
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon dots for photocatalytic H2 production in aqueous media with molecular Co catalysts†
Kalliopi
Ladomenou
 *,
Georgios
Landrou
,
Georgios
Charalambidis
*,
Emmanouil
Nikoloudakis
and
Athanassios G.
Coutsolelos
*
*,
Georgios
Landrou
,
Georgios
Charalambidis
*,
Emmanouil
Nikoloudakis
and
Athanassios G.
Coutsolelos
*
Laboratory of Bioinorganic Chemistry, Chemistry Department, University of Crete, 70013 Heraklion, Crete, Greece. E-mail: kladomenou@uoc.gr; gcharal@uoc.gr; acoutsol@uoc.gr
First published on 16th November 2020
Abstract
Carbon dots and nitrogen doped carbon dots were effectively synthesized and characterized. Their use as light harvesters was examined in the presence of three different cobalt-based catalysts and tris(carboxyethyl)phosphine/ascorbic acid (TCEP/Asc), as the sacrificial electron donor (SED). The electrons are transferred from the valence band (VB) to the conduction band (CB) of the carbon dots and the holes that are formed are filled by electrons transferred from the SED. The best photocatalytic system reported herein produces 17.1 μmol of H2 (TONCAT = 859) under UV radiation for 52 h. The same system can produce 5.3 μmol of H2 (TONCAT = 264) under the Cretan sun for 21 days of solar irradiation.
Introduction
Even though hydrogen is the most abundant element on Earth, it is present in chemical compounds and needs to be extracted with the consumption of energy, in order to use it as a fuel. Therefore, it is of great importance to produce and store H2 using alternative and economically feasible methods in order to replace fossil fuels.1 Hydrogen is believed to have high density, and to be a clean and environmentally friendly energy source, since upon its combustion it produces no pollutants. Therefore, H2 is an attractive solution to resolve the global energy and environmental problems.2,3 Many studies are based on efficient H2 production from which one of the most promising is the photocatalytic splitting of water.4,5 In order to produce an efficient photocatalytic system, three main components must be combined and operated together: a molecule or a material able to absorb the energy of light (light harvester), a catalyst, and a sacrificial electron donor.6 The photosensitizer absorbs light and produce electrons and hole pairs, then it promotes charge transfer to the catalyst that stimulates the reduction of protons. Finally, the sacrificial electron donor fuels the photosensitizer with electrons in order to start the second catalytic cycle. For an efficient photocatalytic system, it is crucial to use a light absorber that is stable, absorbs light in a broad light region, exhibits long lifetimes for effective charge separation, is easily prepared with low-cost and contains noble-metal free elements. Moreover, the redox potentials of the photosensitizer and the catalyst must be well complemented so that the charge transfer process will be thermodynamically and kinetically favored. For that purpose, much research has been done towards the synthesis of numerous light harvesting organic or inorganic molecules and nanomaterials.7–11 The molecular dyes appear to have some disadvantages such as they suffer from photostability in water and their preparation often requires demanding synthetic procedures. On the other hand, among the plethora of nanomaterials, carbon dots exhibit several advantages compared to semiconductor materials that appear to often have toxic metals in their structure.9,12–14 Carbon dots are small carbon nanoparticles with usual size less than 10 nm. The surface of these materials can be passivated with several elements such as nitrogen in order to modify their fluorescence properties and to facilitate charge separation due to the coexistence of p- and n-domains. Moreover, carbon dots appear to have distinctive donor/acceptor properties, exceptional electron transfer characteristics, photostability and low-cost preparation with various techniques. All the aforementioned properties of carbon dots make them promising candidates to be used as light absorbance materials.9 However, the use of carbon dots or nitrogen doped materials solely as light harvesters with molecular catalysts is limited.15,16 In contrast there are reports of their use as photosensitizers through co-sensitization with TiO2 and in the presence of noble metal catalysts.17–19 Therefore, there is a need to develop an efficient system comprised of carbon dot materials with noble metal free molecular catalysts.In this report carbon dots (CDot) and N-doped carbon dots (NCDot) have been synthesized as light harvesters to direct photocatalysis of protons to H2 with the use of molecular cobalt catalysts (Fig. 1). The molecular H2 evolving catalysts that were selected herein are cobalt based, the macrocyclic CatCo(III)1, the cobaloxime CatCo(III)2, and the porphyrin based catalyst CatCo(II)3 (Fig. 6). This molecular approach regarding the catalysts presents the advantage of facile synthetic preparation and future modification of the compounds, in order to investigate a series of molecules. The carbon dot materials were efficiently synthesized and physically and structurally characterized. Additional studies were performed concerning UV-vis, fluorescence properties and electron transfer capability of all carbon dot materials. H2 photocatalytic experiments exhibited a high production rate of 859 TONCAT with NCDot as the light harvesting material and CatCo(III)1 as the molecular catalyst in the presence of a SED. This value is the highest reported in the literature concerning analogue Co based systems.20 The main drawback of this photocatalytic scheme is the use of the SED that inhibits it from being a real sustainable system for H2 evolution. Nevertheless, these materials present a remarkable stability during photocatalysis that makes them able to produce H2 even after a period of 3 weeks under real solar irradiation, the Cretan sunlight. Also, a H2 evolution mechanism of our best working system is proposed.
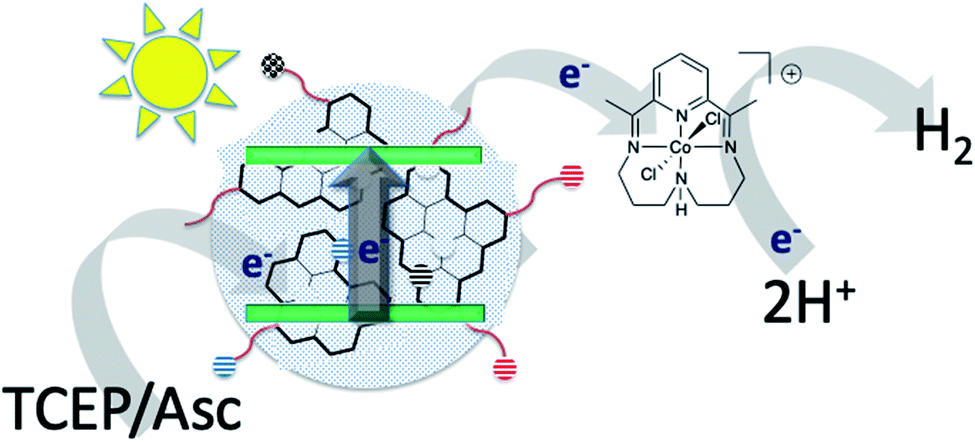 | ||
| Fig. 1 Schematic representation of photo driven H2 production with carbon dots as the photosensitizer in the presence of a cobalt catalyst. | ||
Results and discussion
Synthesis of carbon dots
Various water-soluble carbon dots were synthesized as photosensitizers for efficient hydrogen production using molecular cobalt based catalysts. Bottom-up synthesis was used for the preparation of the carbon dots starting from inexpensive organic materials that were thermally decomposed to form the appropriate carbon core. In this work two different types of materials were prepared CDot and NCDot. The first is designed with sodium carboxylate groups, while the second is doped with nitrogen and contains carboxylic acid and amino terminated groups. In both types of carbon dots citric acid was used as a carbon source. In the case of CDot citric acid was pyrolyzed to form carboxylic acid terminated carbon dots followed by neutralization with NaOH. More specifically, an already published synthetic procedure was followed with slight modification.21 Powder citric acid was heated in an oven under air at 180 °C for 72 h to obtain a brown powder as shown in Scheme 1. | ||
| Scheme 1 Synthetic procedure for the preparation of carbon dots CDots and NCDots. | ||
For the preparation of NCDots, citric acid was hydrothermally treated in the presence of ethylenediamine (Scheme 1).22,23 Citric acid was dissolved in water in a poly(tetrafluoroethylene)-lined autoclave reactor and heated at 180 °C for 8 h. The dark brown products were added in a dialysis membrane bag 1 kDa for 24 h in order to remove residual citric acid and small sized products. The product was added in a round-bottomed flask and freeze-dried in order to obtain NCDot as a dark powder (Scheme 1). Also, NCDotMix was obtained following the same procedure by skipping the step of purification; therefore, the reaction mixture was collected from the reactor and freeze-dried to obtain a brown solid. The yield of all the above synthetic procedures was about 35–40%. The nature and the properties of the synthesized material were studied with several spectroscopic techniques.
Physical characterization of carbon dots
FT-IR spectroscopy was performed in order to obtain information about the surface functionalities of carbon dots. The CDot spectrum (Fig. 2) shows a broad peak in the range of 3335–2900 cm−1 that corresponds to vibration of N–H, O–H, and C–H bonds and peaks at 1560 and 1387 cm−1 that correspond to antisymmetric and symmetric stretches of the carboxylate group, as already referred to in the literature.24 The spectrum of NCDot displays the following peaks: 3241 cm−1 that corresponds to N–H and O–H vibrations, 2920 cm−1 (C–H), 1684 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1646 cm−1 (CO), and 1531 cm−1 (N–H). In the FT-IR spectrum of both materials the CO vibration of citric acid at 1742 cm−1 (CO) is not present, therefore the shift and the broadness of the peaks imply full carbonization of both carbon dot materials of both CDot and NCDot.
C), 1646 cm−1 (CO), and 1531 cm−1 (N–H). In the FT-IR spectrum of both materials the CO vibration of citric acid at 1742 cm−1 (CO) is not present, therefore the shift and the broadness of the peaks imply full carbonization of both carbon dot materials of both CDot and NCDot.
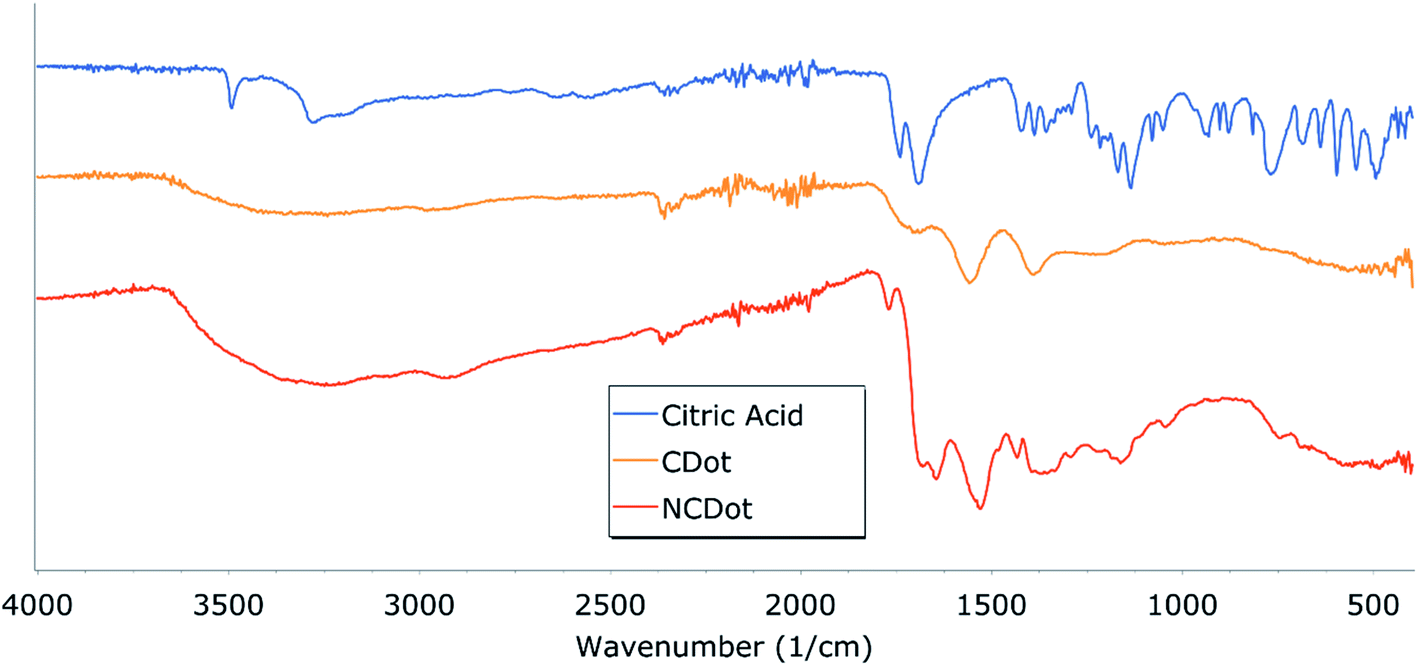 | ||
| Fig. 2 FT-IR spectra of citric acid, CDot and NCDot. | ||
Structural information
The morphology of carbon dots was observed by transmission electron microscopy TEM as shown in Fig. 3b and S1b.† The CDot nanoparticles show a near spherical morphology with a diameter of about 50 nm (Fig. S1b†) where in the case of NCDot the diameter of the material is less than 20 nm (Fig. 3b). In order to further elucidate the structure of carbon dots X-ray powder diffraction (XRD) was performed. NCDot displayed a broad peak at 2θ = 23°, which is attributed to highly defected or disordered carbon atoms as shown in Fig. 3a. The broad peak is a characteristic peak of the long-range disorder structure due to the small size of NCDot as shown in TEM images (Fig. 3b).25 In the case of CDot an amorphous peak at 2θ = 30° was observed in the XRD measurements (Fig. S1a†). | ||
| Fig. 3 (a) XRD pattern and (b) TEM image of NCDots. | ||
UV-vis and fluorescence
The aqueous solutions of carbon dots in all cases were yellow under daylight. All materials emitted blue fluorescence under 360 nm UV lamp radiation. The UV-vis spectrum of CDot in H2O (Fig. 4a) showed an absorption at ca. 200 nm, a typical absorption peak which is assigned to the π → π* transition of the aromatic sp2 areas.26NCDot showed two peaks one at ca. 200 nm and an absorption peak at 340 nm (Fig. 4b). The first peak can be ascribed to the π → π* transition of the unsaturated conjugated nanocarbon structure and the latter is due to the n → π* transition of the CO bond.27 The UV-vis spectrum of NCDotMix is similar to NCDot and is illustrated in Fig. S2b.† The high energy transition of all carbon dots is due to charge transfer from the inner to outer part of the sp2-hybridized core. In addition the lower energy transition is related to the transition from the edge or from the surface groups into the core.28
 | ||
| Fig. 4 UV-vis absorption and fluorescence spectra of (a) CDot and (b) NCDot. | ||
In the fluorescence spectra, CDot displayed a maximum emission at 360 nm, while NCDot and NCDotMix showed a maximum emission at 340 nm as shown in Fig. 4 and S2.† The emission of all carbon dots progressively red shift to a longer wavelength as the excitation increases beyond 320 nm showing an excitation-dependent behavior that is well known in similar carbon dot materials.29
Quantum yield and lifetime
The quantum yield (QY) of carbon dots was calculated using quinine hemisulfate in 0.1 M H2SO4 as a reference. The QY of NCDot was calculated to be 26%, whereas for CDot a much lower QY of 2% was measured indicating that N doping is an effective method to enhance the photoluminescence of these materials.30 Moreover, the QY of NCDotMix, which is the material obtained directly from the reaction with no further purification, was calculated to be 43%. This indicates that the carbon dots with a small molecular weight appear to have larger QY. Lifetime decay fluorescence measurements of NCDot showed τ1 = 3.7 (38%) ns and τ2 = 14.7 (62%) ns whereas the CDot showed τ1 = 2.1 (76%) ns and τ2 = 14.4 (24%) ns. The values were calculated by using a double-exponential function for the satisfactory data fitting. These measurements are according to the literature since it is known that carbon dot materials show multiexponential decay of photoluminescence emission.31Electron transfer assays
The electron transfer ability of carbon dots was investigated by observing the photoreduction of methyl viologen (MV2+) and its capability to quench photoluminescence.24 Therefore, a solution of MV2+ in aqueous ethylenediaminetetraacetic acid (EDTA), at pH = 6 and in the presence of NCDot was bubbled for 15 min under argon in order to remove oxygen. After 2 min of UV irradiation the typical peaks of reduced methyl viologen (MV+.) at 395 and 603 nm appeared as shown in Fig. 5a. During the time, the color of the solution changed from yellow to blue that is indicative of the formation of the reduced species. The same experiments were performed with CDot displaying the same spectra (Fig. 5b). Therefore, the carbon dots have electron transfer properties which are essential for the photocatalysis. | ||
| Fig. 5 UV-vis spectra of (a) NCDot (0.5 mg ml−1) with methyl viologen (1.2 mM) in EDTA 0.1 M at pH = 6 and (b) CDot (0.5 mg ml−1). UV-vis spectra were recorded every 2 min after irradiation with a UV lamp. | ||
Electrochemical measurements
Cyclic voltamograms of all materials (Fig. S3–S5†) were obtained in 0.1 M aqueous solution of sodium sulfate. The oxidation and reduction potentials are presented in the ESI (Table S1).† The oxidation potentials of carbon dots were used as the valence band (VB) values of the photosensitizers, since it has been reported in the literature that for similar systems such as CdTe quantum dots the Eox provides estimated values of the position of the VB.32 Then, the VB values were subtracted from the optical energy values (E00) in order to obtain the potentials of the conduction band (CB). The E00 values were obtained from the intersection between the normalized absorption and emission spectra of carbon dots. In the case of NCDotE00 was estimated at 395 nm that correspond to 3.14 eV. All calculated values are listed in Table 1 and the normalized spectra of CDot and NCDot are shown in Fig. S9 and S10,† respectively.| Carbon dots | E VB (V) | E CB (V) | E 00 (eV) |
|---|---|---|---|
| a Oxidation potentials were measured by cyclic voltammetry and were referenced to NHE by addition of +0.193 V.33 b Values were calculated as the difference of the oxidation potentials and the optical band gap energy E00, calculated in eV. c E 00 values were calculated from the intersection between the normalized absorption and emission spectra. | |||
| NCDot | +1.01 | −2.13 | 3.14 |
| NCDotMix | +1.01 | −2.13 | 3.14 |
| CDot | +1.18 | −1.98 | 3.16 |
Synthesis of catalysts
Cobalt based water-soluble molecular catalysts CatCo(III)1, CatCo(III)2 and CatCo(II)3 were used for our H2 evolution experiments (Fig. 6). The first Co catalyst (CatCo(III)1) was synthesized according to the synthetic procedure described by Busch and later modified by Collomb and coworkers.34,35 The last step of the aforementioned procedure was the treatment of 2,6-diacetylpyridine with CoCl2·6H2O salt, diaminodipropylamine and LiClO4 to afford the desired product in good yield. Then the second cobaloxime based catalyst CatCo(III)2 was prepared from dimethylglyoxime [Co(dmg)2Cl2] with the addition of L-histidine under a N2 atmosphere.36,37 A water-soluble cationic Co-porphyrin catalyst CatCo(II)3 was synthesized as described previously by metalation of tetrapyridylporphyrin with (CH3COO)2Co·4H2O in DMF under reflux. Finally, methylation was achieved by adding excess of iodomethane.38 | ||
| Fig. 6 Molecular structures of cobalt catalysts used in this work for H2 production. | ||
Photocatalytic hydrogen production
Photocatalytic experiments were performed using carbon dots as photosensitizers and different cobalt molecules as catalysts (Fig. 6) in aqueous solution with the use of diverse sacrificial electron donors (SEDs) as shown in Fig. 1. In our first attempts the catalytic system comprised of CDot and N-doped carbon dots pure (NCDot) and impure (NCDotMix) as photosensitizers with CatCo(III)1 in 0.1 M or 1 M ascorbic acid (Asc) as the SED at pH = 5. The system was irradiated under UV or visible white LED light and in any case no H2 was detected. This may be attributed to the well-known formation of dehydroascorbic acid (DHA), which is produced during the catalysis and prevents the ability of Asc to provide electrons to the photosensitizer.39 To overcome this difficulty we used SED as a mixture of TCEP/Asc (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) 0.1 M each at pH = 5 and H2 was produced in all cases in the presence of CatCo(III)1 as a molecular catalyst. The same SED mixture had been previously used by us and others leading to a more efficient hydrogen evolution in all systems.20,40,41 TCEP regenerates the oxidized ascorbic acid and therefore overcomes the limitation of instability of Asc and extends the lifetime of the system. Upon optimization of the system the maximum TONCAT of 859 was reached when N-doped carbon dots (NCDot) were used as the photosensitizer after 52 h of irradiation (Fig. 7). This is the highest TONCAT reported in the literature when carbon dots are used as a photocatalyst with a cobalt molecule as a catalyst. The CatCo(III)1 catalyst was chosen since it shows great stability and activity for H2 evolution in aqueous media.35,42 Moreover it has been used previously in a system of CdTe water soluble quantum dots and reached 650 TONCAT in 1.5 h where it is less efficient compared to our system indicating that for a better performance, CdTe components are not essential. Also, our photosensitizers are easily synthesized from low-cost starting materials.43
:1) 0.1 M each at pH = 5 and H2 was produced in all cases in the presence of CatCo(III)1 as a molecular catalyst. The same SED mixture had been previously used by us and others leading to a more efficient hydrogen evolution in all systems.20,40,41 TCEP regenerates the oxidized ascorbic acid and therefore overcomes the limitation of instability of Asc and extends the lifetime of the system. Upon optimization of the system the maximum TONCAT of 859 was reached when N-doped carbon dots (NCDot) were used as the photosensitizer after 52 h of irradiation (Fig. 7). This is the highest TONCAT reported in the literature when carbon dots are used as a photocatalyst with a cobalt molecule as a catalyst. The CatCo(III)1 catalyst was chosen since it shows great stability and activity for H2 evolution in aqueous media.35,42 Moreover it has been used previously in a system of CdTe water soluble quantum dots and reached 650 TONCAT in 1.5 h where it is less efficient compared to our system indicating that for a better performance, CdTe components are not essential. Also, our photosensitizers are easily synthesized from low-cost starting materials.43
 | ||
| Fig. 7 Photocatalytic hydrogen production plots of NCDot, NCDotMix and CDot. The photocatalytic experiments were conducted in aqueous TCEP/Asc (1:1) 0.1 M each at pH = 5. The amount of carbon dots was 10 mg in each case and the catalyst CatCo(III)1 (20 nmol). All the results presented in the H2 production plots are the average values of three independent measurements (within 5–10% error). | ||
In the case of impure carbon dots (NCDotMix) the maximum TONCAT 334 was attained at 23 h, whereas the maximum TONCAT of CDot was 64 at 23 h. The lower performance of NCDotMix compared to that of the pure ones is possibly due to the presence of citric acid or due to carbonated products with low molecular weight that prevent the absorption of light from the photosensitizer. A characteristic feature of N-doped carbon dots is that they start producing H2 after about 10 h of irradiation at a slower rate compared to CDot. More specifically, after 7 h of irradiation no H2 was detected for NCDot and NCDotMix, while at 7 h CDot attained 52 TONCAT. Therefore, in our system the NCDot chromophore proved to be more efficient compared to CDot. The enhanced TON measured for N-doped carbon dots is well known in the literature, since the presence of nitrogen improves the charge transfer by introducing n-domains in the material.13,44
In all cases the system is stable after UV irradiation for about 6 days. Reisner and co-workers have already reported H2 evolution of carbon dots using water-soluble cobalt catalysts and carboxylate terminated carbon dots, under similar conditions.20 They observed a low rate of H2 production TONCAT ranging from 141 to 56 and a high stability of their system, being active after 4 days of irradiation. In the case of N-doped carbon dots the performance of our catalyst was much better compared to all molecular cobalt catalysts reported in the literature.20 To the best of our knowledge our system shows the highest rates of H2 production when N-doped carbon dots are photosensitized in the presence of a cobalt molecular catalyst in water. Our system proved to be very stable since after about 6 days the carbon dots started to photo bleach and the system stopped producing more H2. The photo bleaching of the photosensitizer was monitored with UV-vis spectroscopy (Fig. S6†). As shown in the spectra the absorption of the material is decreasing overtime indicating the decolorization of the material upon UV irradiation. Therefore, after about 70 h of irradiation the system was not able to absorb more light and the H2 production stopped. In order to investigate further the state of carbon dots during and after the photocatalytic experiment TEM images were obtained in order to investigate if there is a change in the morphology of carbon dots (Fig. S7†). In Fig. S7(a)† the spheres of NCDot are shown after UV irradiation for 50 h and in Fig. S7(b)† are presented in the NCDot after the completion of the photocatalytic experiment. It is evident that the carbon dot materials keep their spherical shape but their diameter gets smaller upon light irradiation. Moreover, the number of the spheres is less upon irradiation. Consequently, it seems that upon irradiation the morphology of the photosensitizer alters. This is in accordance with XRD measurements that were done in NCDot after the photocatalytic experiment (Fig. S8†). The broad peak that was present at 2θ = 23° is the starting material, after 70 h of irradiation the same peak was diminished indicating that the structure of the material has been changed.
Moreover, control experiments were performed in the absence of the catalyst CatCo(III)1, in the absence of the photosensitizer NCDot, in the absence of the SED, TCEP/Asc and in the dark. In any case a negligible amount of H2 was produced. This indicates that all components are essential for H2 production in our system. Also, when 20 nmol of CoCl2 was used as an alternative of the Co molecular catalyst, insignificant H2 was evolved. In addition, control H2 evolution experiments were done using NCDot, CatCo(III)1 and TCEP as the SED (0.1 M at pH 5), where a lower photocatalytic H2 production of 462 TONCAT was observed. This revealed that even though NCDot can oxidize the TCEP, the Asc is the one that quenches the photoinduced holes and the combination of TCEP/Asc is essential for efficient H2 production.20 This finding is in accordance with the emission experiments of NCDot where in the presence of 0.1 M TCEP the carbon dots were slightly quenched (Fig. S11†).
Subsequently, the quantity of the catalyst was evaluated by using four different amounts 10, 20, 50 and 100 nmol of CatCo(III)1 in aqueous solution of TCEP/Asc 0.1 M each (Fig. 8). The maximum TONCAT was obtained when 20 nmol of CatCo(III)1 was used, therefore all the photocatalytic experiments that are reported herein were done with 20 nmol of catalyst.
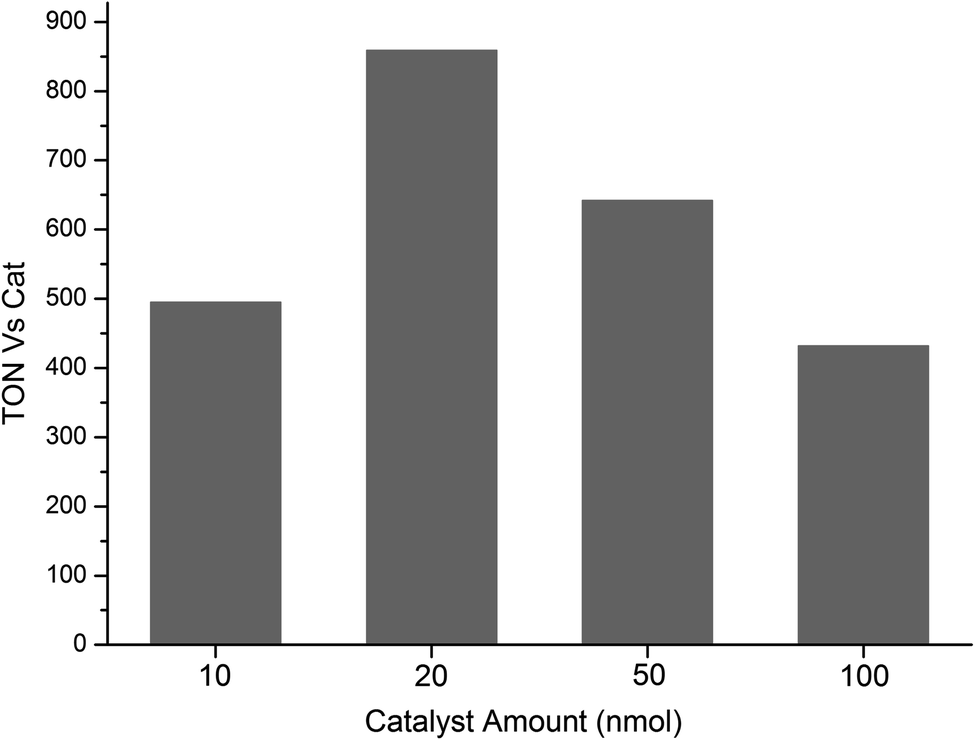 | ||
| Fig. 8 Photocatalytic hydrogen production plots NCDot in four different quantities of the catalyst CatCo(III)1: 10, 20, 50 and 100 nmol. The photocatalytic experiments were conducted in aqueous TCEP/Asc (1:1) 0.1 M each at pH = 5 in the presence of catalyst CatCo(III)1. All the results presented in the H2 production plots are the average values of three independent measurements (within 5–10% error). | ||
In our attempt to find out if our photosensitizer works well with other water soluble catalysts we performed catalytic experiments using NCDot as the photosensitizer and CatCo(III)2 and CatCo(II)3 as catalysts (Fig. 9). The thermodynamics of the system using these two molecules makes them promising catalysts for H2 production.11,45 In our first attempt, when 20 nmol of the cobaloxime-type catalyst CatCo(III)2 was used for H2 evolution experiments 1.01 μmol H2 was obtained after 46 h of UV irradiation. Moreover, when 20 nmol of water-soluble cobalt porphyrin CatCo(II)3 was the catalyst, no H2 was detected after 74 h of UV irradiation. The maximum amount of H2 17.1 μmol was obtained with the stable CatCo(III)1 after 52 h of light irradiation making this Co catalyst superior compared to the other two catalysts. The small H2 production of CatCo(III)2 can be due to its low stability in acidic aqueous media already reported in the literature.43 In addition the undetectable H2 in the case of CatCo(II)3 is possibly once again due to the low stability of the porphyrin molecule under UV irradiation.46
 | ||
| Fig. 9 Photocatalytic hydrogen production plots of 10 mg NCDot with catalysts: CatCo(III)1 (20 nmol), CatCo(III)2 (20 nmol) and CatCo(II)3 (20 nmol). The photocatalytic experiments were conducted in aqueous TCEP/Asc (1:1) 0.1 M each at pH = 5. All the results presented in the H2 production plots are the average values of three independent measurements (within 5–10% error). | ||
In order to understand better the high performance of CatCo(III)1 the ΔG (PS/Cat) were calculated for the three catalysts and the photosensitizer NCDot. The redox potentials of the reported compounds and the thermodynamic driving forces for the electron transfer process are listed in Table 2. The thermodynamic driving forces were calculated according to the literature.47 From these results it is obvious that the driving force of CatCo(III)1 is larger compared to the other two catalysts which is in accordance with the grater H2 production of this catalyst.
| Compounds | E 1/2 Ox | E 1/2 Red |
|---|---|---|
| NCDot | +1.01 | −0.87 |
In all the catalytic systems H2 evolution reaches a plateau after several hours of irradiation and the H2 production stops. After the addition of either the catalyst or the SED no hydrogen production was observed. However, when the photosensitizer, NCDot was added, the catalytic system was effectively regenerated, suggesting that the carbon dots are decomposed after about 6 days of UV light irradiation.
Different irradiation sources were applied to broaden the use of our best performing system as shown in Fig. 10. Visible led light was used to obtain 11.6 μmol in 46 h, slightly less compared to the H2 evolution with UV irradiation (17.1 μmol) possibly due to the higher absorbance of the photosensitizer NCDot at the UV region as shown in the UV-vis spectrum Fig. 4b. Finally, we performed the same experiment under the Cretan sun. During 21 days of irradiation the NCDot remained stable with no observed photo bleaching, making the system very promising for future use obtaining 5.3 μmol of H2 (TONCAT = 264).
 | ||
| Fig. 10 Photocatalytic hydrogen production plots of 10 mg NCDot with CatCo(III)1 (20 nmol) in various solar sources: UV Vis led and Cretan sun. The photocatalytic experiments were conducted in aqueous TCEP/Asc (1:1) 0.1 M each at pH = 5. All the results presented in the H2 production plots are the average values of three independent measurements (within 5–10% error). | ||
H2 evolution mechanism
In order to clarify the mechanism of our photocatalytic system we performed emission spectroscopy experiments of NCDot in the presence of different concentrations of the cobalt catalyst, CatCo(III)1. This experiment will help us understand the favorable pathway that occurs in our system: (a) an oxidative quenching from the chromophore to the catalyst or (b) a reductive quenching from the SED to the chromophore.6,49 The NCDot was excited at 340 nm and upon addition of the catalyst the emission intensity was decreased by about 50% indicating efficient transfer from the CB of the material to the cobalt catalyst (Fig. S12†). The concentration of NCDot was kept constant during the addition of the catalyst. An analogous procedure was followed using an aqueous solution of NCDot at the same concentration as in the previous experiment and we monitored the emission upon excitation at 340 nm when different concentrations of ascorbic acid at pH = 5 were added (Fig. S13†). Once again, the emission intensity was significantly decreased by about 80%. The mixture of TCEP/Asc was not examined since when 0.1 M concentration of TCEP was used, we observed a slight quenching (Fig. S11†). Therefore, the quenching was due to the presence of the ascorbic acid in the solution. Subsequently, the Stern–Volmer constant Ksv was calculated to understand better the mechanism of the photocatalytic reaction. The calculation was done from the fitting of the experimental data according to equation Io/I = 1 + KSV[Q], where Io and I are the fluorescence intensities observed in the absence and in the presence of each quencher, respectively, and [Q] is defined as the quencher concentration (Fig. S14†).50 The KSV in the case of the catalyst was bigger (KSV(cat) = 4612.8 M−1) compared to the sacrificial electron donor ascorbic acid (KSV(Asc) = 13.8 M−1). Moreover, the quenching constant KQ was calculated using KSV = KQτ, where τ stands for the excited state lifetime in the absence of the quencher. In the case of the catalyst CatCo(III)1 the KQ(cat) = 4.38 × 1011 M−1 s−1 and in the case of the sacrificial agent ascorbic acid KQ(Asc) = 1.32 × 109 M−1 s−1. Under similar conditions to the photocatalytic H2 experiments 6 μM of catalyst and 0.1 M of the SED, the quenching is more prominent in the case of ascorbic acid. This finding suggests that a photoinduced electron transfer takes place from the sacrificial electron donor to the VB of carbon dots. Moreover, even though the quenching constant of the catalyst is greater compared to the ascorbic acid the main electron transfer follows the pathway from the ascorbic acid to the CB of carbon dots due to the greater concentration of the SED (0.1 M) compared to the catalyst (0.6 × 10−5 M).51 Therefore, the hydrogen production in our system is done through a reductive quenching by using the SED, leading over the oxidative quenching by the CatCo(III)1.The photocatalytic H2 production seems to be thermodynamically feasible and the energy diagram of the whole process is shown in Fig. 11. The VB and CB potentials of NCDot were calculated and are shown in Table 1, whereas the potentials of the SED ascorbic acid and the catalyst CatCo(III)1 were taken from the literature.43,52 The photo driven mechanism of H2 evolution that is proposed herein is in accordance with the mechanism already mentioned by Llobet and Palomares in a similar system using the same cobalt catalyst.43 In the beginning of the photocatalytic experiment CatCo(III)1 is in the reduced form Co(II), as observed by the UV-vis spectra, where the addition of the SED reduces the catalyst (Fig. S15†). However, the oxidized form of the catalyst Co(III) can be produced during H2 production. In order to produce H2 two electrons are needed to be transferred to the protons. In our system solar irradiation excites the carbon dots and electron transfer occurs from the VB to the CB, then the hole that is formed at the VB of the photosensitizer is filled with an electron transferred from the sacrificial electron donor. This pathway is in accordance with the reductive quenching of our system as discussed previously.
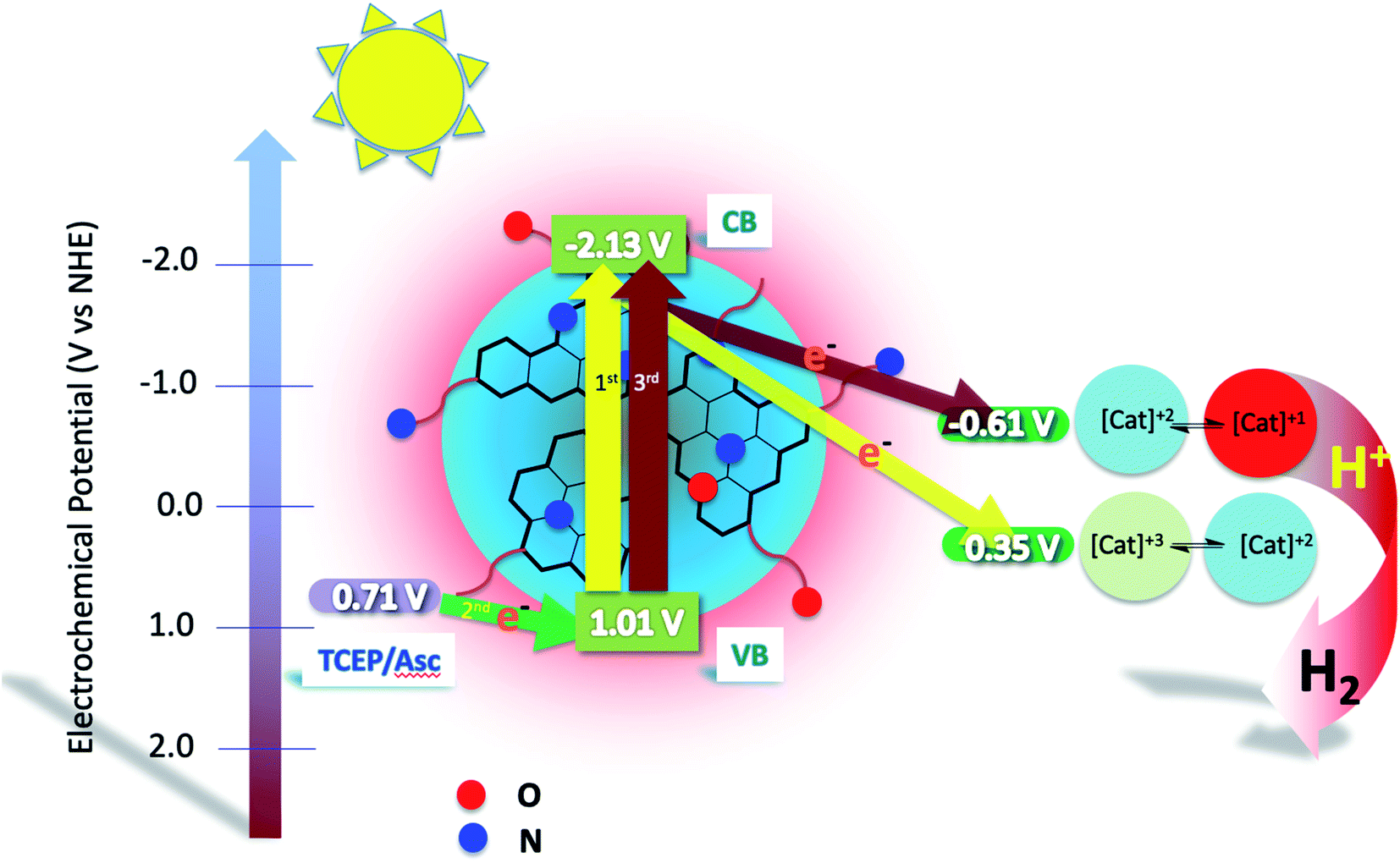 | ||
| Fig. 11 Mechanism of two electron transfer photocatalytic H2 production using N-doped carbon dots, CatCo(III)1 as the catalyst and TCEP/Asc (1:1) 0.1 M each as the SED. | ||
Following this, one electron reduces the cobalt metal of the catalyst from Co(III) to Co(II). The whole process takes place one more time where a second electron reduces the catalyst. According to theoretical calculations from Zhong, Lu, Sakai and co-workers this second reduction is ligand based and not metal-centered.53 Therefore, ligand-reduced species formulated as [CoII(L−˙)]+ are formed, where after their protonation solar H2 evolution is achieved.
Conclusions
In this work we have examined the H2 evolution ability of carbon dots and nitrogen doped materials, in the presence of cobalt catalysts in aqueous media. The key substance for efficient H2 production was TCEP/Asc which acts as the sacrificial electron donor material. TCEP is well known to prevent the formation of dehydroascorbic acid that prevents the ability of ascorbic acid to give electrons to the photosensitizer. The study demonstrated that cobaloxime and porphyrin based cobalt catalysts are not able to promote the photocatalysis. However the quite stable cyclic catalyst CatCo(III)1 was capable of producing a record high H2 production of 17.1 μmol (TONCAT = 859) under UV radiation. The photocatalytic H2 production of our system is performed via a reductive quenching by the SED whereas two electrons are needed to be transferred to the protons for H2 production. This system offers great potential for future work, to explore more water-soluble catalysts and even extend the doping onto the carbon dots with noble-metal free elements in order to improve the light absorption ability. The only drawback of this system is the use of a SED. The improved photocatalytic system can be even applied in the real world since the sun is able to produce H2 and by just adding low-cost carbon dots onto the system it can operate for several weeks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI; project code: 508). This research has been co-financed by the European Union and Greek National Funds through the Regional Operational Program “Crete 2014-2020”, project code OPS: 5029187. In addition the research has been funded by IKY scholarships through the Operational Program “Human Resources Development, Education and Lifelong Learning 2014–2020”, Act: “Strengthening of human research potential through the implementation of doctoral research” – MIS 5000432. Moreover, the European Commission's Seventh Framework Program (FP7/2007-2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIO-SOLENUTI) and the Special Research Account of the University of Crete are gratefully acknowledged for the financial support of this research.References
- M. Pagliaro, A. G. Konstandopoulos, R. Ciriminna and G. Palmisano, Energy Environ. Sci., 2010, 3, 279–287 RSC.
- K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426–10437 CrossRef CAS.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS.
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304, 38–54 CrossRef.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268–9274 RSC.
- J. Gao, M. Zhu, H. Huang, Y. Liu and Z. Kang, Inorg. Chem. Front., 2017, 4, 1963–1986 RSC.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Chem. Soc. Rev., 2017, 46, 6111–6123 RSC.
- M. Wang, K. Han, S. Zhang and L. Sun, Coord. Chem. Rev., 2015, 287, 1–14 CrossRef CAS.
- D. Dolui, S. Ghorai and A. Dutta, Coord. Chem. Rev., 2020, 416, 213335 CrossRef CAS.
- H. Luo, Q. Guo, P. Á. Szilágyi, A. B. Jorge and M.-M. Titirici, Trends Chem., 2020, 2, 623–637 CAS.
- C. Hu, M. Li, J. Qiu and Y.-P. Sun, Chem. Soc. Rev., 2019, 48, 2315–2337 RSC.
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CAS.
- S. Cao and J. Yu, J. Photochem. Photobiol., C, 2016, 27, 72–99 CrossRef CAS.
- Y. Liu, H. Huang, W. Cao, B. Mao, Y. Liu and Z. Kang, Mater. Chem. Front., 2020, 4, 1586–1613 RSC.
- N. C. T. Martins, J. Ângelo, A. V. Girão, T. Trindade, L. Andrade and A. Mendes, Appl. Catal., B, 2016, 193, 67–74 CrossRef CAS.
- R. Shi, Z. Li, H. Yu, L. Shang, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu and T. Zhang, ChemSusChem, 2017, 10, 4650–4656 CAS.
- I. Sargin, G. Yanalak, G. Arslan and I. H. Patir, Int. J. Hydrogen Energy, 2019, 44, 21781–21789 CrossRef CAS.
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 9402–9406 CrossRef CAS.
- C. X. Guo, D. Zhao, Q. Zhao, P. Wang and X. Lu, Chem. Commun., 2014, 50, 7318–7321 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS.
- J. Schneider, C. J. Reckmeier, Y. Xiong, M. von Seckendorff, A. S. Susha, P. Kasák and A. L. Rogach, J. Phys. Chem. C, 2017, 121, 2014–2022 CrossRef CAS.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS.
- F.-D. Han, B. Yao and Y.-J. Bai, J. Phys. Chem. C, 2011, 115, 8923–8927 CrossRef CAS.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS.
- Z. Liang, L. Zeng, X. Cao, Q. Wang, X. Wang and R. Sun, J. Mater. Chem. C, 2014, 2, 9760–9766 RSC.
- C. J. Reckmeier, Y. Wang, R. Zboril and A. L. Rogach, J. Phys. Chem. C, 2016, 120, 10591–10604 CrossRef CAS.
- P. G. Luo, S. Sahu, S.-T. Yang, S. K. Sonkar, J. Wang, H. Wang, G. E. LeCroy, L. Cao and Y.-P. Sun, J. Mater. Chem. B, 2013, 1, 2116–2127 RSC.
- P. Yang, J. Zhao, J. Wang, H. Cui, L. Li and Z. Zhu, ChemPhysChem, 2015, 16, 3058–3063 CrossRef CAS.
- P. M. Gharat, J. M. Chethodil, A. P. Srivastava, P. K. Praseetha, H. Pal and S. Dutta Choudhury, Photochem. Photobiol. Sci., 2019, 18, 110–119 RSC.
- S. K. Haram, A. Kshirsagar, Y. D. Gujarathi, P. P. Ingole, O. A. Nene, G. B. Markad and S. P. Nanavati, J. Phys. Chem. C, 2011, 115, 6243–6249 CrossRef CAS.
- C. G. Zoski, Handbook of Electrochemistry, Elsevier, Amsterdam, 2007 Search PubMed.
- K. M. Long and D. H. Busch, J. Coord. Chem., 1974, 4, 113–123 CrossRef CAS.
- S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544–17552 RSC.
- G. N. Schrauzer, G. W. Parshall and E. R. Wonchoba, Bis(Dimethylglyoximato)Cobalt Complexes: (“Cobaloximes”), Inorganic Syntheses, John Wiley & Sons, Ltd, 2007, pp. 61−70 Search PubMed.
- D. Dolui, S. Das, J. Bharti, S. Kumar, P. Kumar and A. Dutta, Cell Reports Physical Science, 2020, 1, 100007 CrossRef.
- D. Skrzypek, I. Madejska and J. Habdas, Solid State Sci., 2007, 9, 295–302 CrossRef CAS.
- M. Guttentag, A. Rodenberg, R. Kopelent, B. Probst, C. Buchwalder, M. Brandstätter, P. Hamm and R. Alberto, Eur. J. Inorg. Chem., 2012, 59–64 CrossRef CAS.
- N. Queyriaux, E. Giannoudis, C. D. Windle, S. Roy, J. Pécaut, A. G. Coutsolelos, V. Artero and M. Chavarot-Kerlidou, Sustainable Energy Fuels, 2018, 2, 553–557 RSC.
- S. Schnidrig, C. Bachmann, P. Müller, N. Weder, B. Spingler, E. Joliat-Wick, M. Mosberger, J. Windisch, R. Alberto and B. Probst, ChemSusChem, 2017, 10, 4570–4580 CrossRef CAS.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS.
- C. Gimbert-Suriñach, J. Albero, T. Stoll, J. Fortage, M.-N. Collomb, A. Deronzier, E. Palomares and A. Llobet, J. Am. Chem. Soc., 2014, 136, 7655–7661 CrossRef.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2017, 56, 6459–6463 CrossRef CAS.
- J. Ma, J. Wu, J. Gu, L. Liu, D. Zhang, X. Xu, X. Yang and Z. Tong, J. Mol. Catal. A: Chem., 2012, 357, 95–100 CrossRef CAS.
- N. M. Berezina, M. B. Berezin and A. S. Semeikin, J. Mol. Liq., 2019, 290, 111196 CrossRef.
- N. Queyriaux, R. A. Wahyuono, J. Fize, C. Gablin, M. Wächtler, E. Martinez, D. Léonard, B. Dietzek, V. Artero and M. Chavarot-Kerlidou, J. Phys. Chem. C, 2017, 121, 5891–5904 CrossRef CAS.
- S.-M. Chen and S.-W. Chiu, J. Mol. Catal. A: Chem., 2001, 166, 243–253 CrossRef CAS.
- W. T. Eckenhoff, W. R. McNamara, P. Du and R. Eisenberg, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 958–973 CrossRef CAS.
- J. Keizer, J. Am. Chem. Soc., 1983, 105, 1494–1498 CrossRef CAS.
- S. Panagiotakis, G. Landrou, V. Nikolaou, A. Putri, R. Hardré, J. Massin, G. Charalambidis, A. G. Coutsolelos and M. Orio, Front. Chem., 2019, 7, 405 CrossRef CAS.
- E. Benazzi, V. C. Coni, M. Boni, R. Mazzaro, V. Morandi and M. Natali, Dalton Trans., 2020, 49, 10212–10223 RSC.
- J.-W. Wang, K. Yamauchi, H.-H. Huang, J.-K. Sun, Z.-M. Luo, D.-C. Zhong, T.-B. Lu and K. Sakai, Angew. Chem., Int. Ed., 2019, 58, 10923–10927 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se01630f |
| This journal is © The Royal Society of Chemistry 2021 |