
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc-electrocatalyzed hydrogenation of furfural in near-neutral electrolytes†
Manali S.
Dhawan
 b,
Ganapati D.
Yadav
a and
Scott
Calabrese Barton
*b
b,
Ganapati D.
Yadav
a and
Scott
Calabrese Barton
*b
aDepartment of Chemical Engineering, Institute of Chemical Technology, Matunga, Mumbai 400019, India. E-mail: gdyadav@yahoo.com
bDepartment of Chemical Engineering and Material Science, Michigan State University, East Lansing, Michigan 48824, USA. E-mail: scb@msu.edu; Tel: +1 5175759393
First published on 3rd May 2021
Abstract
Electrocatalytic hydrogenation (ECH) of biomass-derived platform chemicals is a sustainable approach to produce value added fuels and chemicals as compared to the chemo-catalytic hydrogenation pathway. In this work, zinc metal was studied as a novel electrocatalyst for potentiostatic ECH of furfural to furfuryl alcohol (FAL) and 2-methylfuran (MF), products having applications in pharmaceutical, polymer and fuel industries. The activity of zinc was compared to that of other well-known catalysts, copper and nickel. The yield and faradaic efficiency (FE) of furfural ECH was studied for varying electrolyte pH, which was found to significantly affect the FE and product profile. Electrolysis in near-neutral electrolyte (pH 6 to 8) exhibited increased yields and FE as compared to under acidic and alkaline conditions. We attribute this result to the optimum proton concentration in neutral electrolytes that restricts the HER while minimizing side reactions. At neutral pH, the reaction was more selective towards FAL formation than MF. The best activity of the zinc catalyst was obtained with 0.5 M sodium bicarbonate (NaHCO3) electrolyte (pH = 8.4) at −0.7 V/RHE, yielding 73% FE for FAL and 86% FE overall. To the best of our knowledge, this is the highest FE for FAL that has been reported to date. Oxidation of zinc was observed during electrolysis, only in the presence of furfural, suggesting that oxidized zinc may play a role in the reaction mechanism.
Introduction
Interest in production of fuels and chemicals from renewable resources continues to grow, as the environmental impact of fossil resources becomes more apparent.1,2 Biomass is an important renewable resource for the production of biofuels and fine chemicals.3,4 Furfural is a bio-derived feedstock obtained by the dehydration of pentose sugars such as xylose, which in turn are obtained from the ligno-cellulosic portion of the biomass such as sugarcane bagasse, wheat straw, corn cobs, corn stover, oat hulls, etc. by acid hydrolysis.2,5–7 Furfural can be valorized by hydrogenation to form furfuryl alcohol (FAL) and 2-methyl furan (MF).6–9 FAL is used as a solvent, for the production of thermostatic resins, in adhesives and coatings and has applications in perfumery, polymer and pharmaceutical industries.2,6,10–13 MF is a promising liquid biofuel or fuel additive because of its low water solubility, high octane number and energy density.2,6,10–14 Further reduction of FAL and MF yields tetrahydrofurfuryl alcohol and 2-methyltetrahydrofuran, respectively.5,8The furfural hydrogenation reactions are typically carried out in a vapor-phase reactor at high temperatures (90–500 °C) and pressures (0.1–2 MPa) using hydrogen gas in the presence of catalysts such as copper, copper chromite, iron, Cu/SiO2, Cu/ZnO, Cu/C, bimetallic Pt–Sn, Cu–Cr, Cu–Co, CuO/CeO2/Al2O3etc.15–25 The studies report almost 98–100% conversion of furfural14,15,20,25 with 70–96% yield of FAL15–17 and 90–98% yield of MF.14,20 The storage and transportation of hydrogen gas required for these processes generate safety issues. Also, catalysts such as chromium and solvents used for conventional furfural hydrogenation are toxic and environmentally hazardous.19,26 Thus, there is a need to design reaction operating conditions that are mild and safe. Electrochemical reactions are emerging as a green alternative to conventional hydrogenation reactions for the production of fuels and chemicals, as they are carried out at room temperature, atmospheric pressure and in the presence of aqueous solvents. The electrocatalytic conversion of biomass-derived platform chemicals into value added fuels and chemicals can be carried out using hydrogenation and oxidation processes.27–32 With decreasing prices of electricity and its increasing production from renewable sources such as solar and wind energy, electrocatalytic reactions provide a sustainable method to store the energy derived from renewable feedstocks in liquid biofuels.5 We aim to convert furfural to FAL and MF by ECH, wherein the source of protons required for hydrogenation is generated in situ at the anode by water splitting rather than remotely supplied molecular hydrogen gas, thereby lowering the operation and energy cost and thus increasing the efficiency.
The ECH of furfural to FAL and MF is depicted by Scheme 1.6,10 Furfural adsorbed on the metal catalyst electrode surface reacts with two moles of protons and electrons to give rise to the hydrogenated product, FAL. FAL further hydrogenates to MF by consuming two protons and electrons with the removal of a water molecule.
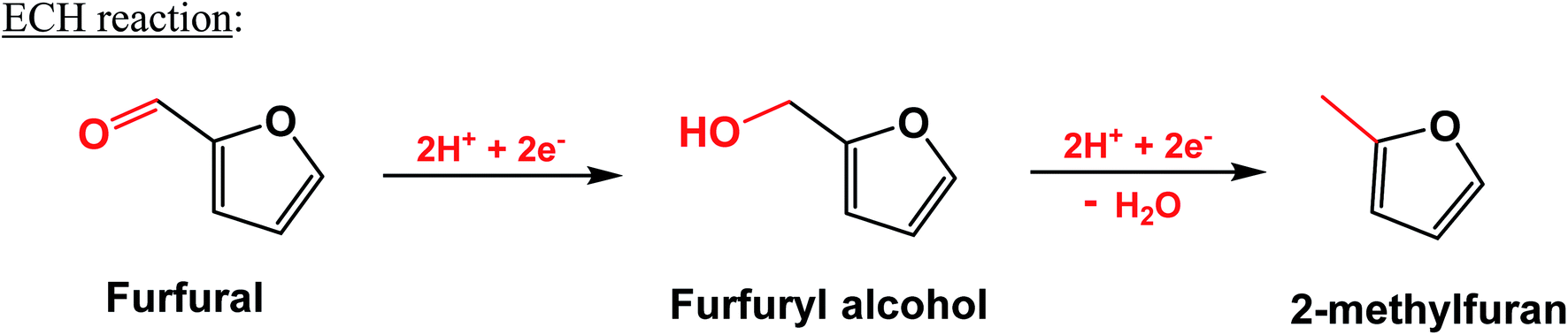 | ||
| Scheme 1 ECH of furfural to FAL and methyl furan. | ||
Dissolved protons may be partially regenerated at the anode, for example by water oxidation (1). The protons may adsorb on the surface of the metal catalyst by the Volmer reaction (2).
Water oxidation:
| 2H2O → 4H+ + 4e− + O2 | (1) |
Volmer reaction:
| H+ + e− + M → (Hads)M | (2) |
The selectivity for ECH may be limited by the HER, which consumes adsorbed hydrogen from the catalyst surface to produce H2 by Tafel or Heyrovsky reactions.2,6,10 Consumption of protons by the HER makes them less available for ECH, thereby lowering the FE of the system. Another side reaction is the dimerization of furfural radicals that are intermediate in FAL generation to produce hydrofuroin (Scheme 2).12,13,33 The choice of a metal catalyst that has high FE for ECH over both the HER and dimerization helps to maximize the efficiency of the reaction system.
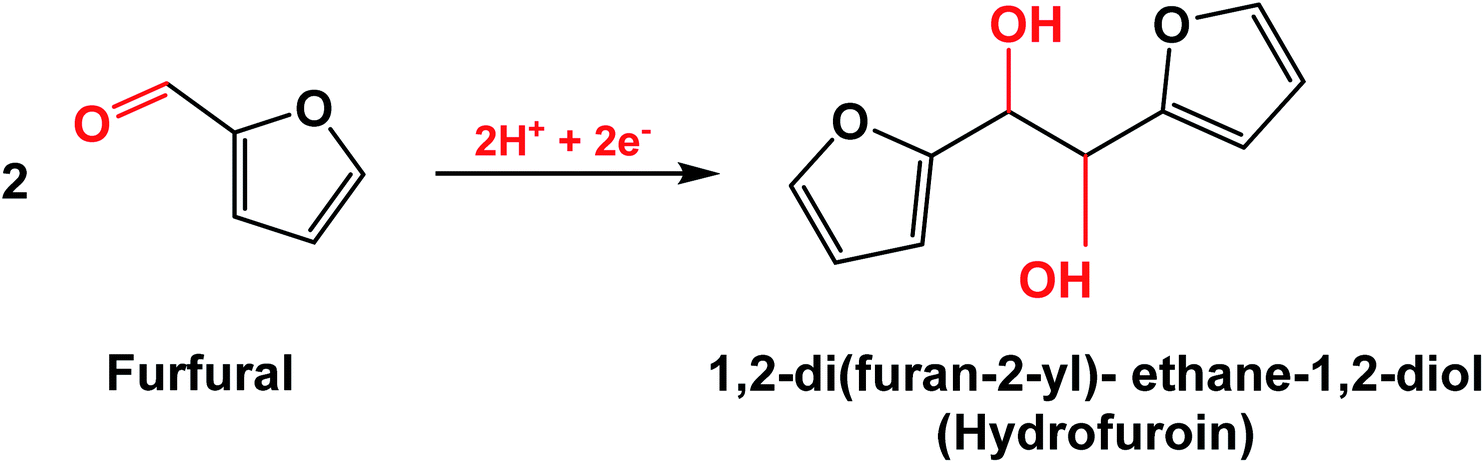 | ||
| Scheme 2 Electrodimerization reaction of furfural to hydrofuroin.33 | ||
There have been a number of reports of furfural ECH by various groups, who studied the impact of reaction conditions and electrode materials on the conversion, FE and product yields.11,34–36 The literature for ECH of furfural has been mainly dominated by copper and nickel as a catalyst at various pH values resulting in good reactant conversion and product yields.2,12,28,34,35,37 Zhao et al. compared the reactivity of copper, nickel, lead and platinum for the ECH of furfural. Pure platinum gave the highest selectivity for FAL (99%), but the conversion was very low. Copper gave high conversion and low selectivity whereas nickel yielded low conversion and high selectivity in basic pH with higher onset potentials.38 Thus, there is a need to find a catalyst that gives both higher conversions and selectivities for the ECH of furfural.
Apart from metal catalyst screening, the effect of pH on FE and product selectivities from ECH of furfural have been studied.12 Li et al. achieved a good yield of FAL (63%) in 0.2 M ammonium chloride (pH 5) using nickel as a catalyst cathode with an electrochemical efficiency of 56% (ref. 6) whereas MF was reported to be formed at higher selectivities (∼80%) at low acidic pH (∼0.5) by Nilges et al.5
Similar findings were achieved by Jung et al. who studied the activity of bulk copper and compared it to higher surface area nano- and microcrystalline copper catalyst particles electrodeposited on copper itself.13,29 The highest selectivity for FAL (25.1%) was obtained at pH 5.13 Thus, the main conclusion of these studies was that MF was formed under strongly acidic conditions (pH = 0.5) and FAL was a preferred product at a pH of 5. Copper was found to promote FAL and MF formation over H2 gas at less negative potentials, but the conversion was low. More negative potentials increased the conversion of furfural but at lower FE.13
The applied potential may determine whether the HER or ECH is favored.10,13,29 In acidic pH, the onset potentials for both ECH and the HER are close, which leaves less room to modify the kinetics of the reactions.10,29 High starting concentrations of furfural (100 mM) have been shown to favor ECH over the HER due to higher catalytic surface coverage and adsorption.6,10 A further increase in the initial furfural concentration to 200 mM has been reported to promote undesired electrodimerization reactions.10 Higher electrolyte concentrations have also been reported to favor ECH, and higher conversion and product yield.13 High electrolyte concentrations provide high ionic conductivity, reducing potential drop across the cell.
It was also shown by Jung and Biddinger that furfural, FAL and MF degrade in acidic solutions by polymerization/oligomerization to give rise to humins.13 Kim et al. used Raman spectroscopy to observe the acid-catalyzed polymerization of FAL in sulfuric acid at room temperature to conjugated diene and diketone species, which were also evident by the color and viscosity changes of FAL within an hour.39 Liu et al. studied the reactivity of various catalysts in basic carbonate buffer at pH 10, and the highest current efficiency of FAL (70%) was achieved with copper at −0.56 V/RHE. However, the reaction time of 10 h was required to achieve these results.40 Thus, neutral or basic pH can be expected to avoid degradative side reactions and increase the FE of furfural ECH.
Other catalysts such as gold,41 silver,30 palladium,42 rhodium43etc. have been reported for ECH in the literature but have not been used for ECH of furfural. Zinc has been reported to effectively catalyze CO2 and 5-hydroxymethylfurfural (HMF) electroreduction with high faradaic efficiencies.41,44,45 Feng et al. observed an 18% increase in FE for the formation of ethylene from CO2 after alloying a copper catalyst with zinc and using it as a bimetallic catalyst.46 HMF is a structural analog of furfural with an additional hydroxyl functional group present at the fifth carbon. Zinc exhibited exceptional catalytic activity for the reduction of HMF to 2,5-hexanedione with a FE of 72.4% and outperformed copper by 70% in terms of FE and selectivity to the desired product.41 Many reports on chemical hydrogenation of furfural have been published using zinc oxide catalysts either as an active phase or as a support.14–16 Thus, it was hypothesized that the zinc catalyst could have good catalytic activity for the ECH of furfural.
In this work, the hydrogenation of furfural to FAL was carried out using various catalysts such as copper, nickel and zinc. This is the first-time zinc has been used as metal catalyst for the hydrogenation of furfural. The present work establishes zinc as a catalyst that is selective for the ECH of furfural while inhibiting the HER. The variables used to assess the activity of the catalyst were the applied potential, achieved current density, reactant conversion, yield and FE for the desired reaction products. Apart from these variables, the electrolyte and the pH, which determine the proton concentration in the solvent and thus affect the selectivity of the reaction and product formation have also been studied.10,13 Preliminary tests to screen the activity of the catalyst under different pH conditions – acidic, basic and neutral were performed on the catalyst wire using transient and steady-state voltammetric techniques such as cyclic voltammetry (CV) and staircase voltammetry (SV). The catalysts and electrolyte pH were screened on the basis of potential windows and the current density for the occurrence of the desired ECH reaction and to avoid the HER. The activity of the catalysts for the ECH was screened on the basis of FE, conversion and selectivity towards the desired products measured by potentiostatic electrolysis.
Experimental
Chemicals
The following chemicals were obtained from reputed vendors and used without further purification: furfural (99%, Sigma-Aldrich), FAL (FAL, 98%, Sigma-Aldrich), MF (MF, 99%, 200–400 ppm BHT as stabilizer, Sigma-Aldrich), acetonitrile (ACN, 99.5%, Mallinckrodt Chemicals), p-xylene (≥99%, Sigma-Aldrich), sulfuric acid (H2SO4, 98%, EMD Chemicals), sodium hydroxide (NaOH, Macron Fine Chemicals), sodium bicarbonate (NaHCO3, 100.3%, J. T. Baker), sodium sulphate (Na2SO4, 99.6%, Fisher Chemical), ammonium chloride (NH4Cl, ≥99.5%, Columbus Chemical Industries), sodium chloride (NaCl, ≥99%, Columbus Chemical Industries), sodium phosphate monobasic (NaH2PO4·H2O, 99.7%, J. T. Baker), and sodium phosphate dibasic (Na2HPO4·7H2O, Jade Scientific). 0.5 M phosphate was prepared by mixing 0.37 M Na2HPO4·7H2O and 0.13 M NaH2PO4·H2O. All the electrolytes were prepared in deionized water (≥18 MΩ cm, Thermo Scientific).Electrodes
The metal wires used as working electrodes for the ECH of furfural are as follows: copper (1.2 mm diameter, 18 AWG, 99.9%, Arcor Electronics), nickel (1.0 mm diameter, 99.98%, Alfa Aesar), and zinc (1.0 mm diameter, 99.95%, Alfa Aesar). The catalyst wire electrodes were rubbed with sandpaper (800 grit (P2400), diameter = 6.5 μm, Buehler) to remove oxides, and rinsed with ethanol and deionized water prior to being used for the reaction. The geometric surface area of the working electrode for cyclic and staircase voltammetry experiments was 1 cm2 and that for electrolysis experiments was 5 cm2. A reversible hydrogen electrode was used as a reference electrode (RE). It was made in-house using a Pt wire (0.5 mm dia., 99.95%, Thermo Fisher Scientific) in a one-end sealed glass tube. The RE tube was filled with working electrolyte before each experiment. A graphite rod (2.5 cm diameter, Pine Research Instrumentation) was used as a counter electrode.Electrochemical set-up
The reactions were conducted using a three-electrode system connected via a VSP Bio-logic SA potentiostat in a divided glass jacketed H-cell. The compliance voltage range of the potentiostat was 20 V. The anodic chamber contained 25 ml of desired electrolyte and the cathodic chamber contained 25 ml of 100 mM furfural in the same electrolyte. The anodic and cathodic compartments were separated by a Nafion 115 cation exchange membrane (Ion Power). Nafion 115 membranes were pretreated by boiling in 1 M solutions of sulfuric acid, sodium sulphate or sodium hydroxide at 80 °C for 1 h depending on the pH to be used for electrolysis i.e. acidic, neutral, and basic respectively. They were subsequently boiled in DI water at 80 °C for 1 h and stored in DI water thereafter. The on-center distance between the two compartments was measured to be 5 cm. The pH of the electrolyte was measured prior to and following every reaction using a pH meter (Fisher Scientific, Accumet Basic AB15). The cathodic chamber was purged with nitrogen throughout the reaction to maintain oxygen-free conditions and to create a positive pressure inside for the removal of evolved gases and MF. MF was collected in a cold trap connected to the H-cell as it is volatile.13 The cold trap was maintained at −15 ± 3 °C using 20 wt% NaCl and ice mixture in Dewar.5 All reactions were carried out at 30 ± 0.1 °C. A rotation speed of 900 rpm was maintained using a magnetic stirrer in the cathode chamber.Electrochemical techniques
Cyclic voltammetry (CV) was used for transient studies to analyze the onset potential and reduction peaks for copper, nickel and zinc electrodes in different pH electrolytes. The onset potential was recorded as the potential where the current reached 0.1 mA. CVs were acquired at a scan rate of 20 mV s−1 with end potential as −1 V/RHE. The starting potential for CV was varied based on the open circuit potential of the electrode in the electrolyte, in order to record the onset potential.Staircase voltammetry (SV) was used as a steady-state technique to analyze the faradaic current density obtained with each electrode in different pH electrolytes while avoiding the background capacitive current to a greater extent as opposed to CV. For SV, a potential step of 100 mV was applied consecutively from −0.6 to −1 V/RHE with a halt at each potential for 1 min and then reversed to the initial potential to check steady state current density.
Potentiostatic electrolysis experiments were run using chronoamperometry at the desired potential for 2 h.
Analysis
During electrolysis, 0.2 ml of reaction samples were collected intermittently at specific time intervals up to 2 h. Reaction analysis was performed with a Varian 450 gas chromatography system equipped with a SolGel-Wax column (30 m, 0.53 mm ID, 1 μm film thickness) using a flame ionization detector and helium as a carrier gas at 1 ml min−1. The temperature program started at an initial temperature of 37 °C (held for 4 min) and then ramped at 10 °C min−1 to 90 °C (held for 3 min) followed by ramp at 10 °C min−1 to 150 °C and ramp at 30 °C min−1 to 230 °C (held for 2 min). The split ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. Injector and detector temperatures were set at 270 °C.47 The furfural, FAL standards and reaction samples were prepared by ten-fold dilution with ACN containing p-xylene as the internal standard. The diluted samples were filtered with a 0.22 μm syringe filter prior to injection on the column. The MF standard was prepared in ACN, and the cold trap sample was directly analyzed without any dilution. The unknown concentration in the reaction samples was calculated by comparing to the calibration curve prepared using the standard samples of furfural, FAL and MF.
:100. Injector and detector temperatures were set at 270 °C.47 The furfural, FAL standards and reaction samples were prepared by ten-fold dilution with ACN containing p-xylene as the internal standard. The diluted samples were filtered with a 0.22 μm syringe filter prior to injection on the column. The MF standard was prepared in ACN, and the cold trap sample was directly analyzed without any dilution. The unknown concentration in the reaction samples was calculated by comparing to the calibration curve prepared using the standard samples of furfural, FAL and MF.
Some diluted reaction samples were also analyzed by gas chromatography with mass spectrometry (GC-MS). We used a Shimadzu QP-5050A gas chromatograph equipped with a Restek Rtx-1701 capillary column (60 m, 0.25 mm ID, 0.25 μm film thickness) coupled with an electron ionization mass spectrometer. The ionization energy was 80 eV and m/z values ranged from 40 to 400.6 The GC program started at 40 °C (held for 1 min) and then ramped at 3 °C min−1 to 121 °C followed by ramp at 8 °C min−1 to 270 °C (held for 1 min). Injector and detector temperatures were set at 270 °C. The mass spectrum of each chromatogram peak was identified by comparing to the mass spectrum of the probable chemical compounds provided by the NIST library, based on the similarity index.
It is possible for furfural to cross over through the Nafion membrane to the anode compartment, resulting in an error in our conversion calculations. We conducted crossover measurements to observe the concentration of furfural transferred to the anode by permeation through the membrane. The concentration profile was fitted to an equation derived for the flux of furfural across the membrane which is proportional to the concentration gradient (Fig. S2†). The fit demonstrated a membrane permeability to furfural of 1 × 10−6 cm2 s−1. Based on this value, we estimate an error of less than 1% in our conversion estimates due to furfural crossover from the cathode to anode during electrolysis.
Calculations
The conversion, yield and FE were calculated as follows: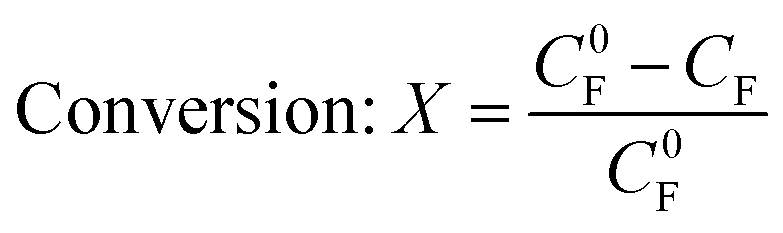 | (3) |
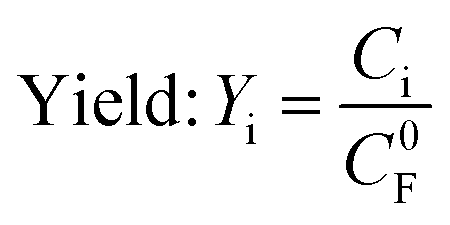 | (4) |
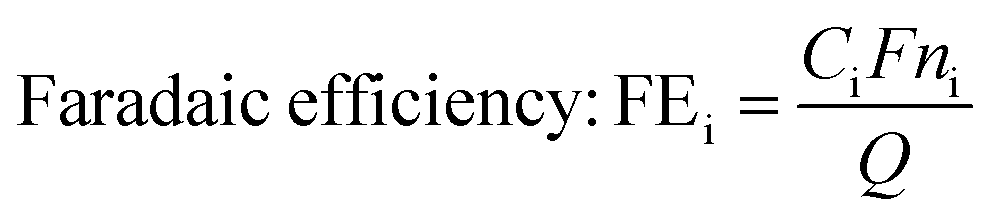 | (5) |
Catalyst characterization
The surface morphology and elemental analysis of zinc wires and precipitates was conducted using scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM/EDS, JEOL 6610LV). Dried wires were mounted on specimen stubs partially coated with a thin film of carbon tape. The wire portion which is not stuck to the carbon tape was analyzed. The precipitates were embedded in a cylinder stub with a hole for sample containment to avoid carbon taping as it would obscure any organic carbonaceous compound present in the precipitates. The samples were analyzed at a low vacuum of 60 Pa and acceleration voltage of 10 kV.Results and discussion
Steady state voltammetry with zinc in different pH electrolytes
To study the catalytic activity of the zinc catalyst for hydrogenation of furfural, we first conducted polarization studies of the zinc electrode in the presence and absence of furfural, and in different electrolytes. Fig. 1 shows steady-state polarization curves of the zinc wire in three 0.5 M electrolytes of widely varying pH: H2SO4 (pH = 0.5), NaOH (pH = 13.9), and NaHCO3 (pH = 8.4), representing acidic, basic and neutral electrolytes. Dashed and bold lines indicate the potential-dependent current density in the absence and presence of 100 mM furfural, respectively.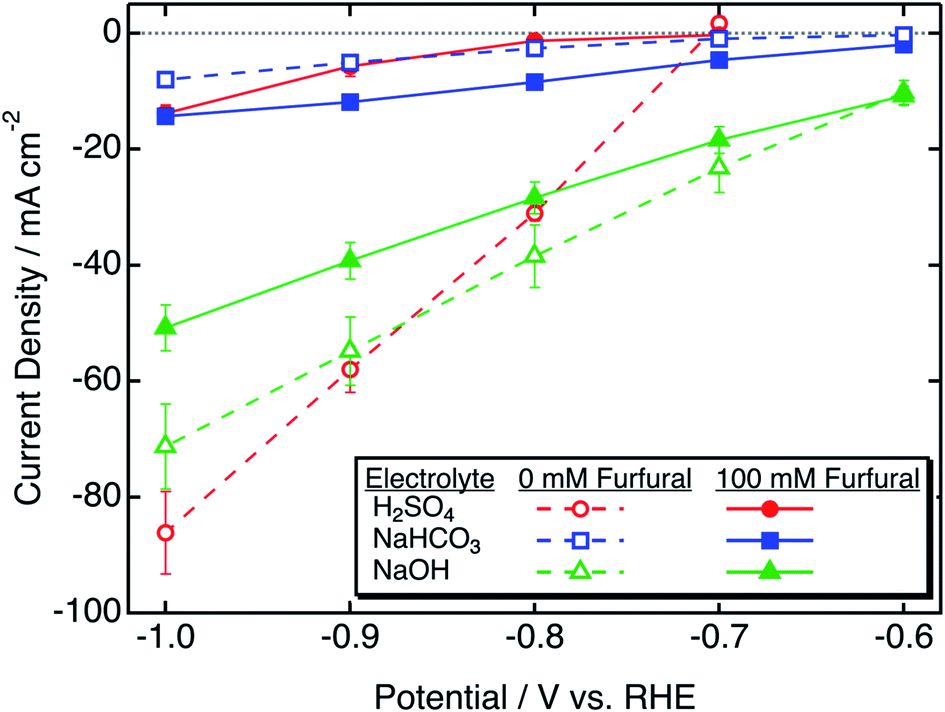 | ||
| Fig. 1 Furfural reduction and HER polarization curves obtained with zinc in electrolytes of varying pH. | ||
With no oxygen or furfural present, only the HER occurs on the catalyst in this potential range. The addition of furfural to the electrolyte leads to increased or decreased current density, depending on the dominance of the ECH reaction and the effect of furfural on the HER.
With zinc at acidic pH, the polarization curve was recorded from −0.7 to −1 V/RHE because of the low onset potential (Table 1). There was a significant decrease in faradaic current density in the presence of furfural. This suggests that the HER was hindered, which could be due to the surface coverage of the electrode with furfural that would affect electron transfer to form H2 gas. The low current density would still suggest less activity and slower kinetics of ECH at acidic pH. Additionally, in the absence of furfural, the linear potential dependence at zero current suggests a mixed reaction involving both the HER and zinc oxidation.
| Electrolyte | Onset potential (V/RHE) | |
|---|---|---|
| HER | ECH | |
| H2SO4 | −0.69 ± 0.003 | −0.69 ± −0.01 |
| NaHCO3 | −0.5 ± 0.005 | −0.56 ± 0.007 |
| NaOH | −0.45 ± 0.001 | −0.39 ± 0.002 |
At basic pH, there was a lesser decrease in current density after the addition of furfural as compared to acidic pH. Thus, we might expect ECH to have higher efficiency in basic electrolytes. In near-neutral pH electrolyte, 0.5 M NaHCO3, at all studied potentials, an increase in current density was observed after the addition of furfural. This strongly suggests that ECH is a more dominant reaction in neutral pH electrolytes and higher efficiency of ECH may be achieved under these conditions.
Table 1 shows the onset potential of the HER and ECH reactions with zinc at different pH values, recorded from their respective cyclic voltammograms (ESI, Fig. S3†). In the case of acidic pH, the onset potential of the HER and ECH was similar, and thus the potential window cannot be altered to favor ECH over the HER, and the HER is the dominant reaction at low pH at all applied potentials.
At basic pH, the onset potential of ECH was less negative than that of the HER, but decreased current density was observed after addition of furfural (Fig. 1) suggesting that ECH selectivity would increase at less negative potentials. In near-neutral pH electrolyte, 0.5 M bicarbonate, although the onset potential of ECH was more negative than that of the HER, higher current density in the presence of furfural (Fig. 1) suggests higher selectivity toward ECH over the HER.
Cyclic voltammograms obtained with zinc in the presence of furfural in different pH electrolytes show features due to various reactions involving metallic zinc (Fig. S3 and S4†). These may include oxidation–reduction, dissolution to form zinc hydroxide, and hydrogenation of FAL to MF. In acidic electrolyte, zinc also shows additional onset peaks in the absence of furfural (Fig. S3†). This could be due to high instability of the zinc catalyst at acidic pH which leads to its oxidation and dissolution.48
Steady state voltammetry with varying catalysts in near-neutral electrolyte
Steady-state polarization performed with zinc in bicarbonate was compared to that performed with well-studied metallic ECH catalysts, copper and nickel.2,6,10,13,40 Steady state polarization curves obtained with these catalysts are shown in Fig. 2.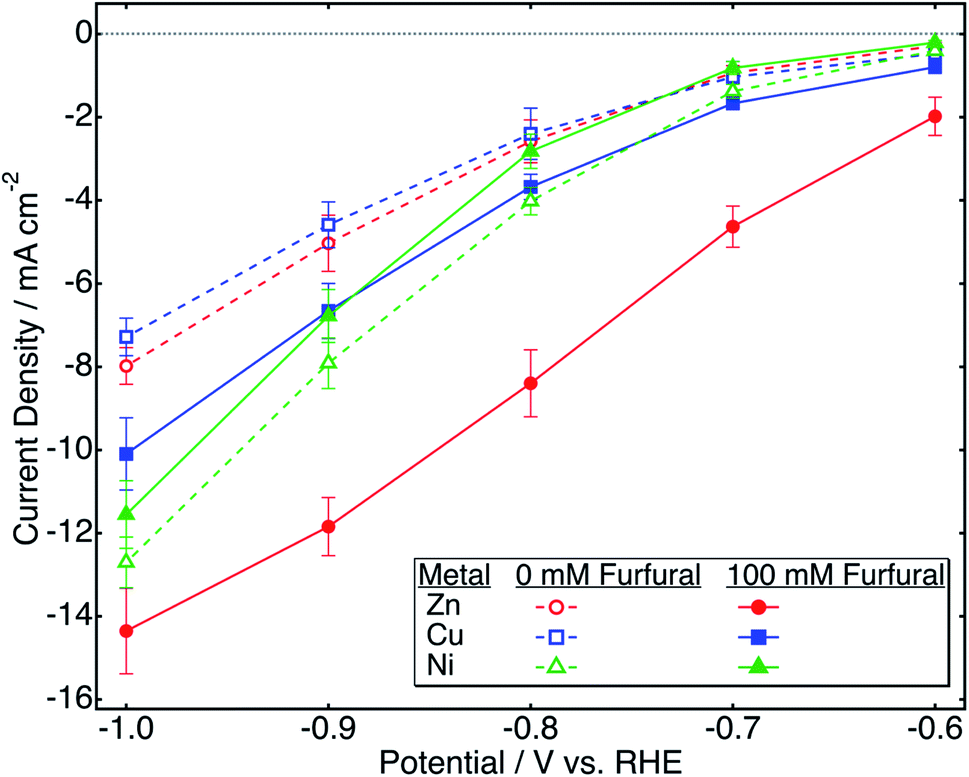 | ||
| Fig. 2 Furfural reduction and HER polarization curves obtained with different catalysts in 0.5 M NaHCO3. | ||
With nickel, there was a decrease in current density after the addition of furfural, which indicates fast kinetics of the HER over ECH. In contrast, both copper and zinc catalysts showed an increase in current density after the addition of furfural, with the increase with zinc being considerably greater as compared to that with copper at all potentials. At −0.7 V/RHE, the increase in current density with zinc was around 398% as opposed to 61% with copper. Thus, zinc catalysis appears to be selective toward furfural ECH.
Table 2 shows ECH and HER onset potentials obtained by CV in NaHCO3 with different catalysts. For the nickel catalyst, the onset potential for ECH was significantly more negative as compared to that of the HER; thus, we might expect the HER to be dominant over ECH at all potentials on nickel. The onset potentials of ECH and the HER were close for both copper and zinc, suggesting competitive kinetics for both reactions. No additional peaks were observed with copper or nickel catalysts in the absence or presence of furfural (Fig. S4†).
| Catalyst | Onset potential (V/RHE) | |
|---|---|---|
| HER | ECH | |
| Copper | −0.31 ± 0.009 | −0.29 ± −0.015 |
| Zinc | −0.5 ± 0.005 | −0.56 ± 0.007 |
| Nickel | −0.24 ± 0.009 | −0.45 ± 0.03 |
Electrolysis with zinc in 0.5 M NaHCO3 at −0.7 V/RHE
Fig. 3a shows the variation of current density with time during a two-hour potentiostatic electrolysis of zinc in bicarbonate electrolyte at −0.7 V/RHE. The initial current density of ∼22 mA cm−2 increases for 3 minutes, reaching 26 mA cm−2. This can be due to an initial period of activation which may involve adsorption of furfural on the electrode, zinc dissolution, or surface oxide formation. After this period, the current density continuously decreases with time, reaching 1.3 mA cm−2 after 120 minutes. | ||
| Fig. 3 Potentiostatic electrolysis of furfural on the zinc catalyst in 0.5 M NaHCO3 at −0.7 V vs. RHE (a) chronoamperometry during electrolysis; (b) evolution of the conversion, yield, and FE of products. | ||
Typical gas chromatograms (GC) of the reaction mixture containing furfural, FAL, and p-xylene (internal standard) are shown in Fig. S1 (ESI†). Data are included for pre- and post-electrolysis along with the post electrolysis cold trap sample containing MF. GC results are interpreted as conversion, yield, and FE in Fig. 3b. A steep increase in conversion occurs during initial electrolysis. In contrast, the relatively small yield and FE of MF were assayed only at the end of electrolysis.
The initial furfural concentration of 100 mM has been well studied in the literature6,10,13,29,34,40 allowing for direct comparison of results. After two hours, furfural conversion of 88% was obtained with 40% yield of FAL. The yield and FE of MF were obtained as 3.5% and 13%, respectively. The zinc-catalyzed reaction maintained nearly constant faradaic efficiency, varying from 72 to 77% throughout the reaction, a demonstration of high selectivity towards furfural ECH over the HER.
Effect of electrolyte pH on the activity of the zinc catalyst
Identifying the pH range that minimizes both the HER and degradative side reactions can maximize product yield and FE for ECH. Potentiostatic electrolysis experiments were conducted in electrolytes of various pH using the zinc catalyst, to observe the stability of furfural and its products at various pH values. A high electrolyte concentration of 0.5 M was used for all the electrolytes, to minimize ionic resistance.Electrolysis was performed at −0.7 V/RHE for 2 h in 0.5 M electrolytes of the following composition and pH: sulfuric acid (H2SO4, pH = 0.5), sodium sulphate (Na2SO4, pH = 6), phosphate (Na2HPO4/NaH2PO4, pH = 7.4), sodium bicarbonate (NaHCO3, pH = 8.4) and sodium hydroxide (NaOH, pH = 13.9).
Fig. 4 shows the conversion of furfural and yield and FE of both FAL and MF after 2 h electrolysis at −0.7 V/RHE in different electrolytes. Yield and FE are displayed as stacked bars, to indicate the overall value as well as the contribution to furfuryl alcohol (FAL) and methyl furan (MF). The pH of the electrolyte significantly affected the product yield and FE. At acidic pH, the conversion of furfural after 2 h was only 11.5% which resulted in very low yields of MF (0.2%). Such low conversion can be explained by a higher proton concentration, favoring the HER. No FAL product was observed in acid. However, the presence of MF, the hydrogenation product of FAL, suggests the degradation of FAL possibly by resinification under acidic conditions.34 MF may escape the degradation reaction in acidic electrolyte due to its lower aqueous solubility.
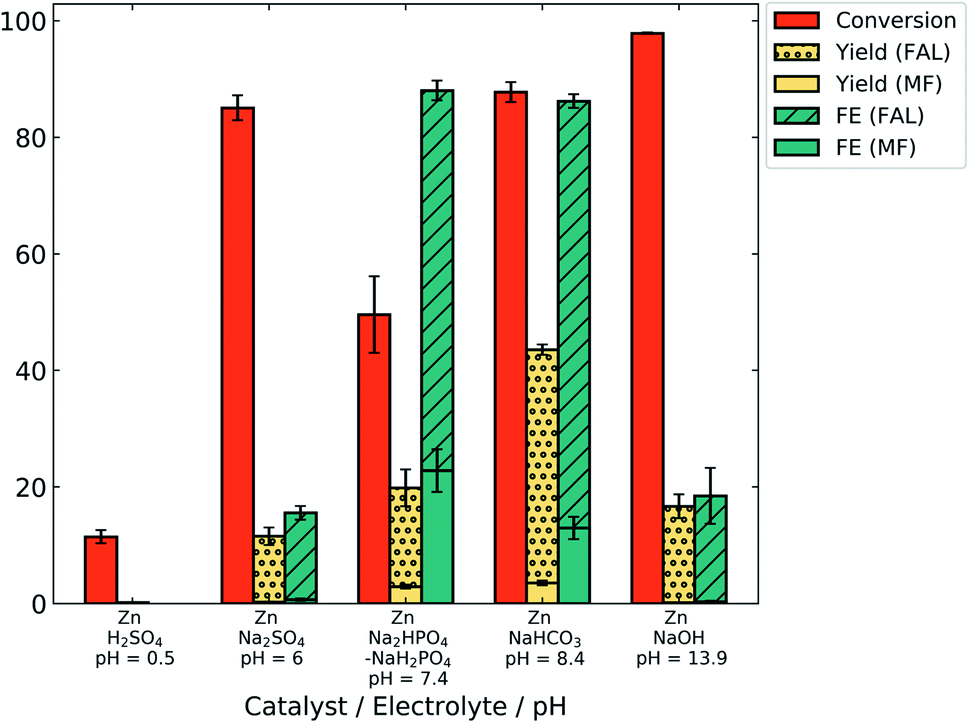 | ||
| Fig. 4 ECH electrolysis results with the zinc catalyst at −0.7 V vs. RHE for 2 h in different pH electrolytes. | ||
The open circuit potential of zinc in acid was measured to be −0.61 ± 0.01 V/RHE (Fig. S6†). Thus, the observed current density obtained at −0.7 V was low (−0.25 ± 0.14 mA cm−2) due to the lower overpotential, leading to imprecise FE calculations. For this reason, FE values are not given for H2SO4 (pH 0.5) in Fig. 4.
In alkaline electrolyte (NaOH, pH 13.9), conversion was very high (98%). Still, the yields of FAL and MF remain low at 16.5% and 0.1%, respectively. High conversion may result from minimal competition by the HER. Low product yield, on the other hand, suggests that either furfural or the generated FAL degraded. Highly acidic and basic conditions have been reported to induce furfural polymerization reactions.13,39,49
However, similar results were observed at pH 6 in Na2SO4 electrolyte, with high conversion (85%) and low yield (11.3% for FAL). This may indicate polymerization of furfural in neutral electrolyte as well. The buffer strength of the electrolyte may also play a role; pH measurements of pre- and post-electrolysis show a significant pH shift for un-buffered Na2SO4 electrolyte (ESI, Fig. S6†), suggesting that polymerization may take place at high pH.
An increase in the yield and FE of products was observed as the pH was increased from 6 to 8 using buffered electrolytes. In 0.5 M phosphate (pH = 7.4) and bicarbonate (pH = 8.4) electrolytes, the yield of FAL was 17% and 40% for a conversion of 49.5% and 88%, respectively, which suggests reduced degradation and higher ECH efficiency in these electrolytes. Additionally, the highest FE for MF (23%) was obtained in phosphate electrolyte (pH = 7.4).
The total FE obtained in phosphate electrolyte (88%) was close to that obtained with bicarbonate electrolyte (86%). This suggests that a pH range of 7–9 could be optimum to favor ECH over the HER with minimal degradation. In our study, the highest FAL yield (40%) and FE (73%) were obtained in bicarbonate electrolyte at pH 8.4. As a control, a negligible change in the furfural concentration (0.03%) was observed over 2 h in bicarbonate electrolyte without any electrode (data not shown), which eliminates the possibility of non-electrochemical or autocatalytic polymerization reactions of furfural at pH 8.4. Thus, bicarbonate electrolyte (pH 8.4) was used for further studies.
In all the neutral pH range electrolytes, we observed that the pH of the reaction mixture increased during electrolysis. The cathode open-circuit potential (OCV) was also observed to shift to more negative potentials during electrolysis. We consider the change in OCV to be related to the increased pH, due to proton depletion. Fig. S6 (ESI†) shows the change of pH and OCV during the two-hour ECH reaction in different pH electrolytes. Strong acidic H2SO4 (pH = 0.5) and basic NaOH (pH = 13.9) electrolytes did not show a significant change in pH during the reaction because of low pKa and pKb values, respectively. H2SO4 (pH = 0.5) electrolyte also did not show a significant change in the OCV. However, the NaOH electrolyte did show a high change in OCV, possibly due to the lower proton concentration. Among the neutral pH range electrolytes, Na2SO4 (pH = 6) showed the largest shift in both pH and OCV, which is expected as it is a non-buffered electrolyte.
ECH in buffered electrolytes (bicarbonate, phosphate) led to positive shifts in pH accompanied by negative shifts in the OCV. We surmise that the lower magnitude of pH increase, and the consequent OCV decrease, during ECH in these electrolytes is related to buffer capacity.
The buffer capacity of 0.5 M sodium bicarbonate and phosphate was calculated analytically using their respective equilibrium equations and disassociation constants and assuming electroneutrality (Fig. S7†). The resulting buffer capacity of phosphate (pH 7.4) was 0.27 M per pH and that of bicarbonate (pH 8.4) was 0.03 M per pH, largely due to the proximity of the nearest pKa (7.2 for phosphate, 6.4 for bicarbonate). The higher buffer capacity of phosphate explains its higher resistance to change in pH and hence, the OCV.
Effect of the applied potential on the activity of the zinc catalyst in NaHCO3
The activity of zinc in bicarbonate electrolyte was studied by potentiostatic electrolysis at the applied potentials of −0.6 and −0.7 V/RHE to determine the potential that would give the maximum conversion, yield, and FE of the products (Fig. 5). Less negative potentials, i.e. lower overpotentials, are preferred to maximize energy efficiency. Potentiostatic electrolysis provides an advantage over galvanostatic experiments in that the kinetics of the system can be studied with control over the potential, a significant thermodynamic driving force for the reaction.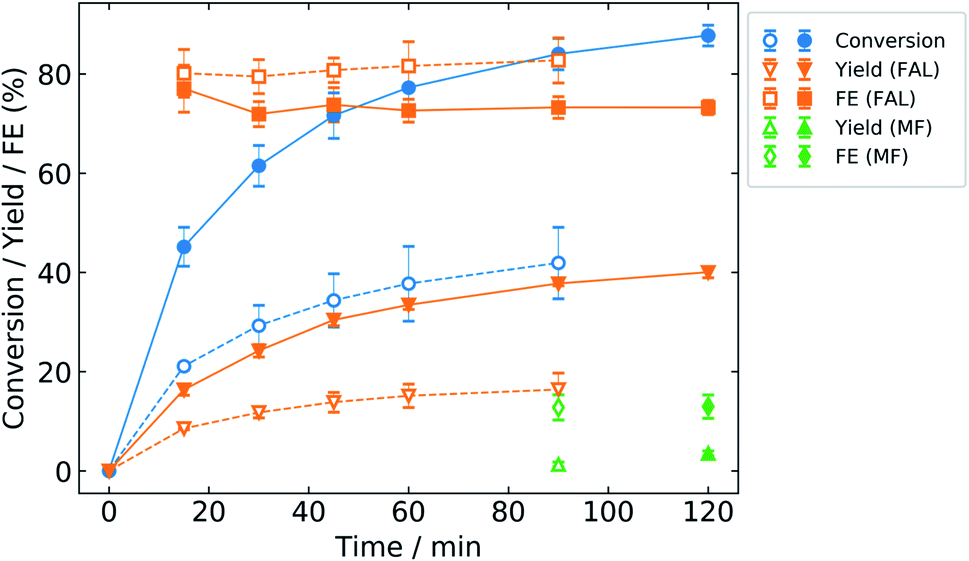 | ||
| Fig. 5 ECH electrolysis results with the zinc catalyst in 0.5 M NaHCO3 at different cathode potentials. Open symbols with dashed lines: −0.6 V vs. RHE. Closed symbols with solid lines: −0.7 V vs. RHE. | ||
As discussed previously, the open circuit potential of zinc during ECH in bicarbonate was observed to shift to more negative potentials as pH increases due to proton depletion (Fig. S6†). Thus, potentiostatic electrolysis at −0.5 V/RHE would have resulted in positive oxidation currents over the course of 2 h resulting in the formation of zinc oxide or hydroxide on the catalyst cathode. Therefore, the potentiostatic electrolysis for ECH of furfural was performed at −0.6 V/RHE.
Even at −0.6 V/RHE, the current density became positive after 1.5 h of reaction. Thus, the reaction at −0.6 V/RHE was terminated after 1.5 h. Satisfactory conversion of furfural (41.9%) was obtained at −0.6 V/RHE potential with 16.4% and 1.3% yields of FAL and MF, respectively. The cell voltage when the ECH was conducted at −0.7 V/RHE was around 19 V, primarily due to the electrolyte resistance (data not shown). We did aim to carry out ECH at −0.8 V/RHE; however, the cell voltage exceeded the 20 V limit of our equipment. Thus, −0.7 V/RHE is the most negative potential reported here. The FE of MF remained almost the same at ∼13% for both applied potentials, but the FE of FAL at −0.6 V/RHE (83%) was higher as compared to −0.7 V/RHE (73%). Also, at both potentials, zinc maintains a nearly constant FE for FAL throughout the reaction. This suggests that zinc is an active catalyst for furfural ECH and does not degrade at these time scales. The higher conversion of furfural (88%) along with the higher yield of FAL (40%) and MF (3.5%) was obtained at −0.7 V/RHE, so this potential was chosen for further studies.
Effect of different catalysts in NaHCO3
After establishing the catalytic activity of zinc for the ECH of furfural at −0.7 V/RHE in 0.5 M bicarbonate (pH = 8.4), it was compared to the electrolytic activity of copper and nickel catalysts under the same reaction conditions (Fig. 6). It was found that the catalyst had a tremendous effect on the reactant conversion, product yield and FE of the reaction. The main product was FAL in all the cases and MF was formed with low yields. Nickel catalysis resulted in 9% yield of FAL with an FE of 18%. The highest yield and FE of MF were obtained with the nickel catalyst as 5.7% and 20% respectively, indicating that nickel is more selective to MF formation in bicarbonate electrolyte as compared to copper and zinc. The yield of both the desired products was tremendously low on copper but the FE was fair (13.5% (FAL) and 18% (MF)). The highest reactant conversion for furfural (88%) and FE for FAL (73%) was obtained with zinc as a catalyst. To our knowledge, this is the highest FE of FAL that has been reported. Zinc was highly selective towards the formation of FAL, with 40% yield as compared to 3.5% yield of MF. This result is consistent with observations made by Jung and Biddinger in mildly acidic electrolyte where the copper catalyst was more selective to FAL production as compared to MF.13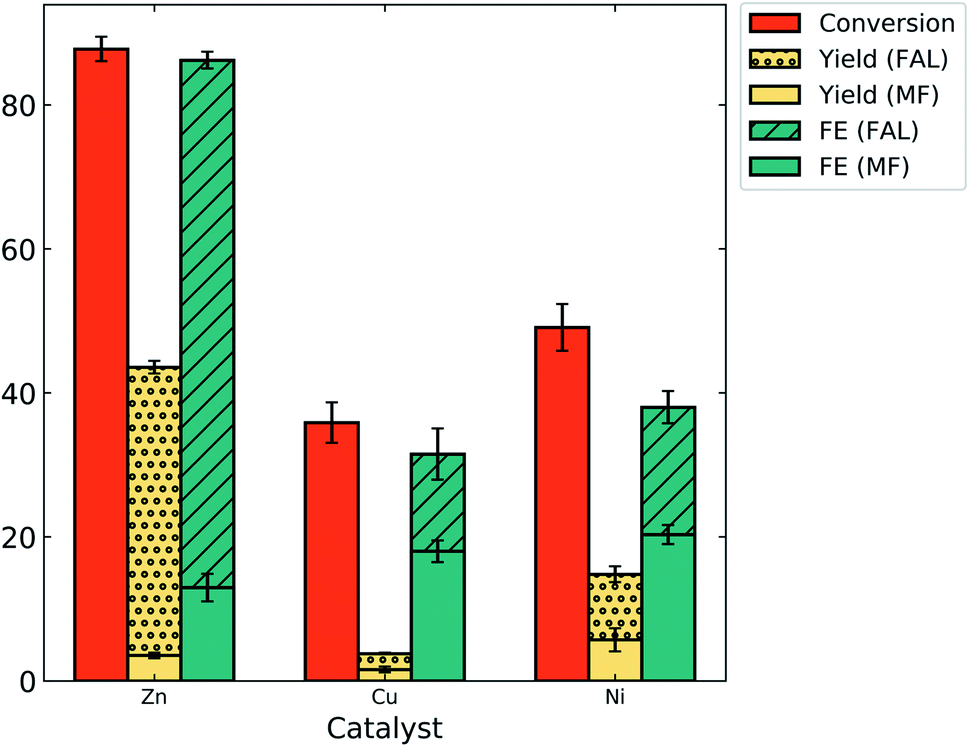 | ||
| Fig. 6 ECH electrolysis results with different catalysts in 0.5 M NaHCO3 at −0.7 V vs. RHE. | ||
The results obtained with zinc were also compared to the results obtained in the literature with the copper and nickel catalyst. The plots of the activity of the zinc catalyst compared to that of the copper and nickel catalyst at varying pH in the literature based on conversion, yield and FE of products are given in the ESI (Fig. S8†). Higher conversions have been observed in the literature at acidic and basic pH (Fig. S8a†).2,6,10,13,29,34 We have obtained 88% conversion with zinc at pH of 8.4 which is comparable to copper and nickel in a broader pH range (Fig. S8a†). Yield and FE for MF have been observed to decrease with increase in pH in the literature (Fig. S8d and e†).6,10,13,29 In the neutral pH range, the highest FE for MF was obtained with zinc at 23% in phosphate (pH 8.4). The nickel catalyst achieved an MF yield of 5.7% at pH of 8.4, nearly the same as that obtained at pH 1 (6%).6 The yields of FAL with zinc, copper and nickel were low at pH 8.4 as compared to the literature (Fig. S8b†).6,13,34 However, the FE of FAL with zinc at pH 8.4 was high as compared to literature results for all catalysts at varying pH (Fig. S8c†).2,6,10,13,29,34,40 This signifies that zinc favors ECH over the HER and suggests that a mechanistic role of zinc favors hydrogen transfer to furfural making it less available for H2 formation.
For all catalysts, the yield of all products is less than the conversion of furfural, and the mole balance is not 100%; thus, there are side products formed during furfural ECH. GC-MS analysis of the reaction mixture revealed the presence of multiple side products formed during ECH. A GC chromatogram of the reaction mixture containing furfural, furfuryl alcohol, p-xylene (internal standard) along with the side product peaks is provided in the ESI (Fig. S9†). In the figure, six side product peaks appear at a high retention time (42–44 min). The mass spectra of each peak were obtained and compared to those of chemical compounds returned by the NIST library, based on the similarity index (SI). All side product peaks generated the same probable products, varying in similarity indices (Table S1†). The most common match was alpha-furoin, with a SI of 80+ for all side product peaks.
A representative mass spectrum of the peak at retention time 43.72 min is given in Fig. S10.† Based on GC-MS data, the side products formed during ECH are alpha-furoin (1,2-di-2-furanyl-2-hydroxyethanone),50 hydrofuroin (1,2-di-2-furanyl-ethane-1,2-diol), 4-methyl-5-(2-methyl-2-propenyl)-2(5H)-furanone51 and 1-(2-furanyl)-3-methyl-3-butene-1,2-diol. It is suspected that alpha-furoin and hydrofuroin are the primary side products that could be formed by electro-dimerization. 4-Methyl-5-(2-methyl-2-propenyl)-2(5H)-furanone could be formed by ring opening and fragmentation of hydrofuroin. The formation of 1-(2-furanyl)-3-methyl-3-butene-1,2-diol during ECH is less likely.
Similar side reactions have been reported in the literature, where for example the formation of a pinacol product (hydrofuroin) has been identified, which occurs via coupling of furfuryl radicals formed after the first electron transfer process.5,10 Such radical coupling inhibits subsequent electron transfer and formation of desired products (FAL and MF). The higher FE for FAL with zinc suggests that the second electron transfer required for the hydrogenation of furfuryl radicals is more facile for zinc as compared to electrodimerization. This also reflects the significant variation in catalytic specificity among the electrode materials.
Catalyst characterization
Precipitates were observed on the electrode surface during electrolysis with zinc in the presence of furfural. The precipitates detached from the electrode and loosely settled at the bottom of the H-cell. Additionally, a mass loss of 0.8 ± 0.08% zinc catalyst was observed after ECH electrolysis. No precipitates or mass loss was observed during ECH electrolysis with copper and nickel catalysts. Additionally, no precipitates or mass loss was observed in the Zn wire during a control electrolysis experiment with no furfural present under the same reaction conditions. This indicates that the formation of precipitates on the zinc surface is observed only during electrolysis in the presence of furfural, even at a negative potential. Similarly, Roylance and Choi observed roughening of the Zn surface in SEM analysis after electrochemical HMF reduction at −0.9 V/RHE, which was attributed to surface restructuring due to the interactions between the Zn surface and HMF.41 Furfural radical or hydroxyl anions formed during the ECH reaction may stabilize the oxidized form of zinc, leading to increased zinc dissolution and subsequent precipitation as zinc oxides.33,40 Furfural has also been reported to act as a stabilizing agent by forming an ion pair with the cationic species due to the lone pair of electrons on carbonyl oxygen.52 There is some question of whether the dissolved zinc participates in the ECH reaction.A control experiment wherein the zinc electrode was polarized to −0.7 V/RHE in bicarbonate electrolyte (pH = 8.4) in the absence of furfural showed a lesser increase in pH (0.6) as compared to that during ECH (1.6, Fig. S6†) over 2 h. This could be due to increased proton consumption as indicated by higher charge transfer during ECH as compared to the HER.40
Metallic zinc is known to easily oxidize in the presence of air and aqueous alkaline electrolytes.45,48,53 Zinc passivates in aqueous bicarbonate solutions by formation of a zinc oxide surface layer with partial dissolution to form zinc hydroxide.54 Zincate ion formation has also been observed in zinc–air cells.55 Thus, we expect these precipitates to comprise zinc oxides or hydroxides.
The surface composition of the pre- and post-electrolysis zinc wire along with zinc precipitates was analyzed by scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM/EDS). The SEM images and EDS spectra are shown in Fig. 7 and S5, ESI,† respectively. The clean zinc wire yielded a surface composition of 91.6 wt% zinc with a minor carbon (6 wt%) and oxide content (2.5 wt%, Table 3). The surface content of the post-electrolysis wire was 27.8 wt% zinc, 19.6 wt% sodium and 39.6 wt% oxygen. A 7 wt% increase in the carbon content was observed in all post-electrolysis samples, which could be due to adsorbed organic compounds from the ECH reaction, contributing to electrode poisoning.
 | ||
| Fig. 7 SEM images of the (a) pre-electrolysis zinc wire, (b) post-electrolysis zinc wire and (c) zinc precipitates. | ||
| Element | Pre-electrolysis | Post-electrolysis | Precipitates | ||
|---|---|---|---|---|---|
| Overall | Triangle particle | Agglomerated particle | |||
| Zinc | 91.6 | 27.8 | 36.3 | 27.3 | 57.4 |
| Sodium | — | 19.6 | 15.7 | 21.1 | 4.7 |
| Oxygen | 2.5 | 39.6 | 37.3 | 39.6 | 30.6 |
| Carbon | 6 | 13 | 10.6 | 12 | 7.3 |
The SEM images of the post-electrolysis catalyst wire showed particles of triangular morphology surrounded by agglomerated fine particles (Fig. 7b). EDS of one triangular particle was compared to that of the agglomerated particles. The triangular particle displayed an almost equal zinc and oxygen content of 36.3 wt% and 37.3 wt% respectively. This may be interpreted as zincate (Zn(OH)42−), based on theoretical wt%. The agglomerated particles on post-electrolysis zinc wire contained 27.3% zinc and 39.6 wt% oxygen, which is still close to the theoretical wt% and is therefore likely zincate as well. In comparison, precipitates showed a higher zinc content (57.4 wt%) with 30.6 wt% oxygen and minor traces of sodium (4.7 wt%). This result suggests that the precipitate is primarily zinc hydroxide (based on theoretical wt%).
The high activity of zinc for ECH over the HER implies that there could be a mechanistic role of adhered oxidized zinc precipitates that favors hydrogen transfer to furfural making it less available for H2 formation. The high concentration of zincate ions in the vicinity of the electrode surface as compared to bulk solution leads to their deposition on the electrode surface.56 At saturated concentrations, soluble zincate ions precipitate out as insoluble zinc oxide.55,57 Burch et al. reported that the zinc oxide catalyst acted as a reservoir for atomic hydrogen and promoted hydrogen spillover towards copper to catalyze methanol synthesis from H2 and CO2.58 The presence of the oxidized layer on the zinc metal surface has been proposed to enable stabilization of adsorbed intermediates, thus promoting higher product selectivity.59
Based on the above discussion, Fig. S11† shows a proposed mechanism, wherein zinc oxides provide adsorption sites for protons, which gets reduced to adsorbed hydrogen on the zinc metal by the Volmer reaction (eqn (2)). Zinc is an oxophilic metal that promotes carbonylic aldehyde group coordination of an organic compound on its surface.60,61 Thus, furfural could adsorb on metallic zinc. Zinc is reported to form a loose passive zinc oxide layer on the surface at alkaline pH,54,55 which could facilitate proton association with electronegative oxygen. From there, hydrogen may adsorb to the metal surface, and react with adsorbed furfural to produce FAL and MF. The remaining steps shown for hydrogenation to FAL and MF are based on the literature for chemical hydrogenation of furfural to FAL and MF.15,24 There is still a question of whether both furfural and hydrogen adsorb on zinc oxides, which requires further mechanistic investigation. Thus, there is scope for further studies to fully elucidate the electrocatalysis of ECH by oxidized zinc.
Kinetic analysis
In principle, potentiostatic electrolysis enables chronological data to be fit to kinetic models, which is more difficult in the case of constant-current data. Initial time-dependent furfural concentration data for different catalysts and varying potential in bicarbonate electrolyte were fitted to a first order rate law using a nonlinear least squares procedure. As shown in Fig. 8, the first-order model does not fit the data over the entire period of electrolysis. This is likely due to catalyst surface modification or poisoning by reaction byproducts, which is consistent with the presence of increased carbon content after electrolysis, as observed by EDS (Table 3). Instead, we took the initial reaction rate by fitting the first order model to the initial data points, between 0 and 30 min.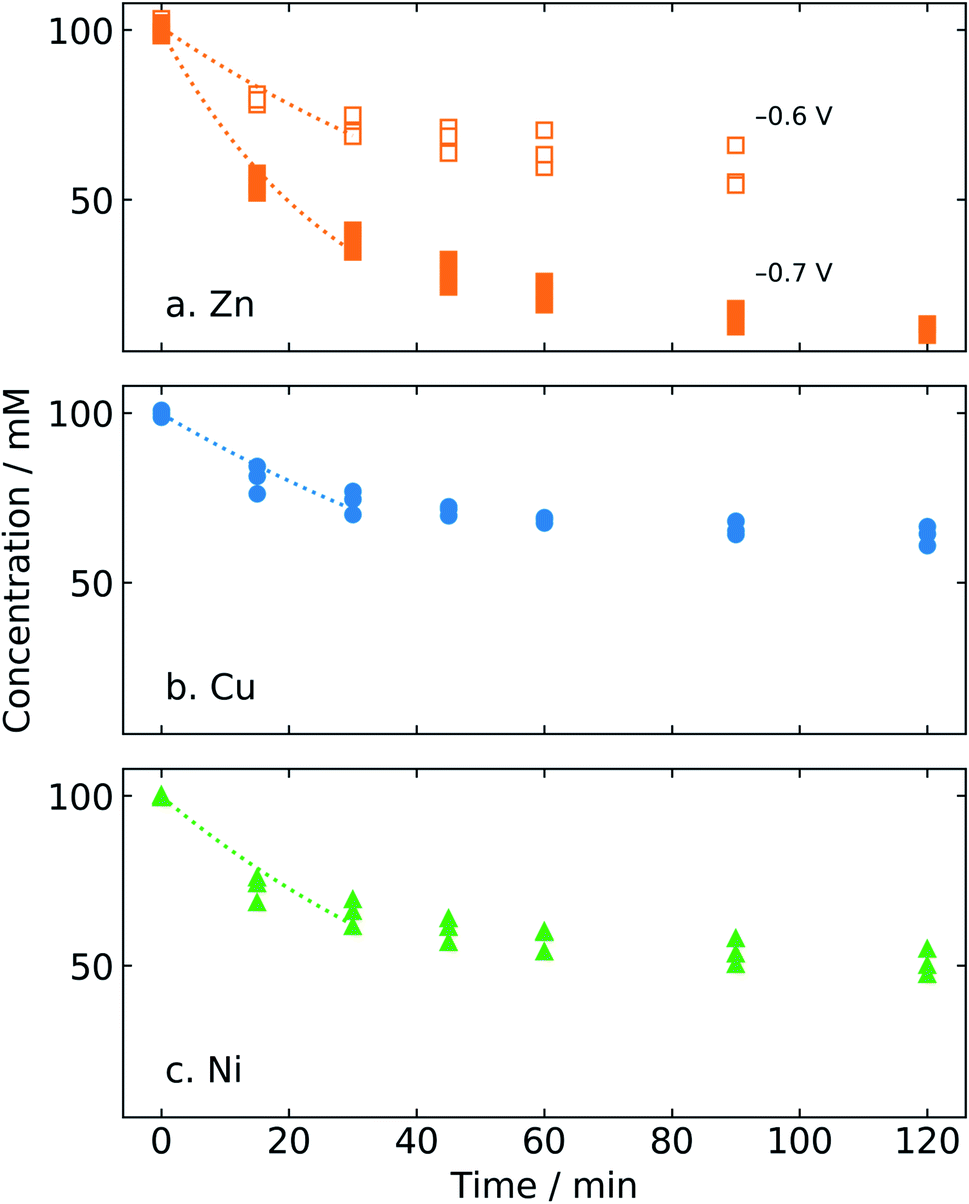 | ||
| Fig. 8 Experimental furfural concentration profiles during electrolysis in 0.5 M NaHCO3 with the (a) zinc catalyst at −0.6 and −0.7 V vs. RHE; (b) copper catalyst at −0.7 V vs. RHE; (c) nickel catalyst at −0.7 V vs. RHE. Curves represent fitting of the initial reaction rate (0–30 min) to a first order rate law. | ||
The experimental and fitted concentration profiles are given in Fig. 8. The fit was found to be good, with an R2 value of varying from 89–97%, for different cases. The highest rate constant for ECH of furfural was obtained for the zinc catalyst at −0.7 V/RHE as 0.42 h−1 cm−2 (Table 4). As expected, the rate constant of zinc at −0.7 V/RHE was over 60% higher than that of zinc at −0.6 V/RHE.
| Catalyst | Potential (V/RHE) | k (h−1 cm−2) | R 2 |
|---|---|---|---|
| Zinc | −0.6 | 0.15 ± 0.01 | 0.93 |
| Zinc | −0.7 | 0.42 ± 0.02 | 0.97 |
| Copper | 0.13 ± 0.01 | 0.89 | |
| Nickel | 0.19 ± 0.02 | 0.90 |
The rate constants for copper and nickel were found to be similar. As seen in Fig. 7, the activity of copper and nickel in terms of conversion of furfural did not show a significant difference. The rate constant of zinc at −0.7 V/RHE was found around 55% higher than that of copper and nickel. By this measure, therefore, zinc is a comparatively better catalyst for ECH of furfural.
Conclusion
Electrochemical hydrogenation of furfural to FAL and MF was studied using various catalysts in multiple electrolytes of varying pH. The nature of the electrode material and electrolyte pH immensely affected the conversion, yield, and FE to the desired products. Higher product yield and FE were obtained at pH 7.4 to 8.4 as compared to acidic and basic pH. FAL yield was high at high pH with MF formed at lower yields. Although, the yields of FAL and MF were low on copper and nickel, the FE was appreciable. The highest yield of MF was achieved with nickel as a catalyst.The best activity was achieved using zinc as a catalyst on the basis 88% conversion, 40% yield and 73% FE of FAL in 0.5 M bicarbonate electrolyte for potentiostatic electrolysis at −0.7 V/RHE over 2 h. The total FE for ECH of furfural was obtained as 86% with the zinc catalyst in bicarbonate electrolyte (pH = 8.4), which suggests that the HER was restricted. The total FE of products obtained with zinc in phosphate (pH = 7.4) was close to that in bicarbonate (pH = 8.4). This emphasizes the role of proton concentration in the neutral pH range electrolytes to inhibit the HER, polymerization and electrodimerization reactions. Additionally, dissolution of zinc during electrolysis, only in the presence of furfural, suggests participation of dissolved zinc in the hydrogenation mechanism. Further investigations on the reaction mechanism for the role of zinc or oxidized zinc for ECH of furfural will help gain better insight into the reason for the high activity of zinc for this reaction. This system may also be applicable to ECH of other aldehydes, such as benzaldehyde or acetaldehyde.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Manali S. Dhawan acknowledges a Research Fellowship under J.C. Bose National Fellowship of Department of Science and Technology, Govt. of India. G. D. Yadav acknowledges support from R.T. Mody Distinguished Professor Endowment and J.C. Bose National Fellowship from Department of Science and Technology, Govt. of India.References
- N. L. Panwar, S. C. Kaushik and S. Kothari, Role of renewable energy sources in environmental protection: A review, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524, DOI:10.1016/j.rser.2010.11.037.
- B. Zhao, M. Chen, Q. Guo and Y. Fu, Electrocatalytic hydrogenation of furfural to furfuryl alcohol using platinum supported on activated carbon fibers, Electrochim. Acta, 2014, 135, 139–146, DOI:10.1016/j.electacta.2014.04.164.
- F. Cherubini, The biorefinery concept: Using biomass instead of oil for producing energy and chemicals, Energy Convers. Manage., 2010, 51, 1412–1421, DOI:10.1016/j.enconman.2010.01.015.
- D. Kim, K. Lee and K. Y. Park, Upgrading the characteristics of biochar from cellulose, lignin, and xylan for solid biofuel production from biomass by hydrothermal carbonization, J. Ind. Eng. Chem., 2016, 42, 95–100, DOI:10.1016/j.jiec.2016.07.037.
- P. Nilges and U. Schröder, Electrochemistry for biofuel generation: production of furans by electrocatalytic hydrogenation of furfurals, Energy Environ. Sci., 2013, 6, 2925, 10.1039/c3ee41857j.
- Z. Li, S. Kelkar, C. H. Lam, K. Luczek, J. E. Jackson, D. J. Miller and C. M. Saffron, Aqueous electrocatalytic hydrogenation of furfural using a sacrificial anode, Electrochim. Acta, 2012, 64, 87–93, DOI:10.1016/j.electacta.2011.12.105.
- C. Chatterjee, F. Pong and A. Sen, Chemical conversion pathways for carbohydrates, Green Chem., 2015, 17, 40–71, 10.1039/c4gc01062k.
- S. K. Green, J. Lee, H. J. Kim, G. A. Tompsett, W. B. Kim and G. W. Huber, The electrocatalytic hydrogenation of furanic compounds in a continuous electrocatalytic membrane reactor, Green Chem., 2013, 15, 1869, 10.1039/c3gc00090g.
- J. B. Binder, J. B. Binder, R. T. Raines and R. T. Raines, Simple chemical transformation of lignocellulosic biomass into furans for fuels and chemicals, J. Am. Chem. Soc., 2009, 131, 1979–1985, DOI:10.1021/ja808537j.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J. P. Tessonnier and W. Li, Mechanisms of Furfural Reduction on Metal Electrodes: Distinguishing Pathways for Selective Hydrogenation of Bioderived Oxygenates, J. Am. Chem. Soc., 2017, 139, 14120–14128, DOI:10.1021/jacs.7b06331.
- F. Wang, M. Xu, L. Wei, Y. Wei, Y. Hu, W. Fang and C. G. Zhu, Fabrication of La-doped TiO2 Film Electrode and investigation of its electrocatalytic activity for furfural reduction, Electrochim. Acta, 2015, 153, 170–174, DOI:10.1016/j.electacta.2014.11.203.
- Y. Cao and T. Noël, Efficient Electrocatalytic Reduction of Furfural to Furfuryl Alcohol in a Microchannel Flow Reactor, Org. Process Res. Dev., 2019, 23, 403–408, DOI:10.1021/acs.oprd.8b00428.
- S. Jung and E. J. Biddinger, Electrocatalytic Hydrogenation and Hydrogenolysis of Furfural and the Impact of Homogeneous Side Reactions of Furanic Compounds in Acidic Electrolytes, ACS Sustainable Chem. Eng., 2016, 4, 6500–6508, DOI:10.1021/acssuschemeng.6b01314.
- F. Dong, Y. Zhu, H. Zheng, Y. Zhu, X. Li and Y. Li, Cr-free Cu-catalysts for the selective hydrogenation of biomass-derived furfural to 2-methylfuran: The synergistic effect of metal and acid sites, J. Mol. Catal. A: Chem., 2015, 398, 140–148, DOI:10.1016/j.molcata.2014.12.001.
- R. V. Sharma, U. Das, R. Sammynaiken and A. K. Dalai, Liquid phase chemo-selective catalytic hydrogenation of furfural to furfuryl alcohol, Appl. Catal., A, 2013, 454, 127–136, DOI:10.1016/j.apcata.2012.12.010.
- C. P. Jiménez-Gómez, J. A. Cecilia, D. Durán-Martín, R. Moreno-Tost, J. Santamaría-González, J. Mérida-Robles, R. Mariscal and P. Maireles-Torres, Gas-phase hydrogenation of furfural to furfuryl alcohol over Cu/ZnO catalysts, J. Catal., 2016, 336, 107–115, DOI:10.1016/j.jcat.2016.01.012.
- M. A. Jackson, M. G. White, R. T. Haasch, S. C. Peterson and J. A. Blackburn, Hydrogenation of furfural at the dynamic Cu surface of CuOCeO2/Al2O3 in a vapor phase packed bed reactor, Mol. Catal., 2018, 445, 124–132, DOI:10.1016/j.mcat.2017.11.023.
- J. Yang, H. Y. Zheng, Y. L. Zhu, G. W. Zhao, C. H. Zhang, B. T. Teng, H. W. Xiang and Y. Li, Effects of calcination temperature on performance of Cu–Zn–Al catalyst for synthesizing γ-butyrolactone and 2-methylfuran through the coupling of dehydrogenation and hydrogenation, Catal. Commun., 2004, 5(9), 505–510, DOI:10.1016/j.catcom.2004.06.005.
- R. S. Rao, R. Terry, K. Baker and M. A. Vannice, Furfural hydrogenation over carbon-supported copper, Catal. Lett., 1999, 60, 51–57, DOI:10.1023/A:1019090520407.
- J. Lessard, J. F. Morin, J. F. Wehrung, D. Magnin and E. Chornet, High yield conversion of residual pentoses into furfural via zeolite catalysis and catalytic hydrogenation of furfural to 2-methylfuran, Top. Catal., 2010, 53(15–18), 1231–1234, DOI:10.1007/s11244-010-9568-7.
- A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Bimetallic PtSn catalyst for the selective hydrogenation of furfural to furfuryl alcohol in liquid-phase, Catal. Commun., 2009, 10(13), 1665–1669, DOI:10.1016/j.catcom.2009.05.005.
- P. D. Vaidya and V. V. Mahajani, Kinetics of liquid-phase hydrogenation of furfuraldehyde to furfuryl alcohol over a Pt/C catalyst, Ind. Eng. Chem. Res., 2003, 42(17), 3881–3885, DOI:10.1021/ie030055k.
- S. Sitthisa, T. Sooknoi, Y. Ma, P. B. Balbuena and D. E. Resasco, Kinetics and mechanism of hydrogenation of furfural on Cu/SiO2 catalysts, J. Catal., 2011, 277, 1–13, DOI:10.1016/j.jcat.2010.10.005.
- S. Srivastava, G. C. Jadeja and J. Parikh, Copper-cobalt catalyzed liquid phase hydrogenation of furfural to 2-methylfuran: An optimization, kinetics and reaction mechanism study, Chem. Eng. Res. Des., 2018, 132, 313–324, DOI:10.1016/j.cherd.2018.01.031.
- X. Yang, Q. Meng, G. Ding, Y. Wang, H. Chen, Y. l. Zhu and Y. W. Li, Construction of novel Cu/ZnO-Al2O3 composites for furfural hydrogenation: The role of Al components, Appl. Catal., A, 2018, 561, 78–86, DOI:10.1016/j.apcata.2018.04.005.
- M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. J. Marchi, Selective liquid-phase hydrogenation of furfural to furfuryl alcohol over Cu-based catalysts, Catal. Today, 2013, 213, 87–92, DOI:10.1016/j.cattod.2013.02.031.
- N. Jiang, X. Liu, J. Dong, B. You, X. Liu and Y. Sun, Electrocatalysis of Furfural Oxidation Coupled with H2 Evolution via Nickel-Based Electrocatalysts in Water, ChemNanoMat, 2017, 3, 491–495, DOI:10.1002/cnma.201700076.
- X. Zhang, M. Han, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Simultaneously high-rate furfural hydrogenation and oxidation upgrading on nanostructured transition metal phosphides through electrocatalytic conversion at ambient conditions, Appl. Catal., B, 2019, 244, 899–908, DOI:10.1016/j.apcatb.2018.12.025.
- S. Jung, A. N. Karaiskakis and E. J. Biddinger, Enhanced activity for electrochemical hydrogenation and hydrogenolysis of furfural to biofuel using electrodeposited Cu catalysts, Catal. Today, 2019, 323, 26–34, DOI:10.1016/j.cattod.2018.09.011.
- J. J. Roylance, T. W. Kim and K. S. Choi, Efficient and Selective Electrochemical and Photoelectrochemical Reduction of 5-Hydroxymethylfurfural to 2,5-Bis(hydroxymethyl)furan using Water as the Hydrogen Source, ACS Catal., 2016, 6, 1840–1847, DOI:10.1021/acscatal.5b02586.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles, Green Chem., 2014, 16, 3778–3786, 10.1039/c4gc00401a.
- S. Jung and E. J. Biddinger, Controlling Competitive Side Reactions in the Electrochemical Upgrading of Furfural to Biofuel, Energy Technol., 2018, 6, 1370–1379, DOI:10.1002/ente.201800216.
- L. Nadjo and J. M. Saveant, Dimerization, disproportionation and e.c.e. mechanisms in the reduction of aromatic carbonyl compounds in alkaline media, J. Electroanal. Chem., 1971, 33, 419–451, DOI:10.1016/S0022-0728(71)80127-4.
- P. Parpot, A. P. Bettencourt, G. Chamoulaud, K. B. Kokoh and E. M. Belgsir, Electrochemical investigations of the oxidation–reduction of furfural in aqueous medium – Application to electrosynthesis, Electrochim. Acta, 2004, 49, 397–403, DOI:10.1016/j.electacta.2003.08.021.
- G. Chamoulaud, D. Floner, C. Moinet, C. Lamy and E. M. Belgsir, Biomass conversion II: Simultaneous electrosyntheses of furoic acid and furfuryl alcohol on modified graphite felt electrodes, Electrochim. Acta, 2001, 46, 2757–2760, DOI:10.1016/S0013-4686(01)00507-2.
- D. Chu, Y. Hou, J. He, M. Xu, Y. Wang, S. Wang, J. Wang and L. Zha, Nano TiO2 film electrode for electrocatalytic reduction of furfural in ionic liquids, J. Nanopart. Res., 2009, 11(7), 1805–1809, DOI:10.1007/s11051-009-9610-5.
- Y. Kwon, K. J. P. Schouten, J. C. van der Waal, E. De Jong and M. T. M. Koper, Electrocatalytic Conversion of Furanic Compounds, ACS Catal., 2016, 6, 6704–6717, DOI:10.1021/acscatal.6b01861.
- B. Zhao, M. Chen, Q. Guo and Y. Fu, Electrocatalytic hydrogenation of furfural to furfuryl alcohol using platinum supported on activated carbon fibers, Electrochim. Acta, 2014, 135, 139–146, DOI:10.1016/j.electacta.2014.04.164.
- T. Kim, R. S. Assary, C. L. Marshall, D. J. Gosztola, L. A. Curtiss and P. C. Stair, Acid-catalyzed furfuryl alcohol polymerization: Characterizations of molecular structure and thermodynamic properties, ChemCatChem, 2011, 3(9), 1451–1458, DOI:10.1002/cctc.201100098.
- L. Liu, H. Liu, W. Huang, Y. He, W. Zhang, C. Wang and H. Lin, Mechanism and kinetics of the electrocatalytic hydrogenation of furfural to furfuryl alcohol, J. Electroanal. Chem., 2017, 804, 248–253, DOI:10.1016/j.jelechem.2017.09.021.
- J. J. Roylance and K. S. Choi, Electrochemical reductive biomass conversion: Direct conversion of 5-hydroxymethylfurfural (HMF) to 2,5-hexanedione (HD): Via reductive ring-opening, Green Chem., 2016, 18, 2956–2960, 10.1039/c6gc00533k.
- A. M. Polcaro, S. Palmas and S. Dernini, Role of Catalyst Characteristics in Electrocatalytic Hydrogenation: Reduction of Benzaldehyde and Acetophenone on Carbon Felt/Pd Electrodes, Ind. Eng. Chem. Res., 1993, 32, 1315–1322, DOI:10.1021/ie00019a005.
- Y. Song, O. Y. Gutiérrez, J. Herranz and J. A. Lercher, Aqueous phase electrocatalysis and thermal catalysis for the hydrogenation of phenol at mild conditions, Appl. Catal., B, 2016, 182, 236–246, DOI:10.1016/j.apcatb.2015.09.027.
- H. S. Jeon, I. Sinev, F. Scholten, N. J. Divins, I. Zegkinoglou, L. Pielsticker and B. R. Cuenya, Operando Evolution of the Structure and Oxidation State of Size-Controlled Zn Nanoparticles during CO2 Electroreduction, J. Am. Chem. Soc., 2018, 140, 9383–9386, DOI:10.1021/jacs.8b05258.
- D. L. T. Nguyen, M. S. Jee, D. H. Won, H. Jung, H. S. Oh, B. K. Min and Y. J. Hwang, Selective CO2 Reduction on Zinc Electrocatalyst: The Effect of Zinc Oxidation State Induced by Pretreatment Environment, ACS Sustainable Chem. Eng., 2017, 5, 11377–11386, DOI:10.1021/acssuschemeng.7b02460.
- Y. Feng, Z. Li, H. Liu, C. Dong, J. Wang, S. A. Kulinich and X. Du, Laser-Prepared CuZn Alloy Catalyst for Selective Electrochemical Reduction of CO2 to Ethylene, Langmuir, 2018, 34, 13544–13549, DOI:10.1021/acs.langmuir.8b02837.
- I. Nezam, L. Peereboom and D. J. Miller, Continuous condensed-phase ethanol conversion to higher alcohols: Experimental results and techno-economic analysis, J. Cleaner Prod., 2019, 209, 1365–1375, DOI:10.1016/j.jclepro.2018.10.276.
- T. F. Fuller and J. N. Harb, Electrochemical Engineering, John Wiley & Sons, Inc, 1st edn, 2018 Search PubMed.
- N. S. Gould, H. Landfield, B. Dinkelacker, C. Brady, X. Yang and B. Xu, Selectivity Control in Catalytic Reductive Amination of Furfural to Furfurylamine on Supported Catalysts, ChemCatChem, 2020, 12, 2106–2115, DOI:10.1002/cctc.201901662.
- NIST MS number: 228113, NIST Chemistry WebBook, NIST Standard Reference Database 69, National Institute of Standards and Technology, (n.d.), https://webbook.nist.gov/cgi/cbook.cgi?ID=C552863%26Units=SI%26Mask=200#Mass-Spec, accessed January 4, 2021.
- NIST MS number: 151114, NIST Chemistry WebBook, NIST Standard Reference Database 69, National Institute of Standards and Technology, (n.d.), https://webbook.nist.gov/cgi/cbook.cgi?ID=C89902249%26Units=SI%26Mask=200#Mass-Spec, accessed January 4, 2021.
- R. Lakra, M. S. Kiran and P. S. Korrapati, Furfural mediated synthesis of silver nanoparticles for photocatalytic reduction of hexavalent chromium, Environ. Technol. Innovation, 2021, 21, 101348, DOI:10.1016/j.eti.2020.101348.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, Electrodeposited Zn Dendrites with Enhanced CO Selectivity for Electrocatalytic CO2 Reduction, ACS Catal., 2015, 5, 4586–4591, DOI:10.1021/acscatal.5b00922.
- H. Kaesche, The passivity of zinc in aqueous solutions of sodium carbonate and sodium bicarbonate, Electrochim. Acta, 1964, 9, 383–394, DOI:10.1016/0013-4686(64)80044-X.
- S. C. Barton and A. C. West, Electrodissolution of Zinc at the Limiting Current, J. Electrochem. Soc., 2001, 148, A490, DOI:10.1149/1.1365141.
- J. Yi, P. Liang, X. Liu, K. Wu, Y. Liu, Y. Wang, Y. Xia and J. Zhang, Challenges, mitigation strategies and perspectives in development of zinc-electrode materials and fabrication for rechargeable zinc–air batteries, Energy Environ. Sci., 2018, 11, 3075–3095, 10.1039/c8ee01991f.
- W. Yu, W. Shang, X. Xiao, P. Tan, B. Chen, Z. Wu, H. Xu and M. Ni, Achieving a stable zinc electrode with ultralong cycle life by implementing a flowing electrolyte, J. Power Sources, 2020, 453, 227856, DOI:10.1016/j.jpowsour.2020.227856.
- R. Burch, S. E. Golunski and M. S. Spencer, The role of copper and zinc oxide in methanol synthesis catalysts, J. Chem. Soc., Faraday Trans., 1990, 86(15), 2683–2691, 10.1039/FT9908602683.
- D. H. Won, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Highly Efficient, Selective, and Stable CO2 Electroreduction on a Hexagonal Zn Catalyst, Angew. Chem., Int. Ed., 2016, 55, 9297–9300, DOI:10.1002/anie.201602888.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, How Catalysts and Experimental Conditions Determine the Selective Hydroconversion of Furfural and 5-Hydroxymethylfurfural, Chem. Rev., 2018, 118(22), 11023–11117 CrossRef CAS PubMed.
- S. Siwal, N. Devi, V. Perla, R. Barik, S. Ghosh and K. Mallick, The influencing role of oxophilicity and surface area of the catalyst for electrochemical methanol oxidation reaction: a case study, Mater. Res. Innovations, 2019, 23, 440–447, DOI:10.1080/14328917.2018.1533268.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se00221j |
| This journal is © The Royal Society of Chemistry 2021 |