
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifactor theoretical modeling of solar thermal fuels built on azobenzene and norbornadiene scaffolds†
Reuben
Szabo‡
,
Khoa N.
Le‡§
and
Tim
Kowalczyk
 *
*
Department of Chemistry, Advanced Materials Science and Engineering Center, Institute for Energy Studies, Western Washington University, Bellingham, WA, USA. E-mail: Tim.Kowalczyk@wwu.edu
First published on 6th April 2021
Abstract
Solar thermal fuels (STFs) offer a unique way of harnessing energy from the sun by absorbing photons and storing the energy in a metastable photoisomerized state. The reverse isomerization process can then be catalytically or thermally triggered to release the stored energy and return the fuel to its stable configuration. Functionalization of these compounds is necessary to reach practical energy storage densities, but substitutions that increase the energy storage density may adversely impact performance at other steps along the fuel cycle. Recent computational screening efforts to identify high-performance STF candidates have focused on properties that can be estimated from ground-state electronic structure methods. Here we argue that computational screening of STF candidates across the full fuel cycle benefits from a multifactor approach with excited-state properties like excitation energies and photoisomerization quantum yields addressed alongside key ground-state properties like energy storage densities and reverse isomerization barriers. As a critical step toward multifactor high-throughput screening and optimization of STFs, in this work we first systematically simulate the specific storage energy and excitation energy of substituted azobenzene- and norbornadiene-based STFs through electronic structure calculations. Density-functional tight-binding (DFTB) predictions are benchmarked against density functional theory (DFT) and experimental measurements where available. To encompass the complete solar thermal fuel cycle in these compounds, we then apply DFT methods to analyze the reverse isomerization process and its relationship to the photoisomerization quantum yield. We find that DFTB provides a useful balance between accuracy and computational efficiency for virtual screening of STF photoabsorption and energy storage, while isomerization barrier and quantum yield predictions require more sophisticated approaches.
1 Introduction
A major challenge in meeting present and future demand for renewable energy, and solar energy in particular, is resource intermittency: without a way to store excess solar energy generated during peak daylight hours, solar energy cannot meet the sharp increase in residential energy demand that occurs after sunset, giving rise to the so-called “duck curve”.1,2 Chemical solutions to the renewable energy storage challenge include advanced battery technologies3–5 and production of solar fuels, i.e. hydrocarbons produced via photoelectrochemical reduction of CO2 through artificial photosynthesis.6–9 Solar thermal fuels (STFs) offer a novel solution by coupling solar energy absorption and storage at the single-molecule level through photoisomerization, potentially bypassing the need to manufacture, operate, and maintain separate solar energy conversion and storage systems. Operating like a nanoscale solar battery or rechargeable solar fuel, STFs compounds convert photons to chemical energy and release it as heat on demand.10–12 The low energy densities and narrow absorption bands of STFs place limits on the scope of potential applications for this technology relative to combustion-based solar fuels; nevertheless, the direct coupling of energy conversion and storage in STFs makes them exciting targets as sustainable fuels, particularly for localized heating applications.13The essential features of a promising STF candidate are summarized in Fig. 1. Candidate compounds should readily absorb visible light and photoisomerize with high quantum yield to a mestastable configuration. Reverse isomerization to the stable configuration then allows the stored energy to be released as heat. Many compounds are capable of performing these steps of the STF cycle, but several potentially conflicting properties must be simultaneously optimized to achieve high-performance STFs. First, the absorption spectrum of the compound must closely match the solar spectrum. Second, the photoisomerization quantum yield must be high. Third, the ground state energy difference between the metastable and stable isomers must be maximized, while the molar mass should be kept as low as possible to create an energy-dense fuel. Finally, the energy barrier for the reverse isomerization must result in a half-life for the metastable configuration that lasts long enough for reliable storage, yet not be great enough to prohibit efficient reclamation of the chemical energy on demand by triggering the reverse isomerization either thermally or catalytically.14
 | ||
| Fig. 1 Processes comprising the solar thermal fuel cycle along ground- and excited-state potential energy curves. | ||
Two classes of STF candidates that have received considerable attention in the literature are the azobenzenes15–18 and the norbornadiene–quadricyclane (NBD–QC) system,19–34 shown in Fig. 2. Without functionalization, the parent compounds both of these photoswitches fall short of meeting the requirements of a high-performance STF. Azobenzene absorbs visible light at around 400 nm, but only stores 41.5 kJ mol−1 of energy and has a lifetime of only 4 days.10 In contrast, the NBD–QC system has a high storage energy at 96 kJ mol−1 and a low molecular weight, but only absorbs UV light.10 For both systems, the photoswitch scaffold can be functionalized with electron-donating and withdrawing groups to tune those properties that fail to meet high-performance STF criteria while hopefully preserving the properties that are already satisfactory.
 | ||
| Fig. 2 Photoisomerization of (a) azobenzene from the trans to cis isomer (b) norbornadiene to quadricyclane. | ||
Unfortunately, the photophysical and thermochemical properties of STFs are not completely independently tunable: functionalization that improves one performance metric may adversely impact another. This is apparent in the balance between functionalization and low molecular weight, and in the estimated efficiency limits for STFs. An ideal absorption energy for our solar spectrum is near 2 eV, but this will limit the best storage energy densitites to around 500 kJ kg−1 while maintaining desirable lifetimes.12 Further trends already identified in the literature are shown in Fig. 3, illustrating in particular the observation by Jorner et al. of a strong negative correlation between photoisomerization quantum yield and molar energy storage density, identified through analysis of complete active space self-consistent field (CASSCF) wavefunctions of related norbornadienes.21 They justified this inverse correlation on the basis of the Hammond postulate: a reduction of energy storage density by raising the energy of the stable isomer should shift the location of the transition state along the reaction coordinate toward the stable isomer. This shift of the transition state toward the stable isomer then leads to a larger excited-state gradient in the vicinity of the conical intersection and consequently a larger photoisomerization quantum yield.21 A motivating goal of our study is to understand the robustness of these trends at the DFT and DFTB levels so that we can attempt to seek compounds that defy them.
 | ||
| Fig. 3 Shifts in the location of the transition state along the reaction coordinate towards the reactants (red) or products (blue) are expected to influence both the storage energy and quantum yield of the STF.21 | ||
To find functionalization patterns that optimize the performance of a given STF scaffold, computational methods provide a useful means of narrowing the scope of possibilities. If a semiemprical method like density-functional tight-binding (DFTB)35–38 is sufficiently accurate for the task, then hundreds of thousands of potential functional groups can be screened for their effect on the thermodynamic and photophysical properties of the resulting STF compounds.
Since the initial DFT-based high-throughput screening for azobenzene-class STFs by Liu et al.,18 a semiempirical-based screening approach has been recently applied to ground-state properties in a different class of STFs, the dihydroazulene/vinylheptafulvene family of compounds, using Grimme's GFN2-xTB method39 coupled to linear regression models.40 When properly benchmarked, these approaches mitigate the limitations of more accurate but computationally intensive approaches rooted in multireference electronic structure theory,21 but unlike multireference methods, they do not directly screen critical excited-state features such as excitation energies and photoisomerization quantum yields. By incorporating excited-state STF properties at an analogous level of approximation to ground-state properties via an excited-state DFTB approach, we can screen for STF candidates that perform well throughout the full cycle of energy conversion, storage, and release.
Another advantage of a semiempirical approach to STF screening is the flexibility afforded by the computational speed of the approach in terms of incorporating environmental factors directly into the screening process. Recent works have established that the thermodynamics of STFs can be substantially influenced by solvent effects,41 conformational effects,42 and intermolecular interactions.43 Incorporating these effects within wavefunction-based or DFT electronic structure models is computationally intensive. Therefore, another benefit of studying the performance of DFTB as a screening tool for STF properties is that the higher potential throughput of DFTB makes it better suited than more computationally intensive electronic structure tools for incorporating conformational sampling, explicit solvent interactions, and intermolecular interactions between STF molecules directly into the screening strategy. In this work we focus especially on the correlation of ground- and excited-state STF properties of isolated compounds estimated by DFT and by DFTB as a building block toward predictive screening strategies.
In this article, we measure the accuracy of ground- and excited-state DFTB in predicting important STF characteristics, including excitation energies and storage energy densities, for two classes of STF compounds. We also estimate reverse isomerization energy barriers and quantum yields for photoisomerization for these candidates using DFT-based approaches. Where the data are available, our calculations are directly compared against experimental measurements.
The remainder of the article is organized as follows. We first introduce two sets of candidate STF compounds based on the azobenzene and norbornadiene scaffolds, including an overview of prior experimental and theoretical analysis of these compounds as STF candidates. We then provide details of our computational approach to determining storage energies, absorption energies, reverse isomerization barriers, and photoisomerization quantum yields in these compounds. Next we report and discuss the performance of our approach simulating each of these four properties through comparison to experimental (where available) and DFT reference data. Finally, we summarize our conclusions regarding the screening potential of DFTB for STFs and implications for the design and further optimization of STFs.
2 Solar thermal fuel candidates
The azobenzene derivatives adopted in this study are selected from among those identified in an earlier screening study by Liu et al. that focused principally on energy storage.18Fig. 4a shows a representative azobenzene from this set in its cis and trans configurations. Due to the presence of multiple local minima for the Z isomer reported for certain compounds in ref. 18, the 45 E isomers in this set correspond to 62 Z structures; therefore, we label the compounds by the index of the Z isomer as assigned in ref. 18. For each E isomer in this set of compounds, only the Z configuration(s) identified in ref. 18 were included in the analysis; that is, no further conformational sampling was pursued. Capping hydrogen atoms were added to the structures where necessary. We excluded compounds 8, 44, 45, and 62 because their isomerizations either involved multiple azo groups or ring-opening reactions, placing them in distinct categories outside the scope of our analysis (see the ESI for structural data†). Isomers of the remaining set of compounds can be described unambiguously as cis or trans, so these terms are also used in this work to distinguish between the metastable and stable isomers. | ||
| Fig. 4 Stable and metastable isomers of representative (a) azobenzene and (b) norbornadiene solar thermal fuel candidates. | ||
The twelve norbornadiene compounds evaluated here were the subject of a 2016 study by Quant et al.20 We label these compounds using the same indices introduced in that study. As shown in the representative NBD–QC structures in Fig. 4b, these compounds have been substituted at both ends of one of the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds of the NBD core. These 10 compounds can be classified into three groups according to the electron-donating and electron-withdrawing character of their substituents. Compounds 3a–e bear different aryl functional groups at the two substitution sites, while compounds 4a–d possess one cyano group and one aryl group tethered through an ethynyl linker, as in Fig. 4b. Compound 5 is related to compound 4a by excision of the ethynyl linker. Although two rotamers were identified for compounds 4b and 4c, their structural RMSD was sufficiently small that only the rotamer with the lower energy is considered in this study. Optimized structures for all compounds are available in the ESI.†
C double bonds of the NBD core. These 10 compounds can be classified into three groups according to the electron-donating and electron-withdrawing character of their substituents. Compounds 3a–e bear different aryl functional groups at the two substitution sites, while compounds 4a–d possess one cyano group and one aryl group tethered through an ethynyl linker, as in Fig. 4b. Compound 5 is related to compound 4a by excision of the ethynyl linker. Although two rotamers were identified for compounds 4b and 4c, their structural RMSD was sufficiently small that only the rotamer with the lower energy is considered in this study. Optimized structures for all compounds are available in the ESI.†
3 Computational methods
Ground-state geometry optimization and vertical excitation energy calculations were performed for all compounds in both stable and metastable configurations. These calculations were used to obtain the storage energy and vertical excitation energy for each STF system. Specific storage energies are calculated by subtracting the ground-state energy of the stable isomer from that of the metastable isomer, and then dividing the result by the mass of the compound. The vertical excitation energy of the stable isomer is calculated by subtracting the ground-state energy from the excited-state energy at the ground-state optimized geometry.The influence of the chemical environment surrounding the STF on its photophysical properties can be substantial; for example, experimental and computational evidence reveals a strong dependence of NBD–QC storage energies and photoisomerization quantum yields on solvent polarity.41 However, because of our focus on comparing DFTB and DFT approaches for high-throughput virtual screening applications, in this study we intentionally focus on comparisons rooted in gas-phase calculations. The DFT and DFTB-based approaches examined here could be coupled to standard force fields through QM/MM molecular dynamics simulations for mechanistic studies incorporating solvent effects,44,45 or a shell of solvent could even be directly incorporated into the calculations at the DFTB level.
Density functional theory (DFT) calculations were carried out in Q-Chem version 5.0.46 We assessed the sensitivity of results to basis set completeness for ground-state energies of stable and metastable isomers of two compounds and for vertical excitation energies. The deviation between ground-state energies obtained with triple-ζ Pople-type basis sets and with the 6-31G* basis set is shown in the ESI, Fig. S1.† The correction factors are small enough to justify use of a double-ζ basis, and therefore DFT calculations in this study use the 6-31G* basis set. For excited states, ESI Fig. S2† shows that the lowest singlet excitation energy in these compounds is only weakly sensitive to basis set completeness beyond the double-zeta level and to the inclusion of diffuse functions.
We adopted the ωB97X-D and PBE0 functionals for ground-state energies and geometry optimizations. Excited state calculations were performed using the restricted open-shell Kohn–Sham (ROKS) excited-state formalism,47 also with the ωB97X-D and PBE0 functionals.
Ground-state DFTB calculations were performed with the DFTB+ program, version 19.1.48 The SCC-DFTB method (also known as DFTB2) was employed with colinear spin polarization.35,49 Additional details are provided in the ESI.†
To assess vertical excitation energies of the STFs, a direct self-consistent field method (ΔSCF) was employed within the DFTB framework.50 This method, which we refer to as ΔDFTB, differs in its approach to excited-state properties from the more frequently adopted linear response time-dependent approach to excited states in DFTB (TD-DFTB).51–54 In ΔDFTB, self-consistent charge (SCC) optimization under orbital occupation constraints yields the target excited-state energy and density directly. The demonstrated accuracy of ΔDFTB for Stokes shifts of organic chromophores,50 its even-handed treatment of ground and excited states, and its computational efficiency motivate its use here for assessing vertical excitation energies of the different STF candidates. The calculations reported here were performed with an in-house modified version of the DFTB+ code; these modifications are available in the most recent version of the DFTB+ package.45
Isomerization pathways and transition state geometries were obtained with the freezing string method (FSM) and intrinsic reaction coordinate (IRC) analysis as implemented in Q-Chem.46,55 For the FSM calculations, input structures corresponding to the stable and metastable isomers were provided, and structures along the reaction coordinate were refined until a minimum energy path was obtained. The highest energy structure along the reaction coordinate then becomes an initial guess for transition-state optimization, yielding a structure with only one imaginary frequency. For the IRC calculations, a guess of the transition state structure was first optimized and validated via identification of a single imaginary frequency, and then stable and metastable structures were obtained by geometry optimization along opposing steepest-descent trajectories from the transition state.
4 Results
4.1 Storage energies
We begin by evaluating the energy storage potential of the STF candidates. Our measure of choice is the specific storage energy, that is, storage energy on a per-unit-mass basis. While molar energy storage densities are also desirable to obtain, these depend more sensitively on the details of the molecular environment; we focus on specific storage energies because our calculations do not explicitly include such interactions.For the norbornadiene compounds, DFTB specific storage energies show a strong correlation with DFT predictions across several different exchange-correlation functionals, as shown in Fig. 5. Table 1 reveals that the slope of the linear regression is closest to unity for the B3LYP functional.56 However, B3LYP specific storage energies are systematically greater than those obtained with DFTB, while PBE0 and ωB97X-D specific storage energies are much lower. Compared against the experimental data available from ref. 20 for system 4d, DFTB specific storage energies are in fact closer to the observed values than any DFT method, while second to the B3LYP functional for system 5 (Table 2). Although the absolute errors in specific storage energy can be large, the correlations shown in Table 1 and Fig. 5 suggest that DFT and DFTB methods can both effectively capture the influence of chemical substitutions on the specific storage energy of norbornadienes.
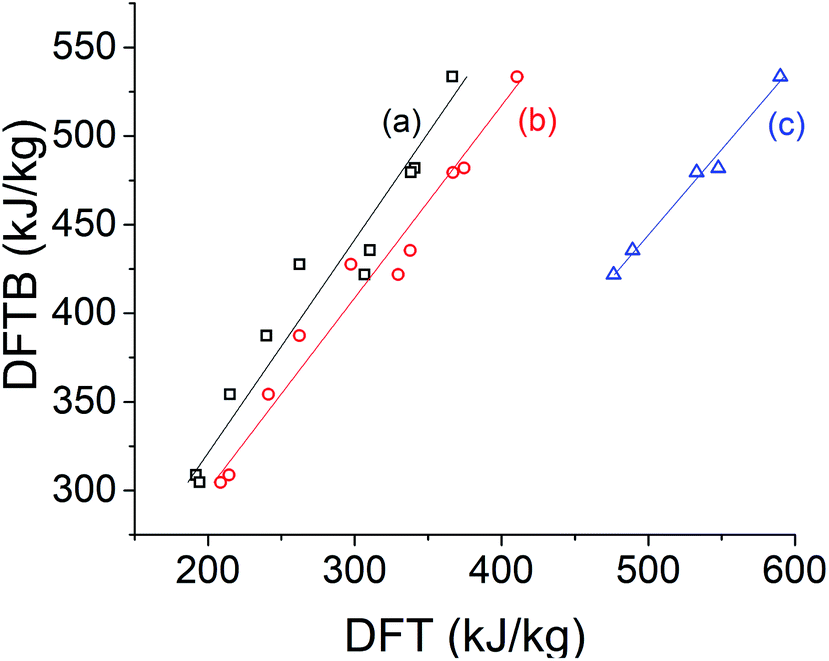 | ||
| Fig. 5 Comparison of DFTB specific storage energy of norbornadienes versus DFT/6-31G* with the (a) PBE0, (b) ωB97X-D, and (c) B3LYP20 exchange-correlation functionals. | ||
| PBE0 | ωB97X-D | B3LYP | |
|---|---|---|---|
| R 2 | 0.944 | 0.969 | 0.989 |
| m | 0.830 | 0.922 | 1.03 |
| b | −66.6 | −76.9 | 40.6 |
| Methodology | SSE (4d) | SSE (5) |
|---|---|---|
| DFTB | 422 | 533 |
| ωB97X-D | 330 | 411 |
| PBE0 | 307 | 366 |
| B3LYP | 476 | 590 |
| Experiment | 396 | 629 |
To more directly probe the question of how well DFTB reproduces the same ranking of norbornadiene STF candidates that would be produced by DFT, we turn to the Spearman rank-order correlation coefficient rs which measures the Pearson correlation between the rank-orderings of the underlying data,57
 | (1) |
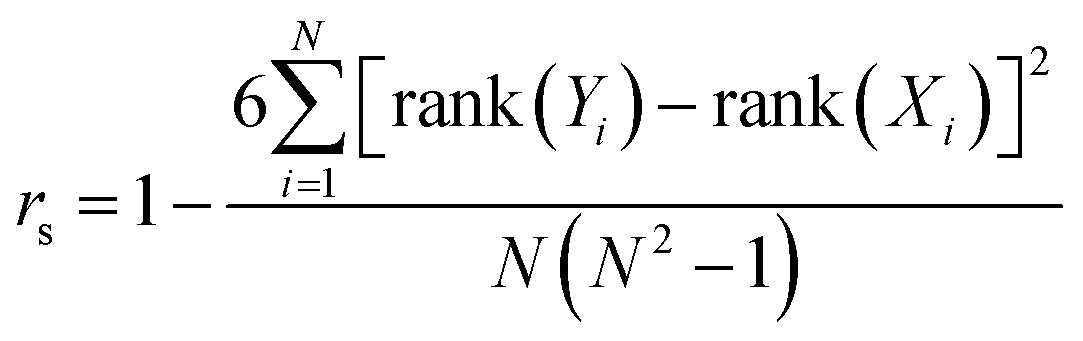 | (2) |
Fig. 6 summarizes the Spearman rank correlations among the four DFT and DFTB methods adopted to compute specific storage energies of the norbornadienes. These correlations are all close to 1, indicating that the rankings produced by the four methods are quite similar.
 | ||
| Fig. 6 Spearman correlation between DFTB and DFT predictions of specific storage energy for norbornadienes with different exchange-correlation functionals. | ||
For the azobenzene systems, DFTB calculations of the storage energy typically underestimate the storage energy compared to DFT (ωB97X-D) calculations. This trend is evident from the slope less than unity in Fig. 7. We ascribe the weaker correlation here between DFTB and DFT (R2 = 0.668), compared to the case of the norbornadienes (R2 > 0.9), to the greater degree of conformational flexibility in these compounds, in contrast with the relatively rigid norbornadiene scaffold. The ranking capability of DFTB for the azobenzenes is also lower than for the NBD–QC compounds, at rs = 0.830 compared to 0.988 for the norbornadienes with the same ωB97X-D functional.
 | ||
| Fig. 7 Calculated storage energies of azobenzenes with DFTB and DFT with ωB97X-D functional. The R2 value is 0.668 and the slope is 0.340. | ||
Table 3 summarizes the mean absolute and mean signed errors (MAE and MSE, respectively) of DFTB against DFT with both ωB97X-D and PBE0 functionals for both classes of STF compounds. DFTB overestimates norbornadiene storage energies, and underestimates those of the azobenzenes, but does so mostly systemtically, as evidenced by near-identical MAE and MSE values.
| Norbornadienes | Azobenzenes | |||
|---|---|---|---|---|
| ωB97X-D | PBE0 | ωB97X-D | PBE0 | |
| MAE (kJ kg−1) | 109.1 | 136.8 | 89.76 | 148.9 |
| MSE (kJ kg−1) | 109.1 | 136.8 | −87.86 | −137.9 |
Overall the results suggest that while DFTB is less reliable for absolute specific storage energies of STFs, it is well-suited for estimating the relative storage energies of STF candidates in the norbornadiene family. It is only slightly less well-suited for the azobenzenes, especially when the ranking capability of DFTB compared to DFT is taken into account. In this light, DFTB methods appear sufficiently accurate for high-throughput virtual screening of storage energies of substituted azobenzenes.
4.2 Excitation energies
We are especially interested in the ability of ΔDFTB to rank STF compounds according to their excitation energy so that screening methods can prioritize overlap with the solar spectrum. The ΔDFTB vertical excitation energies are compared to experimental absorption maxima20 in Fig. 8 and 9 compares the excitation energies predicted by ΔDFTB versus the ROKS approach to excited states in DFT, using the ωB97X-D functional. ΔDFTB excitation energies are well correlated with the predictions of ROKS (R2 = 0.818) and only slightly less strongly correlated with experimental data as shown in Fig. 8 (R2 = 0.723). The systematic underestimation of the excitation energy by ΔDFTB relative to ROKS appears to be more exaggerated for the diphenyl-substituted norbornadienes (3a–3e) and less exaggerated for the parent norbornadiene 5 as well as the push–pull type norbornadienes (4a–4d). | ||
| Fig. 8 Excitation energies for norbornadienes predicted by ΔDFTB versus absorption maxima from ref. 20. The linear regression has a slope of 0.549 and R2 of 0.723. | ||
 | ||
| Fig. 9 Linear regression of DFTB and ROKS excitation energies for norbornadienes. Slope = 0.961 and R2 = 0.818. | ||
A Spearman rank correlation analysis for norbornadiene excitation energies (Fig. 10) shows that the ranking of the compounds predicted by ΔDFTB deviates more from the ROKS predictions with different exchange-correlation functionals than the ROKS predictions deviate from one another. However, the Spearman correlation between ΔDFTB and ROKS exceeds 0.9 in each case examined, demonstrating that ΔDFTB is quite reliable for ranking the first singlet excited states of these norbornadienes.
 | ||
| Fig. 10 Spearman correlation between ΔDFTB and DFT predictions of excitation energies for norbornadienes with different exchange-correlation functionals. | ||
We also compare ΔDFTB vertical excitation energies of the azobenzenes against those predicted by ROKS. Fig. 11 shows that in general, ΔDFTB underestimates the excitation energies relative to ROKS; this outcome is consistent with previous observations of its performance for other small-molecule chromophores.50
 | ||
| Fig. 11 Linear regression of ΔDFTB and ROKS excitation energies for azobenzenes. Slope = 0.342 and R2 = 0.241. | ||
Table 4 summarizes the MAE and MSE for ΔDFTB calculations of norbornadiene and azobenzene excitation energies.
| Norbornadienes | Azobenzenes | |||
|---|---|---|---|---|
| ωB87X-D | PBE0 | ωB97X-D | PBE0 | |
| MAE (eV) | 0.9730 | 0.3277 | 1.105 | 0.5895 |
| MSE (eV) | −0.9730 | −0.3277 | −1.105 | −0.5878 |
Experimental evidence of a relationship between the azo-group dihedral angle of the stable isomer and the absorption maximum for the π–π* transition58 prompted us to examine whether this trend was strong enough in our data to predict excitation energy shifts from the ground-state dihedral angle alone. However, the lack of significant correlation argues for the necessity of a direct evaluation of vertical excitation energies for screening of excited-state features for the case of azobenzenes.
Overall, the results suggest that ΔDFTB should be suitably accurate for high-throughput screening and ranking of norbornadiene STF candidates by excitation energy. ΔDFTB is less effective at reproducing the excitation energy ranking predicted by ROKS for azobenzenes, but it can still serve as a first step for identifying candidates that are likely to be particularly red- or blue-shifted.
5 Discussion
Full-cycle optimization of STFs should extend beyond the specific storage energy and excitation energy reported above to also include the reverse isomerization barrier ΔE‡ and the photoisomerization quantum yield Φiso, which govern the cyclability and efficiency of the STF, respectively. We are especially interested in understanding the extent to which the transition state properties and photoisomerization quantum yield can be tuned independently of the storage energy, potentially defying the trends depicted in Fig. 3.Here we evaluate energies along the reverse isomerization reaction path with the freezing string method (FSM)55,59 and intrinsic reaction coordinate (IRC) approach.60,61 We then introduce suitable reaction coordinates for the norbornadiene–quadricyclane and azobenzene isomerizations for the purpose of identifying the location of the transition state along the reaction path. We analyze the relationship between TS locations, storage energies, and measured quantum yields where available, to determine whether they follow the hypothesized trend depicted in Fig. 3.
5.1 Transition state energies and the reaction path
Calculated reverse isomerization barriers for the norbornadienes, obtained via FSM with the ωB97X-D functional, are plotted against experimental activation enthalpies,20 derived from Eyring analysis of UV/vis kinetics in Fig. 12. Spin densities were also examined to confirm the anticipated biradical nature of the transition state. To facilitate the convergence of the transition state for the different substituted norbornadienes, the transition state of the unsubstituted norbornadiene–quadricyclane ground-state isomerization was optimized and then used as a template for the other compounds. The optimized geometries of the functional groups were preserved from the FSM TS-optimized55 geometries of each compound and appended to this core and reoptimized.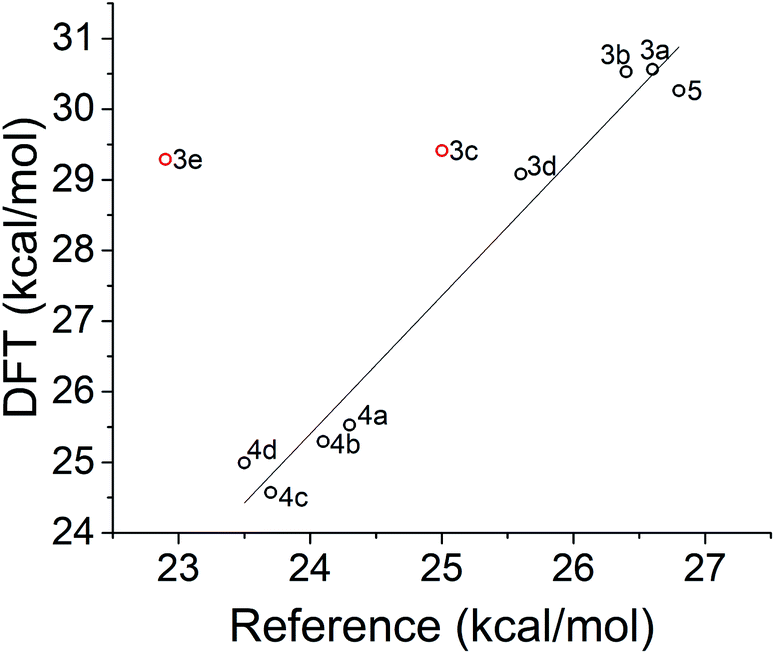 | ||
| Fig. 12 Transition state energies of norbornadiene compounds calculated with DFT and the ωB97X-D functional compared to experimentally derived activation enthalpies from ref. 20. The regression excludes compounds 3c and 3e in red (see text) and yields a coefficient of determination R2 = 0.970. | ||
As shown in Fig. 12, the correlation between predicted and reference transition state energies is strong when the fluorinated compounds, 3c and 3e, are excluded. The origin of the overestimated activation barriers for the fluorinated compounds from FSM is unclear; possible factors include differences in interaction strength with the local environment which are not accounted for in this model. The difference in TS location for the sets of compounds 4a–d and 3a–e is evident, as FSM clusters both sets tightly together. Whether or not the 3a–e series has its own regression with shallower slope is difficult to assess without a larger data set.
With an optimized transition state, the intrinsic reaction coordinate (IRC) procedure can be used to explore the energy landscape along the reaction path. Key features of a reaction path for the norbornadiene–quadricyclane isomerization are presented in Fig. 13. The TS energy is located at the central plateau. The discontinuity in the profile is an expected artifact of the switch from an unrestricted to restricted Kohn–Sham formalism for the ground state optimization; it does not adversely impact the estimation of the reverse isomerization activation barrier.
 | ||
| Fig. 13 Identification of the transition state for reverse isomerization via IRC for NBD–QC compound 5. | ||
The thermally activated reverse isomerization of azobenzenes has been previously examined with a variety of theoretical model chemistries and transition state search strategies such as intrinsic reaction coordinate analysis.62–64 Theoretical work has established the C–NN–C dihedral angle and the NN–C angle as the key degrees of freedom comprising the isomerization reaction coordinate; independently, these degrees of freedom correspond to “rotation” and “inversion” mechanisms for the isomerization, respectively.65 To uncover the details of the reaction coordinate's dependence on functionalization of the azobenzene, sophisticated strategies such as transition path sampling have been employed;66 here, our focus on virtual screening compels us to adopt a simpler approach.
Similar to our analysis of the norbornadienes, we applied both the FSM and IRC procedures to evaluate the transition states of the azobenzenes in our benchmark set. We had mixed success converging optimized TS structures for the azobenzenes via FSM, reflecting both the challenge of locating transition states generally as well as the sensitivity of the isomerization pathway to functionalization of the azobenzene.66 For the TS structures that were succesfully located and identified, we observed a wide variety of C–NN–C dihedral angles and several of the reverse isomerization energy barriers were predicted to be improbably close to zero. As shown in Fig. S7,† the majority of compounds near a dihedral angle corresponding to the cis or trans configuration have isomerization barriers near zero, potentially signaling that most of these isomerizations occur via a rotation mechanism.
There is also significant support in the literature for a rotation-driven mechanism.65,67–69 For this reason, we additionally optimized the azobenzene TS structures starting from an initial guess with a C–NN–C dihedral angle of 90°. Of the reverse isomerization energy barriers obtained by this alternative route, none were near or below zero, and only compound 5 did not have an optimized dihedral angle within 20° of 90°. For representative azobenzene compound 12, the reaction path obtained using IRC and starting from the TS optimized from a 90° dihedral angle initial guess is presented in Fig. 14. With these reaction paths in hand, we can proceed to explore relationships between the TS energy, the location of the TS along the reaction path, and the photoisomerization quantum yield for related STF compounds.
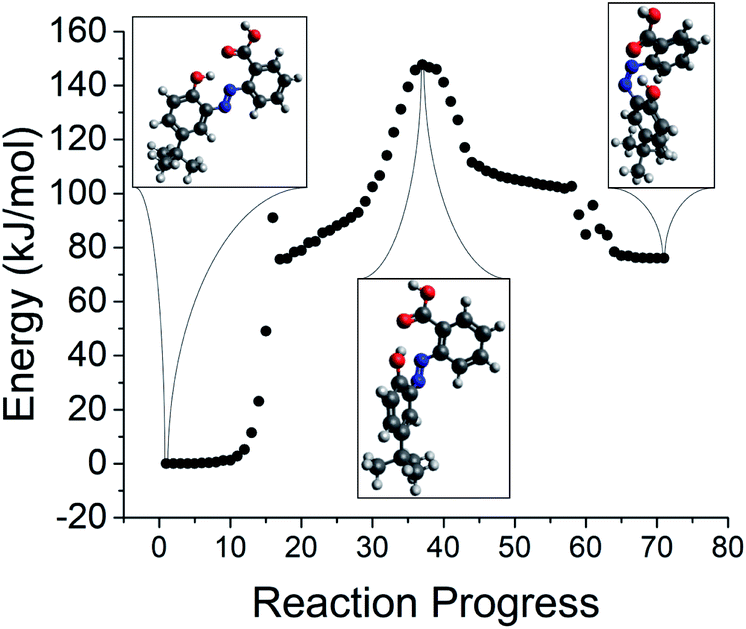 | ||
| Fig. 14 Identification of the transition state for reverse isomerization via IRC for azobenzene compound 12. | ||
5.2 Quantum yields by transition state analysis
Now we turn to the relationship between measured quantum yields for STF candidates and the location of the TS. For this purpose, we will define a reverse isomerization reaction coordinate for each class of STF. A suitable reaction coordinate for norbornadiene isomerization depends primarily on two carbon–carbon distances, b1 and b2 shown in Fig. 15 at the transition state. However, because of the asymmetric nature of these distances at the transition state, where b1 is roughly equal to the distance of a fully formed C–C bond while b2 corresponds to the separation of the two radical carbons, the reaction coordinate may not depend on them equally or even linearly.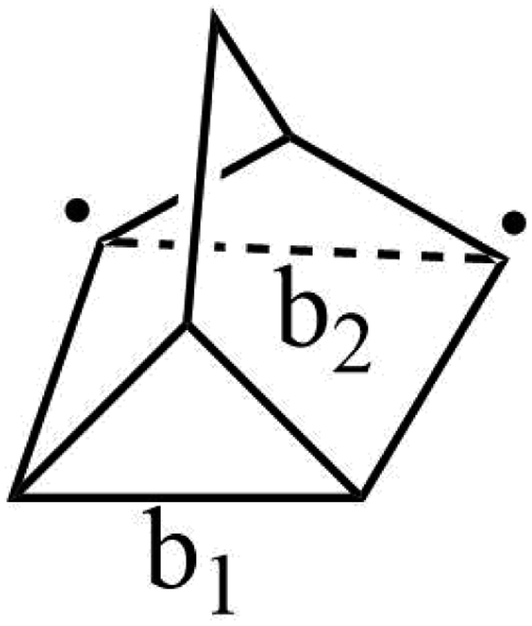 | ||
| Fig. 15 Carbon–carbon distances b1 and b2 that define the reaction coordinate for the transition from norbornadiene to quadricyclane. | ||
Using the Hammond postulate, two linear relationships were anticipated: quantum yield plotted against the transition state reaction coordinate should yield a negative correlation, as quantum yield should decrease as the transition state shifts towards the quadricyclane geometry. In contrast, storage energy plotted against the reaction coordinate of the transition state should yield a positive regression, because a transition state near quadricyclane indicates a more stable norbornadiene conformation.
By plotting b1 and b2 separately against reference quantum yield and DFT storage energy in Fig. 15, we see qualitatively different relationships for the two bond lengths. While b1, the fully formed C–C bond, correlates negatively with quantum yield as we would expect for the isomerization reaction coordinate, b2 is positively correlated with quantum yield. This suggests that b1 increases as the reaction coordinate advances towards a quadricyclane configuration, while b2 decreases. This analysis only holds true in the vicinity of the transition state: the increasing of C–C distance b1 does not hold for the entire reaction path from norbornadiene to quadricyclane. The correlations between these reaction coordinates and the STF properties of quantum yield and storage energy is strongest between the different subgroups (3a–e and 4a–d) of the norbornadienes; the correlation within a single subgroup is much weaker, similar to our findings for the reverse isomerization energy barrier itself. The correlations of b1 and b2 against quantum yield are moderate, with R2 values between 0.55 and 0.6 attributable mostly to the clustering of the two subgroups of compounds. For the storage energy, the correlations of b1 and b2 with specific storage energy are similarly strong, with coefficients of determination R2 of 0.822 and 0.878 for b1 and b2, respectively (Fig. 16). These conclusions appear to be insensitive to the choice of functional used for the TS analysis, as we obtained similar results with the PBE0 functional (Fig. S3–S6†).
 | ||
| Fig. 16 Carbon–carbon distances b1 and b2versus experimental quantum yield20,23 and DFT storage energy calculated with the ωB97X-D functional. Coefficients of determination R2 are (a) 0.598, (b) 0.559, (c) 0.841, and (d) 0.878. | ||
To perform a transition state location analysis on the azobenzene compounds, we determine a reaction coordinate for the rotation mechanism, with an NN dihedral angle of ±180° in the trans configuration and 0° in the cis configuration, shown in eqn (3). At RC = 0, the compound is in the stable trans configuration, while RC = 1 corresponds to the metastable cis configuration.
 | (3) |
For azobenzene compounds converged to the transition state from an initial guess at 90°, we have applied eqn (3) to the dihedral angles to identify its location along the reaction coordinate. For compounds with more than one C–NN–C dihedral angle, the one closest to 90° was chosen. No transition states were found where a second dihedral angle deviated significantly from the cis or trans configuration.
In Fig. 17 the location of each candidate's TS along the isomerization reaction coordinate is plotted against the candidate's computed specific storage energy. There is only a moderate correlation between the reaction coordinate we selected and the DFT-computed storage energies of these STFs with the ωB97X-D functional (R2 = 0.349), and this correlation is more sensitive to the choice of functional than it was for the norbornadienes: the same analysis with PBE0 functional yields R2 = 0.678. We conclude that STFs with more complex, substitution-dependent or environment-dependent isomerization mechanisms like the azobenzenes are less amenable to the high-throughput TS analysis approach envisioned here than STFs with simpler isomerization mechanisms like the norbornadienes. In these cases, more exhaustive automated energy landscape searches such as global reaction route mapping,70 or even molecular dynamics-driven approaches,71 may be necessary to produce robust activation barrier rankings among closely related compounds.
 | ||
| Fig. 17 DFT calculated storage energies for azobenzenes (with the ωB97X-D functional) compared to the position of the (ωB97X-D) transition state along the reaction coordinate. The trans configuration is located at 0 on the reaction coordinate, and the cis configuration is located at 1. R2 = 0.349. | ||
6 Conclusions
The promise of a solar thermal fuel (STF) that can be charged in sunlight; transported like a conventional liquid fossil fuel; and then utilized and recycled motivates our effort to assess the potential role of computational molecular science in the discovery and optimization of STFs as integrated solar energy conversion and storage materials. In this study we benchmarked ground- and excited-state density functional tight binding (DFTB) against experimental and DFT reference data to determine their suitability for high-throughput screening of STFs. We also performed reaction coordinate analyses to explore relationships between the ground-state reverse isomerization barrier and other STF properties with an eye towards strategies for independently tuning these properties.Based principally on the Spearman rank correlations observed between DFTB and DFT predictions of the essential photophysical properties of these representative STFs, we conclude that DFTB and its time-independent extension to excited states, ΔDFTB, are suitable tools for high-throughput computational screening for STFs with strong visible-light absorption and high specific storage energy. A greater degree of success with the norbornadienes relative to the azobenzenes shows that the approach presented here is especially well-suited for structures with relatively low conformational flexibility and weaker interactions with the environment. Our transition state analysis of these compounds demonstrates that DFT-based screening for reverse isomerization barriers and quantum yields can classify groups of similar STF candidates at a coarse level but struggles to discriminate among similarly substituted compounds. However, the weak correlations observed in our reaction coordinate analysis also suggest that it may be possible to identify substitution patterns that sidestep the expected correlation between storage energy and TS location along the reaction coordinate.
Through this work we can identify several areas where simulation has a role to play in accelerating the development of STFs. Although ΔDFTB alone cannot directly probe the photoisomerization quantum yield, it should be possible to predict quantum yields through a two-state configuration interaction constructed from DFTB (ground) and ΔDFTB (excited) diabatic states;72,73 efforts are underway in our lab to realize this possibility. While this work focused on gas-phase simulations for computational efficiency and ease of comparison across different theoretical models, condensed-phase simulations will be essential for accurately reproducing storage energies and isomerization barriers of STFs in the liquid state or in thin films. The gas-phase and condensed-phase data can then be harnessed to develop data-driven, high-throughput virtual screening strategies for identifying exceptional STF candidates. By probing the performance limits of this class of renewable fuels, it becomes possible to better understand their potential role in a low-carbon-intensity society.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Research Corporation for Science Advancement through a Cottrell Scholar Award to TK and by the National Science Foundation under award CHE-1664674. RS acknowledges support from the Energy Ambassadors Program of WWU's Institute for Energy Studies.Notes and references
- P. Denholm, M. O'Connell, G. Brinkman and J. Jorgenson, Overgeneration from solar energy in California: A field guide to the duck chart, National Renewable Energy Laboratory technical report, 2015 Search PubMed.
- J. Kosowatz, Mech. Eng., 2018, 140, 30–35 CrossRef.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- L. Carbone, S. G. Greenbaum and J. Hassoun, Sustainable Energy Fuels, 2017, 1, 228–247 RSC.
- S. Comello and S. Reichelstein, Nat. Commun., 2019, 10, 2038 CrossRef.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- J. Messinger, O. Ishitani and D. Wang, Sustainable Energy Fuels, 2018, 2, 1891–1892 RSC.
- Y. Yang, S. Ajmal, X. Zheng and L. Zhang, Sustainable Energy Fuels, 2018, 2, 510–537 RSC.
- R. Dittmeyer, M. Klumpp, P. Kant and G. Ozin, Nat. Commun., 2018, 10, 1818 CrossRef PubMed.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS.
- T. J. Kucharski, Y. Tian, S. Akbulatov and R. Boulatov, Energy Environ. Sci., 2011, 4, 4449–4472 RSC.
- K. Börjesson, A. Lennartson and K. Moth-Poulsen, ACS Sustainable Chem. Eng., 2013, 1, 585–590 CrossRef.
- A. Dreos, K. Börjesson, Z. Wang, A. Roffey, Z. Norwood, D. Kushnir and K. Moth-Poulsen, Energy Environ. Sci., 2017, 10, 728–734 RSC.
- C.-L. Sun, C. Wang and R. Boulatov, ChemPhotoChem, 2019, 3, 268–283 CrossRef CAS.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- M. L. Tiago, S. Ismail-Beigi and S. G. Louie, J. Chem. Phys., 2005, 122, 094311 CrossRef PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- Y. Liu and J. C. Grossman, Nano Lett., 2014, 14, 7046–7050 CrossRef CAS PubMed.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- M. Quant, A. Lennartson, A. Dreos, M. Kuisma, P. Erhart, K. Börjesson and K. Moth-Poulsen, Chem.–Eur. J., 2016, 22, 13265–13274 CrossRef CAS.
- K. Jorner, A. Dreos, R. Emanuelsson, O. El Bakouri, I. Fernández Galván, K. Börjesson, F. Feixas, R. Lindh, B. Zietz, K. Moth-Poulsen and H. Ottosson, J. Mater. Chem. A, 2017, 5, 12369–12378 RSC.
- M. J. Kuisma, A. M. Lundin, K. Moth-Poulsen, P. Hyldgaard and P. Erhart, J. Phys. Chem. C, 2016, 120, 3635–3645 CrossRef CAS.
- V. Gray, A. Lennartson, P. Ratanalert, K. Börjesson and K. Moth-Poulsen, Chem. Commun., 2014, 50, 5330–5332 RSC.
- U. Bauer, S. Mohr, T. Döpper, P. Bachmann, F. Späth, F. Düll, M. Schwartz, O. Brummel, L. Fromm, U. Pinkert, A. Görling, A. Hirsch, J. Bachmann, H.-P. Steinrück, J. Libuda and C. Papp, Chem.–Eur. J., 2017, 23, 1613–1622 CrossRef CAS PubMed.
- C. Philippopoulos, D. Economou, C. Economou and J. Marangozis, Ind. Eng. Chem. Prod. Res. Dev., 1983, 22, 627–633 CrossRef CAS.
- S. Miki, Y. Asako and Z. Yoshida, Chem. Lett., 1987, 195–198 CrossRef CAS.
- R. D. Bach, I. L. Schilke and H. B. Schlegel, J. Org. Chem., 1996, 61, 4845–4847 CrossRef CAS PubMed.
- V. A. Bren, A. D. Dubonosov, V. I. Minkin and V. A. Chernoivanov, Russ. Chem. Rev., 1991, 60, 913–948 CAS.
- R. R. Hautala, R. B. King and C. Kutal, Solar Energy: Chemical Conversion and Storage, The Humana Press, Clifton, New Jersey, 1979, pp. 331–370 Search PubMed.
- F. Franceschi, M. Guardigli, E. Solari, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1997, 36, 4099–4107 CrossRef CAS.
- Z. Yoshida, J. Photochem., 1985, 29, 27–40 CrossRef CAS.
- Y. Harel, A. W. Adamson, C. Kutal, P. A. Grutsch and K. Yasufuku, J. Phys. Chem., 1987, 91, 901–904 CrossRef CAS.
- N. J. Turro, W. R. Cherry, M. F. Mirbach and M. J. Mirbach, J. Am. Chem. Soc., 1977, 99, 7388–7390 CrossRef CAS.
- X. S. Wang, B. W. Zhang and Y. Cao, J. Photochem. Photobiol., A, 1996, 96, 193–198 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifer, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260 CrossRef CAS.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- M. Elstner and G. Seifert, Philos. Trans. R. Soc., A, 2014, 372, 20120483 CrossRef PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- M. Koerstz, A. S. Christensen, K. V. Mikkelsen, M. B. Nielsen and J. H. Jensen, High Throughput Virtual Screening of 230 Billion Molecular Solar Heat Battery Candidates, 2019, https://chemrxiv.org/articles/preprint/High_Throughput_Virtual_Screening_of_200_Billion_Molecular_Solar_Heat_Battery_Candidates/8003813/3 Search PubMed.
- M. Quant, A. Hamrin, A. Lennartson, P. Erhart and K. Moth-Poulsen, J. Phys. Chem. C, 2019, 123, 7081–7087 CrossRef CAS.
- M. Jevric, A. U. Petersen, M. Mansø, S. Kumar-Singh, Z. Wang, A. Dreos, C. Sumby, M. B. Nielsen, K. Börjesson, P. Erhart and K. Moth-Poulsen, Chem.–Eur. J., 2018, 24, 12767–12772 CrossRef CAS PubMed.
- A. Kunz, A. H. Heindl, A. Dreos, Z. Wang, K. Moth-Poulsen, J. Becker and H. A. Wegner, ChemPlusChem, 2019, 84, 1145–1148 CrossRef CAS PubMed.
- G. d. M. Seabra, R. C. Walker, M. Elstner, D. A. Case and A. E. Roitberg, J. Phys. Chem. A, 2007, 111, 5655–5664 CrossRef PubMed.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrică, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W.-z. Yu and T. Frauenheim, J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kus, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio Jr, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stueck, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- T. Kowalczyk, T. Tsuchimochi, P.-T. Chen, L. Top and T. Van Voorhis, J. Chem. Phys., 2013, 138, 164101 CrossRef PubMed.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- C. Köhler, G. Seifert, M. Elstner, H. Overhof and T. Frauenheim, Phys. Chem. Chem. Phys., 2001, 3, 5109–5114 RSC.
- T. Kowalczyk, K. Le and S. Irle, J. Chem. Theory Comput., 2016, 12, 313–323 CrossRef CAS PubMed.
- T. Niehaus, J. Mol. Struct.: THEOCHEM, 2009, 914, 38–49 CrossRef CAS.
- F. Trani, G. Scalmani, G. Zheng, I. Carnimeo, M. J. Frisch and V. Barone, J. Chem. Theory Comput., 2011, 7, 3304–3313 CrossRef CAS PubMed.
- A. Domínguez, B. Aradi, T. Frauenheim, V. Lutsker and T. A. Niehaus, J. Chem. Theory Comput., 2013, 9, 4901–4914 CrossRef PubMed.
- V. Barone, I. Carnimeo and G. Scalmani, J. Chem. Theory Comput., 2013, 9, 2052–2071 CrossRef CAS.
- A. Behn, P. M. Zimmerman, A. T. Bell and M. Head-Gordon, J. Chem. Phys., 2011, 135, 224108 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. L. Myers, A. D. Well and R. F. Lorch, in Research Design and Statistical Analysis, Routledge, 3rd edn, 2010, pp. 485–487 Search PubMed.
- L. De Boni, C. Toro, S. C. Zilio, C. R. Mendonca and F. E. Hernandez, Chem. Phys. Lett., 2010, 487, 226–231 CrossRef CAS.
- A. L. Dewyer, A. J. Argüelles and P. M. Zimmerman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1354 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- M. W. Schmidt, M. S. Gordon and M. Dupuis, J. Am. Chem. Soc., 1985, 107, 2585–2589 CrossRef CAS.
- T. Ikegami, N. Kurita, H. Sekino and Y. Ishikawa, J. Phys. Chem. A, 2003, 107, 4555–4562 CrossRef CAS.
- C. Rietze, E. Titov, S. Lindner and P. Saalfrank, J. Phys.: Condens. Matter, 2017, 29, 314002 CrossRef PubMed.
- X.-M. Liu, X.-Y. Jin, Z. X. Zhang, J. Wang and F. Q. Bai, RSC Adv., 2018, 8, 11580–11588 RSC.
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188–8203 CrossRef CAS.
- A. Muždalo, P. Saalfrank, J. Vreede and M. Santer, J. Chem. Theory Comput., 2018, 14, 2042–2051 CrossRef PubMed.
- S. Monti, G. Orlandi and P. Palmieri, Chem. Phys., 1982, 71, 87–99 CrossRef CAS.
- A. A. Blevins and G. J. Blanchard, J. Phys. Chem. A, 2004, 108, 4962–4968 CrossRef CAS.
- P. Tavadze, G. A. Franco, P. Ren, X. Wen, Y. Li and J. P. Lewis, J. Am. Chem. Soc., 2018, 140, 285–290 CrossRef CAS PubMed.
- K. Ohno and S. Maeda, J. Phys. Chem. A, 2006, 110, 8933–8941 CrossRef CAS PubMed.
- L.-P. Wang, A. Titov, R. McGibbon, F. Liu, V. S. Pande and T. J. Martinez, Nat. Chem., 2014, 6, 1044–1048 CrossRef CAS.
- B. Kaduk and T. Van Voorhis, J. Chem. Phys., 2010, 133, 061102 CrossRef PubMed.
- R. Scholz, R. Luschtinetz, G. Seifert, T. Jägeler-Hoheisel, C. Körner, K. Leo and M. Rapacioli, J. Phys.: Condens. Matter, 2013, 25, 473201 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and table; additional computational details; optimized geometries of all compounds. See DOI: 10.1039/d1se00041a |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemistry, University of Oregon, Eugene, OR 97403. |
| This journal is © The Royal Society of Chemistry 2021 |