
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ synthesis of methane using Ag–GDC composite electrodes in a tubular solid oxide electrolytic cell: new insight into the role of oxide ion removal†
Saheli
Biswas
b,
Aniruddha P.
Kulkarni
 *a,
Daniel
Fini
a,
Sarbjit
Giddey
a and
Sankar
Bhattacharya
b
*a,
Daniel
Fini
a,
Sarbjit
Giddey
a and
Sankar
Bhattacharya
b
aCSIRO Energy, Private Bag 10, Clayton South 3169, Victoria, Australia. E-mail: Aniruddha.Kulkarni@csiro.au
bDepartment of Chemical Engineering, Monash University, VIC 3800, Australia
First published on 4th March 2021
Abstract
The conversion of waste CO2 into energy carrier fuels (“electrofuels”) using renewable energy (RE) in solid oxide electrolytic cells (SOECs) is a fast-emerging technology. Methane is one such potential electrofuel under consideration for the transport and local storage of RE. Most of the synthetic methane generation routes under investigation are two-step processes utilizing SOECs as a source of either H2 or syngas (H2/CO mixture) that undergoes methanation in a subsequent thermochemical reactor. However, the technology for direct one-step in situ synthesis of methane in SOECs is still at an early stage. This work demonstrates, for the very first time, purely electrolytic one-step methane generation in a symmetric, tubular SOEC in the temperature range of 500–700 °C in the absence of any methanation catalyst simply by electrolysing a mixture of H2 and CO2. The non-attainment of methane at OCV and in contrast methane generation under applied potential indicate that the phenomenon is completely driven by electrochemical processes. Interestingly, at all temperatures, the first trace of methane was detected at a certain minimum value of current density, and that value of current density increased non-linearly with temperature. The extent of methane generation appears to be effectively shifted by increasing the rate of oxide ion removal from the cell. Thus, we hypothesize that the electrochemical oxygen pumping phenomenon is a facilitator of such a direct methane synthesis reaction envisaged during in situ methanation in SOECs.
1. Introduction
The gradual depletion of fossil fuels, along with the catastrophic levels of greenhouse gas emissions, has made a compelling case for the exploration and deployment of renewable energy-powered electrochemical pathways of fuel synthesis. One such route is known as Power-to-X, where X can be either hydrogen or a hydrogen carrier, such as ammonia, methane or methanol. The electrolytic synthesis of methane in solid oxide electrolytic cells (SOECs) is of great interest for various reasons. For example, an energy efficiency (defined by the energy content of methane produced to the energy input) of ∼95% is theoretically well predicted at the cell level.1 This is primarily due to lower overpotential losses when using SOECs. In addition, SOECs can be used for in situ methane synthesis by electrolysing CO2 in the presence of H2 (from renewable sources) with the right combination of the electrocatalyst and process conditions.However, this route of methane synthesis is an early stage technology, with very limited studies on the fundamental mechanism or development of materials tailored for the methanation process. Xie et al.2 were one of the first to perform in situ methanation using a composite of lanthanum-doped strontium titanate (La0.2Sr0.8TiO3+δ, or LST) and gadolinia-doped ceria (Gd0.2Ce0.8O1.95, or GDC) as a cathode with 8 mol% yttria (Y2O3)-stabilised zirconia (ZrO2) (YSZ) as the electrolyte, and a lanthanum strontium manganite (La0.8Sr0.2MnO3−δ, or LSM) and YSZ composite anode. They also used an additional layer of an iron catalyst placed in direct contact with the cathode. At 650 °C, about 2.8% methane was generated at atmospheric pressure. Bierschenk et al.3 obtained 2% methane from H2/CO2 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) electrolysis in a single-zone SOEC comprising an Ni–YSZ cathode, YSZ electrolyte and LSM–YSZ anode, where the entire cell was operated at 600 °C under a current of 0.4 A. Li et al.4 reported a methane yield of 0.02% at 2 V and 650 °C by co-electrolysing H2O/CO2 (2:1) using an Ni–YSZ cathode, scandia (Sc2O3)-stabilised zirconia (ScSZ) electrolyte and LSM–ScSZ anode. Recently, Luo et al.12 studied the effect of pressure on steam/CO2 co-electrolysis in a single-zone SOEC comprising an Ni–ScSZ cathode, ScSZ electrolyte and LSM–ScSZ anode at 650 °C. They concluded that increasing the pressure from 1 bar to 4 bars increased the methane yield from 2.8% to 28.7%. However, all three groups used a Ni-cermet cathode, which has significant catalytic activity towards methanation. Some researchers have shown evidence of in situ methane synthesis in a dual-temperature-zone SOEC, where the first zone (SOEC) is kept at a high temperature (650–800 °C) that gradually drops to ∼250 °C in the subsequent Fischer–Tropsch (F–T) zone. The rationale here is that CO2 electrolysis, being an endothermic reaction, is favoured at higher temperatures, whereas methanation is highly exothermic and therefore favourable at lower temperatures (250–350 °C). Thus, the SOEC zone being operated between 650 and 800 °C generates sufficient CO, both from CO2 electrolysis and the reverse water gas shift reaction (RWGS), to undergo methanation in the low-temperature F–T zone in the presence of H2. The H2 can either be produced in situ from steam electrolysis or fed directly to the fuel electrode as a H2/CO2 mixture. In one such design, Chen et al.5 conducted in situ methanation using a Ni-cermet cathode, YSZ electrolyte and LSM–YSZ anode. The SOEC zone was operated at 800 °C at an applied potential of 1.3 V, and the temperature was gradually decreased to 250 °C in the F–T regime. They obtained a maximum methane yield of 11.8%. In another study, Chen et al.6 showed that increasing the cell pressure to 3 bar while maintaining the SOEC part at 800 °C and the F–T zone at 250 °C increased the methane yield to 17%. Further increases in pressure had no significant effect on the methane yield, due to the synergistic effect of pressure on the methanation reaction rate and current density. Note that all such experiments have been conducted with Ni or Fe-based methanation catalysts, either present within the cathode (Ni-cermet) or kept as a separate layer in the F–T zone.5,6 Thus, to the best of our knowledge, no trace of in situ methane has yet been reported, where H2/CO2 electrolysis or steam/CO2 co-electrolysis has been carried out in a single-zone, high-temperature SOEC without a subsequent F–T zone and in the absence of any methanation catalyst. Such an investigation is essential to better comprehend the rate-determining step/s of in situ methanation in a single-zone, high or intermediate-temperature SOEC.
:1) electrolysis in a single-zone SOEC comprising an Ni–YSZ cathode, YSZ electrolyte and LSM–YSZ anode, where the entire cell was operated at 600 °C under a current of 0.4 A. Li et al.4 reported a methane yield of 0.02% at 2 V and 650 °C by co-electrolysing H2O/CO2 (2:1) using an Ni–YSZ cathode, scandia (Sc2O3)-stabilised zirconia (ScSZ) electrolyte and LSM–ScSZ anode. Recently, Luo et al.12 studied the effect of pressure on steam/CO2 co-electrolysis in a single-zone SOEC comprising an Ni–ScSZ cathode, ScSZ electrolyte and LSM–ScSZ anode at 650 °C. They concluded that increasing the pressure from 1 bar to 4 bars increased the methane yield from 2.8% to 28.7%. However, all three groups used a Ni-cermet cathode, which has significant catalytic activity towards methanation. Some researchers have shown evidence of in situ methane synthesis in a dual-temperature-zone SOEC, where the first zone (SOEC) is kept at a high temperature (650–800 °C) that gradually drops to ∼250 °C in the subsequent Fischer–Tropsch (F–T) zone. The rationale here is that CO2 electrolysis, being an endothermic reaction, is favoured at higher temperatures, whereas methanation is highly exothermic and therefore favourable at lower temperatures (250–350 °C). Thus, the SOEC zone being operated between 650 and 800 °C generates sufficient CO, both from CO2 electrolysis and the reverse water gas shift reaction (RWGS), to undergo methanation in the low-temperature F–T zone in the presence of H2. The H2 can either be produced in situ from steam electrolysis or fed directly to the fuel electrode as a H2/CO2 mixture. In one such design, Chen et al.5 conducted in situ methanation using a Ni-cermet cathode, YSZ electrolyte and LSM–YSZ anode. The SOEC zone was operated at 800 °C at an applied potential of 1.3 V, and the temperature was gradually decreased to 250 °C in the F–T regime. They obtained a maximum methane yield of 11.8%. In another study, Chen et al.6 showed that increasing the cell pressure to 3 bar while maintaining the SOEC part at 800 °C and the F–T zone at 250 °C increased the methane yield to 17%. Further increases in pressure had no significant effect on the methane yield, due to the synergistic effect of pressure on the methanation reaction rate and current density. Note that all such experiments have been conducted with Ni or Fe-based methanation catalysts, either present within the cathode (Ni-cermet) or kept as a separate layer in the F–T zone.5,6 Thus, to the best of our knowledge, no trace of in situ methane has yet been reported, where H2/CO2 electrolysis or steam/CO2 co-electrolysis has been carried out in a single-zone, high-temperature SOEC without a subsequent F–T zone and in the absence of any methanation catalyst. Such an investigation is essential to better comprehend the rate-determining step/s of in situ methanation in a single-zone, high or intermediate-temperature SOEC.
In this work, we aimed to evaluate a composite of Ag and 10 mol% GDC (Gd0.1Ce0.9O2−δ) as a potential electrode for in situ methane synthesis using a H2/CO2 mixture and to qualitatively determine the role of thermodynamic equilibrium in this process. A porous composite of Ag and Gd0.2Ce0.8O2−δ previously demonstrated superior performance in CO2 electroreduction7 at 800 °C, but has not been tested for in situ methanation using an H2/CO2 (4:1) mixture. GDC has the following advantages: (1) it shows mixed ionic electronic conductivity under a reducing atmosphere;8 (2) it possesses sufficient ionic conductivity between 500 and 700 °C; (3) as ceria is highly reducible, it increases CO2 adsorption as evidenced by heterogeneous catalysis.9,10 We added Ag because it improves the uniformity of the cathode microstructure,7,11 which reduces polarisation losses and improves cell performance. Ag also improves adhesion between GDC and the most common electrolyte, 8 mol% YSZ,7,11 which implies an increment in the effective triple phase boundary (TPB) area that is likely to improve CO2 electroreduction.
Most importantly, we avoided the use of any methanation catalyst in this work to ensure a better understanding of the electrocatalytic activity of Ag–GDC towards in situ methanation. With the electrode chosen in this work, in situ methanation from H2/CO2 electrolysis is expected to follow a complex pathway consisting primarily of four different steps occurring at the cathode, as mentioned elsewhere.12 They include CO2 hydrogenation to methane via methanation (eqn (1)) and to CO via RWGS (eqn (2)); CO2 electroreduction to CO (eqn (3)); and CO hydrogenation to methane (eqn (4)). The anodic reaction involves oxygen evolution (eqn (5)).
At the cathode:
| CO2 + 4H2 = CH4 + 2H2O (ΔH1023K = −164 kJ mol−1) | (1) |
| CO2 + H2 = CO + H2O (ΔH1023K = 41.2 kJ mol−1) | (2) |
| CO2 + 2e− = CO + O2− (ΔH1023K = 282 kJ mol−1) | (3) |
| CO + 3H2 = CH4 + H2O (ΔH1023K = −206.1 kJ mol−1) | (4) |
At the anode:
| O2− = ½O + 2e− | (5) |
Appreciable methane generation can be ascertained only when the cathode exhibits sufficient catalytic activity for eqn (1)–(4) to occur. This is especially the case for CO2 electroreduction, the kinetics of which are very sluggish.13–15 Moreover, the accurate control of the ratio of H2 to CO in steam/CO2 co-electrolysis is quite difficult as opposed to H2/CO2 electrolysis, especially considering the large area of our cell (33 cm2). Thus, we restricted our initial experiments to the electrolysis of dry CO2 only, followed by a mixture of H2/CO2 using electrolyte-supported, symmetric tubular cells. We used the state-of-the-art electrolyte YSZ with an Ag–GDC composite electrode for both dry CO2 and H2/CO2 electrolysis at 500–700 °C, as detailed in Section 2.
2. Results and discussion
2.1 Electrochemical performance of the Ag–GDC composite electrode for dry CO2 electrolysis
Fig. 1A shows the current–voltage (V–I) curves (corrected for lead wire resistances) for an A-GDC/YSZ symmetric tube cell tested at 500, 600, 700 and 800 °C with dry CO2; Fig. S1B in the ESI† shows the corresponding impedance spectra under open-circuit conditions.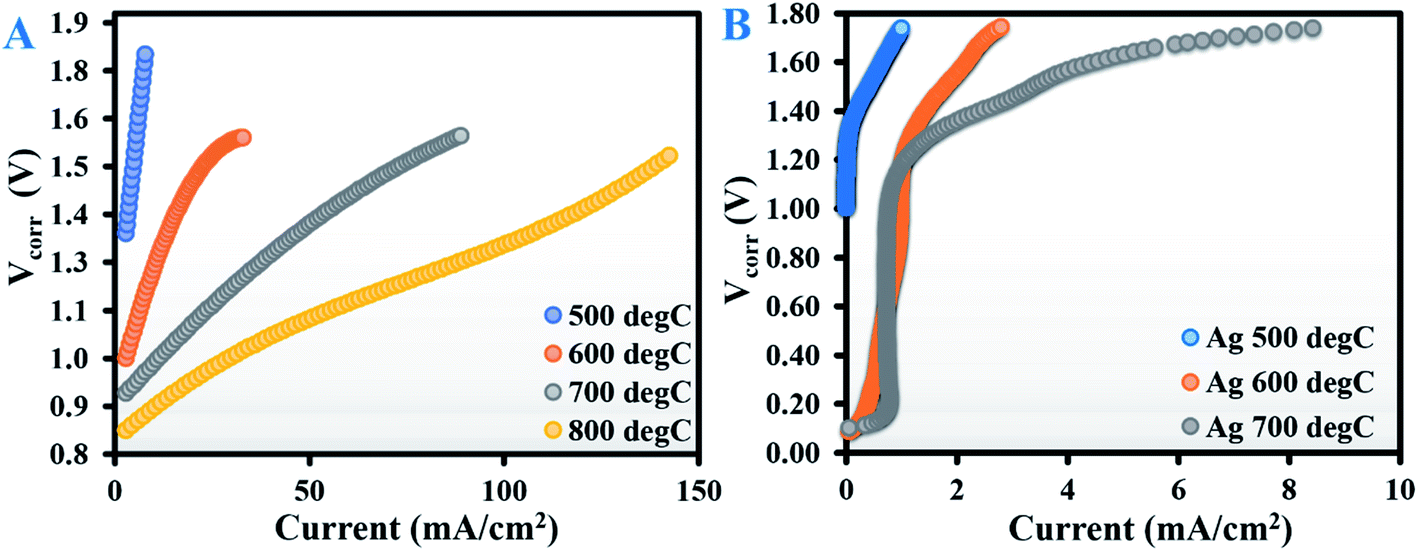 | ||
| Fig. 1 Current–voltage curves (corrected for lead wire resistance) of a tubular solid oxide electrolysis cell, recorded during electrochemical reduction of dry CO2 at (A) 500, 600, 700 and 800 °C with the Ag–GDC composite electrode, and (B) 500, 600 and 700 °C with the bare Ag electrode. | ||
As expected, the current increased as the temperature rose, with a maximum current density of about 140 mA cm−2 obtained at an applied potential of 1.5 V at 800 °C. The increase in current density can be attributed to the synergistic effect of decreased electrolyte ohmic resistance (Rohm) and reduced electrode polarisation resistance (Rpol) due to improved kinetics. The electrochemical impedance spectroscopy (EIS) spectra at all test temperatures (Fig. S1B in the ESI†) were composed of two distinct arcs, which can be nominally separated into high frequency (HF, ranging from 1000 to 5 Hz) and low frequency (LF, ranging from 5 to 0.1 Hz). Based on previous reports,8,13,14,16–19 the LF arc can be ascribed to slower processes related to diffusion-limited mass transfer, dissociative adsorption of CO2, and desorption of CO or the even surface-exchange reaction of CO/CO2 at the gas–cathode interface. The HF arc is usually related to the less energy-intensive oxygen evolution reaction (eqn (5)) occurring at the anode.
The analysis of Rohm values (calculated from the intercepts of the HF arcs on the real axis) in conjunction with the V–I curves showed that beyond 1.4 V, the increase in current as a function of temperature was roughly proportional to the decrease in Rohm. This indicates that in the high-temperature regime, there was a dominating contribution from ohmic losses above 1.4 V, whereas below 1.4 V the process might be controlled by polarisation. The mechanism of the reaction in SOECs is a complex phenomenon and is not yet well understood for operation in electrolytic mode with CO2. Considering the relatively large cell area (33 cm2) in tubular geometry and fact that no reference electrode was used, a deconvolution of the EIS curve would be challenging and hence was not attempted.
At each test temperature, the tube cell was operated at 1.5 V in a dry CO2 environment for 20 min, and the amount of CO produced was measured online using gas chromatography (GC). As expected, CO2 to CO conversion gradually increased with temperature, attaining a maximum value of 70% at 800 °C (Fig. S2 in the ESI†) with a corresponding faradaic efficiency of 97%. Such a high conversion can be attributed to reduced polarisation and activation losses that created a more reducing environment for CO2, and possibly also due to greater adsorption of CO2 on GDC at higher temperatures. GDC is a redox-stable, mixed ionic electronic conductor (MIEC). It is believed18,20–22 that in a reducing environment, Ce4+ is reduced to Ce3+, leading to the formation of oxygen vacancies in the GDC lattice. These vacancies are then occupied by oxygen from CO2 dissociation.
Based on the above results, Ag–GDC appeared to be a promising candidate for effective CO2 electrolysis: the first crucial step for in situ methanation. Note that Ag has been previously reported to be catalytically inactive towards high-temperature CO2 electroreduction;7,11,23 the Ag in our composite electrode is no exception. To confirm our conjecture, CO2 electrolysis was performed with bare Ag electrodes instead of the Ag–GDC composite in the same temperature range of 500–700 °C. The V–I curves are provided in Fig. 1B, and the EIS spectra at open-circuit voltage (OCV) are shown in Fig. S3 of the ESI.†Fig. 1B shows that although the V–I curves were similar in nature to those of the composite electrode, the current was almost an order of magnitude lower; the corresponding CO production rates at 1.5 V were also ∼10 times less than when using Ag–GDC. Such poor performance of the Ag electrode can be attributed to the catalytic inertness of Ag as well as a stipulated reduction in the TPB. Being a pure electronic conductor, Ag alone cannot build up the electron–ion–gas TPB, which is a key to electrochemical reactions. In contrast, Ag–GDC is a mixed ionic electronic conductor that significantly increases the TPBs, thereby enhancing the electrochemical performance of the cell.
The experimental data obtained with Ag electrodes corroborate the idea that Ag did not contribute to the enhanced CO2 electroreduction envisaged when using the Ag–GDC composite electrodes and that the cardinal role was played by the versatile material GDC. Based on these initial results, we concluded that the Ag–GDC composite electrode was worthy of investigation for in situ methanation.
2.2 Performance of the Ag–GDC composite electrode for methanation
An identical tube cell was tested at 500, 600 and 700 °C with a H2/CO2 (4:1) mixture. Fig. 2A–C present the open-circuit EIS spectra at 500, 600 and 700 °C, respectively. Fig. 2D and E demonstrate the corresponding V–I and V–I curves corrected for wire resistance.
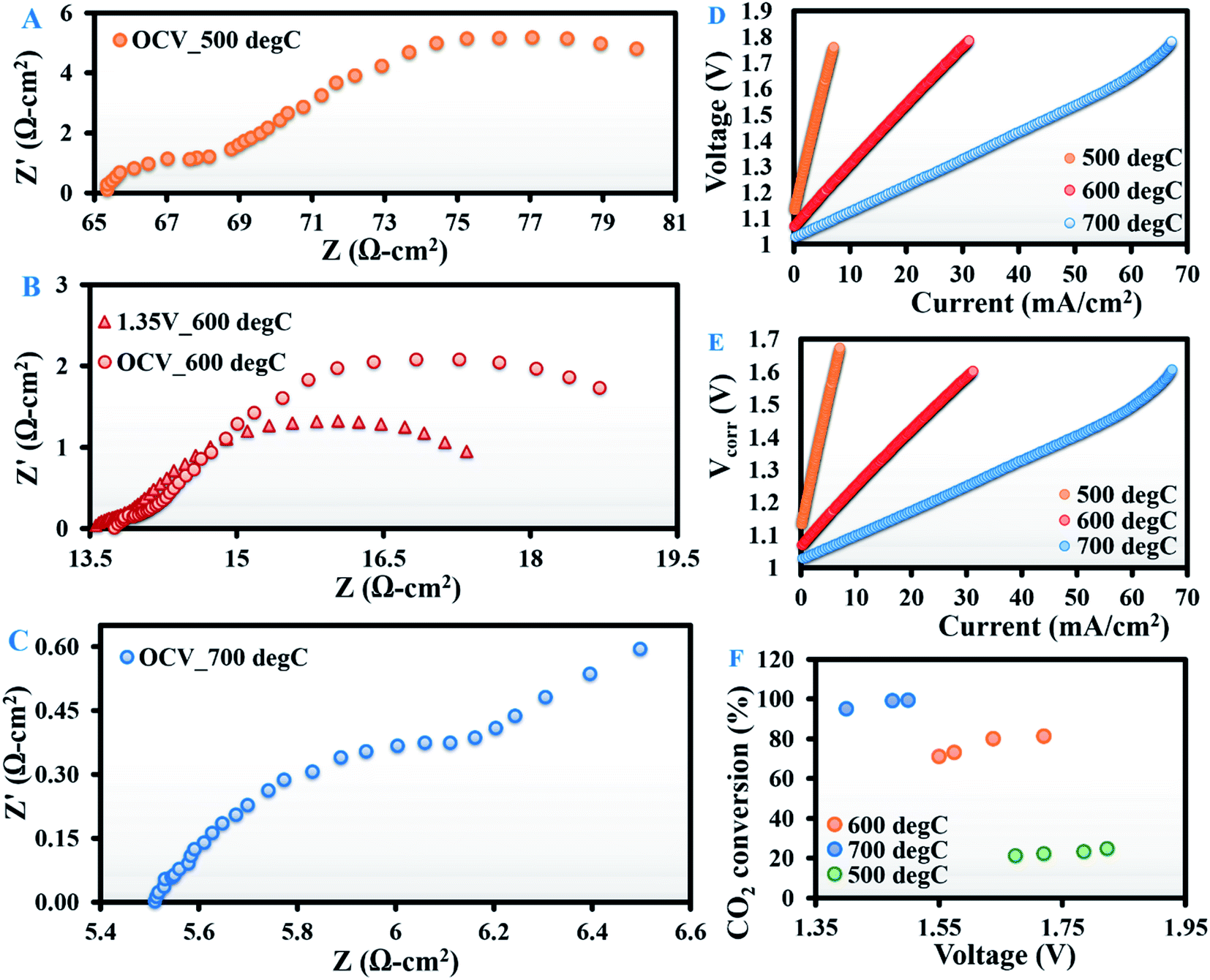 | ||
| Fig. 2 Electrochemical impedance spectra at open-circuit voltage of a tubular solid oxide electrolysis cell with the Ag–GDC composite electrode recorded during electrochemical reduction of H2/CO2 in a ratio of 4:1 at (A) 500, (B) 600 and (C) 700 °C, with the corresponding (D) actual and (E) current–voltage characteristics (wire-resistance corrected). (F) CO2 conversion as a function of applied voltage at 500, 600 and 700 °C. | ||
The magnitude of both HF (∼1000 to 5 Hz) and LF (∼5 to 0.1 Hz) arcs of the impedance spectra decreased with a rise in temperature (Fig. 2A–C). As predicted from the curve fitting of the EIS spectra, the Rpol at OCV was 17.72, 6.50 and 1.02 Ω cm2 at 500, 600 and 700 °C respectively. At all temperatures, the bulk of Rpol came from the LF arc, as noted by Yue et al.19 With an increase in applied potential from OCV to 1.35 V at 600 °C, Rpol decreased from 6.5 to 4 Ω cm2 due to a major reduction in the LF arc (Fig. 2B). This can be attributed to improved cell kinetics at higher potentials.
Another interesting observation from the EIS spectra was a substantial drop in the open-circuit Rpol from dry CO2 to H2/CO2 atmospheres (Table 1). At OCV, when dry CO2 is fed to the cathode, only CO2 electroreduction (eqn (3)) occurs, whereas, when a H2/CO2 mixture with H2 concentration as high as 80% is introduced at the cathode, the dominant reaction is the oxidation of H2 by the oxide ions (eqn (6)) generated from CO2 electroreduction. The latter is kinetically more active than the former, resulting in lower Rpol.
| H2 + O2− = H2O + 2e− | (6) |
| Gas composition | Temperature (°C) | R ohm (Ω cm2) | R pol (Ω cm2) |
|---|---|---|---|
| CO2 | 500 | 68.15 | 63.79 |
| H2/CO2 (4:1) |
500 | 68.15 | 17.72 |
| CO2 | 600 | 13.74 | 50.95 |
| H2/CO2 (4:1) |
600 | 13.74 | 6.50 |
| CO2 | 700 | 5.63 | 11.12 |
| H2/CO2 (4:1) |
700 | 5.515 | 1.02 |
Another possibility is that the presence of a reducing gas such as H2 increases the rate of Ce4+ to Ce3+ reduction, which makes CO2 surface adsorption thermodynamically more favourable and faster, as noted in recent density functional theory studies.24 This helps to reduce the overall cell polarisation resistance. However, we surmise that the lower surface diffusion resistance of smaller H2 molecules compared with CO2 molecules might also contribute to the drop in Rpol. Previously, Matsuzaki et al.25 observed higher impedance with CO than with H2 while studying the electrochemical oxidation of these two gases in fuel cell mode (solid oxide fuel cell). They concluded that CO encounters higher diffusion resistance than H2 while moving across the electrode surface.
GC analysis of the outlet gases evolving at the cathode revealed that at OCV, CO2 conversions were 20, 51 and 66% at 500, 600 and 700 °C, respectively, with the corresponding CO production rates of 3.2, 7.9 and 9.8 ml min−1. At OCV, zero electroreduction of CO2 is plausible; therefore, the source of this CO must be the RWGS reaction (eqn (2)). However, with an increase in applied potential, more CO2 was converted (Fig. 2F) due to enhanced electroreduction. At 500 °C, the increase was minimal (from 21% at 1.67 V to 24.5% at 1.82 V). However, at 600 °C, CO2 conversion increased significantly from 71% at an applied potential of 1.55 V to 81% at 1.72 V. At 700 °C, the effect once again decreased, possibly due to the complete consumption of CO2 by H2 itself for the RWGS reaction. This is also clear from the fact that at 700 °C, CO2 conversion reached ∼99% at an applied potential of 1.48 V. This observation is in line with literature reports26–30 for H2/CO2 electrolysis conducted with different electrode materials, but has been shown here for the first time with an Ag–GDC composite.
Based on GC analysis, under all test conditions, the faradaic efficiency was ∼99% and carbon balance fairly matched 100% within the limits of experimental accuracy, signifying trivial or no carbon deposition. This is consistent with previous reports confirming the anti-coking properties of ceria.16,21,22,31–33
One of the key findings of this research is the in situ generation of methane between 500 and 700 °C under an H2/CO2 (4:1) environment (Table 2) in the absence of any specific methanation catalyst, such as Ni or Fe. Also, according to the literature, Ag has absolutely no catalytic activity towards either CO2 or CO methanation although it catalyses CO2 and CO hydrogenation to methanol to a limited extent.34–36 The most commonly studied catalysts for methanation carried out in a thermochemical reactor at 1 bar and at 200–500 °C are Ni or its bimetallic substitutes (Ni/Ru or Ni/Co or Ni/Mn), on oxide supports such as alumina, silica, titania or zirconia.37–41
| Temp (°C) | Voltage (V) | Current (mA cm−2) | CO (%) | CH4 (%) | CO (millimole s−1 cm−2) | CH4 (millimole s−1 cm−2) | S CH4 (%) | Y CH4 (%) |
|---|---|---|---|---|---|---|---|---|
| 500 | 1.10 (OCV) | 0.00 | 5.80 | 0.000 | 6.54 × 10−5 | 0 | 0 | 0 |
| 1.79 | 6.06 | 6.36 | 0.016 | 7.17 × 10−5 | 1.80 × 10−7 | 0.25 | 0.06 | |
| 1.82 | 6.45 | 6.61 | 0.020 | 7.45 × 10−5 | 2.25 × 10−7 | 0.29 | 0.07 | |
| 600 | 1.06 (OCV) | 0.00 | 12.40 | 0.000 | 1.40 × 10−4 | 0 | 0 | 0 |
| 1.58 | 19.03 | 15.4 | 0.010 | 1.74 × 10−4 | 1.13 × 10−7 | 0.09 | 0.07 | |
| 1.72 | 25.36 | 16.80 | 0.030 | 1.89 × 10−4 | 3.38 × 10−7 | 0.15 | 0.13 | |
| 700 | 1.02 (OCV) | 0.00 | 14.80 | 0.000 | 1.67 × 10−4 | 0 | 0 | 0 |
| 1.50 | 40.91 | 19.90 | 0.020 | 2.24 × 10−4 | 2.25 × 10−7 | 0.09 | 0.08 | |
| 1.90 | 60.61 | 19.80 | 0.096 | 2.23 × 10−4 | 1.08 × 10−6 | 0.50 | 0.49 |
At all test temperatures, no methane was detected at OCV. This clearly establishes that methane evolution is a purely electrochemical phenomenon under the influence of applied potential. The maximum methane generation of 1.08 × 10−7 millimole s−1 cm−2 was recorded at 700 °C under an applied potential of 1.9 V with respect to OCV. Such low methane production can be attributed to the synergistic effect of the thickness (0.9 cm) of the electrolyte and the insufficient catalytic activity of the electrode towards H2 dissociation and H+ ion spillover,42–45 which play pivotal roles in methanation. Thinning down the electrolyte would increase the current density, and thereby the methane yield. However, our intention here was not to maximise the methane yield, but to investigate whether Ag–GDC is electrocatalytically conducive to in situ methanation and determine how such in situ methane synthesis is governed purely electrochemically.
The first trace of methane was attained at different applied currents and correspondingly different voltages for the three different test temperatures, as shown in Table 2 in blue font. This indicates that the current at which methane starts evolving is a function of the operating temperature and hence of thermodynamic equilibrium. The sources of in situ generated methane include CO2 hydrogenation (eqn (1)) and CO hydrogenation (eqn (4)), both of which are exothermic reactions and thus less favourable at higher temperatures. On the other hand, the methane can dissociate by two possible endothermic reactions: cracking (eqn (8)) or steam reforming (eqn (7)). Steam is also generated in situ as a methanation by-product. Thus, it is highly likely that methane is being produced even at currents lower than where it is initially detected, but it undergoes thermodynamic equilibrium-dictated dissociation until the current is sufficiently high to electrolyse and remove steam from the system. Since the tube was almost 30 cm long, the residence time of nascent methane was sufficient to increase the probability of such dissociation. What further corroborated this idea was the observation that with an increase in temperature, the minimum current (Imin) at which methane started evolving gradually increased, which was consistent with the gradual increase in the equilibrium constants (Keql)46 of the methane-dissociation reactions (eqn (7) and (8)) as shown in Table 3.
| CH4 + 2H2O = CO2 + 4H2 (ΔH1023K = 164 kJ mol−1) | (7) |
| CH4 = 2H2 + C (ΔH1023K = 74.8 kJ mol−1) | (8) |
| Reaction | 500 °C | 600 °C | 700 °C | |||
|---|---|---|---|---|---|---|
| K eql | Conversion (%) | K eql | Conversion (%) | K eql | Conversion (%) | |
| CO2 methanation | 62.40 | 83.0 | 3.35 | 71.9 | 0.33 | 60.0 |
| CO methanation | 376.00 | 90.5 | 9.56 | 78.0 | 0.52 | 60.0 |
| CH4 steam reforming | 0.02 | 7.3 | 0.30 | 12.5 | 3.04 | 19.0 |
| CH4 cracking | 0.15 | 17.4 | 0.60 | 30.8 | 1.58 | 46.1 |
However, beyond Imin, the increase in methane production did not exhibit a linear relationship with current density. For complex materials such as Ag–GDC endowed with mixed ionic and electronic (MIEC) properties, a higher applied potential both increases the current density and remarkably improves the electrocatalytic activity towards CO2 electrolysis, as explained in Section 2.1. This results in higher production of CO, which further increases methane generation. Thus, at current densities greater than Imin, the increase in the methane production rate was not directly proportional to the increment in current density; rather, it was higher than that.
Based on all our aforementioned observations, it can be rightly concluded that the in situ methane synthesis observed here is a completely electrochemical phenomenon, governed by the overall thermodynamic equilibrium of the methanation and methane-dissociation reactions taking place while the cell is in operation. The minimum current (Imin) at which methane begins evolving, as well as the overall methane generation, is also dictated by thermodynamic equilibrium. Similar observations have never been previously reported with an Ag–GDC composite electrode or any other electrode. X-ray diffraction (XRD) and scanning electron microscopy (SEM) results are discussed in the ESI (Fig. S5†), since they revealed no significant morphological or phase changes between fresh and used electrodes.
2.3 Possible impact of oxygen stripping on the thermodynamic equilibrium governing electrochemical methane synthesis
The mixed ionic electronic conductivity of GDC makes the Ag–GDC composite a complex material. To investigate how the thermodynamic equilibrium could be shifted to increase methane generation, we deemed it necessary to eliminate any catalytic and/or electrocatalytic effects induced by GDC on CO2 electrolysis by virtue of its redox ability because such effects might impact the methane generation rate under varying conditions of temperature and applied potential. For this evaluation, H2/CO2 (4:1 v/v) electrolysis was carried out with bare Ag electrodes on the YSZ electrolyte in the same temperature range of 500 to 700 °C. We aimed to determine whether methane generation was affected by the rate of oxide ion removal from the cell.
The EIS curves recorded at OCV are provided in Fig. S4 of the ESI.† The V–I data (Fig. 3A) show that for all three test temperatures, the current at any particular voltage was almost an order of magnitude lower when using bare Ag than the corresponding values obtained with the Ag–GDC composite. This resulted in scanty CO2 electroreduction with no methanation observed at 500 °C. The first trace of methane was obtained at an applied potential of 2.3 V (1.69 × 10−7 millimole s−1 cm−2) and 1.85 V (2.25 × 10−7 millimole s−1 cm−2) at 600 and 700 °C, respectively. The corresponding currents (Imin) recorded were 7.88 and 9.7 mA cm−2, respectively.
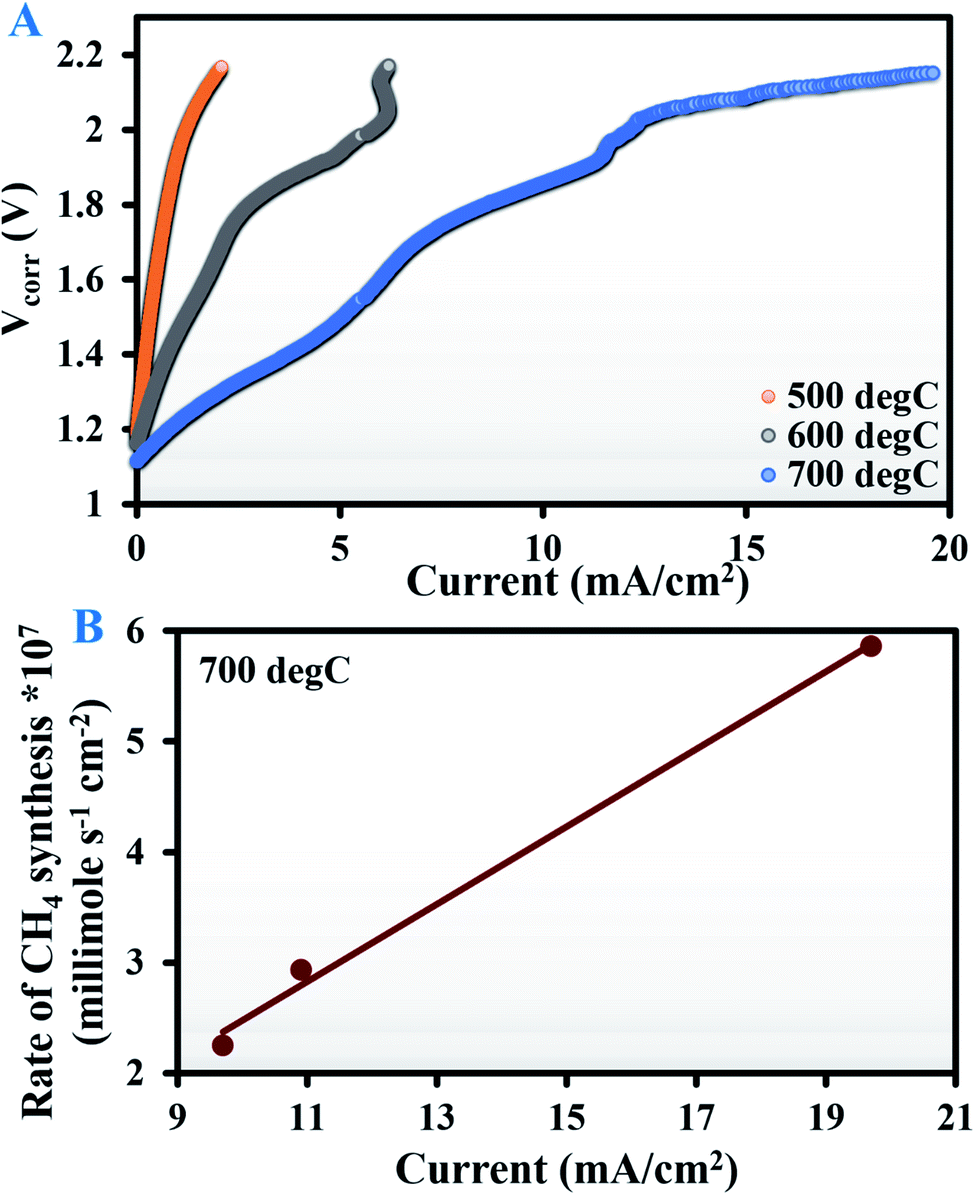 | ||
| Fig. 3 (A) Voltage–current curves (corrected for lead wire resistance) for a tubular solid oxide electrolysis cell with the Ag electrode recorded during electrochemical reduction of a H2/CO2 mixture (4:1 v/v) at 500, 600, and 700 °C. (B) The rate of methane synthesis (millimole s−1 cm−2) at 700 °C shows a linear trend as a function of current beyond an applied potential of 1.85 V. | ||
Interestingly, at 700 °C, when the current was increased beyond 9.7 mA cm−2 (Imin), methane generation increased proportionally (Fig. 3B). This indicates that above Imin, methane generation increases in proportion with the increment in O2− removal, since in solid oxide ion conducting electrolytes, oxygen removal varies proportionally with current, as determined from Faraday's law.47
This is a novel observation that has never been reported in the literature. Similar observations at 600 °C would require cell operation at potentials above 2.3 V, which might cause rapid removal of O2− ions from the electrolyte (YSZ) itself, leading to zirconia blackening.48,49 A plausible alternative is the use of a thin YSZ electrolyte that guarantees less ohmic resistance and thus higher current densities at lower voltages.
It was clear from our results that the extent of methane synthesis was directly related to the rate of oxygen removal from the system. However, for further verification, we carried out H2/CO2 (4:1) hydrogenation in a thermochemical methanation reactor in the absence of oxygen stripping at 1 bar and two different temperatures of 450 and 500 °C. A packed bed of glass wool and Ag–GDC powder (0.2 g) was housed inside the reactor (detailed set-up is described in the experimental section). The Ag–GDC powder had the same composition as the Ag–GDC electrodes used in our symmetric cells discussed in Sections 2.1 and 2.2. The H2/CO2 gas mixture was purged at a constant flow rate of 50 ml min−1 through the reactor. As detected through the online GC connected to the reactor outlet, the amounts of CO obtained at 450 and 500 °C were ∼3 and 4.5%, respectively. The value at 500 °C was similar to that observed with Ag–GDC composite electrodes (5.8%) at the same temperature under OCV conditions. Interestingly, at both 450 and 500 °C, the methane produced was only around 0.001%, which was almost an order of magnitude less than the minimum amount of methane (0.016%) obtained at 500 °C using Ag–GDC composite electrodes (Table 2).
All the above observations hint towards the fact that if oxygen from the in situ generated steam (a by-product of methanation reactions) can be removed very quickly by rapid O2− stripping across the electrolyte, then the forward methanation reactions (eqn (1) and (4)) will possibly be favoured according to Le Chatelier's principle,50 and the dissociation of in situ evolved methane (eqn (6) and (7)) will be inhibited. Such a shift of thermodynamic equilibrium will be manifested by a directly proportional increase in methane production upon increasing current under the same applied potential.
3. Conclusion
We have tested for the first time an Ag–GDC composite electrode as a potential candidate for in situ methane synthesis in SOECs. This electrode composition was electrocatalytically very active at 500–700 °C towards both CO2 electroreduction (during dry CO2 electrolysis) and the RWGS reaction (H2/CO2 (4:1 v/v) electrolysis under no-load conditions, i.e., at OCV), which are the first crucial steps for effective in situ methane generation. GDC, by virtue of its mixed ionic electronic conductivity and high reducibility, imparts a high electrocatalytic activity to the Ag–GDC composite, whereas Ag only improves the electrode's adhesion properties and electronic conductivity.
When H2/CO2 (4:1 v/v) electrolysis was carried out with Ag–GDC electrodes at 500, 600 and 700 °C, some methane was obtained, but only under load conditions. The generation of methane in the absence of any methanation catalyst, such as Ni/Fe/Ru, leads us to conclude that the in situ methane synthesis envisaged in this work is a purely electrochemical phenomenon. The maximum methane production rate was 1.08 × 10−7 millimole s−1 cm−2 at 700 °C under an applied potential of 1.9 V. Interestingly, at any temperature, the first trace of methane was detected at a minimum value of current density (Imin), which increased monotonically with a rise in temperature. Based on this, we propose that H2/CO2 electrolysis is a dynamic process during which both methanation (exothermic) and methane-dissociation (endothermic) reactions take place simultaneously. However, the thermodynamically opposite nature of these reactions gradually makes methane evolution less favourable at higher temperatures. Thus, nascent methane produced in the SOEC environment undergoes thermodynamic equilibrium-dictated dissociation until the current is sufficiently high to electrolyse steam. The steam is a by-product of the methanation reaction and leads to methane dissociation via its reforming. Steam electrolysis essentially implies the removal of oxygen from the cell. Therefore, we can conclude that the extent of methane generation can be effectively shifted by increasing the rate of oxide ion removal through the electrolyte. Further strengthening this hypothesis is the observation that when H2/CO2 (4:1 v/v) electrolysis was carried out using bare Ag electrodes, methane generation beyond Imin increased in direct proportion with current density, or in other words, with the rate of oxygen removal from the cell. This is a completely new concept that is worthy of future investigation.
4. Materials and methods
4.1 Cell fabrication
Electrolyte-supported, open-ended tube cells were fabricated through isostatic pressing of 8 mol% yttria-stabilised zirconia (8-YSZ) powder procured from Tosoh Corporation Japan at 170 MPa using an in-house designed die followed by sintering in air at 1500 °C for 2 h. The final dimensions of the tube cell were OD 11 mm, length 340 mm and electrolyte thickness 0.45 mm. The electrode ink was prepared by mixing Ag powder (Alfa Aesar) and gadolinia-doped ceria (Gd0.1Ce0.9O2−δ) purchased from Fuel Cell Materials Inc. (FCM) in a weight ratio of 3:7 along with a terpinol-based ink vehicle (FCM), followed by ball milling the mixture for 2 h.51 The as-prepared ink was brush-painted on the inside and outside active areas of the sintered electrolyte tubes over a length of 12 cm and sintered at 825 °C for 2 h at heating and cooling rates of 180 °C min−1. The final thickness of each electrode was between 30 and 50 μm, with an equivalent active area of 33 cm2. The current was collected through silver wires that were spirally wound on both sides of the tube spanning the electrode area. Silver paste was then used to improve further adhesion and contact of the current collector with the electrode.
4.2 Physical and electrochemical characterisation
Powder XRD of the Ag–GDC composite cathode after cell testing was performed using a benchtop XRD (Bruker D2 Phaser) with a Cu-Kα radiation source. The diffractogram was recorded between 10 and 80° at a scan rate of 2° min−1. The microstructure of the electrode was imaged using a ZEISS SEM. All electrochemical measurements, such as V–I curves, chronoamperometry and impedance spectroscopy (EIS curves), were conducted using a Princeton Applied Research VERSAStat. V–I measurements were recorded at 5 mV s−1, and impedance measurements were carried out in the frequency range of 1000–0.1 Hz at an amplitude of 70 mV.4.3 Cell performance testing
All electrochemical measurements were carried out using symmetric tube cells fabricated in-house. In the present set-up (Fig. 4), the test gases (dry CO2 or an 80% H2/20% CO2 mixture) were purged into the fuel electrode (cathode), while the air electrode (anode) was exposed to the atmosphere. The first set of measurements were performed at four different temperatures of 500, 600, 700 and 800 °C by purging dry CO2 (industrial grade, BOC) at a constant flow rate of 50 ml min−1 using Ag–GDC composite electrodes and an 8-YSZ electrolyte. Next, the same experiments were repeated under identical conditions of temperature and flow rate, but with bare Ag electrodes. In the next phase, electrochemical measurements for methanation tests were performed using the Ag–GDC composite electrode and 8-YSZ electrolyte at 500–700 °C by purging an 80% H2/20% CO2 mixture (industrial grade, BOC) at a constant flow rate of 50 ml min−1. The same experiments were repeated for bare Ag electrodes. | ||
| Fig. 4 Tube cell test set-up comprising a furnace, VersaSTAT and Micro-GC. The inset at the top right corner shows a magnified view of the tube cell positioned inside the furnace. | ||
The mass-flow meters used were calibrated using separate, certified flow meters. The temperature at the centre of the cathode was monitored using a single K-type thermocouple and is designated henceforth as the operating temperature. At each temperature, after recording the V–I curves and EIS spectra at OCV, the cell was loaded at different voltages for 30 min and the composition of the gases evolving at the cathode was analysed using a GC (Agilent Micro-GC). The impedance plots were recorded under no-load conditions (OCV). All V–I curves were recorded against OCV obtained under the particular cell testing conditions of temperature and gas mixture.
4.4 Ag–GDC testing in a thermochemical methanation reactor
Ag powder (Alfa Aesar) and GDC (Gd0.1Ce0.9O2−δ) purchased from FCM were mixed in a weight ratio of 3:7 followed by ball milling for 2 h. The mixture was then sintered at 825 °C for 2 h at heating and cooling rates of 180 °C min−1. The as-prepared Ag–GDC composite powder was subsequently tested for the CO2 hydrogenation reaction using a fixed-bed, custom-designed, stainless-steel reactor lined internally with brass sleeves. The powder (0.2 g) was evenly dispersed into a bed of glass wool plugged inside the reactor. The reaction temperature was measured with a K-type thermocouple buried into the glass wool bed. The reactor was held within a furnace equipped with a temperature controller. The flow rates were controlled using Bronkhorst High-Tech Series mass-flow controllers. The Ag–GDC bed was heated at 300 °C (ramp rate ∼ 3 °C min−1) and 1 bar pressure for 2 h under a 100 ml min−1 Ar (Air Liquide 99.9% purity) flow. Following this, the temperature was further increased to the desired set point while keeping the argon flow rate constant. Subsequently, the gas flow was changed to 50 ml min−1 of H2/CO2 (4:1 v/v, purchased from BOC) and 50 ml min−1 of Ar. CO2 hydrogenation was conducted and the performance of the catalysts was tested under steady-state conditions. The reaction products were analysed online using gas chromatography using an Agilent 7890A GC-TCD/FID fitted with HayeSep N, MolSieve 5A and Porapak Q columns. Nitrogen was used as an internal standard for chromatographic analyses. The gas was sampled online at intervals of ∼35 min for a period of 2 h. Subsequently, the reactor was cooled under a 100 ml min−1 Ar flow.
Funding
This work received funding from the Australian Renewable Energy Agency (ARENA) as part of ARENA's Research and Development Program—Renewable Hydrogen for Export, and also from the CSIRO Hydrogen Energy Systems Future Science Platform, CSIRO Research Office, and a PhD scholarship from Monash University. The funder was not involved in the study design, collection, analysis, and interpretation of data, the writing of the manuscript, or the decision to submit it for publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This Activity received funding from ARENA as part of ARENA's Research and Development Program – Renewable Hydrogen for Export, and also from CSIRO Hydrogen Energy Systems Future Science Platform. The authors would like to thank Daniel Roberts and Shambhu Singh Rathore for an internal review of the paper.References
- S. Biswas, A. Kulkarni, S. Giddey and S. Bhattacharya, A Review on Synthesis of Methane as a Pathway for Renewable Energy Storage with a Focus on Solid Oxide Electrolytic Cell-Based Processes, Frontiers in Energy Research, 2020, 8, 570112 CrossRef.
- K. Xie, Y. Zhang, G. Meng and J. T. Irvine, Direct synthesis of methane from CO2/H2O in an oxygen-ion conducting solid oxide electrolyser, Energy Environ. Sci., 2011, 4(6), 2218–2222 RSC.
- D. M. Bierschenk, J. R. Wilson and S. A. Barnett, High efficiency electrical energy storage using a methane–oxygen solid oxide cell, Energy Environ. Sci., 2011, 4(3), 944–951 RSC.
- W. Li, H. Wang, Y. Shi and N. Cai, Performance and methane production characteristics of H2O–CO2 co-electrolysis in solid oxide electrolysis cells, Int. J. Hydrogen Energy, 2013, 38(25), 11104–11109 CrossRef CAS.
- L. Chen, F. Chen and C. Xia, Direct synthesis of methane from CO2–H2O co-electrolysis in tubular solid oxide electrolysis cells, Energy Environ. Sci., 2014, 7(12), 4018–4022 RSC.
- B. Chen, H. Xu and M. Ni, Modelling of SOEC-FT reactor: pressure effects on methanation process, Appl. Energy, 2017, 185, 814–824 CrossRef CAS.
- Y. Xie, J. Xiao, D. Liu, J. Liu and C. Yang, Electrolysis of carbon dioxide in a solid oxide electrolyzer with silver-gadolinium-doped ceria cathode, J. Electrochem. Soc., 2015, 162(4), F397–F402 CrossRef CAS.
- R. D. Green, C.-C. Liu and S. B. Adler, Carbon dioxide reduction on gadolinia-doped ceria cathodes, Solid State Ionics, 2008, 179(17–18), 647–660 CrossRef CAS.
- C. de Leitenburg, A. Trovarelli and J. Kašpar, A temperature-programmed and transient kinetic study of CO2 activation and methanation over CeO2 supported noble metals, J. Catal., 1997, 166(1), 98–107 CrossRef CAS.
- P. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk and M. Daturi, et al., Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy, Catal. Today, 2013, 215, 201–207 CrossRef CAS.
- J. Zhang, Y. Ji, H. Gao, T. He and J. Liu, Composite cathode La0.6Sr0.4Co0.2Fe0.8O3–Sm0.1Ce0.9O1.95–Ag for intermediate-temperature solid oxide fuel cells, J. Alloys Compd., 2005, 395(1–2), 322–325 CrossRef CAS.
- Y. Luo, Y. Shi, Y. Chen, W. Li, L. Jiang and N. Cai, Pressurized tubular solid oxide H2O/CO2 coelectrolysis cell for direct power-to-methane, AIChE J., 2020, 66(5), e16896 CrossRef CAS.
- X. Yue and J. T. Irvine, Impedance studies on LSCM/GDC cathode for high temperature CO2 electrolysis, Electrochem. Solid-State Lett., 2012, 15(3), B31 CrossRef CAS.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, High-Temperature CO2 Electrolysis in Solid Oxide Electrolysis Cells: Developments, Challenges, and Prospects, Adv. Mater., 2019, 31(50), 1902033 CrossRef CAS PubMed.
- L. Spiridigliozzi, E. Di Bartolomeo, G. Dell'Agli and F. Zurlo, GDC-Based Infiltrated Electrodes for Solid Oxide Electrolyzer Cells (SOECs), Appl. Sci., 2020, 10(11), 3882 CrossRef.
- G. Kaur, A. P. Kulkarni, S. Giddey and S. P. S. Badwal, Ceramic composite cathodes for CO2 conversion to CO in solid oxide electrolysis cells, Appl. Energy, 2018, 221, 131–138 CrossRef CAS.
- L. Chen, F. Chen and C. Xia, Direct synthesis of methane from CO2–H2O co-electrolysis in tubular solid oxide electrolysis cells, Energy Environ. Sci., 2014, 7(12), 4018–4022 RSC.
- A. Kulkarni, S. Giddey and S. P. S. Badwal, Efficient conversion of CO2 in solid oxide electrolytic cells with Pd doped perovskite cathode on ceria nanofilm interlayer, J. CO2 Util., 2017, 17, 180–187 CrossRef CAS.
- X. Yue and J. T. S. Irvine, Alternative cathode material for CO2 reduction by high temperature solid oxide electrolysis cells, J. Electrochem. Soc., 2012, 159(8), F442 CrossRef CAS.
- O. A. Marina, C. Bagger, S. Primdahl and M. Mogensen, A solid oxide fuel cell with a gadolinia-doped ceria anode: preparation and performance, Solid State Ionics, 1999, 123(1–4), 199–208 CrossRef CAS.
- C. Neofytidis, V. Dracopoulos, S. Neophytides and D. K. Niakolas, Electrocatalytic performance and carbon tolerance of ternary Au-Mo-Ni/GDC SOFC anodes under CH4-rich internal steam reforming conditions, Catal. Today, 2018, 310, 157–165 CrossRef CAS.
- J. B. Goodenough and Y.-H. Huang, Alternative anode materials for solid oxide fuel cells, J. Power Sources, 2007, 173(1), 1–10 CrossRef CAS.
- F.-Y. Wang, S. Cheng and B.-Z. Wan, Porous Ag–CGO cermets as anode materials for ITSOFC using CO fuel, Catal. Commun., 2008, 9(7), 1595–1599 CrossRef CAS.
- N. Kumari, M. A. Haider, M. Agarwal, N. Sinha and S. Basu, Role of reduced CeO2 (110) surface for CO2 reduction to CO and methanol, J. Phys. Chem. C, 2016, 120(30), 16626–16635 CrossRef CAS.
- Y. Matsuzaki and I. Yasuda, Electrochemical Oxidation of H2 and CO in a H2-H2O-CO-CO2 System at the Interface of a Ni-YSZ Cermet Electrode and YSZ Electrolyte, J. Electrochem. Soc., 2000, 147(5), 1630 CrossRef CAS.
- E. Ioannidou, S. Neophytides and D. K. Niakolas, Experimental clarification of the RWGS reaction effect in H2O/CO2 SOEC co-electrolysis conditions, Catalysts, 2019, 9(2), 151 CrossRef.
- T.-R. Lee, H.-N. Im, S.-Y. Jeon, Y.-S. Yoo, A. U. Chavan and S.-J. Song, Dependence of H2O/CO2 co-electrolysis performance of SOEC on microstructural and thermodynamic parameters, J. Electrochem. Soc., 2016, 163(7), F728–F736 CrossRef CAS.
- W. Zhang, Y. Zheng, B. Yu, J. Wang and J. Chen, Electrochemical characterization and mechanism analysis of high temperature co-electrolysis of CO2 and H2O in a solid oxide electrolysis cell, Int. J. Hydrogen Energy, 2017, 42(50), 29911–29920 CrossRef CAS.
- Y. Wang, T. Liu, L. Lei and F. Chen, High temperature solid oxide H2O/CO2 co-electrolysis for syngas production, Fuel Process. Technol., 2017, 161, 248–258 CrossRef CAS.
- S. Hou and K. Xie, Enhancing the performance of high-temperature H2O/CO2 co-electrolysis process on the solid oxide Sr2Fe1.6Mo0.5O6-δ-SDC/LSGM/Sr2Fe1.5Mo0.5O6-δ-SDC cell, Electrochim. Acta, 2019, 301, 63–68 CrossRef CAS.
- J. G. Lee, O. S. Jeon, H. J. Hwang, J. Jang, Y. Lee and S. H. Hyun, et al., Durable and high-performance direct-methane fuel cells with coke-tolerant ceria-coated Ni catalysts at reduced temperatures, Electrochim. Acta, 2016, 191, 677–686 CrossRef CAS.
- A. Elleuch and K. Halouani, Intermediate-temperature solid oxide fuel cell fueled by biofuels, in Intermediate Temperature Solid Oxide Fuel Cells, Elsevier, 2020. pp. 427–476 Search PubMed.
- M. Cimenti and J. M. Hill, Importance of pyrolysis and catalytic decomposition for the direct utilization of methanol in solid oxide fuel cells, J. Power Sources, 2010, 195(1), 54–61 CrossRef CAS.
- D. J. Goodman, Methanation of carbon dioxide, UCLA, 2013 Search PubMed.
- J. Wambach, A. Baiker and A. Wokaun, CO2 hydrogenation over metal/zirconia catalysts, Phys. Chem. Chem. Phys., 1999, 1(22), 5071–5080 RSC.
- K. Stangeland, D. Kalai, H. Li and Z. Yu, CO2 methanation: the effect of catalysts and reaction conditions, Energy Procedia, 2017, 105, 2022–2027 CrossRef CAS.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló and U. De-La-Torre, et al., Isotopic and in situ DRIFTS study of the CO2 methanation mechanism using Ni/CeO2 and Ni/Al2O3 catalysts, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- W. L. Vrijburg, G. Garbarino, W. Chen, A. Parastaev, A. Longo and E. A. Pidko, et al., Ni-Mn catalysts on silica-modified alumina for CO2 methanation, J. Catal., 2020, 382, 358–371 CrossRef CAS.
- C. Lv, L. Xu, M. Chen, Y. Cui, X. Wen and C.-e. Wu, et al., Constructing highly dispersed Ni based catalysts supported on fibrous silica nanosphere for low-temperature CO2 methanation, Fuel, 2020, 278, 118333 CrossRef CAS.
- L. Zeng, Y. Wang, Z. Li, Y. Song, J. Zhang and J. Wang, et al., Highly Dispersed Ni Catalyst on Metal–Organic Framework-Derived Porous Hydrous Zirconia for CO2 Methanation, ACS Appl. Mater. Interfaces, 2020, 12(15), 17436–17442 CrossRef CAS PubMed.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, A review of recent catalyst advances in CO2 methanation processes, Catal. Today, 2020, 471–489 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu and C.-H. Yan, et al., Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect, ACS Catal., 2018, 8(7), 6203–6215 CrossRef CAS.
- X. Jia, X. Zhang, N. Rui, X. Hu and C.-j. Liu, Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- H. Jiang, Q. Gao, S. Wang, Y. Chen and M. Zhang, The synergistic effect of Pd NPs and UiO-66 for enhanced activity of carbon dioxide methanation, J. CO2 Util., 2019, 31, 167–172 CrossRef CAS.
- W. J. Lee, C. Li, H. Prajitno, J. Yoo, J. Patel and Y. Yang, et al., Recent trend in thermal catalytic low temperature CO2 methanation: a critical review, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.02.017.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu and F. Gu, et al., A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas, RSC Adv., 2012, 2(6), 2358–2368 RSC.
- F. C. Walsh, The overall rates of electrode reactions: Faraday's laws of electrolysis, Trans. IMF, 1991, 69(4), 155–157 CrossRef CAS.
- X. Vendrell and A. R. West, Induced p-type semiconductivity in yttria-stabilized zirconia, J. Am. Ceram. Soc., 2019, 102(10), 6100–6106 CrossRef CAS.
- J. Janek and C. Korte, Electrochemical blackening of yttria-stabilized zirconia–morphological instability of the moving reaction front, Solid State Ionics, 1999, 116(3–4), 181–195 CrossRef CAS.
- J. De Heer, The principle of le chatelier and braun, J. Chem. Educ., 1957, 34(8), 375 CrossRef CAS.
- D. Fini, S. P. Badwal, S. Giddey, A. P. Kulkarni and S. Bhattacharya, Evaluation of Sc2O3–CeO2–ZrO2 electrolyte-based tubular fuel cells using activated charcoal and hydrogen fuels, Electrochim. Acta, 2018, 259, 143–150 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se01887b |
| This journal is © The Royal Society of Chemistry 2021 |