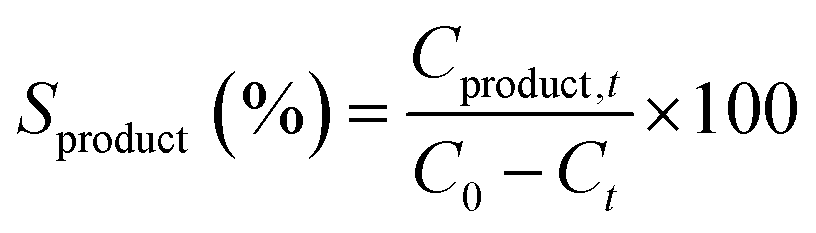Role of transition metals on MoS2-based supported catalysts for hydrodeoxygenation (HDO) of propylguaiacol†
Received
4th February 2021
, Accepted 6th March 2021
First published on 8th March 2021
Abstract
Transition metal sulfides (TMSs) are typically used in the traditional petroleum refining industry for hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) applications. Bio-oils require an upgrading process like catalytic hydrodeoxygenation (HDO) to produce advanced biofuels and chemicals. Herein, MoS2/γ-Al2O3 promoted by transition metals like nickel (Ni), copper (Cu), zinc (Zn), and iron (Fe) was evaluated for the HDO of a bio-oil model compound, 4-propylguaiacol (PG) in a batch reactor at 340 °C under 50 bar H2 pressure. The catalyst screening results showed that the sulfided Ni-promoted catalyst gave a high 94% yield of deoxygenated cycloalkanes, however for the sulfided Cu-promoted catalyst, 42% of phenolics remain in the reaction medium after 5 h. The results also revealed that the sulfided Zn and Fe-promoted catalysts gave a final yield of 16% and 19% at full PG conversion, respectively, for deoxygenated aromatics. A kinetic model considering the main side reactions was developed to elucidate the reaction pathway of demethoxylation and dehydroxylation of PG. The developed kinetic model was able to describe the experimental results well with a coefficient of determination of 97% for the Ni-promoted catalyst system. The absence of intermediates like 4-propylcyclohexanone and 4-propylcyclohexanol during the reaction implies that direct deoxygenation (DDO) is the dominant pathway in the deoxygenation of PG employing sulfided catalysts. The current work also demonstrated that the activity of the transition metal promoters sulfides for HDO of PG could be correlated to the yield of deoxygenated products from the hydrotreatment of Kraft lignin.
1. Introduction
It is of utmost importance to increase the utilization of sustainable liquid biofuels as one of the solutions to halt global warming. Catalytic pyrolysis possesses great potential for producing bio-oils, a viscous brown liquid mainly comprising phenols and aromatic compounds with oxygen-containing functional groups.1 The high oxygen content, high thermal instability, low calorific values, and acidity of raw bio-oil make it difficult to use directly as a transportation fuel in combustion engines.2 Hence, an upgrading process is required to enhance the quality of the bio-oil for transportation fuel production.3 The upgrading process can be achieved by catalytic hydrodeoxygenation (HDO). It is an emerging and efficient catalytic process to remove oxygen from the bio-feeds in the form of water in the presence of hydrogen.4
Several simple bio-oil model compounds, such as guaiacol,5 eugenol,6 isoeugenol,7–9 anisole,10p-cresol,11 and vanillin,12 have been examined for the HDO of phenolic compounds and provided fundamental insight into developing active and selective catalysts for HDO. These model substrates contain oxygenate hydroxyl and methoxy groups, which commonly exist in the lignin-derived components present in bio-oils. The employment of phenolic model substrates allows us to further elucidate and understand the reaction mechanism and kinetics for HDO. There are several studies on catalytic HDO of phenolic compounds.13–17 The catalytic HDO of phenolics involves a range of complex reactions including hydrodeoxygenation, hydrogenolysis, dehydration, isomerization, alkylation, and hydrogenation. In theory, the removal of the oxygen atom from phenolics can occur via two pathways: (i) the direct deoxygenation (DDO) route where the hydroxyl (Csp2–OH) and methoxy (Csp2–OCH3) group from the aromatic ring will cleave by C–O bond scission, and (ii) the hydrogenation and dehydration (HYD) route where aromatic ring hydrogenation first occurs and followed by removal of a water molecule. The former route is preferred in the deoxygenation process in terms of hydrogen economy.
The catalytic performance and the progression of the entire reaction involved in HDO depend largely on the type of catalysts, the support used, and operating conditions (reactor types, reactants, reaction temperature and time, solvent system, and pressure). Transition metal sulfide catalysts are usually employed in hydrodesulfurization (HDS) and hydrodenitrogenation (HDN).18 The traditional catalyst system used is molybdenum or tungsten supported on alumina promoted by cobalt or nickel.18 These as-prepared catalysts are in oxidized form and need to be sulfided and this can in research studies be done using a sulfiding agent, such as dimethyl disulfide to create an active sulfide phase.
Lately, much focus has been placed on HDO reactions employing the conventional NiMo and CoMo catalysts in sulfided form due to the shift of using renewable bio-feedstocks. Table S1 (in ESI†) presents the state of the art sulfided catalysts employed in HDO of phenolic monomers with details like catalysts type, reaction parameters, reactant conversion, and product selectivities. For example, Wu et al. prepared hydrophobic unsupported MoS2, NiS2–MoS2, and CoS2–MoS2 catalysts using a hydrothermal method with silicomolybdic acid.14 Results showed that the unsupported sulfided catalysts gave full conversion to HDO of PG and high selectivity of 83.7% towards 4-propylbenzene as the main product after 6 h with the reaction temperature of 250 °C.14 Templis and coworkers also studied phenol HDO over a sulfided and reduced NiMo catalyst.19 They found that the commercial sulfided NiMo γ-alumina supported catalysts exhibited 90% selectivity for cyclohexane production and suggested that phenol HDO proceeded via two parallel pathways, DDO and HYD.19 Additionally, the reduced NiMo catalyst was more active as compared to the sulfided catalysts.19 On the other hand, Romero and colleagues applied the same sulfided (Ni or Co)Mo/Al2O3 supported catalysts on 2-ethylphenol HDO and observed that both promoters improved the deoxygenation rate, where Ni only promotes the HYD route, while Co promotes both HYD and DDO routes.20 Recently, Song et al. applied an efficient CoMoS nanosulfide catalyst on p-cresol. The effective unsupported catalysts demonstrated a 98% arene yield.21 These studies show the importance of the incorporation of a promoter, such as Co, which can significantly enhance the performance of HDO.21
Other than the promoter playing a role in the hydrotreating catalysts, the support also affects the catalytic activity. Tavizón-Pozos et al. studied the effect of mixed Al2O3–TiO2 as support for MoS2.22 They observed that the use of this mixed oxide improved the HDO activity for phenols by improving the metal-support interaction and eventually increasing the active phase of the catalysts.22 A new approach, such as using a different sulfided catalyst like Ni/W/TiO2 has been examined also for the HDO of guaiacol.23 The best performing catalyst was 2 wt% Ni/12 wt% W/TiO2 anatase, giving full guaiacol conversion, and 16% yield cyclohexane. The study also showed that Ni was a better promoter compared to Co for the HDO reaction.23 Non-traditional sulfided catalysts like ReS2 supported on SiO2 and γ-Al2O3 were investigated in the co-processing of 4,6-dimethyldibenzothiophene and guaiacol.24 Both Re-based sulfided catalysts showed high HDS and HDO activities and ReS2-SiO2 shows a higher HDO rate giving a high deoxygenated product yield via the demethoxylation (DMO) route.24 There is also research reporting on the use of noble metals as promoters for metal sulfides, which provides promising results for HDO reactions.25 A small amount of Ir and Pt was doped on RuS2/SBA-15 and examined for HDO of phenols, and both catalysts demonstrated better phenol conversion (37–41%) and cyclohexane selectivity (62–63%) as compared to the non-promoted RuS2/SBA-15.25 Although it is worth mentioning that the use of noble metals involves high costs for catalyst production.
Ternan26 compared the activity parameter calculated in the work of Harris and Chianelli27 and concluded that there is a correlation between the HDS promotional effect for the first-row transition metal sulfides and calculated activity parameters. These correlations show that the properties of the same catalysts giving the same influences on the promotion of HDS, HDN, and hydrogenation. Moreover, Eijsbouts et al. discussed the economic and technical aspects and the possibility of replacing Co and Ni (3d transition metals) promoters in hydrotreating catalysts in a comprehensive review.28 They mentioned the ability of Co and Ni for donating electrons to Mo while increasing the number of the d electrons on Mo which would weaken the interaction between Mo and sulfur resulting in the creation of sulfur vacancies and eventually influence the HDS activity.27,28 Several studies have been carried out to understand different transition metals that would have the same or better effect as that of Co and Ni in HDS applications.26,27,29–31 It is also worth pointing out that none of these transition metals performed like Ni and Co, but instead changed the selectivity of the catalyst at a lower activity.
However, according to our knowledge, there are no studies available where different transition metals like Cu, Zn, and Fe have been added to sulfided catalytic systems used for HDO of bio-oil compounds. The exploratory work for finding a correlation between the promotional effect of the first-row transition metal in sulfided catalysts on HDO is also lacking. Therefore, in this paper, for the first time, we performed a screening of Ni, Fe, Zn, and Cu on Mo-based sulfided catalysts for the HDO of PG in a batch reactor set-up. Also, the effect of these transition metals was studied and compared with the conventional NiMo catalyst for the development of HDO catalysts. The catalysts were characterized with X-ray diffraction (XRD), nitrogen physisorption (BET), inductively coupled plasma (ICP)-sector field mass spectroscopy, X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). Moreover, modeling was performed in the current work to explore the reaction networks for HDO of PG over sulfided catalyst systems, also considering the side reactions. Finally, sulfided Mo/Al2O3 promoted by Ni, Fe, and Zn were examined for Kraft lignin hydrotreatment, and the results were compared with the data obtained from HDO of PG.
2. Experimental
2.1 Catalyst synthesis
The preparation of unpromoted Mo alumina supported catalyst has been described earlier.32 The alumina-supported NiMo, CuMo, ZnMo, and FeMo catalysts were prepared by the conventional incipient wetness impregnation method. For the Ni-promoted catalyst, 3 wt% of Ni(C5H7O2)2 was dissolved in 20 mL of ethanol. The unpromoted Mo on alumina catalyst was then dissolved in 25 mL of ethanol. The nickel precursor solution was then added dropwise to the Mo catalyst solution. 10 mL of ethanol was used to wash off the residue in the beaker. The catalyst slurry was then stirred overnight to let the ethanol evaporate. The dry catalyst was then calcined at 400 °C for 4 h in air. The same procedure was employed for FeMo, where iron(III) acetylacetonate (99%), Fe(C5H7O2)3 was used as the Fe precursor. Zinc acetylacetonate hydrate (99.995%) and copper(II) nitrate hemi pentahydrate (98%) were used as Zn and Cu precursors, respectively. Before all catalytic reactions, the synthesized catalysts underwent a sulfidation treatment following a procedure reported earlier.33 0.5 g of catalyst was loaded in the batch reactor followed by 0.5 mL of DMDS, dimethyl disulfide (≥99.5%, Sigma-Aldrich) with 20 bar of H2 at 340 °C for 4 h. An excess amount of DMDS was added to ensure complete sulfidation of the Mo-based catalysts. The catalysts in the sulfided form were denoted as Mo, NiMo, CuMo, ZnMo, and FeMo.
2.2 Catalytic reaction and product analysis for PG experiments
The kinetic experiments for HDO of PG were performed in a 300 mL parr batch reactor. The reactor was charged with 1 g of reactant, 0.5 g of sulfided catalyst, 0.1 mL of DMDS, and 100 mL of dodecane for each experiment. The reaction temperature and pressure were set at 300 °C and 50 bar of H2 pressure, respectively with a 1000 rpm stirring rate. The reaction liquid samples were collected periodically (0.5 h, 1 h, 2 h, 3 h, 4 h, and 5 h) and filtrated before analysis. The sampling line was purged with N2 before collecting reaction samples. A small pressure drop of ca. 1 bar was observed after collecting the reaction sample. The pressure drop was compensated by repressurizing the system to 50 bar with additional H2. When the reaction experiment was completed, the reactor was cooled to room temperature, and the catalyst was recovered, filtered, and washed with acetone to remove adhering reactants and products. It was further dried under atmospheric conditions and used for characterization. The liquid samples were analyzed by GC-MS (Agilent 7890-5977A). The GC-MS was equipped with an HP-5 column (30 m × 250 μm × 0.25 μm), and a Flame Ionization Detector (the setpoint was 335 °C). External calibration curves were obtained for 4-propylguaiacol, propylcyclohexane, 4-propylphenol, propylbenzene, 4-propylresorcinol, 1,2-dimethoxy-4-propenylbenzene, gamma terpene, 4-tertbutylanisole, and 2-methyl-6-propyl phenol using commercial chemicals.
The unit for the concentration of reactant and reaction products is expressed in molar percent (mol%). The following definitions are used in this study:
PG conversion was calculated as
| |  | (1) |
where
C0 is the initial concentration of PG and
Ct is the concentration of PG at reaction time equal to
t.
Reaction product yields were calculated as
| |  | (2) |
where
Cproduct,t is the concentration of the reaction product at reaction time
t.
Reaction product selectivities were calculated as
| |  | (3) |
The molar balance was calculated to evaluate the material balance in the liquid phase. The molar balance was calculated by dividing the sum of the concentration of all identified reaction products and initial feed at reaction time t by the concentration of the initial feed at time zero. The carbon balance based on the liquid phase analysis was checked for all reported experiments and was found in the range between 94.8% and 98.7%. The missing carbon could be attributed to experimental errors and small amounts of light hydrocarbons and carbon oxide byproducts in the gas phase after the reaction. Since the compound groups in this work are mainly found in bio-oils, the focus is put on the evaluation of the liquid phase reaction products.
2.3 Kraft lignin hydrotreatment and product analysis
The hydrotreatment experiment for Kraft lignin was performed in the same batch reactor described for the PG model reaction. 2.25 g of Kraft lignin, 0.75 g of sulfided catalyst, and 75 mL of hexadecane were used in the experiment. Reaction parameters were set as 340 °C, 40 bar initial H2 pressure, 1000 rpm, and 5 h. The liquid products were subjected to 2D GC × GC analysis. The liquid product analysis and selectivities calculation are described in detail in ESI.†
2.4 Catalyst characterization
The specific surface area and pore size volume were determined by nitrogen physisorption (BET) isotherms using a TriStar 3000 gas adsorption analyzer measured at isothermal conditions in liquid nitrogen (77 K). The catalyst (approximately 0.3 g) was weighed in a quartz tube and dried at 250 °C for 2 h in a flow of N2 before the measurement. The specific surface area was calculated from the adsorption isotherm according to the Brunauer–Emmett–Teller (BET) equation. Pore size distribution, mean pore diameter, and total pore volume were calculated using the Barrett–Joyner–Halenda (BJH) equation. The metal content in the fresh as-synthesized catalysts, catalysts were analyzed by Inductively Coupled Plasma (ICP)-sector Field Mass Spectroscopy by ALS Scandinavia AB, Luleå, Sweden. The X-ray diffraction (XRD) pattern of the sulfided catalysts was analyzed using a Bruker AXSD8 Advance X-ray powder diffractometer with CuKα radiation (λ = 1.542 Å). The measured 2θ angle range was 20° to 80°.
Transmission Electron Microscopy (TEM) images were acquired using an FEI Titan 80-300 transmission electron microscope (TEM) operated at an accelerating voltage of 300 kV. A high-angle annular dark-field (HAADF) was used to acquire scanning TEM (STEM) images. Energy dispersive X-ray (EDX) analysis was performed using an Oxford X-sight detector under STEM mode to identify the chemical elements in the catalyst samples. TEM Imaging & Analysis (TIA) software was used for data analysis and spectrum acquisition. 15–25 representative images were used for data analysis. Approximately 300–350 MoS2 slabs were measured and processed by ImageJ software to calculate the average MoS2 slab length (ΔL) and stacking number (Δn) using the following equations:34
| |  | (4) |
| |  | (5) |
where
i is the total number of MoS
2 slabs.
xi is the number of MoS
2 slabs with
Ni layers of length
li.
Ni is the stacking number and
li is the MoS
2 slab length.
We also further calculate the MoS2 dispersion (fmo) with the following equation reported in the literature:34
| |  | (6) |
where Mo
edge is the number of Mo atoms that are located on the edges of the MoS
2 slabs while Mo
total is the total number of Mo atoms.
ni is the number of Mo atoms along the edge of MoS
2 slabs with its length obtained by (
L = 3.2(2
ni − 1) Å), and
m is the total number of MoS
2 slabs obtained from the TEM images of different catalysts.
The edge-to-corner ratio of MoS2 slabs was calculated based on the following equation:35
| |  | (7) |
X-ray Photoelectron Spectroscopy (XPS) in this study was performed using a PerkinElmer PHI5000 VersaProbe III Scanning XPS microscope. The X-ray source was a monochromated Al-Kα X-ray with a binding energy of 1486.6 eV. The vacuum chamber was at less than 2 × 10−8 torr and the beam size diameter was 100 μm. The freshly sulfided samples underwent a survey scan first to evaluate the overall composition with a step size of 1.0 eV. This was followed by a narrow scan to analyze the chemical state of each element on the selected region with a step size of 0.1 eV. High-resolution spectra for Ni, Mo, O, S, Cu, C, and Al core levels were obtained. All measurements were aligned with the adventitious carbon peak (C 1s) at 284.8 eV. Multipack software was then used to analyze the raw data obtained from XPS.
3. Results and discussion
3.1 Catalyst characterization
The textural and compositional properties of the synthesized catalysts are presented in Table 1. The specific surface area and pore volume of the catalysts decreased when doping the catalyst with a second transition metal, indicating pore blockage.
Table 1 Elemental composition (wt%) and BET results for the as-synthesized catalysts
| Catalyst |
Elemental composition, (wt%) |
N2 physisorption |
| Mo |
Ni |
Cu |
Fe |
Zn |
S
a
|
V
p
|
d
p
|
|
S
a = BET surface area (m2 g−1), Vp = pore volume (cm3 g−1), dp = average pore size (Å).
|
| Mo |
13.2 |
— |
— |
— |
— |
155 |
0.36 |
93.2 |
| NiMo |
13.4 |
3.47 |
— |
— |
— |
133 |
0.29 |
87.2 |
| CuMo |
12.4 |
— |
3.32 |
— |
— |
144 |
0.32 |
89.2 |
| FeMo |
12.3 |
— |
— |
2.47 |
— |
139 |
0.34 |
97.7 |
| ZnMo |
11.0 |
— |
— |
— |
2.23 |
141 |
0.34 |
96.3 |
The XRD patterns of the freshly sulfided alumina supported catalyst and γ-alumina are shown in Fig. 1. Prominent diffraction peaks of γ-Al2O3 characterized at 2θ = 32.3°, 37.0°, 39.2°, 45.7°, 60.7°, and 66.6°, corresponds to (220), (311), (222), (400), (511) and (440) orientation, can be distinguished in all catalysts.36 Since all synthesized catalysts are pre-sulfided prior to XRD analysis, the main diffraction peaks for MoS2 were expected. However, the diffraction peaks that correspond to MoS2 (2θ = 33.4°, 39.5°, 49.5°, 58.6°, and 68.5°) were absent for all studied catalysts.37 This could be due to the MoS2 peaks being masked by the alumina peaks, which are in the same region, for instance, 2θ = 33.4° and 39.5°.37 Another possibility is that the MoS2 phase was well dispersed on the support and exhibiting low crystallinity. The metal sulfide phases like NiS, FeS, FeS2, and CuS were also not distinguished in the XRD pattern which could be attributed to the relatively low metal loading that is below the XRD detection limit. However, for the sulfided ZnMo catalyst, three characteristic peaks at 2θ = 28.6°, 47.6°, and 56.5° which corresponds to (111), (220) and (311) planes were observed, indicating the presence of the ZnS phases.38
 |
| | Fig. 1 Diffractograms of the freshly sulfided catalysts and γ-alumina from XRD measurements. | |
XPS analysis was carried out to understand the chemical and electronic state of the prepared sulfided catalysts. The XPS spectra for each catalyst are shown in ESI (Fig. S1†) and Table 2 shows a summary of the Mo 3d XPS results. The two characteristic peaks for Mo 3d core-level spectra at 228.7 eV and 231.8 eV were found, which is attributed to the doublet of Mo 3d5/2 and Mo 3d3/2, respectively.39 These two peaks are associated with the Mo4+ state in MoS2. The other two peaks at 232.4 eV and 235.5 eV are attributed to the Mo6+ state of MoO3.40 Besides, peaks at 230.6 eV and 233.7 eV, corresponding to the Mo 3d5/2 and Mo 3d3/2 levels, respectively, for Mo5+ species was present in the CuMo catalyst.40 The Mo sulfidation degree was calculated based on the ratio of the Mo4+ (MoS2) contribution over the total Mo species (Mo4+, Mo5+, and Mo6+) identified in the Mo 3d spectra (see Table 2.). It should be noted that the presence of Mo5+ and Mo6+ can be attributed to the incomplete sulfidation of Mo oxide species, or possibly also due to surface re-oxidation of the present MoS2 while transferring the samples under air to the XPS analysis. For S 2p core-level spectra, characteristic peaks at 161.6 eV and 162.8 eV corresponding to S 2p1/2 and S 2p3/2, respectively, were observed (see Fig. S1a†), which represent S2− species.40 Besides, SO42− species also exist in FeMo and ZnMo catalysts with one doublet at binding energies of 168.7 eV and 169.9 eV in S 2p spectra (Fig. S1e and f†).41
Table 2 Mo 3d XPS characterization results for the measured sulfided catalysts and TEM analysis
| Sulfided catalyst |
Mosulfidation (%) |
Binding energy (eV) |
Average slab length (ΔL), nm |
Average stacking degree (Δn) |
MoS2 dispersion (fMo) |
Mo edge-to-corner ratio (fedge/fcorner)Mo |
| Mo4+ |
Mo5+ |
Mo6+ |
| 3d5/2 |
3d3/2 |
3d5/2 |
3d3/2 |
3d5/2 |
3d3/2 |
| Mo |
69.2 |
228.7 |
231.8 |
— |
— |
232.4 |
235.5 |
5.64 |
1.95 |
0.139 |
7.32 |
| NiMo |
57.9 |
229.3 |
232.4 |
— |
— |
232.5 |
235.6 |
5.10 |
2.16 |
0.146 |
6.47 |
| CuMo |
28.4 |
229.3 |
232.4 |
230.6 |
233.7 |
233.7 |
236.9 |
5.02 |
1.96 |
0.153 |
6.34 |
| FeMo |
92.3 |
229.1 |
232.5 |
— |
— |
233.7 |
236.1 |
5.15 |
1.87 |
0.149 |
6.54 |
| ZnMo |
83.1 |
228.9 |
232.1 |
— |
— |
233.7 |
235.6 |
4.23 |
2.07 |
0.168 |
5.11 |
Different Ni species can be identified from the Ni 2p spectra for sulfided NiMo catalyst (Fig. S1b†). The binding energies at 853.9 eV and 856.5 refer to Ni 2p3/2, while 871.1 eV and 874.2 eV to Ni 2p1/2, indicating the presence of Ni2+ in NiMo catalyst.42 Among these, NiSx exhibits a minor peak at 853.9 eV, and NiMoS shows a distinct peak at 856.5 eV.42 Besides, two satellite peaks at 863.4 eV and 880.9 eV can be determined in the Ni 2p spectra.43 For CuMo catalyst (Fig. S1c†), two distinct peaks at 937.2 eV and 957 eV corresponding to the Cu 2p3/2 and Cu 2p1/2, respectively demonstrate the existence of Cu+ species.44 Besides that, other clear peaks at 933.1 eV and 952.9 eV, were both attributed to the pure Cu metal.44 Cu2+ species is excluded as no satellite peak can be seen in the Cu 2p spectra. However, it should be noted that Cu can be reduced by the X-rays in XPS, which could reduce the amount of Cu2+.45 The Fe 2p spectrum (Fig. S1d†) for sulfided FeMo catalyst shows three doublets, which correspond to the binding energies of Fe2+, Fe3+, and satellite characteristics.46 Two peaks with binding energies at 708.3 eV and 721.4 eV are attributed to Fe2+ species.46 While the peaks at 711.5 eV and 725.1 eV are referred to as Fe3+ species.46 These results suggest the presence of both Fe2+ and Fe3+ in the sulfided FeMo catalyst. The peaks at 717.3 eV and 731 eV are the satellite components for Fe 2p3/2 and Fe 2p1/2, respectively. For the ZnMo sample (Fig. S1e†), two characteristic peaks can be observed at 1021.7 eV and 1044.7 eV for Zn 2p3/2 and Zn 2p1/2, respectively.47 Both are attributed to Zn2+ species. This corroborated the results obtained from XRD showing the presence of ZnS.
The morphologies of all studied catalysts were examined by TEM. Typical linear and curvy MoS2 black fringes scattered around can be observed in all TEM images as shown in Fig. 2(a–e). The interplanar distance of 0.64 nm corresponds to the characteristic basal plane of MoS2, which can be identified in the TEM images. The statistical results for the average slab length and average stacking layer for MoS2, MoS2 dispersion, and edge-to-corner ratio for a MoS2 slab are presented in Table 2.
 |
| | Fig. 2 TEM images of (a) Mo, (b) NiMo (c) CuMo, (d) ZnMo, (e) FeMo, and (f) HAADF STEM-EDX images of NiMo. | |
The distributions for the number of MoS2 stack layers and slab lengths are shown in Fig. 3. The number of stacks in all catalysts was in the range from one to six, with one being the most frequent. While the slab lengths were mostly between 4 nm and 6 nm. It is clear that the doping of different metals on the Mo catalyst reduced the slab length as can be seen in Table 2. In addition, the stacking layer of MoS2 increased for NiMo and ZnMo. This could result from the metal species hindering the growth of MoS2 fringes during the sulfidation. Overall, the increase in the average stacking layer of the metal-doped catalysts and the reduction in slab lengths improved the MoS2 dispersion. A good dispersion of MoS2 can also explain the absence of MoS2 peaks in the XRD analysis. The improvement in MoS2 dispersion also increases the exposure of active edges for the catalytic reaction. Moreover, an elemental mapping was performed on a selected area at the edge of the sulfided NiMo catalyst shown in Fig. 2(f) and the results indicated that there is an even distribution of Ni, Mo, and S elements on the catalyst surface. The higher dispersion of the active particles may contribute to the better HDO activities demonstrated in the case of the sulfided NiMo catalyst shown in Section 3.2.
 |
| | Fig. 3 The (a) number of stacks and (b) MoS2 slab lengths distribution for all sulfided catalysts. | |
3.2 HDO of 4-propylguaiacol (PG)
The role of Ni, Fe, Zn, and Cu on MoS2/γ-Al2O3 catalysts in sulfided form were investigated for HDO of PG at 300 °C for 5 h. The reactivity of PG and catalyst activity was studied in a batch reactor setup. The complete conversion was achieved in 2–3 h for all studied catalysts at 300 °C (Fig. 4). About 91% conversion for PG was achieved in 1 h for the Mo catalyst. It is worth noting that the catalysts doped with Ni, Fe, Cu, and Zn show a lower conversion, and the NiMo catalyst gives the lowest conversion after 1 h (74%). Using 1 h as a reference time, PG conversion decreased in the order of Mo > ZnMo > CuMo > FeMo > NiMo. The lower conversion for these metal impregnated catalysts can be reasoned by the slower rate in cleaving the methoxy group of PG forming 4-propylphenol as the first step in the deoxygenation route. A mixture of partially deoxygenated compounds (phenolics), deoxygenated aromatics, and cycloalkanes compounds are formed from PG undergoing demethoxylation, dehydroxylation, hydrogenolysis, hydrogenation, transalkylation, and isomerization at different times.
 |
| | Fig. 4 PG conversion for all sulfided catalysts. | |
For the sake of simplicity, all products and intermediates are grouped into classes of compounds with two oxygen atoms, phenolics, aromatics, and deoxygenated cycloalkane hydrocarbons. The main intermediates and products are shown in Fig. 5. The distribution of different products and intermediates from the HDO of PG over all studied catalysts is illustrated by plotting the yield of the identified compounds versus reaction time (Fig. 6). As can be seen in Fig. 6, the yield of phenolics (mainly 4-propylphenol) increased to a maximum yield after 1–2 h and dropped slowly afterward. This can be explained by the demethoxylation of PG forming 4-propylphenol as the first step in the deoxygenation route, which then further undergoes dehydroxylation and hydrogenation forming propylcyclohexene and propylcyclohexane. It is noteworthy to mention that propylcyclohexanone was not observed for any of the catalysts in this study, but it has been reported in the literature.7,8 The primary route for the formation of propylcyclohexane is from the dehydroxylation of 4-propylphenol and hydrogenation of propylbenzene when sulfided catalysts are used. However, in the studies by Lindfors et al. and Tieuli et al. where non-sulfided catalysts were used, the formation of 4-propylcyclohexanone from tautomerization was observed.7,8 Hence in the current study using sulfided catalysts, the tautomerization of a keto–enol is non-existing or only minor, since these intermediates occur in an insignificant amount and were undetectable. Trace amounts of compounds that contain two oxygen atoms (4-propylcatechol and 1,2-dimethoxy-4-propylbenzene) were detected after 30 min for all catalysts and ceased later after 2–3 h as shown in Fig. 6. For example, at 30 min, 9% of such compounds were found for Mo catalyst, and in contrast, sulfided NiMo catalyst gave 19%.
 |
| | Fig. 5 Reaction product classes (oxygenates, hydrogenation, and hydrodeoxygenation products) for HDO of PG at a total pressure of 50 bar and 300 °C over Mo, NiMo, FeMo, ZnMo, and CuMo sulfided catalysts. | |
 |
| | Fig. 6 Product distribution over time for HDO of PG over sulfided (a) Mo, (b) NiMo, (c) CuMo, (d) ZnMo, and (e) FeMo sulfided catalysts. Reaction conditions: 300 °C, 50 bar, and 1000 rpm. Markers present the experimental points. | |
The yields of propylcyclopentane, 1-methyl-2-propylcyclopentane, 1-methyl-2-propylcyclohexane, propylcyclohexane, and propylcyclohexene (see Fig. 6) were grouped to better understand the production of these deoxygenated cycloalkanes from each catalyst. The major compound among all cycloalkanes was propylcyclohexane for all studied catalysts. The cycloalkane compounds dominated in the latter part of the reaction for all catalysts, except CuMo. For instance, 70.2% cycloalkanes were formed over the Mo catalyst after 5 h as shown in (Fig. 6(a)). It is worth mentioning that 4.5% of 1-methyl-2-propylcyclopentane was formed at 5 h suggesting that ring contraction occurred during the reaction in addition to deoxygenation and ring hydrogenation. The same results were obtained for NiMo, ZnMo, and FeMo catalysts giving a small yield of 3%, 4.6%, and 4.5%, respectively, for such cyclopentane-derived compounds. The results showed that the NiMo catalyst gave the highest total cycloalkane yield of 94%. While for FeMo, ZnMo, and CuMo catalysts, the total cycloalkane yield, were 58.1%, 67.2%, and 44.4%, respectively. In comparison, the Mo catalyst gave a final deoxygenated cycloalkane yield of 70%. The results suggest that the sulfided NiMo catalyst was the most efficient among the examined catalysts for the removal of oxygen atoms giving the highest total cycloalkane yields, while the addition of doping metals such as Fe, Zn, and Cu lowered the deoxygenation rate with Cu being the most inefficient. The better HDO activity for NiMo catalyst could be attributed to the high dispersion of active particles as shown from the TEM analysis (Table 2). On the other hand, the lower HDO activity of ZnMo, FeMo, and CuMo suggested that a better MoS2 dispersion was not always the deciding factor for a better HDO activity, because for example, the ability of the added metal to interact with Mo to create sulfur vacancy sites may be of primary importance.48
The components produced from HDO were considered while designing the study. It is worth highlighting the importance of aromatic compounds as the blending of such components in gasoline could increase the octane number.49 Besides, aromatics can be used as a feedstock for bulk chemical production.50 A final 12% aromatic hydrocarbon yield, which is mainly composed of propylbenzene and 1-methyl-3-propylbenzene was observed at the end of the reaction for the Mo catalyst. As illustrated in (Fig. 6(d) and (e)), the deoxygenated aromatics were formed already after 1–2 h and increased as the reaction proceeded, giving a final yield of 16% and 19% for sulfided FeMo and ZnMo catalysts, respectively. The lower amount of deoxygenated aromatics for the NiMo catalyst, which only accounted for 7% of the product can be due to the high hydrogenation activity of the Ni-based catalyst. Interestingly, the incorporation of Fe and Zn to modify the conventional hydrotreating catalysts was able to suppress the hydrogenation activity and produce more deoxygenated aromatic compounds. The comparison of the final yields of deoxygenated compounds (aromatics and cycloalkanes) was made. The Mo catalyst gives a total yield of deoxygenated compounds of 82.1%. For NiMo catalyst, a complete deoxygenation was achieved after 5 h. While for ZnMo, FeMo and CuMo catalysts, total yield of deoxygenated compounds was 86.6%, 74.3% and 50.1%, respectively. These results demonstrated that NiMo catalyst was efficient in deoxygenation and ZnMo catalyst was able to improve the PG deoxygenation as compared to the base Mo catalyst. On the other hand, FeMo and CuMo catalysts inhibited the production of deoxygenated compounds.
The phenolics were first observed to reach a maximum and later decrease for all catalysts. As can be seen in (Fig. 6(b)), the same trend can be observed for the NiMo catalyst, with the highest phenolics deoxygenation giving the highest cycloalkanes yields of 94% and about 6% deoxygenated aromatics at the end of the reaction. Interestingly, for the NiMo catalyst, the phenolics yield which mainly consists of 4-propylphenol gave a maximum yield of 34% after 2 h of reaction whereas, for the Mo catalyst, the phenolics yield reached a maximum of 44% after 1 h. This can be explained by a faster rate of cleavage of the methoxy group in PG for the Mo catalyst compared to the NiMo catalyst. This can also be observed for the ZnMo catalyst in (Fig. 6(d)), where a maximum phenolics yield of 36% was achieved after 1 h. For the CuMo catalyst (Fig. 6(c)), the phenolics yield increased steadily to 48% in 3 h and then experienced a drop giving the final yield of 42%. On the other hand, for the FeMo catalyst (Fig. 6(e)), it took 2 h to achieve 54% phenolics yield which then decreased to 26% after 5 h. The highest phenolics yield at the end of the reaction was for the CuMo catalyst, indicating its lowest deoxygenation activity among all studied catalysts.
The yield of phenolics with 4-propylphenol being the dominant intermediate, cycloalkanes, and deoxygenated aromatics at 5 h was compared for all catalysts as presented in Fig. 7. As expected, PG was fully hydrodeoxygenated by the NiMo catalyst as results showed no phenolics present at the end of the experiment. However, the phenolics deoxygenation activity of the CuMo catalyst was the least compared to the other catalysts, giving the largest phenolic yields. This result suggests that the addition of the Cu precursor to the conventional MoS2/Al2O3 catalyst was able to give a high selectivity towards phenolics. Other than that, the higher aromatic yield for FeMo and ZnMo catalysts among all sulfided catalysts suggests that both catalysts possess a greater ability to hinder the saturation of the benzene ring to retain its aromaticity. In contrast, the NiMo catalyst is active in the hydrogenation of aromatic rings with its well-known high hydrogenation capability and hence the major product obtained from this NiMo catalyst was deoxygenated cycloalkanes.
 |
| | Fig. 7 (a) Comparison of the yield of phenolics, deoxygenated cycloalkanes and aromatic compounds at 5 h reaction, (b) selectivity to aromatic compounds, (c) phenolics compounds, and (d) cycloalkanes compounds as a function of PG conversion. Reaction conditions: 300 °C, 50 bar, and 1000 rpm. Lines are present only to indicate trends. | |
Selectivity towards phenolic compounds, deoxygenated aromatic and cycloalkane compounds as a function of PG conversion is illustrated in Fig. 7(b)–(d). As explained earlier, the dominant phenolics, 4-propylphenol, an intermediate was formed via the demethoxylation of PG. The results depicted in Fig. 7(c) show an increase in the selectivity for phenolics during low conversion, which decreases drastically at higher PG conversion, indicating that consecutive reactions occurred. The selectivity for deoxygenated cycloalkane compounds (Fig. 7(d)) follows a general trend for all catalysts, increasing slowly at low PG conversion. This can be attributed to the formation of 4-propylphenol at the beginning of the reaction. Therefore, a higher selectivity of phenolics can be observed at lower PG conversion. The selectivity for cycloalkane compounds at high conversion (90–100%) decreased in the following order: NiMo > Mo > ZnMo > FeMo > CuMo. This suggests that the NiMo catalyst has the highest hydrogenation activity while the CuMo catalyst has the least. Interestingly, there was no selectivity for the deoxygenated aromatics at a low conversion level (Fig. 7(b)). The selectivity for deoxygenated aromatics increases at 60% and 80% conversion for ZnMo and FeMo catalysts, respectively. The selectivity for deoxygenated aromatics increases at higher conversion following the order: ZnMo > FeMo > Mo > NiMo > CuMo.
3.3 Modeling the reaction pathways for hydrodeoxygenation of 4-propylguaiacol with sulfided catalysts
Based on our results and various research articles on HDO of phenolic compounds,7,9,17 a reaction route for HDO of PG was established in this study as shown in Scheme 1. At the studied reaction conditions, PG undergoes demethoxylation giving 4-propylphenol as a major intermediate. A trace amount of 4-propylcatechol was observed to increase slowly in the first 30 min, but during the following hour, it decreased in yield. The formation of 4-propylcatechol can be rationalized by the demethylation of the methoxy group in PG.16 Among the two oxygen atoms containing compounds that have been lumped together as a sum, 1,2-dimethoxy-4-propylbenzene was also observed at the beginning of the reaction. The formation of 1,2-dimethoxy-4-propylbenzene can be explained by the intermolecular transfer of the methyl group to the hydroxyl group.16,51 4-Propylphenol was further converted to propylbenzene by hydrogenolysis/deoxygenation reactions forming propylcyclohexane. A partially hydrogenated compound like propylcyclohexene was detected during the reaction, and it likely underwent further hydrogenation giving propylcyclohexane as one of the final products. Some alkylation reactions could be observed, as trace amounts of alkylated products such as 4-(1-methylpropyl)phenol and 1-methyl-3-propylbenzene were detected.15 The drastic formation of 4-propylphenol as an intermediate from all catalysts also suggests that demethoxylation is favored as compared to dehydroxylation. This is well explained in the review article by Mäki-Arvela and Murzin,2 where the cleavage of the hydroxyl group from the phenyl ring is more thermodynamically demanding as compared to the cleavage of the methoxy group. The reaction products formed during the complex reaction routes for HDO of PG were grouped into oxygenate intermediates which consist of partially deoxygenated products and hydrogenation and fully deoxygenated products (deoxygenated cyclic compounds and aromatics) as shown in Fig. 5.
 |
| | Scheme 1 Proposed reaction routes of the HDO of PG over sulfided catalysts. | |
Modeling for hydrodeoxygenation of PG was performed based on the experimental results for the sulfided catalysts in the batch reactor set-up. The main objective of the kinetic modeling of HDO of PG in the current study is to better understand the reaction routes of PG forming different main and side products while employing a sulfided catalyst. Also, we aimed to study how the impregnation of different metals on the Mo catalyst can influence the rates of different reactions in the HDO of PG. There are several literature studies reported for the kinetics for hydrodeoxygenation of phenolic model compounds over zirconia-supported Rh catalysts,52 Pt, and Ir-modified bifunctional catalysts,9 carbon-supported metal catalysts,12 and Ni promoted sulfated zirconia on SBA-15.8 However, studies of reaction behavior using sulfided catalysts for hydrodeoxygenation of phenolics are less frequent.
A simplistic pseudo-first-order kinetic model was employed to fit the kinetic data for HDO of PG obtained in the batch mode. The model was selected based on its low complexity, and low number of parameters to be estimated, and considering its purpose in this context is to allow an evaluation of the reaction pathways with the different promoted sulfided catalysts. The hydrogen concentration and catalyst mass were assumed to be constant throughout the reaction period. Their influence on the reaction rates is therefore lumped into the apparent rate constants as listed in the equations below. All reaction conditions were strictly kept the same during the evaluation of catalytic activities to ensure comparable kinetic constants.
We have developed a model by considering a simplified route as “a: 4-propylguaiacol → b: 4-propylphenol → c: propylbenzene → d: propylcyclohexane” based on the proposed reaction scheme as illustrated in Scheme 1. The rate equations for this simplified route were defined as the following:
A set of ordinary differential equations (ODE) representing the batch reactor mass balances was considered:
| |  | (11) |
| |  | (12) |
| |  | (13) |
| |  | (14) |
The ordinary differential equations were solved numerically by the MATLAB ode15s function. In the estimation of the kinetic constants for all major and side reactions, fitting experimental data to the kinetic model was performed. The residual sum of squares (SSres) was minimized and defined as:
| | | SSres = ∑(Cexp,t − Cmodel,t)2 | (15) |
where
Cexp,t is the concentration of different reaction products obtained from experimental values and
Cmodel,t is the estimated concentration from the kinetic model.
The coefficient of determination (R2) is defined as the following:
| |  | (16) |
where
Cmean is the mean value of the parameter (concentration or yield). The coefficient of determination is used as an indication of the feasibility of the kinetic model by comparing it with the average concentrations.
The data fitting results with the simplified kinetic model representing the concentration trend of PG, 4-propylphenol, propylbenzene, and propylcyclohexane over the NiMo catalyst are illustrated in Fig. 8. It can be observed from the plot that the simplified kinetic model was able to describe the general trend of the deoxygenation route for PG. The deoxygenation route for PG in the current study involves the direct scission of Caryl–OCH3 bond (DDO) giving first 4-propylphenol and then further cleavage of C–OH bond yielding propylbenzene. The drastic formation of propylcyclohexane can be explained by the rapid hydrogenation of the aromatic ring of propylbenzene. The coefficient of determination for this model is 90.5%, reflecting a rather good description of the experimental values. However, as can be seen from Fig. 8, there are some clear deviations between predictions from this simplified kinetic model and the experimental results. This could be attributed to the side reactions that were not considered.
 |
| | Fig. 8 Concentration profiles for HDO of PG over NiMo catalyst at a total pressure of 50 bar and 300 °C. The solid line denotes the modeling results and the symbols correspond to the experimental values. Notation: A = propylguaiacol, B = 4-propylphenol, C = propylbenzene, D = propylcyclohexane. | |
The simplified model was then further improved by including the main side reactions as shown in Scheme 1. An additional set of rate equations were defined as:
where
ki is corresponding to the apparent rate constants of the reaction steps in
Scheme 1. The notations for all compounds in the rate equation were as the following: a: 4-propylguaiacol, b: 4-propylphenol, c: propylbenzene, d: propylcyclohexane, e: 4-propylcyclohexene, f: 4-propylcatechol, g: 1,2-dimethoxyl-4-propylbenzene, h: 1-methyl-3-propylbenzene, and i: side products. The concentrations of propylcyclopentane, 1-methyl-2-propylcyclopentane, and 1-methyl-2-propylcyclohexane were low and hence have been lumped as one and labeled as
ci. The complete mass balance equations for the improved kinetic model include the following:
| |  | (25) |
| |  | (26) |
| |  | (27) |
| |  | (28) |
| |  | (29) |
| |  | (30) |
| |  | (31) |
| |  | (32) |
| |  | (33) |
where
ca is the concentration of the initial feed (4-propylguaiacol) expressing in mol L
−1,
cx is the concentration of compound x (4-propylphenol or any side products) and
t is the reaction time.
Hence, another model with the inclusion of all the main side reactions from the intermediates and reactants was proposed. As shown in Fig. 9, the reaction kinetics for all studied catalysts were generally improved and the experiments agreed well with the pseudo-first-order kinetic model. The best description for the concentration profile was obtained with NiMo catalyst, with a coefficient of determination of 97%. Overall, it can be concluded that the experimental data for all catalysts and kinetic trends were well described by the proposed model. The estimated parameters for the apparent kinetic rate constants (k1−k11) together with their 95% confidence interval are presented in Table S3.† It can be observed that the estimated confidence intervals are in general quite high, which could be attributed to the small number of experimental sets and that the parameters are highly correlated.
 |
| | Fig. 9 HDO of PG over sulfided (a) Mo, (b) NiMo, (c) CuMo, (d) ZnMo, and (e) FeMo at a total pressure of 50 bar and 300 °C. The solid line denotes the modeling results and the symbols correspond to the experimental values. Notation: A = propylguaiacol, B = 4-propylphenol, C = propylbenzene, D = propylcyclohexane, E = propylcyclohexene and I = sum of propylcyclopentane, 1-methyl-2-propylcyclopentane and 1-methyl-2-propylcyclohexane. | |
The analysis of results presented in Fig. 9 and Table S3,† for the HDO of PG revealed that the same reaction routes can in general be applied for all studied catalysts. Also, the results showed that adding promoters to the Mo catalyst did not change the reaction routes significantly. The rate constant k1 represents the rate for the demethoxylation step of PG, and with the Mo catalyst, its value (k1 = 1.86 × 10−2 min−1) was the highest among all catalysts. This result explains the faster demethoxylation rate for the unpromoted Mo catalyst as compared to the others and it can also relate to the faster initial PG conversion for Mo catalyst during the first 1–2 h of the reaction. It can be seen that the kinetic rate constant k3 (8.50 × 10−2 min−1) and k9 (9.92 × 10−2 min−1) for NiMo catalyst were the highest. These results correlate with the highest rate of hydrogenation of propylbenzene to propylcyclohexane and propylcyclohexene giving the highest yield of deoxygenated products as shown previously for the NiMo catalyst. Moreover, it can be observed that for CuMo and FeMo catalysts, both displayed a lower rate constant k2 compared to the Mo catalyst suggesting that they inhibit the dehydroxylation of 4-propylphenol. While the ZnMo catalyst has a higher rate constant k2 (1.05 × 10−2 min−1) than the Mo catalyst, but a lower k3 (1.65 × 10−2 min−1) and k9 (6.73 × 10−2 min−1) rate constant relative to the Mo catalyst, hence corroborating its highest aromatics production. The lowest rate constant k3 for the FeMo also verified its low rate of propylbenzene hydrogenation which resulted in a 16% aromatic yield at the end of the reaction.
3.4 Comparison of sulfided NiMo, ZnMo, and FeMo for Kraft lignin hydrotreatment
In the current work, 4-propylguaiacol (PG) was used as a model compound for the study of HDO reaction using the Mo sulfided catalysts promoted by transition metal (Ni, Fe, Cu, and Zn). The purpose was the assessment of the activity of catalysts and selectivity for different products based on their functionalities like phenolics, deoxygenated cyclic compounds, and aromatics before investigating their activities for more complex substrates like lignin.
To verify the representability of PG as an appropriate model compound for more complex bio-feedstocks. The evaluation of the sulfided NiMo, ZnMo, and FeMo was further performed for the hydrotreatment of Kraft lignin. Kraft lignin is a complex biopolymer that consists of methoxylated monolignol monomers, for instance, coniferyl, sinapyl, and p-coumaryl alcohol which are interlinked by recalcitrant C–C and C–O bonds.53 Since lignin accounts for 15–30% of the dry weight of lignocellulosic biomass and biomass-derived bio-oils is an attractive alternative fuel and chemical feedstock.54
Fig. 10 presents a comparison of different product selectivities (in terms of relative MS blob volume %) for sulfided ZnMo, FeMo, and NiMo catalysts in the hydrotreatment of Kraft lignin at 340 °C and 40 bar initial H2 pressure using the same batch reactor system as described for HDO of PG. The focus was on the analysis of the upgraded lignin oil and the product selectivities when using different sulfided catalysts. It should be noted that besides lignin oil formation, also char was observed. The reaction samples were subjected to 2D GC × GC analysis for in-depth product analysis and the respective chromatograms are shown in Fig. S4.† An array of products like deoxygenated aromatics and cycloalkanes, dimers and polyaromatics, and also oxygenated compounds were obtained from the simultaneous depolymerization and hydrotreatment of lignin. A noticeable difference in selectivity for deoxygenated monomeric cycloalkanes of 26% and 13% were obtained for ZnMo and FeMo sulfided catalysts, respectively. While a 62% monomeric cycloalkane selectivity was obtained for NiMo catalyst. The higher deoxygenation ability of the NiMo catalyst can be clearly observed here in the case of hydrotreatment of Kraft lignin in agreement with the results obtained from the model reaction.
 |
| | Fig. 10 2D GC × GC analysis for the comparison of detectable liquid phase product selectivities using sulfided ZnMo, FeMo, and NiMo catalysts for hydrotreatment of Kraft lignin. Reaction conditions: 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 lignin to catalyst mass ratio, 340 °C, 40 bar initial H2 pressure, 5 h, and 1000 rpm. :1 lignin to catalyst mass ratio, 340 °C, 40 bar initial H2 pressure, 5 h, and 1000 rpm. | |
While for deoxygenated monomer aromatic products, sulfided NiMo catalyst was seen to possess a higher selectivity of 12% as compared to ZnMo (8%) and FeMo catalysts (4%). Oxygenate products like monomeric and dimeric phenolic compounds could be observed for all the catalyst systems with FeMo (50.9%) giving the highest selectivity. One of the main differences in the product distribution between the model reaction and lignin hydrotreatment was the presence of dimeric and trimeric like naphthalene and anthracene-derived products in the lignin hydrotreatment experiments. This was mainly attributed to the depolymerization of lignin at the onset of the reaction producing the monomeric, dimeric, and polymeric phenolic fragments from lignin as observed in the GC × GC analysis results. The higher deoxygenation ability of NiMo catalysts also resulted in the absence of naphthol-derived products in the product distribution after a 5 h hydrotreatment.
The reactivity of lignin appeared to follow first the depolymerization of lignin fragments yielding compounds with hydroxy- and methoxy-groups. These oxygen-contained lignin fragments further underwent different upgrading reactions like HDO and partial HDO yielding deoxygenated products and alkylphenols. The use of PG as a model compound could qualitatively indicate the reactivity scale of the sulfided catalysts towards desired products and facilitate the search for the probable reaction network towards the upgrading of complex lignin compounds to different valuable products. Further studies on the upgrading of lignin, including in-depth product and kinetic analysis, are underway considering the formation of larger molecules, such as e.g. dimers and trimers in biomass upgrading reactions.
4. Conclusions
The effect of impregnation of 3d transition metals like Ni, Fe, Zn, and Cu on sulfided Mo-based catalysts on the conversion of 4-propylguaiacol (PG) and the selectivities for phenolics, deoxygenated aromatics, and cycloalkanes was studied. In terms of PG conversion, the addition of Ni, Fe, Cu, and Zn to Mo catalyst lowered the rate of demethoxylation giving a decreasing initial conversion in the order Mo > ZnMo > CuMo > FeMo > NiMo.
For deoxygenated cycloalkanes, the sulfided NiMo catalyst showed a final yield of 94%. On the contrary, final cycloalkane yields of 58.1%, 67.2%, and 44.4% were obtained for FeMo, ZnMo, and CuMo sulfided catalysts, respectively. For the non-promoted Mo catalyst, the cycloalkane yield was 70%. The results suggest that Ni is a promoter for the Mo catalyst while doping metals such as Fe, Zn, and Cu acted as inhibitors for the formation of deoxygenated cycloalkanes. Interestingly, the selectivity of deoxygenated aromatics increased at higher conversion following the order: ZnMo > FeMo > Mo > NiMo > CuMo, with 19% and 16% aromatics for ZnMo and FeMo catalysts, respectively. Both Zn and Fe had a negative impact on the HDO activity for PG but changed the selectivity towards aromatics like propylbenzene at full conversion. Moreover, a pseudo-first-order kinetic modeling analysis of PG hydrodeoxygenation was performed, and it clarified the deoxygenation routes and reaction network. Also, the inclusion of side reactions was critical for the kinetic model. The model was able to well explain the experimental results, with over a 90% coefficient of determination for all catalysts in this study. The direct deoxygenation of PG is the major pathway for the removal of oxygen-containing groups with 4-propylphenol being the major intermediate. Hence, the model developed in this study shows that the proposed reaction routes can be adapted for all studied catalysts. The influence of promoters on the Mo catalyst is also indicated in the modeling for HDO of PG. For Ni promoted catalyst, it resulted in higher hydrogenation rates of aromatic rings yielding cycloalkanes. On the other hand, Fe and Zn promoted catalysts inhibited the hydrogenation of the aromatic ring and facilitated the formation of aromatics. The current results are important from the industrial perspective for a better understanding of the reaction mechanism for the HDO of oxygen-containing compounds that can largely be found in biomass-derived feedstock using sulfided catalysts. To further study the effect of these transition metal-promoted sulfided catalysts on the hydrotreatment of biomass, experiments with Kraft lignin were performed. Our results show that the activity for HDO of PG qualitatively corresponded to their yields of deoxygenated products from the hydrotreatment of Kraft lignin.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project is a collaboration between Chemical Engineering at Chalmers, Preem AB, and RISE Energy Technology Center (ETC). We would like to acknowledge the Swedish Energy Agency (2017-010890) and Preem for the research funding. We would also like to acknowledge the Chalmers Material Analysis Laboratory (CMAL) for SEM, TEM, XPS, and XRD analysis.
References
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- P. Mäki-Arvela and D. Murzin, Catalysts, 2017, 7(9), 265 CrossRef.
- W. Jin, L. Pastor-Pérez, D. K. Shen, A. Sepúlveda-Escribano, S. Gu and T. Ramirez Reina, ChemCatChem, 2019, 11, 924–960 CrossRef CAS.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- A. Bjelić, M. Grilc and B. Likozar, Chem. Eng. J., 2018, 333, 240–259 CrossRef.
- C. Lindfors, P. Mäki-Arvela, P. Paturi, A. Aho, K. Eränen, J. Hemming, M. Peurla, D. Kubička, I. L. Simakova and D. Y. Murzin, ACS Sustainable Chem. Eng., 2019, 7, 14545–14560 CrossRef CAS.
- S. Tieuli, P. Mäki-Arvela, M. Peurla, K. Eränen, J. Wärnå, G. Cruciani, F. Menegazzo, D. Y. Murzin and M. Signoretto, Appl. Catal., A, 2019, 580, 1–10 CrossRef CAS.
- L. Bomont, M. Alda-Onggar, V. Fedorov, A. Aho, J. Peltonen, K. Eränen, M. Peurla, N. Kumar, J. Wärnå, V. Russo, P. Mäki-Arvela, H. Grénman, M. Lindblad and D. Y. Murzin, Eur. J. Inorg. Chem., 2018, 24, 2841–2854 CrossRef.
- S. Wang, D. Xu, J. Zhao, W. Zheng, C. Hu, X. Wen, Y. Yang and Y. Li, Catal. Sci. Technol., 2019, 9, 5712–5724 RSC.
- W. Wang, L. Li, K. Wu, G. Zhu, S. Tan, Y. Liu and Y. Yang, RSC Adv., 2016, 6, 31265–31271 RSC.
- J. L. Santos, M. Alda-Onggar, V. Fedorov, M. Peurla, K. Eränen, P. Mäki-Arvela, M. Centeno and D. Y. Murzin, Appl. Catal., A, 2018, 561, 137–149 CrossRef CAS.
- X. Liu, W. Jia, G. Xu, Y. Zhang and Y. Fu, ACS Sustainable Chem. Eng., 2017, 5, 8594–8601 CrossRef CAS.
- K. Wu, Y. Liu, W. Wang, Y. Huang, W. Li, Q. Shi and Y. Yang, Mol. Catal., 2019, 477, 110537 CrossRef CAS.
- S. Song, J. Zhang and N. Yan, Fuel Process. Technol., 2020, 199, 106224 CrossRef CAS.
- A. Vuori and J. B. Bredenberg, Holzforschung, 1984, 38, 253–262 CrossRef CAS.
- N. Joshi and A. Lawal, Ind. Eng. Chem. Res., 2013, 52, 4049–4058 CrossRef CAS.
- R. R. Chianelli, G. Berhault and B. Torres, Catal. Today, 2009, 147, 275–286 CrossRef CAS.
- C. C. Templis, C. J. Revelas, A. A. Papastylianou and N. G. Papayannakos, Ind. Eng. Chem. Res., 2019, 58, 6278–6287 CrossRef CAS.
- Y. Romero, F. Richard and S. Brunet, Appl. Catal., B, 2010, 98, 213–223 CrossRef CAS.
- W. Song, S. Zhou, S. Hu, W. Lai, Y. Lian, J. Wang, W. Yang, M. Wang, P. Wang and X. Jiang, ACS Catal., 2019, 9, 259–268 CrossRef CAS.
- J. A. Tavizón-Pozos, V. A. Suárez-Toriello, P. Del Ángel and J. A. De Los Reyes, Int. J. Chem. React. Eng., 2016, 14, 1211–1223 Search PubMed.
- Y. K. Hong, D. W. Lee, H. J. Eom and K. Y. Lee, J. Mol. Catal. A: Chem., 2014, 392, 241–246 CrossRef CAS.
- C. Sepúlveda, N. Escalona, R. García, D. Laurenti and M. Vrinat, Catal. Today, 2012, 195, 101–105 CrossRef.
- A. Infantes-Molina, B. Pawelec, J. L. G. Fierro, C. V. Loricera, A. Jiménez-López and E. Rodríguez-Castellón, Top. Catal., 2015, 58, 247–257 CrossRef CAS.
- M. Ternan, J. Catal., 1987, 104, 256–257 CrossRef CAS.
-
S. Harris and R. R. Chianelli, Catalysis by Transition Metal Sulfides: A Theoretical and Experimental Study of the Relation between the Synergic Systems and the Binary Transition Metal Sulfides, 1986, vol. 98 Search PubMed.
- S. Eijsbouts, G. H. Anderson, J. A. Bergwerff and S. Jacobi, Appl. Catal., A, 2013, 458, 169–182 CrossRef CAS.
- J. Shabtai, Q. Guohe, K. Balusami, N. K. Nag and F. E. Massoth, J. Catal., 1988, 113, 206–219 CrossRef CAS.
- J. A. Rodriguez, J. Phys. Chem. B, 1997, 101, 7524–7534 CrossRef CAS.
- A. M. Mastral and J. M. Palacios, Fuel Process. Technol., 1993, 36, 161–167 CrossRef CAS.
- P. Arora, H. Ojagh, J. Woo, E. Lind Grennfelt, L. Olsson and D. Creaser, Appl. Catal., B, 2018, 227, 240–251 CrossRef CAS.
- H. Ojagh, D. Creaser, S. Tamm, P. Arora, S. Nyström, E. Lind Grennfelt and L. Olsson, Ind. Eng. Chem. Res., 2017, 56, 5547–5557 CrossRef CAS.
- M. A. Salam, P. Arora, H. Ojagh, Y. W. Cheah, L. Olsson and D. Creaser, Sustainable Energy Fuels, 2019, 4, 149–163 RSC.
- D. Ferdous, A. K. Dalai, J. Adjaye and L. Kotlyar, Appl. Catal., A, 2005, 294, 80–91 CrossRef CAS.
- J. Lian, J. Ma, X. Duan, T. Kim, H. Li and W. Zheng, Chem. Commun., 2010, 46, 2650–2652 RSC.
- W. Wang, L. Li, K. Zhang, Z. Qiao, P. Liu and Y. Yang, React. Kinet., Mech. Catal., 2014, 113, 417–429 CrossRef CAS.
- M. S. Akhtar, S. Riaz, R. F. Mehmood, K. S. Ahmad, Y. Alghamdi, M. A. Malik and S. Naseem, Mater. Chem. Phys., 2017, 189, 28–34 CrossRef CAS.
- T. Weber, J. C. Muijsers and J. W. Niemantsverdriet, J. Phys. Chem., 1995, 99, 9194–9200 CrossRef CAS.
- L. Benoist, D. Gonbeau, G. Pfister-Guillouzo, E. Schmidt, G. Meunier and A. Levasseur, Solid State Ionics, 1995, 76, 81–89 CrossRef CAS.
- S. Tresintsi, K. Simeonidis, N. Pliatsikas, G. Vourlias, P. Patsalas and M. Mitrakas, J. Solid State Chem., 2014, 213, 145–151 CrossRef CAS.
- W. Han, H. Nie, X. Long, M. Li, Q. Yang and D. Li, Catal. Today, 2017, 292, 58–66 CrossRef CAS.
- G. M. Bremmer, L. van Haandel, E. J. M. Hensen, J. W. M. Frenken and P. J. Kooyman, Appl. Catal., B, 2019, 243, 145–150 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- N. Wilken, R. Nedyalkova, K. Kamasamudram, J. Li, N. W. Currier, R. Vedaiyan, A. Yezerets and L. Olsson, Top. Catal., 2013, 56, 317–322 CrossRef CAS.
- X. Guo, W. Wang, K. Wu, Y. Huang, Q. Shi and Y. Yang, Biomass Bioenergy, 2019, 125, 34–40 CrossRef CAS.
- A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev, I. G. Danilova, I. P. Prosvirin, V. V. Kriventsov, V. I. Zaikovskii, D. Freude and A. G. Stepanov, ACS Catal., 2017, 7, 1818–1830 CrossRef CAS.
- L. S. Byskov, J. K. Nørskov, B. S. Clausen and H. Topsøe, J. Catal., 1999, 187, 109–122 CrossRef CAS.
-
M. A. Fahim, T. A. Alsahhaf and A. Elkilani, Fundam. Pet. Refin., 2010, pp. 11–31 Search PubMed.
- R. W. Thring, S. P. R. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 2000, 62, 17–30 CrossRef CAS.
- A. Vuori, A. Helenius and J. B. S. Bredenberg, Appl. Catal., 1989, 52, 41–56 CrossRef CAS.
- Y. He, Y. Bie, J. Lehtonen, R. Liu and J. Cai, Fuel, 2019, 239, 1015–1027 CrossRef CAS.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Cheng, M. Addy, Q. Lu, M. M. Omar, Y. Liu, Y. Wang, L. Dai, E. Anderson, P. Peng, H. Lei and R. Ruan, Bioresour. Technol., 2017, 241, 1118–1126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se00184a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a