 Kauffman et al. presented a lab-scale prototype of a solar cell powered CO2 electrochemical device.270 The configuration was provided a bias of 6 V by a solar panel and is able to generate 412 L h−1 CO with 96% selectivity for 12 h in two days. A much higher production rate of 781 L h−1 was detected by using a rechargeable 6 V battery which provides more stable and higher power density electricity. They also estimated the conversion capacity of CO2 is 1 tonne per acre per day and 1.6 tonnes per turbine per day for solar and wind plants, respectively. ECO2R based on a SOEC is also regarded as an efficient approach of energy storage for intermittent renewable energy with energy storage cost around 3 kW−1 h−1.271 The integrated system of ECO2R and following treatment sections like Fischer–Tropsch (F–T) was investigated to provide a technical and economic feasibility study of converting CO2 back into diesel fuel.272 It is suggested that this pathway to synthesize fuel can be economically competitive with fossil fuel.
3.2 Operating condition of ECO2R
Pressurizing CO2 has been proven an efficient way to promote the electrochemical performance and selectivity of hydrocarbons.251,275,276 It is considered as a method to improve the solubility and mass transfer of CO2 at the surface of the electrode.277 Besides, the reaction rate of ECO2R can also be accelerated through increasing the concentration of reagent. A desirable high carbon product selectivity is also obtained at pressurized conditions,278 which means the local concentration of CO2 plays a critical role in C2+ generation, as shown in Fig. 7. The study of Kas et al. indicated that the FE of C2 rises by about three times to 43.7% at a pressure of 9 atm.279 It is also suggested that high pressure can result in the polymerization of low carbon products.280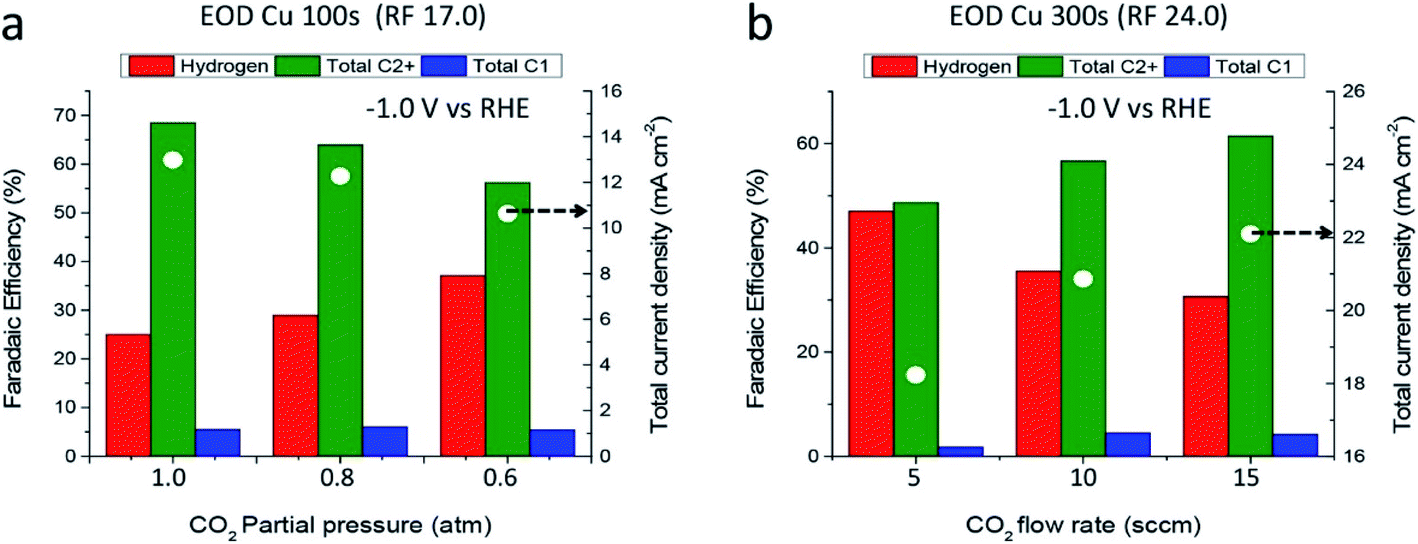 | ||
| Fig. 7 Products distribution and current performance with CO2 pressure (a) and flow rate (b).278 Reproduced from ref. 278 with permission from American Chemical Society, copyright 2017. | ||
The temperature effect on ECO2R depends on the type of electrolyte. In organic electrolytes, like CH3OH, a low temperature to maintain a higher solubility of CO2.281 In contrast, rising in temperature leads to performance improvement in aqueous electrolyte due to the improvement on kinetics, despite modest reductions in the solubility of CO2.38 Dufek et al. reported a rise and fall effect of temperature in current performance and product selectivity.282 The results from the study of Gutiérrez-Guerra et al. suggested that higher operating temperature is preferred for high carbon products generation.283
3.3 Electrolyte
As already shown in this review, the selection of electrolytes is a crucial factor for ECO2R. Product distribution is heavily affected by the properties of electrolyte e.g. pH, protic vs. aprotic and CO2 solubility. In addition, to conventional electrolytes, ionic liquids (ILs) have garnered much attention for ECO2R in recent years.ILs are a novel class of liquids, made up solely of ions which possess a unique set of properties. These properties, such as chemical tunability, high stability, low vapour pressure, large liquidus temperature range and wide electrochemical window make them useful in a number of applications.284 ILs have also been shown to exhibit superior gas sorption/capture capacity in comparison to organic solvents.285 The tunability of ILs has allowed the design of ILs to capture greater than equal molar amounts of gas.286 For ECO2R the large CO2 capture potential of ILs is advantageous and, additionally, the ability to chemically absorb CO2 can in some cases lead to activation of CO2 for ECO2R.287
Many reports of ILs used for electrocatalytic reduction of CO2 exist but products reported are mainly C1 type compounds.288 Only a small number of papers report the production of C2+ compounds and in most cases the IL is acting as a co-catalyst rather than an electrolyte. In all of the cases reported the IL has been paired with a copper type catalyst to produce C2+ products. Sun et al. showed the conversion of CO2 to acetic acid at 80.3% FE when Cu(I) and a carbon-doped boron nitride catalyst was used alongside a LiI-water-IL electrolyte where the IL used was 1-ethyl-3-methylimidazolium tetrafluoroborate ([Emim][BF4]).289 Hui et al. showed the incorporation of IL (1-octyl-3-methylimidazolium chloride, OmimCl) into a Cu2O supported on a graphite sheet catalyst could more than double the FE of C2H4.290 Another group reports the production of C2H5OH at a rate of 24.42 μmol cm−2 h−1 at a potential of −1.6 V vs. Ag/AgCl (KCl sat.) using a combination of copper nanofoam and an electrolyte of 0.1 M KHCO3 + 0.04 M 1-butyl-3-methyl-imidazolium bromide (BmimBr).291
The limited reports in the literature would suggest that the utilisation of ILs to produce C2+ products has been largely unexplored. However, these reports also outline the potential of ILs to enhance production and selectivity to C2+ products. Most reports on C2+ conversion to date focus on imidazolium based ILs, therefore, there is a large variety of ILs which remain unstudied. It is estimated that there are potentially a million different ILs available284 with diverse properties including those which reactively solubilise CO2. Therefore, it is highly likely that ILs could hold the key to going beyond C1 products and should be studied more closely for this application.
4. Perspectives
4.1 Catalyst design and development
Understanding the catalytic mechanism and catalyst design if one wants to improve the yields of C2+ products, then there should be efficient C–C coupling steps. Perhaps the most efficient manner to advance in the intelligent design of catalysts is to understand the reaction mechanism and identify and analyze the limiting factors.Despite computational tools being extremely useful to describe reaction mechanisms, they are still far from accurately considering all the processes included in an electrochemical reaction. There are several intrinsic shortcomings of DFT methods (commonly used) in describing the energies of several reaction intermediates, for example, (i) overestimation or underestimation of CO and CO2 adsorption energies on transition metal-based catalysts when using DFT instead of DFT+U, especially in semiconductors;300 (ii) overestimation of CO2 hydrogenation energy.301–303 Improved methods to describe electrochemical reactions in-depth need to consider the following aspects within a dynamic characteristics of the Helmholtz double-layer: (i) an electrochemical phase diagram as a function of the applied potential;304 (ii) the modified Poisson–Boltzmann approach for Tafel lines generation;305 (iii) constant-charge or constant-potential, which is better in modeling the electrolyte–catalyst interface (electrochemical double layer); and (iv) cyclic voltammetry curves simulation using KMC and microkinetic methods.17
4.2 Eliminating energy loss through process intensification and device innovation
Many efforts have been done on designing and optimizing ECO2R devices. For room temperature membrane-based devices, the mass transfer limitation of CO2 is basically resolved through adjusting GDE design.308 However, the mono ion-exchange membrane (CEM or AEM) restrains the application of different pH environment on cathode and anode. As discussed in Section 3.1, bipolar membrane offer a bright prospect on the manipulation of electrolyte of half-reactions, which enables low cost and flexible choice of electrocatalysts via pH differential operations, as suggested by recent numerical studies.307 Advanced bipolar membranes should be developed to reduce the ion transfer resistance with acceptable performance degradation. For high-temperature SOEC, more research attention should be paid on promoting the product selectivity. We believe pressurized CO2 and optimized temperature operation guides the future direction of CRR device design, not only because of better mass transfer performance, but also due to the potential to produce high carbon products. Additionally, alternative electrolytes, such as ILs have the ability in catalyzing ECO2R. With beneficial properties such as high CO2 solubility and intrinsic electrical conductivity, ILs are prime candidates to enhance the activity for C2+ products, and due to the lack of scientific reports in this area, it is clear there is much work that can still be done.Abbreviations
| CCU | Carbon capture and utilization |
| ECO2R | Electrochemical CO2 reduction |
| OD | Oxide derived |
| MES | Microbial electro-synthesis |
| BES | Bio-electrochemical system |
| VFA | Volatile fatty acids |
| EF | Electro-fermentation |
| W–L | Wood–Ljungdahl |
| SBR | Sequencing batch reactor |
| VSS | Volatile suspended solids |
| RVC | Reticulated vitreous carbon |
| MWCN | Multi-walled carbon nanotubes |
| EPD | Electrophoretic deposition |
| ABE | Acetone–butanol–ethanol |
| IBE | Isopropanol–butanol–ethanol |
| HRT | Hydraulic retention time |
| CEM | Cation-exchange membrane |
| AEM | Anion exchange membrane |
| BPM | Bipolar membrane |
| OER | Oxygen reduction reaction |
| GDE | Gas diffusion electrode |
| SOEC | Solid oxide electrolyzer cell |
| F–T | Fischer–Tropsch |
| IL | Ionic liquid |
| PHAs | Polyhydroxyalkanoates |
| PHB | Polyhydroxybutyrate |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support from the UKRI Interdisciplinary Centre for Circular Chemical Economy (EP/V011863/1). E. Yu, D. Li, S. Rasul, J.-M. Fontmorin, P. Izadi and H. Xiang thank EPSRC LifesCO2R project (EP/N009746/1), EPSRC NECEM (EP/R021503/1), NERC MeteoRR (NE/L014246/1), NBIC 002POC19034 and P. Izadi and H. Xiang thank the Doctoral Training Awards (SAgE DTA, 2015 cohort) from Faculty of Science, Agriculture and Engineering, Newcastle University for supporting their PhD studies. The project was also supported by the Open Fund Project for State Key Laboratory of Clean Energy Utilization with Zhejiang University, project number ZJUCEU2019004. A. Cowan thanks EPSRC for support (EP/N010531/1).Notes and references
- E. R. Delaria, J. Kim, H. L. Fitzmaurice, C. Newman, P. J. Wooldridge, K. Worthington and R. C. Cohen, Atmos. Meas. Tech., 2021, 14, 5487–5500 CrossRef.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- F. Sanchez, D. Motta, A. Roldan, C. Hammond, A. Villa and N. Dimitratos, Top. Catal., 2018, 61, 254–266 CrossRef CAS PubMed.
- F. Sanchez, M. H. Alotaibi, D. Motta, C. E. Chan-Thaw, A. Rakotomahevitra, T. Tabanelli, A. Roldan, C. Hammond, Q. He, T. Davies, A. Villa and N. Dimitratos, Sustainable Energy Fuels, 2018, 2, 2705–2716 RSC.
- Q. Fan, M. Zhang, M. Jia, S. Liu, J. Qiu and Z. Sun, Mater. Today Energy, 2018, 10, 280–301 CrossRef.
- R. M. Arán-Ais, D. Gao and B. Roldan Cuenya, Acc. Chem. Res., 2018, 51, 2906–2917 CrossRef PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355 Search PubMed.
- Y. Hori, Modern Aspects of Electrochemistry, ed. C. Vayenas, Springer, New York, 2008 Search PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS PubMed.
- Z. P. Jovanov, H. A. Hansen, A. S. Varela, P. Malacrida, A. A. Peterson, J. K. Nørskov, I. E. L. Stephens and I. Chorkendorff, J. Catal., 2016, 343, 215–231 CrossRef CAS.
- E. Bertheussen, Y. Abghoui, Z. P. Jovanov, A.-S. Varela, I. E. L. Stephens and I. Chorkendorff, Catal. Today, 2017, 288, 54–62 CrossRef CAS.
- Y. Hori, A. Murata and Y. Yoshinami, J. Chem. Soc., Faraday Trans., 1991, 87, 125–128 RSC.
- J. Yoshihara, S. C. Parker, A. Schafer and C. T. Campbell, Catal. Lett., 1995, 31, 313–324 CrossRef CAS.
- J. Yoshihara and C. T. Campbell, J. Catal., 1996, 161, 776–782 CrossRef CAS.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- A. Roldan, Curr. Opin. Electrochem., 2018, 10, 1–6 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- M. J. Janik, C. D. Taylor and M. Neurock, J. Electrochem. Soc., 2009, 156, B126–B135 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- M. Ebaid, K. Jiang, Z. Zhang, W. S. Drisdell, A. T. Bell and J. K. Cooper, Chem. Inf., 2020, 32, 3304–3311 CAS.
- P.-P. Yang, X.-L. Zhang, F.-Y. Gao, Y.-R. Zheng, Z.-Z. Niu, X. Yu, R. Liu, Z.-Z. Wu, S. Qin, L.-P. Chi, Y. Duan, T. Ma, X.-S. Zheng, J.-F. Zhu, H.-J. Wang, M.-R. Gao and S.-H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS PubMed.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- T. H. Phan, K. Banjac, F. P. Cometto, F. Dattila, R. García-Muelas, S. J. Raaijman, C. Ye, M. T. M. Koper, N. López and M. Lingenfelder, Nano Lett., 2021, 21, 2059–2065 CrossRef CAS PubMed.
- Q. Fan, X. Zhang, X. Ge, L. Bai, D. He, Y. Qu, C. Kong, J. Bi, D. Ding, Y. Cao, X. Duan, J. Wang, J. Yang and Y. Wu, Adv. Energy Mater., 2021, 2101424 CrossRef CAS.
- J. Santatiwongchai, K. Faungnawakij and P. Hirunsit, ACS Catal., 2021, 11, 9688–9701 CrossRef CAS.
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, ACS Catal., 2021, 11, 4456–4463 CrossRef CAS.
- P. Shao, W. Zhou, Q.-L. Hong, L. Yi, L. Zheng, W. Wang, H.-X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef CAS PubMed.
- L. Ou, Z. He, H. Yang and Y. Chen, ACS Omega, 2021, 6, 17839–17847 CrossRef CAS PubMed.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- J. Zhang, Z. Li, S. Xia, T. Zhang, Y. Wang, Y. Wu and J. Wu, Chem. Commun., 2021, 57, 8276–8279 RSC.
- J. J. Kim, D. P. Summers and K. W. Frese, J. Electroanal. Chem. Interfacial Electrochem., 1988, 245, 223–244 CrossRef CAS.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1987, 728–729 RSC.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- P. Zhu, C. Xia, C.-Y. Liu, K. Jiang, G. Gao, X. Zhang, Y. Xia, Y. Lei, H. N. Alshareef, T. P. Senftle and H. Wang, Proc. Natl. Acad. Sci., 2021, 118, e2010868118 CrossRef CAS PubMed.
- Y. Li, S. H. Chan and Q. Sun, Nanoscale, 2015, 7, 8663–8683 RSC.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS PubMed.
- J. Li, A. Xu, F. Li, Z. Wang, C. Zou, C. M. Gabardo, Y. Wang, A. Ozden, Y. Xu, D.-H. Nam, Y. Lum, J. Wicks, B. Chen, Z. Wang, J. Chen, Y. Wen, T. Zhuang, M. Luo, X. Du, T.-K. Sham, B. Zhang, E. H. Sargent and D. Sinton, Nat. Commun., 2020, 11, 3685 CrossRef CAS PubMed.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- Y. Zhou and B. S. Yeo, J. Mater. Chem. A, 2020, 8, 23162–23186 RSC.
- H. Bai, T. Cheng, S. Li, Z. Zhou, H. Yang, J. Li, M. Xie, J. Ye, Y. Ji, Y. Li, Z. Zhou, S. Sun, B. Zhang and H. Peng, Sci. Bull., 2021, 66, 62–68 CrossRef CAS.
- H. Li, Y. Li, M. T. Koper and F. Calle-Vallejo, J. Am. Chem. Soc., 2014, 136, 15694–15701 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- Y. Wang, J. Liu and G. Zheng, Adv. Mater., 2021, 2005798 CrossRef PubMed.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- M. Gonçalves, A. Gomes, J. Condeço, R. Fernandes, T. Pardal, C. Sequeira and J. Branco, Energy Convers. Manage., 2010, 51, 30–32 CrossRef.
- M. Goncalves, A. Gomes, J. Condeço, T. Fernandes, T. Pardal, C. Sequeira and J. Branco, Electrochim. Acta, 2013, 102, 388–392 CrossRef CAS.
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- J.-F. Xie, Y.-X. Huang, W.-W. Li, X.-N. Song, L. Xiong and H.-Q. Yu, Electrochim. Acta, 2014, 139, 137–144 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- H. Song, M. Im, J. T. Song, J.-A. Lim, B.-S. Kim, Y. Kwon, S. Ryu and J. Oh, Appl. Catal., B, 2018, 232, 391–396 CrossRef CAS.
- F. Scholten, I. Sinev, M. Bernal and B. Roldan Cuenya, ACS Catal., 2019, 9, 5496–5502 CrossRef CAS PubMed.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 824–830 RSC.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- J. Gao, D. Ren, X. Guo, S. M. Zakeeruddin and M. Grätzel, Faraday Discuss., 2019, 215, 282–296 RSC.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- X. Chen, D. A. Henckel, U. O. Nwabara, Y. Li, A. I. Frenkel, T. T. Fister, P. J. Kenis and A. A. Gewirth, ACS Catal., 2019, 10, 672–682 CrossRef.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li and F. P. G. de Arquer, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer III, M. Chi, C. Ma and B. G. Sumpter, ChemistrySelect, 2016, 1, 6055–6061 CrossRef CAS.
- Q. Li, W. Zhu, J. Fu, H. Zhang, G. Wu and S. Sun, Nano Energy, 2016, 24, 1–9 CrossRef CAS.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei and A. Zitolo, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- Z. Han, R. Kortlever, H.-Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- Y. Song, S. Wang, W. Chen, S. Li, G. Feng, W. Wei and Y. Sun, ChemSusChem, 2019, 293–297 Search PubMed.
- G. Lu, H. Wang, Z. Bian and X. Liu, Sci. World J., 2013, 2013, 8 Search PubMed.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS PubMed.
- M. Wang, Q. Zhang, Q. Xie, L. Wan, Y. Zhao, X. Zhang and J. Luo, Nanoscale, 2020, 12, 17013–17019 RSC.
- W. Fu, Z. Liu, T. Wang, J. Liang, S. Duan, L. Xie, J. Han and Q. Li, ACS Sustainable Chem. Eng., 2020, 8, 15223–15229 CrossRef CAS.
- Z. Tan, T. Peng, X. Tan, W. Wang, X. Wang, Z. Yang, H. Ning, Q. Zhao and M. Wu, ChemElectroChem, 2020, 7, 2020–2025 CrossRef CAS.
- B. Liu, C. Cai, B. Yang, K. Chen, Y. Long, Q. Wang, S. Wang, G. Chen, H. Li, J. Hu, J. Fu and M. Liu, Electrochim. Acta, 2021, 388, 138552 CrossRef CAS.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- S. Wang, T. Kou, S. E. Baker, E. B. Duoss and Y. Li, Mater. Today Nano, 2020, 12, 100096 CrossRef.
- C. Chen, X. Yan, Y. Wu, S. Liu, X. Sun, Q. Zhu, R. Feng, T. Wu, Q. Qian, H. Liu, L. Zheng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 5938–5943 RSC.
- F. Yang, P. Deng, Q. Wang, J. Zhu, Y. Yan, L. Zhou, K. Qi, H. Liu, H. S. Park and B. Y. Xia, J. Mater. Chem. A, 2020, 8, 12418–12423 RSC.
- D. Cheng, Z.-J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- F. Dattila, R. García-Muelas and N. López, ACS Energy Lett., 2020, 5, 3176–3184 CrossRef CAS.
- P. Qi, L. Zhao, Z. Deng, H. Sun, H. Li, Q. Liu, X. Li, Y. Lian, J. Cheng, J. Guo, Y. Cui and Y. Peng, J. Phys. Chem. Lett., 2021, 12, 3941–3950 CrossRef CAS PubMed.
- Y.-J. Zhang and A. A. Peterson, Phys. Chem. Chem. Phys., 2015, 17, 4505–4515 RSC.
- C.-C. Chang, E. Y. Li and M.-K. Tsai, Phys. Chem. Chem. Phys., 2018, 20, 16906–16909 RSC.
- P. Sirkiä, T. Saario, K. Maekelae, T. Laitinen and M. Bojinov, Electric and Electrochemical Properties of Surface Films Formed on Copper in the Presence of Bicarbonate Anions, Radiation and Nuclear Safety Authority, 1999 Search PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo and J. W. Ager, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS PubMed.
- Q. Lei, H. Zhu, K. Song, N. Wei, L. Liu, D. Zhang, J. Yin, X. Dong, K. Yao, N. Wang, X. Li, B. Davaasuren, J. Wang and Y. Han, J. Am. Chem. Soc., 2020, 142, 4213–4222 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- A. Eilert, F. Cavalca, F. S. Roberts, J. r. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel and L. G. Pettersson, J. Phys. Chem. Lett., 2016, 8, 285–290 CrossRef PubMed.
- C. Liu, M. P. Lourenço, S. Hedström, F. Cavalca, O. Diaz-Morales, H. A. Duarte, A. Nilsson and L. G. Pettersson, J. Phys. Chem. C, 2017, 121, 25010–25017 CrossRef CAS.
- H. Xiang, S. Rasul, K. Scott, J. Portoles, P. Cumpson and H. Y. Eileen, J. CO2 Util., 2019, 30, 214–221 CrossRef CAS.
- M. Watanabe, M. Shibata, A. Kato, M. Azuma and T. Sakata, J. Electrochem. Soc., 1991, 138, 3382–3389 CrossRef CAS.
- P. Hirunsit, J. Phys. Chem. C, 2013, 117, 8262–8268 CrossRef CAS.
- A. Dutta, I. Z. Montiel, R. Erni, K. Kiran, M. Rahaman, J. Drnec and P. Broekmann, Nano Energy, 2020, 68, 104331 CrossRef CAS.
- L. R. L. Ting, O. Piqué, S. Y. Lim, M. Tanhaei, F. Calle-Vallejo and B. S. Yeo, ACS Catal., 2020, 10, 4059–4069 CrossRef CAS.
- X.-G. Zhang, S. Feng, C. Zhan, D.-Y. Wu, Y. Zhao and Z.-Q. Tian, J. Phys. Chem. Lett., 2020, 11, 6593–6599 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- S. Shen, X. Peng, L. Song, Y. Qiu, C. Li, L. Zhuo, J. He, J. Ren, X. Liu and J. Luo, Small, 2019, 15, 1902229 CrossRef PubMed.
- W. Zhu, K. Zhao, S. Liu, M. Liu, F. Peng, P. An, B. Qin, H. Zhou, H. Li and Z. He, J. Energy Chem., 2019, 37, 176–182 CrossRef.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- A. N. Kuhn, H. Zhao, U. O. Nwabara, X. Lu, M. Liu, Y.-T. Pan, W. Zhu, P. J. A. Kenis and H. Yang, Adv. Funct. Mater., 2021, 31, 2101668 CrossRef CAS.
- A. Herzog, A. Bergmann, H. S. Jeon, J. Timoshenko, S. Kühl, C. Rettenmaier, M. Lopez Luna, F. T. Haase and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2021, 60, 7426–7435 CrossRef CAS PubMed.
- X. Zhang, J. Li, Y.-Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS PubMed.
- S. Dongare, N. Singh, H. Bhunia and P. K. Bajpai, Electrochim. Acta, 2021, 392, 138988 CrossRef CAS.
- Z. Li, R. M. Yadav, L. Sun, T. Zhang, J. Zhang, P. M. Ajayan and J. Wu, Appl. Catal., A, 2020, 606, 117829 CrossRef CAS.
- X. Su, Y. Sun, L. Jin, L. Zhang, Y. Yang, P. Kerns, B. Liu, S. Li and J. He, Appl. Catal., B, 2020, 269, 118800 CrossRef CAS.
- Y. Song, J. R. C. Junqueira, N. Sikdar, D. Öhl, S. Dieckhöfer, T. Quast, S. Seisel, J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 9135–9141 CrossRef CAS PubMed.
- A. H. M. da Silva, S. J. Raaijman, C. S. Santana, J. M. Assaf, J. F. Gomes and M. T. M. Koper, J. Electroanal. Chem., 2021, 880, 114750 CrossRef CAS.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- Y. Feng, Z. Li, H. Liu, C. Dong, J. Wang, S. A. Kulinich and X. Du, Langmuir, 2018, 34, 13544–13549 CrossRef CAS PubMed.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min and R. Quintero-Bermudez, Nat. Catal., 2018, 1, 421 CrossRef CAS.
- Q. Wan, J. Zhang, B. Zhang, D. Tan, L. Yao, L. Zheng, F. Zhang, L. Liu, X. Cheng and B. Han, Green Chem., 2020, 22, 2750–2754 RSC.
- K. Lv, Y. Fan, Y. Zhu, Y. Yuan, J. Wang, Y. Zhu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 5025–5031 RSC.
- Q. Zhang, S. Tao, J. Du, A. He, Y. Yang and C. Tao, J. Mater. Chem. A, 2020, 8, 8410–8420 RSC.
- L. Ji, L. Li, X. Ji, Y. Zhang, S. Mou, T. Wu, Q. Liu, B. Li, X. Zhu, Y. Luo, X. Shi, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2020, 59, 758–762 CrossRef CAS PubMed.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. Koper, Chem. Commun., 2016, 52, 10229–10232 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- E. Boutin and M. Robert, Trends Chem., 2021, 3, 359–372 CrossRef CAS.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 1–28, 2102151 CrossRef.
- K. Tanaka, Y. Kushi, K. Tsuge, K. Toyohara, T. Nishioka and K. Isobe, Inorg. Chem., 1998, 37, 120–126 CrossRef CAS PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef CAS PubMed.
- M. Meser Ali, H. Sato, T. Mizukawa, K. Tsuge, M.-a. Haga and K. Tanaka, Chem. Commun., 1998, 249–250 Search PubMed.
- H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1994, 33, 3415–3420 CrossRef CAS.
- K. Toyohara, H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1995, 34, 5399–5400 CrossRef CAS.
- T. Mizukawa, K. Tsuge, H. Nakajima and K. Tanaka, Angew. Chem., Int. Ed., 1999, 38, 362–363 CrossRef CAS.
- H. Nakajima, Y. Kushi, H. Nagao and K. Tanaka, Organometallics, 1995, 14, 5093–5098 CrossRef CAS.
- K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS.
- K. Tanaka and T. Mizukawa, Appl. Organomet. Chem., 2000, 14, 863–866 CrossRef CAS.
- G. Neri, I. M. Aldous, J. J. Walsh, L. J. Hardwick and A. J. Cowan, Chem. Sci., 2016, 7, 1521–1526 RSC.
- T. V. Magdesieva, T. Yamamoto, D. A. Tryk and A. Fujishima, J. Electrochem. Soc., 2002, 149, D89 CrossRef CAS.
- N. Sonoyama, M. Kirii and T. Sakata, Electrochem. Commun., 1999, 1, 213–216 CrossRef CAS.
- K. Ogura, K. Mine, J. Yano and H. Sugihara, J. Chem. Soc., Chem. Commun., 1993, 20–21 RSC.
- K. Ogura, M. Higasa, J. Yano and N. Endo, J. Electroanal. Chem., 1994, 379, 373–377 CrossRef.
- K. Ogura, N. Endo, M. Nakayama and H. Ootsuka, J. Electrochem. Soc., 1995, 142, 4026–4032 CrossRef CAS.
- K. Ogura, M. Nakayama and C. Kusumoto, J. Electrochem. Soc., 1996, 143, 3606–3615 CrossRef CAS.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- M. Balamurugan, H. Y. Jeong, V. S. K. Choutipalli, J. S. Hong, H. Seo, N. Saravanan, J. H. Jang, K. G. Lee, Y. H. Lee, S. W. Im, V. Subramanian, S. H. Kim and K. T. Nam, Small, 2020, 16, 1–10 CrossRef PubMed.
- N. Sakamoto, Y. F. Nishimura, T. Nonaka, M. Ohashi, N. Ishida, K. Kitazumi, Y. Kato, K. Sekizawa, T. Morikawa and T. Arai, ACS Catal., 2020, 10, 10412–10419 CrossRef CAS.
- J. X. Wu, S. Z. Hou, X. D. Zhang, M. Xu, H. F. Yang, P. S. Cao and Z. Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- F. Yang, A. Chen, P. L. Deng, Y. Zhou, Z. Shahid, H. Liu and B. Y. Xia, Chem. Sci., 2019, 10, 7975–7981 RSC.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer, M. Chi, C. Ma, B. G. Sumpter and A. J. Rondinone, ChemistrySelect, 2016, 1, 6055–6061 CrossRef CAS.
- M. Hammouche, D. Lexa and J. M. Saveant, J. Electroanal. Chem., 1988, 249, 347–351 CrossRef CAS.
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS PubMed.
- X. Wang, Z. Wang, F. P. García de Arquer, C. T. Dinh, A. Ozden, Y. C. Li, D. H. Nam, J. Li, Y. S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T. T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S. F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- W. A. Thompson, E. Sanchez Fernandez and M. M. Maroto-Valer, ACS Sustainable Chem. Eng., 2020, 8, 4677–4692 CrossRef CAS.
- E. M. Cedeño Morales, B. I. Kharisov and M. A. Méndez-Rojas, Mater. Today: Proc., 2021, 46, 2982–2997 Search PubMed.
- Y. Zhang, B. Xia, J. Ran, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 1903879 CrossRef CAS.
- S. Matavos-Aramyan, S. Soukhakian, M. H. Jazebizadeh, M. Moussavi and M. R. Hojjati, Appl. Mater. Today, 2020, 18, 100499 CrossRef.
- J. Wang, S. Lin, N. Tian, T. Ma, Y. Zhang and H. Huang, Adv. Funct. Mater., 2021, 31, 2008008 CrossRef CAS.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- T. J. LaTempa, S. Rani, N. Bao and C. A. Grimes, Nanoscale, 2012, 4, 2245–2250 RSC.
- C. Wang, R. L. Thompson, P. Ohodnicki, J. Baltrus and C. Matranga, J. Mater. Chem., 2011, 21, 13452–13457 RSC.
- X. Zhang, F. Han, B. Shi, S. Farsinezhad, G. P. Dechaine and K. Shankar, Angew. Chem., Int. Ed., 2012, 51, 12732–12735 CrossRef CAS PubMed.
- M. Sayed, L. Zhang and J. Yu, Chem. Eng. J., 2020, 397, 125390 CrossRef CAS.
- A. E. Nogueira, G. T. S. T. da Silva, J. A. Oliveira, J. A. Torres, M. G. S. da Silva, M. Carmo and C. Ribeiro, Catal. Commun., 2020, 137, 105929 CrossRef CAS.
- J. Zhou, Y. Li, L. Yu, Z. Li, D. Xie, Y. Zhao and Y. Yu, Chem. Eng. J., 2020, 385, 123940 CrossRef.
- D. Giusi, C. Ampelli, C. Genovese, S. Perathoner and G. Centi, Chem. Eng. J., 2021, 408, 127250 CrossRef CAS.
- C. An, J. Yuan and J. Zhu, J. Electrochem. Soc., 2018, 165, H1066–H1071 CrossRef CAS.
- H. Homayoni, W. Chanmanee, N. R. de Tacconi, B. H. Dennis and K. Rajeshwar, J. Electrochem. Soc., 2015, 162, E115–E122 CrossRef CAS.
- S. Jiao, X. Fu, L. Zhang, L. Zhang, S. Ruan, Y.-J. Zeng and H. Huang, Nano Today, 2021, 36, 101028 CrossRef CAS.
- T. Li, X. Dong, W. Chen, X. Zhao, G. Li, G. Feng, Y. Song, W. Wei and Y. Sun, Appl. Surf. Sci., 2020, 526, 146578 CrossRef CAS.
- X. Wang, C. Gao, J. Low, K. Mao, D. Duan, S. Chen, R. Ye, Y. Qiu, J. Ma, X. Zheng, R. Long, X. Wu, L. Song, J. Zhu and Y. Xiong, Sci. Bull., 2021, 66, 1296–1304 CrossRef CAS.
- X.-D. Zhang, K. Liu, J.-W. Fu, H.-M. Li, H. Pan, J.-H. Hu and M. Liu, Front. Phys., 2021, 16, 63500 CrossRef.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CrossRef CAS PubMed.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 RSC.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- K. Hirota, D. A. Tryk, T. Yamamoto, K. Hashimoto, M. Okawa and A. Fujishima, J. Phys. Chem. B, 1998, 102, 9834–9843 CrossRef CAS.
- K. Zhao, X. W. Nie, H. Z. Wang, S. Chen, X. Quan, H. T. Yu, W. Y. Choi, G. H. Zhang, B. Kim and J. G. G. Chen, Nat. Commun., 2020, 11, 10 CrossRef PubMed.
- B. Bian, S. Bajracharya, J. J. Xu, D. Pant and P. E. Saikaly, Bioresour. Technol., 2020, 302, 12 CrossRef PubMed.
- L. Jourdin and T. Burdyny, Trends Biotechnol., 2021, 39, 359–369 CrossRef CAS PubMed.
- L. Jourdin, S. Freguia, V. Flexer and J. Keller, Environ. Sci. Technol., 2016, 50, 1982–1989 CrossRef CAS PubMed.
- D. Zang, Q. Li, G. Dai, M. Zeng, Y. Huang and Y. Wei, Appl. Catal., B, 2021, 281, 119426 CrossRef CAS.
- X. Li, S. Chen, D. Liang and M. Alvarado-Moralesa, J. Power Sources, 2020, 477, 228990 CrossRef CAS.
- S. Srikanth, D. Singh, K. Vanbroekhoven, D. Pant, M. Kumar, S. K. Puri and S. S. V. Ramakumar, Bioresour. Technol., 2018, 265, 45–51 CrossRef CAS PubMed.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- J. Annie Modestra, R. Katakojwala and S. Venkata Mohan, Chem. Eng. J., 2020, 394, 124759 CrossRef CAS.
- S. Bajracharya, S. Srikanth, G. Mohanakrishna, R. Zacharia, D. P. B. T. B. Strik and D. Pant, J. Power Sources, 2017, 356, 256–273 CrossRef CAS.
- A. G. Fast and E. T. Papoutsakis, Curr. Opin. Chem. Eng., 2012, 1, 380–395 CrossRef.
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. H. Ou, T. L. Woodard, O. L. Snoeyenbos-West and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882–2886 CrossRef CAS PubMed.
- V. G. Debabov, Microbiology, 2021, 90, 273–297 CAS.
- G. Merli, A. Becci, A. Amato and F. Beolchini, Sci. Total Environ., 2021, 798, 149292 CrossRef CAS PubMed.
- D. R. Lovley and K. P. Nevin, Curr. Opin. Biotechnol., 2013, 24, 385–390 CrossRef CAS PubMed.
- B. Bian, S. Bajracharya, J. Xu, D. Pant and P. E. Saikaly, Bioresour. Technol., 2020, 302, 122863 CrossRef CAS PubMed.
- H.-Y. Yang, N.-N. Hou, Y.-X. Wang, J. Liu, C.-S. He, Y.-R. Wang, W.-H. Li and Y. Mu, Sci. Total Environ., 2021, 790, 148128 CrossRef CAS PubMed.
- M. Roy, R. Yadav, P. Chiranjeevi and S. A. Patil, Bioresour. Technol., 2021, 320, 124289 CrossRef CAS PubMed.
- S. Das, S. Das and M. M. Ghangrekar, Process Biochem., 2021, 101, 237–246 CrossRef CAS.
- Y. Jiang, Q. Liang, N. Chu, W. Hao, L. Zhang, G. Zhan, D. Li and R. J. Zeng, Sci. Total Environ., 2020, 741, 140198 CrossRef CAS PubMed.
- P. Izadi, J.-M. Fontmorin, A. Godain, E. H. Yu and I. M. Head, npj Biofilms Microbiomes, 2020, 6, 40 CrossRef CAS PubMed.
- J. Philips, Front. Microbiol., 2020, 10, 2997 CrossRef PubMed.
- I. Vassilev, P. A. Hernandez, P. Batlle-Vilanova, S. Freguia, J. O. Kromer, J. Keller, P. Ledezma and B. Virdis, ACS Sustainable Chem. Eng., 2018, 6, 8485–8493 CrossRef CAS.
- R. Ganigue, P. Sanchez-Paredes, L. Baneras and J. Colprim, Front. Microbiol., 2016, 7, 702 Search PubMed.
- H. D. May, P. J. Evans and E. V. LaBelle, Curr. Opin. Biotechnol., 2016, 42, 225–233 CrossRef CAS PubMed.
- J. B. A. Arends, S. A. Patil, H. Roume and K. Rabaey, J. CO2 Util., 2017, 20, 141–149 CrossRef CAS.
- E. V. LaBelle, C. W. Marshall, J. A. Gilbert and H. D. May, PLoS One, 2014, 9, e109935 CrossRef PubMed.
- E. Perona-Vico, L. Feliu-Paradeda, S. Puig and L. Bañeras, Sci. Rep., 2020, 10, 19852 CrossRef CAS PubMed.
- P. Izadi, J.-M. Fontmorin, S. S. Lim, I. Head and E. H. Yu, Faraday Discuss., 2021, 230, 344–359 RSC.
- A. Prévoteau, J. M. Carvajal-Arroyo, R. Ganigué and K. Rabaey, Curr. Opin. Biotechnol., 2020, 62, 48–57 CrossRef PubMed.
- F. Aulenta, L. Catapano, L. Snip, M. Villano and M. Majone, ChemSusChem, 2012, 5, 1080–1085 CrossRef CAS PubMed.
- K. W. Meereboer, M. Misra and A. K. Mohanty, Green Chem., 2020, 22, 5519–5558 RSC.
- F. A. El-malek, H. Khairy, A. Farag and S. Omar, Int. J. Biol. Macromol., 2020, 157, 319–328 CrossRef CAS PubMed.
- J. Medeiros Garcia Alcântara, F. Distante, G. Storti, D. Moscatelli, M. Morbidelli and M. Sponchioni, Biotechnol. Adv., 2020, 42, 107582 CrossRef PubMed.
- M. Liu, M. Arshadi, F. Javi, P. Lawrence, S. M. Davachi and A. Abbaspourrad, J. Cleaner Prod., 2020, 276, 123353 CrossRef CAS.
- B. Yadav, A. Pandey, L. R. Kumar and R. D. Tyagi, Bioresour. Technol., 2020, 298, 122584 CrossRef CAS PubMed.
- T. Pepè Sciarria, P. Batlle-Vilanova, B. Colombo, B. Scaglia, M. D. Balaguer, J. Colprim, S. Puig and F. Adani, Green Chem., 2018, 20, 4058–4066 RSC.
- M. Stöckl, S. Harms, I. Dinges, S. Dimitrova and D. Holtmann, ChemSusChem, 2020, 13, 4086–4093 CrossRef PubMed.
- V. Sivalingam, V. Ahmadi, O. Babafemi and C. Dinamarca, Catalysts, 2021, 11, 40 CrossRef CAS.
- Z. Gong, H. Yu, J. Zhang, F. Li and H. Song, Synth. Syst. Biotechnol., 2020, 5, 304–313 CrossRef PubMed.
- Y. Jiang, H. D. May, L. Lu, P. Liang, X. Huang and Z. J. Ren, Water Res., 2019, 149, 42–55 CrossRef CAS PubMed.
- O. Choi, T. Kim, H. M. Woo and Y. Um, Sci. Rep., 2014, 4, 6961 CrossRef CAS PubMed.
- S. Zhang, J. Jiang, H. Wang, F. Li, T. Hua and W. Wang, J. CO2 Util., 2021, 51, 101640 CrossRef CAS.
- O. Cabau-Peinado, A. J. J. Straathof and L. Jourdin, Front. Microbiol., 2021, 12, 669218 CrossRef PubMed.
- N. Chu, Q. Liang, W. Zhang, Z. Ge, W. Hao, Y. Jiang and R. J. Zeng, ACS Sustainable Chem. Eng., 2020, 8, 8773–8782 CrossRef CAS.
- G. Jabeen and R. Farooq, J. Biosci., 2016, 41, 367–380 CrossRef CAS PubMed.
- I. Vassilev, F. Kracke, S. Freguia, J. Keller, J. O. Krömer, P. Ledezma and B. Virdis, Chem. Commun., 2019, 55, 4351–4354 RSC.
- I. Vassilev, P. A. Hernandez, P. Batlle-Vilanova, S. Freguia, J. O. Krömer, J. Keller, P. Ledezma and B. Virdis, ACS Sustainable Chem. Eng., 2018, 6, 8485–8493 CrossRef CAS.
- L. Jourdin, S. M. T. Raes, C. J. N. Buisman and D. P. B. T. B. Strik, Front. Energy Res., 2018, 6, 7 CrossRef.
- P. L. Tremblay and T. Zhang, Front. Microbiol., 2015, 6, 201 Search PubMed.
- B. Schiel-Bengelsdorf and P. Durre, FEBS Lett., 2012, 586, 2191–2198 CrossRef CAS PubMed.
- C. W. Marshall, D. E. Ross, E. B. Fichot, R. S. Norman and H. D. May, Environ. Sci. Technol., 2013, 47, 6023–6029 CrossRef CAS PubMed.
- P. Batlle-Vilanova, S. Puig, R. Gonzalez-Olmos, M. D. Balaguer and J. Colprim, J. Chem. Technol. Biotechnol., 2016, 91, 921–927 CrossRef CAS.
- P. Izadi, J.-M. Fontmorin, B. Virdis, I. M. Head and E. H. Yu, Appl. Energy, 2021, 283, 116310 CrossRef CAS.
- S. Gildemyn, K. Verbeeck, R. Slabbinck, S. J. Andersen, A. Prevoteau and K. Rabaey, Environ. Sci. Technol. Lett., 2015, 2, 325–328 CrossRef CAS.
- L. Jourdin, T. Grieger, J. Monetti, V. Flexer, S. Freguia, Y. Lu, J. Chen, M. Romano, G. G. Wallace and J. Keller, Environ. Sci. Technol., 2015, 49, 13566–13574 CrossRef CAS PubMed.
- H. Liang, X. Ma, W. Ning, Y. Liu, A. J. Sinskey, G. Stephanopoulos and K. Zhou, Metab. Eng., 2021, 65, 223–231 CrossRef CAS PubMed.
- F. V. Gambacorta, J. J. Dietrich, Q. Yan and B. F. Pfleger, Curr. Opin. Chem. Biol., 2020, 59, 182–192 CrossRef CAS PubMed.
- R. Miao, H. Xie, X. Liu, P. Lindberg and P. Lindblad, Curr. Opin. Chem. Biol., 2020, 59, 69–76 CrossRef CAS PubMed.
- P. N. Ashani, M. Shafiei and K. Karimi, Bioresour. Technol., 2020, 308, 123267 CrossRef CAS PubMed.
- X. Zhen, Y. Wang and D. Liu, Renewable Energy, 2020, 147, 2494–2521 CrossRef CAS.
- J. Lee, Y.-S. Jang, S. J. Choi, J. A. Im, H. Song, J. H. Cho, D. Y. Seung, E. T. Papoutsakis, G. N. Bennett and S. Y. Lee, Appl. Environ. Microbiol., 2012, 78, 1416–1423 CrossRef CAS PubMed.
- S. Y. Lee, Y. S. Jang, J. Y. Lee and J. Lee, J. Biotechnol., 2010, 150, S557 Search PubMed.
- I. Vassilev, F. Kracke, S. Freguia, J. Keller, J. O. Kromer, P. Ledezma and B. Virdis, Chem. Commun., 2019, 55, 4351–4354 RSC.
- C. M. Spirito, H. Richter, K. Rabaey, A. J. M. Stams and L. T. Angenent, Curr. Opin. Biotechnol., 2014, 27, 115–122 CrossRef CAS PubMed.
- C. Delacourt, P. L. Ridgway, J. B. Kerr and J. Newman, J. Electrochem. Soc., 2008, 155, B42–B49 CrossRef CAS.
- O. Aschenbrenner and P. Styring, Energy Environ. Sci., 2010, 3, 1106–1113 RSC.
- F. Proietto, B. Schiavo, A. Galia and O. Scialdone, Electrochim. Acta, 2018, 277, 30–40 CrossRef CAS.
- Z. M. Yang, D. Li, L. Xing, H. Xiang, J. Xuan, S. A. Cheng, E. H. Yu and A. D. Yang, ACS Sustainable Chem. Eng., 2021, 9, 351–361 CrossRef CAS.
- A. Del Castillo, M. Alvarez-Guerra, J. Solla-Gullón, A. Sáez, V. Montiel and A. Irabien, J. CO2 Util., 2017, 18, 222–228 CrossRef CAS.
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, J. Appl. Electrochem., 2014, 44, 1107–1116 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS.
- D. A. Vermaas and W. A. Smith, ACS Energy Lett., 2016, 1, 1143–1148 CrossRef CAS.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109–B111 CrossRef CAS.
- H. Wang, D. Y. C. Leung and J. Xuan, Appl. Energy, 2013, 102, 1057–1062 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- G. Kaur, A. P. Kulkarni, S. Giddey and S. P. S. Badwal, Appl. Energy, 2018, 221, 131–138 CrossRef CAS.
- Y. Tao, S. D. Ebbesen and M. B. Mogensen, J. Electrochem. Soc., 2014, 161, F337–F343 CrossRef CAS.
- L. Zhang, S. Hu, X. Zhu and W. Yang, J. Energy Chem., 2017, 26, 593–601 CrossRef.
- S. Perathoner and G. Centi, ChemSusChem, 2014, 7, 1274–1282 CrossRef CAS PubMed.
- D. R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P. R. Ohodnicki, C. Zeng and R. Jin, ACS Appl. Mater. Interfaces, 2015, 7, 15626–15632 CrossRef CAS PubMed.
- S. H. Jensen, C. Graves, M. Mogensen, C. Wendel, R. Braun, G. Hughes, Z. Gao and S. A. Barnett, Energy Environ. Sci., 2015, 8, 2471–2479 RSC.
- X. Li, P. Anderson, H.-R. M. Jhong, M. Paster, J. F. Stubbins and P. J. A. Kenis, Energy Fuels, 2016, 30, 5980–5989 CrossRef CAS.
- C. Delacourt, P. L. Ridgway and J. Newman, J. Electrochem. Soc., 2010, 157, B1902 CrossRef CAS.
- J. Wu, F. G. Risalvato, P. P. Sharma, P. J. Pellechia, F.-S. Ke and X.-D. Zhou, J. Electrochem. Soc., 2013, 160, F953–F957 CrossRef CAS.
- O. Scialdone, A. Galia, G. L. Nero, F. Proietto, S. Sabatino and B. Schiavo, Electrochim. Acta, 2016, 199, 332–341 CrossRef CAS.
- M. A. Méndez, P. Voyame and H. H. Girault, Angew. Chem., Int. Ed., 2011, 50, 7391–7394 CrossRef PubMed.
- T. Burdyny, P. J. Graham, Y. Pang, C.-T. Dinh, M. Liu, E. H. Sargent and D. Sinton, ACS Sustainable Chem. Eng., 2017, 5, 4031–4040 CrossRef CAS.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- R. Kas, R. Kortlever, H. Yılmaz, M. T. M. Koper and G. Mul, ChemElectroChem, 2015, 2, 354–358 CrossRef CAS.
- D. Chu, G. Qin, X. Yuan, M. Xu, P. Zheng and J. Lu, ChemSusChem, 2008, 1, 205–209 CrossRef CAS PubMed.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Energy Fuels, 2006, 20, 409–414 CrossRef CAS.
- E. J. Dufek, T. E. Lister, S. G. Stone and M. E. McIlwain, J. Electrochem. Soc., 2012, 159, F514–F517 CrossRef CAS.
- N. Gutiérrez-Guerra, L. Moreno-López, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas-Consuegra, Appl. Catal., B, 2016, 188, 272–282 CrossRef.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- J. F. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS.
- S. F. R. Taylor, C. McCrellis, C. McStay, J. Jacquemin, C. Hardacre, M. Mercy, R. G. Bell and N. H. de Leeuw, J. Solution Chem., 2015, 44, 511–527 CrossRef CAS.
- N. Hollingsworth, S. F. R. Taylor, M. T. Galante, J. Jacquemin, C. Longo, K. B. Holt, N. H. de Leeuw and C. Hardacre, Angew. Chem., Int. Ed., 2015, 54, 14164–14168 CrossRef CAS PubMed.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang and B. Han, Green Chem., 2017, 19, 2086–2091 RSC.
- H. Ning, W. Wang, Q. H. Mao, S. R. Zheng, Z. X. Yang, Q. S. Zhao and M. B. Wu, Acta Phys.-Chim. Sin., 2018, 34, 938–944 CAS.
- R. F. Zarandi, B. Rezaei, H. S. Ghaziaskar and A. A. Ensafi, J. Environ. Chem. Eng., 2019, 7, 103141 CrossRef CAS.
- H. Xiao, T. Cheng, W. A. Goddard and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, J. Phys. Chem. Lett., 2015, 6, 4767–4773 CrossRef CAS PubMed.
- Z. J. Zhao, S. H. Liu, S. J. Zha, D. F. Cheng, F. Studt, G. Henkelman and J. L. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- F. Studt, Front. Catal., 2021, 1, 658965 CrossRef.
- A. A. Latimer, A. R. Kulkarni, H. Aljama, J. H. Montoya, J. S. Yoo, C. Tsai, F. Abild-Pedersen, F. Studt and J. K. Nørskov, Nat. Mater., 2017, 16, 225–229 CrossRef CAS PubMed.
- X. Zhang, J. X. Liu, B. Zijlstra, I. A. W. Filot, Z. Y. Zhou, S. G. Sun and E. J. M. Hensen, Nano Energy, 2018, 43, 200–209 CrossRef CAS.
- L. Lin, H. B. Li, C. C. Yan, H. F. Li, R. Si, M. R. Li, J. P. Xiao, G. X. Wang and X. H. Bao, Adv. Mater., 2019, 31, 7 Search PubMed.
- Y. F. Song, W. Chen, C. C. Zhao, S. G. Li, W. Wei and Y. H. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS PubMed.
- R. Zhang, G. Wang, B. Wang and L. Ling, J. Phys. Chem. C, 2014, 118, 5243–5254 CrossRef CAS.
- I. M. B. Nielsen and K. Leung, J. Phys. Chem. A, 2010, 114, 10166–10173 CrossRef CAS PubMed.
- K. Leung, I. M. B. Nielsen, N. Sai, C. Medforth and J. A. Shelnutt, J. Phys. Chem. A, 2010, 114, 10174–10184 CrossRef CAS PubMed.
- L. I. Bendavid and E. A. Carter, J. Phys. Chem. C, 2013, 117, 26048–26059 CrossRef CAS.
- Q.-J. Hong and Z.-P. Liu, Surf. Sci., 2010, 604, 1869–1876 CrossRef CAS.
- J.-S. Filhol and M. Neurock, Angew. Chem., Int. Ed., 2006, 45, 402–406 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- H. Zhang, H. Wang, K. Jiao and J. Xuan, Appl. Energy, 2020, 268, 115053 CrossRef CAS.
- J.-M. Fontmorin, P. Izadi, D. Li, S. S. Lim, S. Farooq, S. S. Bilal, S. A. Cheng and E. H. Yu, Electrochim. Acta, 2021, 372, 137853 CrossRef CAS.
Footnote |
| † Authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2021 |