Chemical and electrochemical water oxidation mediated by bis(pyrazol-1-ylmethyl)pyridine-ligated Cu(I) complexes†
Received
18th March 2021
, Accepted 22nd April 2021
First published on 23rd April 2021
Abstract
Herein a series of novel bis(pyrazol-1-ylmethyl)pyridine-ligated Cu(I) complexes, C1–C4, bearing different donating groups [[H(C1), Me(C2), t-Bu(C3), Ph(C4)])] on the pyrazole rings, were synthesized and investigated as pre-catalysts in chemical and electrocatalytic water oxidation reactions. Ligands, 2,6-bis((1H-pyrazol-1-yl)methyl)pyridine (L1), 2,6-bis((1H-pyrazol-1-yl)methyl)pyridine (L2), 2,6-bis((3,5-di-tert-butyl-1H-pyrazol-1-yl)methyl)pyridine (L3), and 2,6-bis((3,5-diphenyl-1H-pyrazol-1-yl)methyl)pyridine (L4) were reacted with Cu(MeCN)4PF6 to form complexes C1–C4 respectively. Cerium ammonium nitrate (CAN), sodium m-periodate, and sodium persulfate were investigated as chemical oxidants in chemical water oxidation. Complexes C1–C4 showed catalytic activity towards chemical water oxidation in the presence of CAN as the primary oxidant at 25 °C. Complex C2 was the most active with a turnover number (TON) of 4.6 and a turnover frequency (TOF) of 0.31 s−1. The least active catalyst was complex C4, with a TON of 2.3 and a TOF of 0.0086 s−1. This observed difference in catalytic activity between the complexes illustrated the key role that electronic effects play during catalysis. Other oxidants evaluated with C2 were sodium m-periodate (TON, 3.77; TOF 0.14 s−1) and sodium persulfate (TON, 4.02; TOF 0.044 s−1) however, CAN exhibited the greatest activity. Complexes C1–C4 were investigated in electrocatalytic water oxidation at a neutral pH of 6.5. Complex C2 was the most active in electrocatalytic water oxidation as well, exhibiting an overpotential of 674 mV and TOF of 9.77 s−1 (at 1.7 V vs. NHE), which is better than most reported copper(II) complexes. These Cu(I) complexes C1–C4 show potential as efficient chemical and electrocatalytic water oxidation catalysts, which can be achieved by fine-tuning the steric and electronic properties of the catalysts.
Introduction
Hydrogen is a clean and renewable energy source, or rather an energy store, which provides an attractive alternative to fossil fuels. An environmentally friendly method to produce molecular hydrogen is through the oxidation/splitting of water, an abundant natural resource.1 Different approaches to water oxidation have been considered, which include chemically driven and electrocatalytic water oxidation. As water oxidation is a multi-electron process, with a multi-proton transfer, that is associated with an uphill energy transformation, the use of catalysts is therefore required.2
The oxidation of water using chemical oxidants has been extensively studied over the years.3 To be effective, chemical oxidants, also referred to as sacrificial oxidants, must be able to oxidize the pre-catalysts to form the active catalyst intermediates. Cerium ammonium nitrate (CAN) has been the sacrificial oxidant of choice amongst others, which include sodium peroxodisulfate, Ru(III) tris(bipyridine) cation, potassium peroxymonosulfate (oxone), and sodium m-periodate. The preference for CAN over other oxidants is due to its stability in aqueous solutions and it being readily available.4 Numerous Ru and Ir metal complexes have been reported as efficient homogeneous molecular catalysts for water oxidation.3b,3e,5 The major drawbacks associated with the development of catalysts derived from these metals are high cost, rarity, and toxicity. It is therefore advantageous to develop water oxidation catalysts (WOCs) based on cost-effective, nontoxic, and abundant 1st row transition metals. This holds great benefits to a green and renewable energy environment.
Over the years, there has been a growing number of reports on chemically driven homogeneous water oxidation employing first-row transition metals such as Fe, Co, Cu, and Mn as catalysts.6 One such study was conducted by Fillol et al.2 who reported that efficient water oxidation was achieved by tetradentate nitrogen-based 1-(2′-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane (Me2Pytacn) iron catalysts with turnover numbers (TON) reaching 360 when using CAN and >1000 when sodium m-periodate was used as the oxidant. Das et al.7 explored the water oxidation activity of two Fe-complexes bearing a pentapyridyl ligand (pyridine-2,6-diylbis[di(pyridin-2-yl)methanol]) in CeIV induced water oxidation (TON 16) under acidic conditions (pH ∼1.5) and could identify high valent iron oxo intermediates as the active species. They also proposed two different mechanisms for water oxidation that are largely dependent on pH. Panchbhai et al.8 evaluated tetrazadentate (N,N′-diisopropyl-N,N′-bis(2-pyridylmethyl)-1,2-diaminoethane and N-methyl-N-(2-pyridinylmethyl)-2,2′-bipyridine-6-methanamine) ligated iron complexes using CAN as the primary oxidant, obtaining a TON of 14. Reports on chemical water oxidation employing copper(I or II) complexes are scarce.9 To date, most of the reports on copper(II) water oxidation has been studied electrocatalytically. Electrocatalytic water splitting involves the production of oxygen and protons under applied potential difference. Water oxidation, i.e., the oxygen evolution reaction (OER), occurs at the anode while the hydrogen evolution reaction (HER), i.e. proton reduction, takes place at the cathode in an electrochemical cell set up. Most of the reports of electrocatalytic water oxidation catalysts are on metal oxides rather than molecular complexes.10 Irrespective of the focus on metal oxides, the development of molecular electrocatalytic water oxidation catalysts based on abundant environmentally friendly metals such as nickel, iron, cobalt, and copper has been on the rise due to the possibility of structural and electronic tuning to obtain improved catalysts.11 Amongst these metals, copper(II) has been studied extensively in electrocatalytic water oxidation.12 The first report on copper-based molecular electrocatalysts for water oxidation was Mayer and coworkers'13 application of a very simple Cu–bipyridine system. The electrocatalyst was very efficient with a TOF of 100 s−1 in highly basic media (pH range 11.8–13.3) and at a quite high overpotential (750 mV). Electron paramagnetic resonance spectroscopy (EPR) was used to determine the identity of the species in solution, with (bpy)Cu(OH)2 proposed as the active form of the catalyst above pH 12. Praneeth et al.12d reported a multinuclear copper complex ligated by 1,3-bis(6-hydroxy-2-pyridyl)-1H-pyrazole active in electrocatalytic water oxidation at an overpotential of 500 mV at pH 12.5. UV-vis spectroscopy and energy-dispersive X-ray (EDX) spectroscopy were used to demonstrate that the catalyst operates as a molecular homogeneous catalyst. Najafpour et al.14 reported a copper(II) complex [(L)Cu (NO3)], (L = deprotonated (E)-3-(pyridin-2-yldiazenyl)-naphthalen-2-ol) evaluated in both electrocatalytic and chemical water oxidation. The complex was not active in chemical water oxidation using CAN due to its instability in acidic solution. Electrocatalytic water oxidation at pH 11 led to the decomposition of the molecular complex to form CuO nanoparticles. Scanning electron microscopy (SEM) and EDX analysis confirmed that the copper complex is not a molecular homogeneous water oxidation catalyst. Li et al.15 evaluated a tetranuclear chair like copper(II) complex [Cu4(bpy)4(μ2-OH)2(μ3-OH)2(H2O)2][C8H4O4]2 (bpy = 2,2′-bipyridine, C8H4O4 = terephthalate dianion) containing aqua- and bridging μ-hydroxo ligands in electrocatalytic water oxidation at neutral pH. The complex oxidised water at an overpotential of 730 mV in phosphate buffer at pH 7. Homogeneity of the catalyst during the electrocatalysis was confirmed. The working pH plays a vital role in the stability of homogeneous electrocatalytic water oxidation catalysts. Lu et al.16 observed an entirely homogeneous water oxidation at pH 8, however, formation of heterogeneous species was observed at pH 12 in electrocatalytic water oxidation catalysed by diamine Cu(II) complexes. Copper, an inexpensive and abundant metal has shown a lot of potential in the development of efficient electrocatalytic water oxidation catalysts, however, there is still a lack in chemically driven water oxidation processes employing copper complexes.
Herein we report the preparation of novel bis(pyrazol-1-ylmethyl)pyridine copper(I) complexes bearing different donating groups on the pyrazole ring, which are the first Cu(I) complexes that display activity for chemically driven water oxidation under acidic conditions (pH 1) and electrocatalytic water oxidation under a neutral pH of 6.5.
Results & discussion
Ligand and complex synthesis
Bis(pyrazol-1-ylmethyl)pyridine Cu(I) complexes, C1–C4, were synthesized following previously reported procedures for bis(pyrazol-1-ylmethyl)pyridine ligands17 and [Cu(MeCN)4PF6] (Scheme 1). The synthesis resulted in the preparation of four complexes bearing different groups on the pyrazolyl moiety, namely H (C1), Me (C2), t-Bu (C3), and Ph (C4), which were isolated in moderate to good yields (65–88%). The complexes were characterized by a variety of spectroscopic and analytical techniques (Fig. S15–S44†), including Fourier-transform infrared (FTIR) spectroscopy, magnetic susceptibility, nuclear magnetic resonance (NMR) spectroscopy, electrospray ionization mass spectrometry (ESI-MS), elemental analysis, and single crystal XRD.
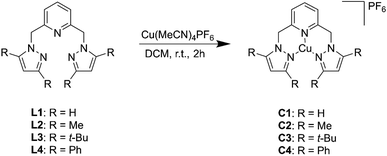 |
| | Scheme 1 Preparation of the bis(pyrazol-1-ylmethyl)pyridine Cu(I) complexes, C1–C4. | |
Magnetic susceptibility studies were conducted in acetonitrile at room temperature. The μeff-values for C1–C4 were zero, which confirmed that these complexes are diamagnetic in the +1 oxidation state.18 As such, they were further characterized by NMR (1H and 13C) spectroscopy. In the 1H NMR spectra of complexes C1–C4, the methylene and pyrazole protons were observed in the range of δ 5.21–5.71 and 6.02–7.09 ppm respectively, coinciding with a downfield shift in comparison to the free ligands, which is characteristic of the coordination of the ligand to the metal centre.19 Analogous shifts of the methylene and pyrazole carbon resonances were observed in the 13C NMR spectra of C1–C4. It should be noted that C1 oxidised rapidly in solution, leading to NMR spectral data which is characteristic of a paramagnetic species (Fig. S23 and S24†). This is attributed to the destabilising effect of the non-coordinating PF6− anion. NMR spectral data of the chloro-analogue of C1 was consistent with a diamagnetic Cu(I) system (Fig. S25 and S26†). FTIR analysis of C1–C4 showed a slight shift in the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N absorption band to lower wave numbers in the range 1597–1604 cm−1, further confirming successful complexation.20 Complexes C1–C4 were also characterized by ESI-MS operating in positive ion mode. The molecular ion peak was easily identified as the cationic species at m/z 302.05, 358.11, 526.30, 606.17 for complexes C1–C4 respectively. Finally, elemental analysis of C1–C4 confirmed the bulk purity of the isolated complexes.
N absorption band to lower wave numbers in the range 1597–1604 cm−1, further confirming successful complexation.20 Complexes C1–C4 were also characterized by ESI-MS operating in positive ion mode. The molecular ion peak was easily identified as the cationic species at m/z 302.05, 358.11, 526.30, 606.17 for complexes C1–C4 respectively. Finally, elemental analysis of C1–C4 confirmed the bulk purity of the isolated complexes.
The molecular structures of complexes C2, C3, and C4 were established with single crystal XRD analysis. Diffraction quality crystals were obtained by slow diffusion of diethyl ether into saturated dichloromethane solutions of the complexes at 4 °C. Ellipsoid diagrams for C2 (Fig. 1), C3 and C4 (Fig. S1†) are provided while crystallographic data and selected bond lengths and angles are provided in Tables S1 and S2† respectively. The coordination geometry around the metal centre for C2, C3, and C4 is a distorted T-shape, with N1–Cu1–N3 and N3–Cu1–N3′ bond angles in the range 89–98° and 173° respectively. The Cu1–N1 (pyridine) bond lengths were in the range 2.075–2.110 Å. In the case of complex C2, the Cu1–N3/3′ (pyrazole) bond lengths were in the range 2.058–2.076 Å. In contrast, the Cu1–N3/3′ (pyrazole) bond lengths were slightly shorter for C3, in the range of 1.924–1.940 Å. The shortest bond lengths for Cu1–N3/3′ (pyrazole) in the range of 1.910–1.907 Å were observed for complex C4. The introduction of bulkier groups reduces the Cu1–N3/3′ (pyrazole) bond length as a result of steric effects.21 The observed bond lengths are within range for analogous Cu(I) complexes reported in literature.22
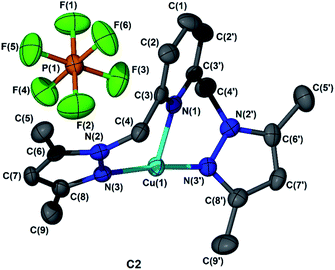 |
| | Fig. 1 Ellipsoid diagrams of C2 drawn at 50% probability. Hydrogen atoms were omitted for clarity. | |
Cu(I)-catalysed chemically driven water oxidation
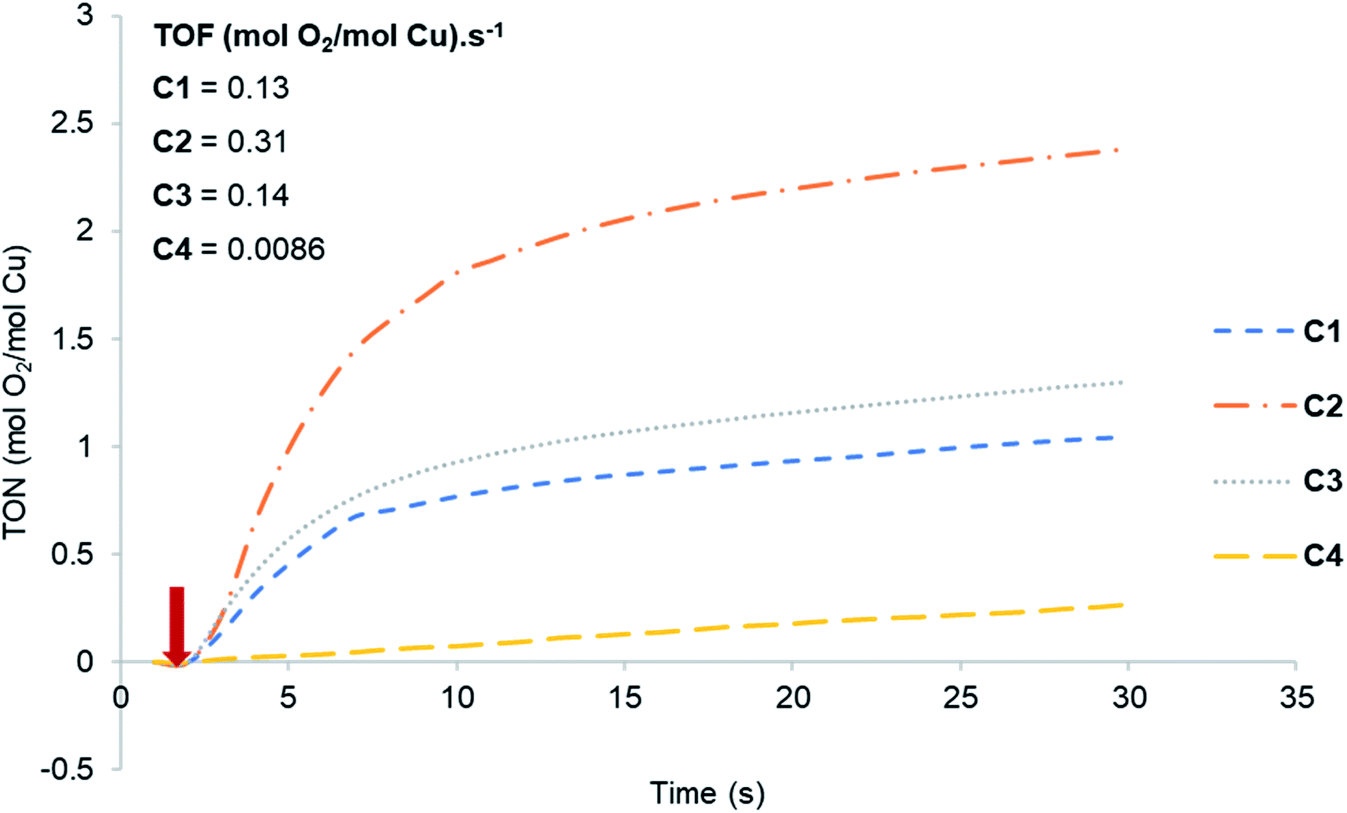
The copper(I) complexes C1–C4 were investigated as pre-catalysts for chemically driven water oxidation (Scheme 2). Oxygen evolution was recorded as a function of time on a Hansatech Oxygraph (Hansatech Instruments Ltd, Norfolk, England), with the oxygen evolution curves shown in Fig. 2.
 |
| | Scheme 2 Chemically driven water oxidation catalysed by C1–C4. | |
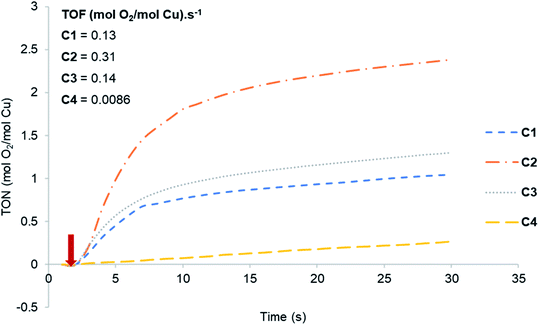 |
| | Fig. 2 Oxygen evolution curves for Cu(I) complexes C1–C4. The arrow corresponds to the point of addition of the pre-catalyst solution. | |
The activity of C1–C4 towards water oxidation was investigated in the presence of CAN at pH 1. The arrow indicates the point of addition of the catalyst to the reaction mixture. From the slope of the curves, it is evident that all of them are catalytically active. Complex C2 was the most active followed by C3 with TOF-values of 0.31 and 0.14 s−1 respectively, whereas C4 is the least active with a TOF of 0.0086 s−1. The results obtained in these reactions are summarized in Table 1. Copper(I) complexes having electron-donating substituents such as Me (C2) and t-Bu (C3) showed higher activity with TON of 4.6 and 3.96, and TOF of 0.31 and 0.14 s−1 respectively, while complexes containing unsubstituted pyrazole (C1) and the phenyl substituent (C4) resulted in lower activities with TON of 2.67 and 2.30, and TOF of 0.13 s−1 and 0.0086 s−1 respectively. Comparatively higher activity of C2 and C3 can be attributed to the electron-donating ability of the substituents on the ligand framework. The presence of electron-donating groups in the ligand framework lowers the oxidation potential of the metal centre, which in turn increases the observed water oxidation efficiency.23 Das et al.24 made a similar observation with their two structurally similar heteroleptic Ru-complexes having t-Bu and NO2 groups on the terpyridine ligand as electron-donating and electron-withdrawing substituents. The results from Fig. 2 and Table 1 are also supported by cyclic voltammetry (CV) studies (Fig. S2†).
Table 1 Summary of results of different copper(I) complexes on chemical water oxidation with CANa
| Entry |
Catalyst |
TONb (mol O2/mol Cu) |
TOFc (mol O2/mol Cu) s−1 |
Reaction conditions: 25 μM, [CAN]: 110 mM, solvent: MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1, 2 mL).
TON after 5 minutes reaction time.
TOF was calculated from the steepest slope of the TON curve over a 5 second period. :H2O (1:1, 2 mL).
TON after 5 minutes reaction time.
TOF was calculated from the steepest slope of the TON curve over a 5 second period.
|
| 1 |
C1
|
2.67 |
0.13 |
| 2 |
C2
|
4.60 |
0.31 |
| 3 |
C3
|
3.96 |
0.14 |
| 4 |
C4
|
2.30 |
0.0086 |
Fig. S2† shows an irreversible wave assigned to the first oxidation of complex C2 from Cu(I) to Cu(II) at Epa +0.224 V (vs. normal hydrogen electrode (NHE)).15 Complexes C1–C4 showed a reversible Cu(III)/Cu(II) couple with E1/2-values in the range +0.56 to +0.83 V (vs. NHE).25 Complex C2 had the lowest oxidation potential (E1/2 = +0.56 V) and was the most efficient pre-catalyst for chemically driven water oxidation (TON and TOF of 4.6 and 0.31 s−1 respectively).
The most positive oxidation potential (E1/2 = +0.83 V) was observed for the least active phenyl (acting as an electron-withdrawing group) substituted complex C4. The oxidation potentials of the unsubstituted complex C1 and the t-butyl substituted complex C3 were E1/2 = +0.59 V and +0.82 V respectively. Although C3 had a more positive oxidation potential than C1 it was more stable under the highly acidic conditions, as it had a TON of 3.96. Steric effects can be a contributing factor to the stability of the metal-bound substrate intermediate during a catalytic cycle.23b Therefore, the activity of these catalysts is influenced by both electronic and steric effects on the pyrazolyl moiety of the catalyst.
Complex C2 was chosen for further investigations towards water oxidation due to its high activity. The rate dependency on [Cu] was studied and the data is illustrated in Fig. 3. The initial rate was determined from the first 5 seconds of the reaction time after the addition of the catalyst. At low concentrations of 2.5–10 μM, a non-linear dependency between the catalyst concentration and the dioxygen evolution was observed, which is characteristic of a second order-reaction. This suggests the formation of copper dimers at low catalyst concentration prior to catalyst activation.5,26 As the concentration increases, the initial reaction rate becomes first order as a function of [Cu], indicative that only one Cu complex is involved in the O–O bond formation step, therefore water oxidation proceeds via water nucleophilic attack.2,5,8 The similar change in reaction order as the concentration increases was also observed by Shaffer et al.27 where ruthenium bipyridine–dicarboxylate complexes were investigated in CAN driven water oxidation.
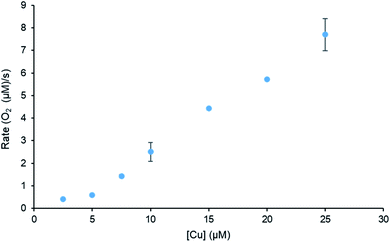 |
| | Fig. 3 Initial rate data of the catalyst system studied using C2 and 110 mM CAN as the primary oxidant showing second-order dependence at low concentrations and first-order dependence at higher concentrations. | |
The effect of [CAN] on the activity was studied at high [Cu] associated with mononuclear Cu(I) species using C2 (25 μM) as shown in Fig. S3.† At low [CAN] the rate shows a linear dependency on the concentration of CAN, attributed to the formation of a Cu–CAN complex at low [CAN].28 At a higher [CAN] (>10 mM) the reaction order is observed to be zero indicating saturation behaviour. Therefore, the rate-determining step at low Ce(IV) concentrations could be viewed as an interaction between the oxidant and the catalyst forming a Cu–CAN active complex. At high CAN concentrations, the Cu–CAN interaction is reversible and independent of [CAN] hence a zero reaction order is observed.2,28 The addition of excess amounts of CAN does not lead to an increase in the activity of the catalyst.8 This could be due to catalyst decomposition caused by ligand oxidation or hydrolysis of the metal centre due to the highly acidic medium. Other chemical oxidants such as sodium m-periodate (NaIO4) and sodium persulfate (Na2S2O8) were studied in chemically driven water oxidation using C2 (Table S3†). The initial pH of the solution was dependent on the oxidant used, with CAN having the lowest pH of 1, while the use of Na2S2O8 exhibited a pH of 2.23. and NaIO4 a pH of 4.50. CAN resulted in the highest activity with the highest TON (4.6) and TOF (0.31 s−1) amongst the three investigated oxidants. Although Na2S2O8 had the lowest TOF (0.044 s−1), the catalyst is more stable exhibiting the second highest TON of 4.02. These results demonstrate that the catalyst is most active in highly acidic conditions with CAN as the primary oxidant and Cu–CAN is the active form of the catalyst, as was proposed by Codolà et al.28 with their highly active Fe-complex.
Cu(I)-catalysed electrocatalytic water oxidation
Cu(I) complexes C1–C4 were evaluated as pre-catalysts for electrocatalytic water oxidation in phosphate buffer (Scheme 3) using CV (Fig. 4). The Cu(I) complexes are insoluble in aqueous solutions; therefore, the complexes were first dissolved in a solution of acetonitrile and 0.1 M NBu4PF6 (supporting electrolyte) followed by the addition of 20% (v/v) 0.2 M phosphate buffer (pH 6.5). The solution was degassed with nitrogen for 15 minutes, prior to each experiment. The CVs were obtained under air using a glassy carbon working electrode (GCE), Ag/AgCl reference electrode, and a platinum wire counter electrode. Potentials were measured versus a Ag/AgCl electrode and are reported versus NHE through the addition of +0.199 V to the measured potential. On scanning at 100 mV s−1 from 0 to +1.7 V, solutions of 0.42 mM in MeCN and 0.2 M phosphate buffer at pH 6.5, all complexes C1–C4 exhibited a reversible oxidation peak between E1/2 = +0.56 V and +0.83 V assigned to the Cu(III)/Cu(II) redox couple.25 This was followed by a large irreversible pH-dependent oxidation wave with an onset potential from +1.52 V to +1.63 V attributed to the oxidation of water. This irreversible wave is absent on the CV of the complex in a solution of MeCN and 0.1 M NBu4PF6, however, the wave emerges upon the addition of 20% (v/v) 0.2 M phosphate buffer (pH 6.5) (Fig. S4†).
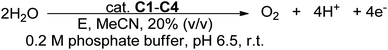 |
| | Scheme 3 Electrocatalytic water oxidation using Cu(I) complexes C1–C4. | |
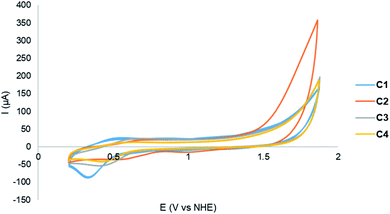 |
| | Fig. 4 CVs of Cu(I) complexes C1–C4 [0.42 mM] in a mixture of MeCN and 20% (v/v) phosphate buffer (0.2 M, pH 6.5) recorded at a scan rate of 100 mV s−1. | |
The activities of complexes C1–C4 were compared based on the overpotential at pH 6.5 (Table 2). Complex C2 had the lowest overpotential of 674 mV and a TOF of 9.77 s−1 making it the most active complex amongst the four catalysts used in this study. In comparison to literature, complex C2 has a lower overpotential and higher TOF than most reported copper pre-catalysts (Table 2 entries 7–12). However, our most active pre-catalyst, C2, still falls short in comparison to the current state of the art in copper-catalysed electrocatalytic water oxidation (Table 2, entries 5, 6 and 13). The most active copper electrocatalyst is the recently reported tetra-amidate macrocycle-ligated copper(II) complex which efficiently catalysed water oxidation at very low overpotential of 200 mV and the highest TOF of 140 s−1 in phosphate buffer at pH 7.29 Complex C1 had the second-lowest overpotential of 744 mV followed by complex C3 at 774 mV. The least active complex, with the highest overpotential of 784 mV, was complex C4. The overpotentials of the Cu(I) complexes are within the range of reported copper complexes studied at near-neutral pH (500–1000 mV).15,30 The order of overpotentials from lowest to highest is dependent on the oxidation potentials (CuIII/CuII) of the complexes. Complex C2 with the lowest oxidation potential of E1/2 = +0.56 V resulted in the lowest overpotential of 670 mV, whereas complex C4 with the highest oxidation potential of E1/2 = +0.83 V resulted in the highest overpotential of 780 mV. The order of activity based on TOF for the electrocatalytic water oxidation was as follows: C2 > C4 > C1 > C3, whereas in CAN driven water oxidation the order of activity based on TOF was: C2 > C3 > C1 > C4. Complex C2 is the most active in both the CAN-driven chemical water oxidation and electrocatalytic water oxidation, however, the least active complex C4 in CAN-driven water oxidation had the second-highest TOF of 1.47 s−1 for electrocatalytic water oxidation. The difference in activity trends between chemical water oxidation and electrocatalytic water oxidation was also observed by Olivares et al.31 with iridium complexes containing a C,N-bidentate chelating triazolylidene-pyridyl ligands as catalysts. They observed enhanced activity for CAN-driven water oxidation in the presence of electron-donating groups on the ligand scaffold whereas in electrocatalytic water oxidation the activity is highest when the triazolylidene ligand is unsubstituted. It indicates an I2M (binuclear) type mechanism where steric plays an important role, instead of a WNA (water nucleophilic attack) mechanism in electrocatalytic water oxidation with these systems.11b As observed in Table 2, there is no clear correlation between overpotential and TOF, since a catalyst can have a low overpotential and a low TOF. However, complex C2, which had the lowest overpotential in this study also had the highest TOF amongst the four complexes.
Table 2 Calculated overpotentials of copper complexes C1–C4 at pH 6.5
| Entry |
Catalyst |
Catalyst conc. (mM) |
pH |
Over-potentiala/mV |
TOFb/s−1 |
Reference |
|
The overpotential was calculated as follows: overpotential = Eonset − (1.23 − (0.059 pH)). Eonset is extrapolated from the CV using the tangent method.
TOF was calculated from the slope of icat/ipvs. 1/v1/2. Abbreviations used in this table: porphyrin = tetrakis(4-N-methylpyridyl)porphyrin, Py3P = N,N-bis(2-(2-pyridyl)ethyl)pyridine-2,6-dicarboxamidate, bpman = 2,7-[bis(2-pyridylmethyl)aminomethyl]-1,8-naphthyridine, en = 1,2-ethylenediamine, bpy = 2,2′-bipyridine, TPA = tris-(pyridylmethyl) amine, cyclam = 1,4,8,11-tetraazacyclotetradecane, Me4cyclam = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane, TAML = tetra-amidate macrocyclic ligand.
|
| 1 |
C1
|
0.42 |
6.5 |
744 |
0.33 |
This work |
| 2 |
C2
|
0.42 |
6.5 |
674 |
9.77 |
This work |
| 3 |
C3
|
0.42 |
6.5 |
774 |
0.30 |
This work |
| 4 |
C4
|
0.42 |
6.5 |
784 |
1.47 |
This work |
| 5 |
Cu porphyrin |
1.0 |
7.0 |
300 |
30 |
32
|
| 6 |
[(Py3P)Cu(OH)]− |
0.7 |
8.0 |
400 |
20 |
33
|
| 7 |
[Cu2(BPMAN)(m-OH)]3+ |
1.0 |
7.0 |
800 |
0.60 |
30b
|
| 8 |
[Cu(en)2]2+ |
1.0 |
8.0 |
540 |
0.40 |
16
|
| 9 |
[Cu4(bpy)4(μ2-OH)2 (μ3-OH)2(H2O)2]2+ |
1.0 |
7.0 |
730 |
— |
15
|
| 10 |
[Cu(TPA)(OH2)]2+ |
1.0 |
7.0 |
970 |
0.10 |
34
|
| 11 |
Cucyclam |
1.0 |
7.0 |
880 |
— |
30a
|
| 12 |
CuMe4cyclam |
1.0 |
7.0 |
880 |
— |
30a
|
| 13 |
CuTAML |
1.0 |
7.0 |
200 |
140 |
29
|
Kinetic studies
The effect of catalyst concentration on the catalytic current was studied using complex C2 in 0.2 M phosphate buffer at pH 6.5 (Fig. 5). The catalytic current at 1.7 V shows a linear dependence on the catalyst concentration at low concentrations (<0.1 mM). This indicates that water oxidation occurs at a single-site and that O–O bond formation proceeds via a water nucleophilic attack mechanism.33,35 Although this is the most common O–O bond formation pathway in electrocatalytic water oxidation involving copper complexes, it is disputed by Koepke et al.12b who suggests that molecular copper-catalysed water oxidation O–O bond formation does not proceed via the WNA mechanism, instead the I2M mechanism is thermodynamically more favourable. Intermolecular WNA and redox isomerization of the dimer were concluded energetically accessible pathways. It should be noted that their results were obtained at pH 12.5, significantly higher than the operating pH in this study. Kafentzi et al.36 studied electrocatalytic water oxidation using a copper(II) complex, [(RPY2)Cu(OTf)2] (RPY2 = N-substituted bis[2-pyridyl(ethylamine)] ligands; R = indane; OTf = triflate). At neutral pH 6–8, the complex was suggested to catalyse water oxidation at a single site with O–O bond formation proceeding via the WNA mechanism. At higher pH catalysis ceased and can be attributed to the formation of dinuclear copper(II) species. Changes in copper complex speciation induced by pH changes was also observed by Barnett et al.13 where a copper bipyridine complex was studied in electrocatalytic water oxidation. EPR spectroscopic studies showed that at pH-values lower than 8, a monomeric aquo complex [(bpy)Cu(H2O)2]2+ is observed whereas at pH-values 8–12 the dimeric form of the complex [(bpy)Cu(μ-OH)22+] is dominant. At pH higher than 12, a monomeric bis-hydroxide (bpy)Cu(OH)2 is observed. The literature reports suggest that at near neutral pH, monomeric species are most likely to be responsible for water oxidation catalysis therefore, O–O bond formation proceeds via the WNA mechanism. The effect of scan rate on the catalytic current was evaluated for C1–C4. The catalytic oxidation wave at 1.7 V was scan rate dependent as shown for C2 (Fig. S5†). The current at 1.7 V was linearly dependent on the square root of the scan rate, which is indicative of a diffusion-limited process thus indicating a homogeneous water oxidation.37 A graph of catalytic current/peak current as a function of 1/(scan rate)1/2 was plotted as shown in Fig. 6. From eqn (1)–(4)kcat, which is the catalytic TOF, was calculated from the slope of the graph.37,38| | | Icat = ncatFA[Cu](kcatDCu)1/2 | (1) |
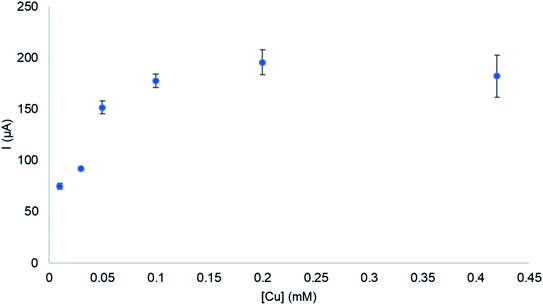 |
| | Fig. 5 The effect of concentration of the catalytic current at 1.7 V studied using complex C2 at pH 6.5. | |
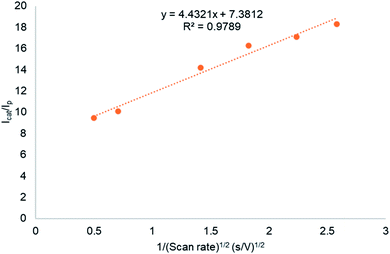 |
| | Fig. 6 A graph of catalytic current/peak current (Icat/Ip) as a function of 1/(ν)1/2 studied using complex C2. | |
In eqn (1)ncat = 4, which is the number of electrons transferred during water oxidation, F is the Faraday constant, A is the electrode surface area in cm2, [Cu] is the concentration of the catalyst (in mol L−1), and DCu is the diffusion coefficient of the catalyst.
The peak current for the non-catalytic redox couple Cu(III/II) at E1/2 = +0.56 vs. NHE varies linearly with the square root of the scan rate. The result is consistent with the Randles–Sevcik equation,
| | | ip = 0.4633nFA[Cu](nFνDCu/RT)1/2 | (2) |
At 25 °C eqn (2) becomes (3):
| | | ip = 2.69 × 105n3/2ADCu1/2[Cu] ν1/2 | (3) |
The ratio of eqn (1)/(2), icat/ip results in eqn (4)
| | | icat/ip = (0.359 nc/np3/2kcat1/2)1/ν1/2 | (4) |
where
nc is the number of electrons transferred in water oxidation (4 electrons),
np is the number of electrons transferred in a non-catalytic redox event for the Cu(
III/
II) couple (1 electron), and
ν is the scan rate. Catalytic turnover frequency,
kcat (TOF) is calculated from the slope of the plot
icat/
ipvs. 1/
ν1/2.
pH dependence studies
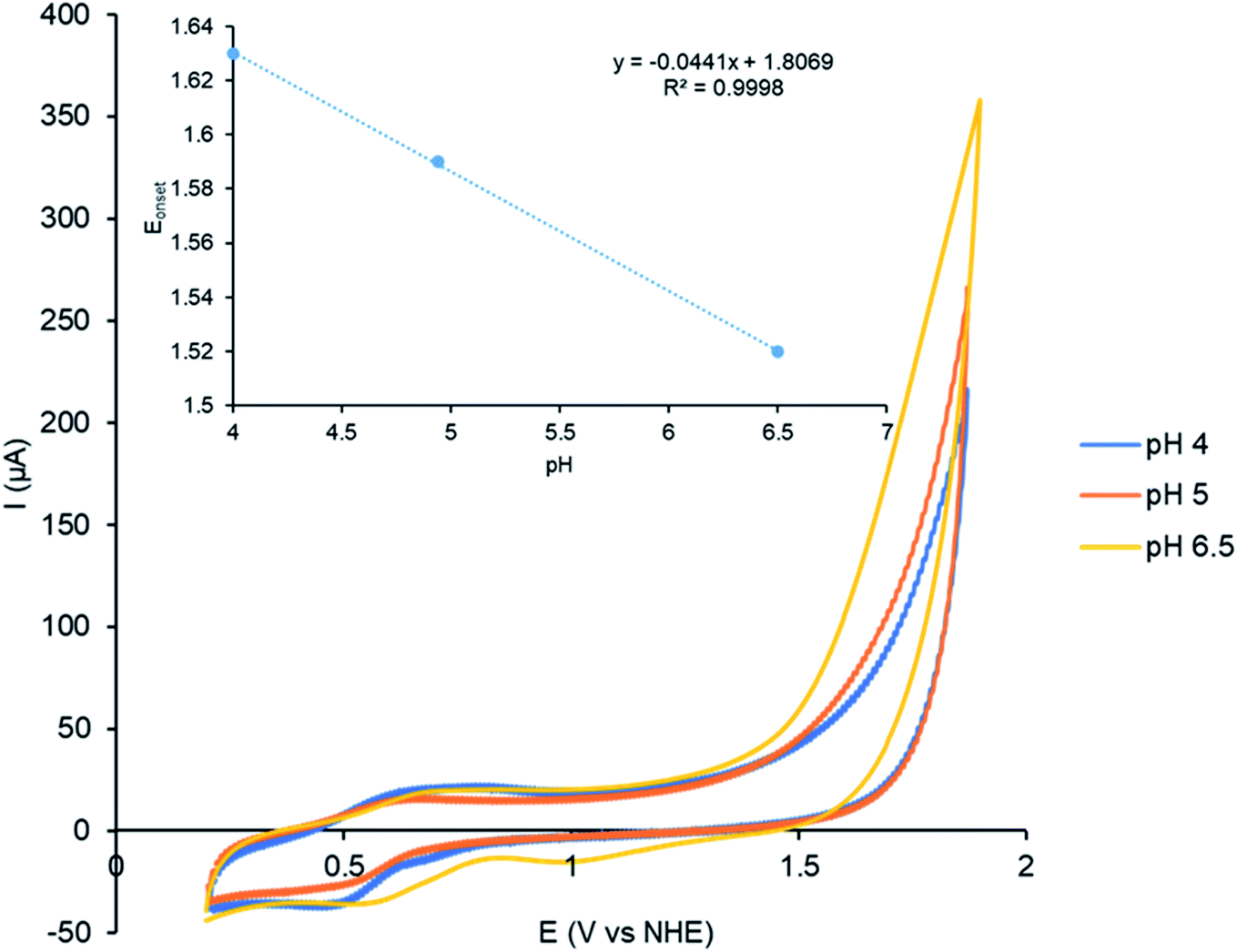
The effect of pH on the Cu(I) catalysed electrocatalytic water oxidation was investigated using complex C2 as shown in Fig. 7. A decrease in the onset potential was observed upon increasing the pH from 4–6.5 consistent with a Proton Coupled Electron Transfer (PCET) process.25,35b The onset potential for water oxidation displays pH dependence with a slope of 44 mV/pH (inset, Fig. 7).
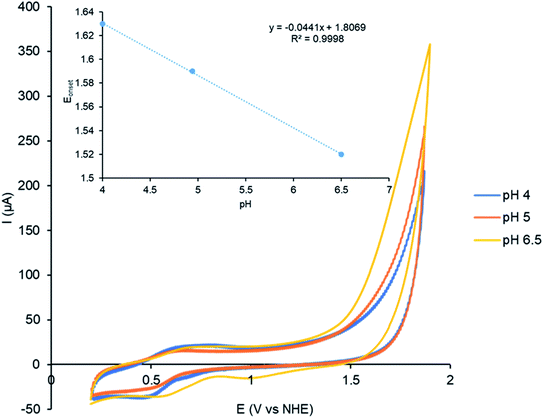 |
| | Fig. 7 CVs showing the effect of pH on the activity of the catalyst studied by complex 0.42 mM C2 in a mixture of MeCN and 20% (v/v) phosphate buffer (0.2 M) pH 4–6.5 recorded at a scan rate of 100 mV s−1. The onset potential was determined using the tangent method. | |
Evidence for homogeneous catalysis
Investigating the integrity of molecular water oxidation catalysts is often necessary as there is a possibility of catalyst degradation and nanoparticle formation. In such cases, nanoparticles are the active species for water oxidation instead of the molecular catalyst.39 During electrocatalysis, no discolouration of the electrodes was observed and suggests that no electrodeposition of nanomaterials took place on the electrode. The absence of electrodeposition after 6 cycles was confirmed by using a rinsed electrode in a blank solution as shown in Fig. 8. The current observed was the same as that of the new polished electrode, therefore no electrodeposition was observed after 6 cycles. The post-electrolysis solution of 0.42 mM C2 in a mixture of MeCN and 20% (v/v) phosphate buffer (0.2 M, pH 6.5) was analysed by Dynamic Light Scattering (DLS). The DLS results shows that there are no copper oxide nanoparticles generated in solution (Fig. S6†). All the above-mentioned methods, which probed the true nature of the catalyst, proved that the catalyst is entirely homogeneous, and no heterogeneous species were observed.
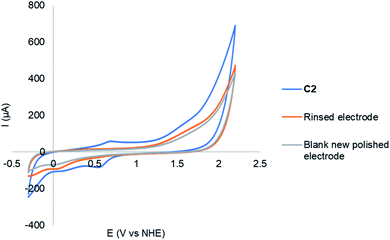 |
| | Fig. 8 CVs in a mixture of MeCN and 20% (v/v) phosphate buffer (0.2 M, pH 6.5) of 0.42 mM C2 (blue) after 6 cycles, followed by rinsed electrode in a fresh blank solution (orange) and a new polished electrode in blank solution (grey), recorded at a scan rate of 100 mV s−1. | |
Stability of the catalyst
The stability of the catalyst was investigated by scanning the electrolyte 150 times in the CV from −0.5 to +2 V (ESI, Fig. S6 and S7†). The shape of the curve changed slightly as the number of cycles increased due to the formation of oxygen bubbles on the surface of the glassy carbon electrode, which interferes with the analysis. The current increased slightly after 33 cycles, which hinted at the formation of additional active species. Therefore, electrodeposition on the glassy carbon after 150 cycles was investigated by using a rinsed electrode in a fresh blank solution (Fig. S8†). A catalytic current was observed, which suggested that the electrodeposition of nanoparticles was occurring on the electrode surface. To probe the integrity of the catalyst the electrode scanning electron microscopy (SEM) analysis was conducted on a glassy carbon electrode post-electrolysis using C2 in a mixture of MeCN and supporting electrolyte and 20% (v/v) 0.2 M phosphate buffer at pH 6.5 after 33 and 150 cycles respectively (Fig. S9 and S11†). The EDX maps (Fig. S10 and S12†) do not show the presence of CuO nanomaterials, but only the crystalline form of the supporting electrolyte (NBu4PF6). In addition, ultra-violet/visible (UV-vis) spectroscopic analysis of C2 pre- and post-electrolysis (33 and 150 cycles respectively, Fig. S13 and S14†) showed broad absorption bands from 350–450 nm, which is characteristic of π–π* and metal-to-ligand charge-transfer (MLCT) in Cu(I) complexes, albeit that the intensity of these bands increase in the post-electrolysis spectra.40 This indicates that nanoparticle formation may be occurring after prolonged electrolysis (150 cycles), but with very low quantities being electrodeposited.
Conclusions
Novel bis(pyrazol-1-ylmethyl)pyridine copper(I) complexes bearing different groups on the pyrazolyl moiety, C1–C4, were successfully synthesized and characterized. Complexes C1–C4 show activity towards chemically driven water oxidation with CAN as the primary chemical oxidant. The complexes show stability under extremely acidic conditions of pH ∼1. Complex C2 bearing a methyl group on the pyrazole was the most active catalyst with a TON and TOF of 4.6 and 0.31 s−1, respectively. The substituents on the pyrazolyl moiety had a significant influence on catalytic activity, with electron-donating groups resulting in higher activities. The initial rate order changes with catalyst concentration as the reaction is second-order at low [Cu], whereas at high [Cu] concentration the reaction is first order. Therefore, depending on [Cu], O–O bond formation proceeds via both the I2M and WNA mechanistic pathways. Complexes C1–C4 were active for electrocatalytic water oxidation under applied potential differences in solutions of MeCN and phosphate buffer at near-neutral pH of 6.5. Complex C2 resulted in the lowest overpotential of 674 mV and the highest TOF of 9.77 s−1 amongst the four complexes, which, to the best of our knowledge, is better than most of the reported copper complexes that operate at neutral pH. The catalytic current at 1.7 V showed a linear dependence on the concentration of the catalyst, which suggests single-site water oxidation catalysis taking place via the WNA mechanism. A PCET process is involved in water oxidation since the onset potential decreased with increasing pH. The order of reactivity of the complexes between chemical and electrocatalytic water oxidation differed slightly due to different reaction conditions and corresponding different water oxidation mechanisms. Regardless, complex C2 was the most active among the catalysts evaluated in this study for both chemical and electrocatalytic water oxidation.
General procedure for the synthesis of ligands and complexes
Synthesis of 2,6-bis((1H-pyrazol-1-yl)methyl)pyridine (HPzPy) (Pz = pyrazole, Py = pyridine) (L1).
A mixture of 2,6-bis(chloromethyl)pyridine (0.5 g, 2.84 mmol) and pyrazole (0.38 g, 5.68 mmol) was dissolved in toluene (20 mL). To the mixture, 12 mL of 40% NaOH was added followed by 10 drops of tetrabutylammonium hydroxide (TBAOH). The reaction mixture was stirred at 80–90 °C for 18 hours. The reaction work up involved the separation of the organic layer from the aqueous layer. The organic layer was washed with water (2 × 20 mL) and dried over MgSO4. The solvent was evaporated under reduced pressure resulting in an oily clear product. Yield: 0.64 g (94%). 1H NMR: (600 MHz, ppm, CDCl3): δ 5.42 (s, 4H, CH2), 6.29 (s, 2H, 4-H (pz)), 6.80 (d, 2H, Hβ (py), 2JHH = 6.0 Hz) 7.50–7.55 (m, 5H, Hγ (py), 5-H (pz), 3-H (pz)). 13C{1H} NMR: (150 MHz, ppm, CDCl3): δ 57.29 (CH2), 106.22 (4-C (pz)), 120.47 (Cβ (py)) 129.9 (5-C (pz) 138.08 (Cγ (py)) 139.93 (3-C) (pz) 156.49 (Cq). FTIR (ATR) ν cm−1: 1596 (CN) (py). Experimental details for L2–L4 provided in the ESI.†
Synthesis of [Cu(I)(HPzPy)]PF6 (C1).
A solution of 2,6-bis(1H-pyrazol-1-yl)methyl)pyridine (L1, 0.060 g, 0.25 mmol) in 2.5 mL dichloromethane was added to a solution of tetrakis(acetonitrile)copper(I) hexafluorophosphate (Cu(MeCN)4PF6) (0.094 g, 0.25 mmol) in 2.5 mL dichloromethane. The resulting green solution was stirred under argon for 2 hours. The addition of diethyl ether resulted in the precipitation of the complex. The complex was isolated as a yellow solid, which undergoes slow oxidation in the solid state to form a green solid. Oxidation is rapid in solution, yielding a green solution. Yield: 0.0978 g (88%). FTIR (ATR) ν cm−1: 1600 (CN). ESI-MS (m/z): 302.05 [M]+. Analysis calc. (found) for C13H13CuF6N5P: C 34.87 (35.0); H 2.93 (3.21); N 15.64 (14.78). NMR spectral data of the chloro-analogue, [Cu(I)(HPzPy)]Cl: 1H NMR: (600 MHz, ppm, CDCl3): δ 5.44 (s, 4H, CH2), 6.30 (s, 2H, 4-H (pz)), 6.88 (d, 2H, Hβ (py), 2JHH = 6.0 Hz) 7.51–7.57 (m, 5H, Hγ (py), 5-H (pz), 3-H (pz)). 13C{1H} NMR: (150 MHz, ppm, CDCl3): δ 57.46 (CH2), 106.50 (4-C (pz)), 121.07 (Cβ (py)) 130.29 (5-C (pz) 138.38 (Cγ (py)) 140.25 (3-C) (pz) 156.42 (Cq). Experimental details for C2–C4 provided in the ESI.†
Chemical water oxidation
Oxygen evolution reactions were performed using a standard Clark-type oxygraph electrode (Hansatech Instruments) placed in a thermostated cell (25 °C) for all measurements. The signal was recorded for the entire duration of the experiment at 1.0 s intervals using the Oxygraph+ software (Hansatech Instruments). The signal was calibrated using a mixture (1:1) of acetonitrile and water ([O2] = 261 μM, 25 °C).41 Stock solutions of the catalysts were freshly prepared by dissolving the complex in acetonitrile. The stock solution was then degassed with argon to ensure an oxygen-free solution. Following the addition of the CAN, the cell was purged with argon before catalyst addition. The oxidants, CAN, sodium m-periodate and sodium persulfate were purchased from Sigma-Aldrich and they were used as received.
Electrocatalytic water oxidation
Cyclic voltammetry (CV) was carried out for electrochemical characterization of the electrocatalysts employing a conventional water-jacketed three-electrode electrochemical cell connected to a refrigerated temperature controller (Julabo F-12 ED). A platinum wire (Bio-Logic) and Ag/AgCl (Bio-Logic) electrode were used as counter and reference electrodes, respectively. A glassy carbon electrode disc insert (Sigradur G, HTW Germany) with a geometrical area of 0.196 cm2 was used as the working electrode. Before performing experiments, the glassy carbon was polished with 0.05 μM alumina suspension and was subsequently washed with Milli-Q water, ethanol, and isopropanol. The working electrode was assembled into a rotating disc electrode set up. Current potential curves of the samples were recorded using a VSP double-channel potentiostat from BioLogic Science Instruments.
Author contributions
AJS, HCMV, DB and RJK contributed to conceptualisation of the project at various stages. AJS, as principal investigator, was responsible for funding acquisition and project administration. AJS, HCMV and RJK provided supervision to TM, as a doctoral candidate. TM investigated and executed the experimental work reported here. TM wrote the original draft, with review and editorial input from AJS, BD, HCMV and RJK.
Conflicts of interest
The authors have no conflict of interest to declare.
Acknowledgements
The work reported here is supported by the National Research Foundation of South Africa, Unique Grant No. 117869, 127291 and 121224. Any opinion, finding and conclusion or recommendation expressed in this material is that of the author(s) and the NRF does not accept any liability in this regard. The infrastructure support provided by the Focus Area for Chemical Resource Beneficiation (CRB) and in particular the Catalysis and Synthesis Research Group, Electrochemistry for Energy & Environment Group and Laboratory for Analytical Services (LAS), at the NWU, is gratefully acknowledged. Prof. Jan Steenekamp of NWU Centre of Excellence for Pharmaceutical Sciences (Pharmacen™) is gratefully acknowledged for providing assistance with DLS analysis.
References
- G. W. Crabtree and M. S. Dresselhaus, MRS Bull., 2008, 33, 421–428 CrossRef CAS.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gàmez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
-
(a) C. Baffert, S. Romain, A. Richardot, J. C. Lepretre, B. Lefebvre, A. Deronzier and M. N. Collomb, J. Am. Chem. Soc., 2005, 127, 13694–13704 CrossRef CAS PubMed;
(b) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed;
(c) B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451–1467 CrossRef CAS;
(d) J. Limburg, J. S. Vrettos, H. Chen, J. C. De Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS PubMed;
(e) M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–35 CrossRef CAS PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247–2252 RSC.
- D. W. Shaffer, Y. Xie and J. J. Concepcion, Chem. Soc. Rev., 2017, 46, 6170–6193 RSC.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178–1186 CrossRef CAS PubMed.
- G. Panchbhai, W. M. Singh, B. Das, R. T. Jane and A. Thapper, Eur. J. Inorg. Chem., 2016, 2016, 3262–3268 CrossRef CAS.
- W.-B. Yu, Q.-Y. He, X.-F. Ma, H.-T. Shi and X. Wei, Dalton Trans., 2015, 44, 351–358 RSC.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
-
(a) A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257, 2607–2622 CrossRef CAS;
(b) B. Das, A. Thapper, S. Ott and S. B. Colbran, Sustainable Energy Fuels, 2019, 3, 2159–2175 RSC;
(c) T. Sun and N. Xia, Int. J. Electrochem., 2018, 13, 4601–4612 Search PubMed;
(d) H. Yamazaki, A. Shouji, M. Kajita and M. Yagi, Coord. Chem. Rev., 2010, 254, 2483–2491 CrossRef CAS.
-
(a) K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS;
(b) S. J. Koepke, K. M. Light, P. E. Vannatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586–8600 CrossRef CAS PubMed;
(c) D. Lukács, Ł. Szyrwiel and J. S. Pap, Catalysts, 2019, 9, 83 CrossRef;
(d) V. K. K. Praneeth, M. Kondo, P. M. Woi, M. Okamura and S. Masaoka, ChemPlusChem, 2016, 81, 1123–1128 CrossRef CAS PubMed;
(e) B. Rudshteyn, K. J. Fisher, H. M. Lant, K. R. Yang, B. Q. Mercado, G. W. Brudvig, R. H. Crabtree and V. S. Batista, ACS Catal., 2018, 8, 7952–7960 CrossRef CAS;
(f) J. Shen, M. Wang, P. Zhang, J. Jiang and L. Sun, Chem. Commun., 2017, 53, 4374–4377 RSC.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- M. M. Najafpour, F. Ebrahimi, R. Safdari, M. Z. Ghobadi, M. Tavahodi and P. Rafighi, Dalton Trans., 2015, 44, 15435–15440 RSC.
- T.-T. Li and Y.-Q. Zheng, Dalton Trans., 2016, 45, 12685–12690 RSC.
- C. Lu, J. Du, X.-J. Su, M.-T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77–83 CrossRef CAS.
-
(a) S. O. Ojwach, I. A. Guzei, L. L. Benade, S. F. Mapolie and J. Darkwa, Organometallics, 2009, 28, 2127–2133 CrossRef CAS;
(b) A. A. Watson, D. A. House and P. J. Steel, Inorg. Chim. Acta, 1987, 130, 167–176 CrossRef CAS.
-
(a) M. S. Arayne, N. Sultana, M. A. Khan and A. Malik, Pak. J. Pharm. Sci., 2002, 15, 69–79 Search PubMed;
(b) X. Jaramillo-Fierro, C. Zambrano, F. Fernández, R. Saez-Puche, C. Costa, V. Guerrero and S. González, Univ. Sci., 2018, 23, 241–266 CrossRef.
-
(a) J. N. Hamann, B. Herzigkeit, R. Jurgeleit and F. Tuczek, Coord. Chem. Rev., 2017, 334, 54–66 CrossRef CAS;
(b) A. Mobinkhaledi, N. Forughifar, B. Hogabry and K. Zamani, J. Chem. Soc. Pak., 2002, 24, 149–151 Search PubMed;
(c) M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077 RSC.
-
(a) J. M. Sánchez-Viveros, J. Bucio-Ortega, N. Ortiz-Pastrana and J. Olguín, New J. Chem., 2019, 43, 9776–9783 RSC;
(b) S. Wang, S. Du, W. Zhang, S. Asuha and W. H. Sun, ChemistryOpen, 2015, 4, 328–334 CrossRef CAS PubMed.
- M. A. Halcrow, Crystals, 2016, 6, 58 CrossRef.
-
(a) Y. Chen, J. S. Chen, X. Gan and W. F. Fu, Inorg. Chim. Acta, 2009, 362, 2492–2498 CrossRef CAS;
(b) C. P. Lutz, M. Engelhardt and H. W. Allan, J. Chem. Soc., Dalton Trans., 1985, 1, 117–123 Search PubMed;
(c) J. S. Uber, Y. Vogels, D. Van Den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 4197–4206 CrossRef CAS.
-
(a) A. F. Abdel-Magied, W. A. A. Arafa, T. M. Laine, A. Shatskiy, M. D. Kärkäs, B. Åkermark and E. V. Johnston, ChemCatChem, 2017, 9, 1583–1587 CrossRef CAS;
(b) M. Navarro, C. A. Smith, M. Li, S. Bernhard and M. Albrecht, Chem.–Eur. J., 2018, 24, 6386–6398 CrossRef CAS PubMed;
(c) D. C. Wanniarachchi, M. J. Heeg and C. N. Verani, Inorg. Chem., 2014, 53, 3311–3319 CrossRef CAS PubMed.
- B. Das, L. Ezzedinloo, M. Bhadbhade, M. P. Bucknall and S. B. Colbran, Chem. Commun., 2017, 53, 10006–10009 RSC.
- M. T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS PubMed.
-
(a) J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS PubMed;
(b) L. Duan, C. M. Araujo, M. S. Ahlquist and L. Sun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15584–15588 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, Inorg. Chem., 2016, 55, 12024–12035 CrossRef CAS PubMed.
- Z. Codolà, L. Gómez, S. T. Kleespies, L. Que Jr, M. Costas and J. Lloret-Fillol, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- P. Garrido-Barros, D. Moonshiram, M. Gil-Sepulcre, P. Pelosin, C. Gimbert-Suriñach, J. Benet-Buchholz and A. Llobet, J. Am. Chem. Soc., 2020, 142, 17434–17446 CrossRef CAS PubMed.
-
(a) A. Prevedello, I. Bazzan, N. Dalle Carbonare, A. Giuliani, S. Bhardwaj, C. Africh, C. Cepek, R. Argazzi, M. Bonchio and S. Caramori, Chem.–Asian J., 2016, 11, 1281–1287 CrossRef CAS PubMed;
(b) X. J. Su, M. Gao, L. Jiao, R. Z. Liao, P. E. Siegbahn, J. P. Cheng and M. T. Zhang, Angew. Chem., 2015, 127, 4991–4996 CrossRef.
- M. Olivares, C. J. van der Ham, V. Mdluli, M. Schmidtendorf, H. Müller-Bunz, T. W. Verhoeven, M. Li, H. J. Niemantsverdriet, D. G. Hetterscheid and S. Bernhard, Eur. J. Inorg. Chem., 2020, 2020(10), 801–812 CrossRef CAS.
- Y. Liu, Y. Han, Z. Zhang, W. Zhang, W. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC.
- M. K. Coggins, M. T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed.
- X.-J. Su, C. Zheng, Q.-Q. Hu, H.-Y. Du, R.-Z. Liao and M.-T. Zhang, Dalton Trans., 2018, 47, 8670–8675 RSC.
-
(a) F. Chen, N. Wang, H. Lei, D. Guo, H. Liu, Z. Zhang, W. Zhang, W. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368–13375 CrossRef CAS PubMed;
(b) S. Nestke, E. Ronge and I. Siewert, Dalton Trans., 2018, 47, 10737–10741 RSC;
(c) J. Wang, H. Huang and T. Lu, Chin. J. Chem., 2017, 35, 586–590 CrossRef CAS.
- M. C. Kafentzi, R. Papadakis, F. Gennarini, A. Kochem, O. Iranzo, Y. Le Mest, N. Le Poul, T. Tron, B. Faure and A. J. Simaan, Chem.–Eur. J., 2018, 24, 5213–5224 CrossRef CAS PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods, 2001, 2, pp. 580–632 Search PubMed.
-
(a) J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed;
(b)
P. Zanello, C. Nervi and F. F. de Biani, Practice and Application, RSC, Cambridge, UK, 2003 Search PubMed.
-
(a) N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473–10481 CrossRef CAS PubMed;
(b) J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS PubMed.
-
(a) H. Chen, L.-X. Xu, L.-J. Yan, X.-F. Liu, D.-D. Xu, X.-C. Yu, J.-X. Fan, Q.-A. Wu and S.-P. Luo, Dyes Pigm., 2020, 173, 108000 CrossRef CAS;
(b) N.-Y. Chen, L.-M. Xia, A. J. J. Lennox, Y.-Y. Sun, H. Chen, H.-M. Jin, H. Junge, Q.-A. Wu, J.-H. Jia, M. Beller and S.-P. Luo, Chem.–Eur. J., 2017, 23, 3631–3636 CrossRef CAS PubMed.
- G. A. Truesdale and A. L. Downing, Nature, 1954, 173, 1236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2034903–2034905. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1se00402f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
R. J.
Kriek
b,
R. J.
Kriek