
Hydrogen-efficient non-oxidative transformation of methanol into dimethoxymethane over a tailored bifunctional Cu catalyst†
Ruiyan
Sun
 a,
Chalachew
Mebrahtu
a,
Jan Philipp
Hofmann
bd,
Dominik
Bongartz
c,
Jannik
Burre
c,
Christian Henning
Gierlich
a,
Peter J. C.
Hausoul
a,
Alexander
Mitsos
c and
Regina
Palkovits
*a
a,
Chalachew
Mebrahtu
a,
Jan Philipp
Hofmann
bd,
Dominik
Bongartz
c,
Jannik
Burre
c,
Christian Henning
Gierlich
a,
Peter J. C.
Hausoul
a,
Alexander
Mitsos
c and
Regina
Palkovits
*a
aInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
bLaboratory for Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cProcess Systems Engineering (AVT.SVT), RWTH Aachen University, 52074 Aachen, Germany
dSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany
First published on 28th September 2020
Abstract
Dimethoxymethane (DMM), a promising synthetic fuel enabling clean combustion, is usually produced by condensation of methanol and formaldehyde, where the latter stems from methanol oxidation. Here, we report the hydrogen efficient non-oxidative DMM synthesis over a bifunctional Cu/zeolite catalyst in a continuous gas-phase fixed bed reactor. Methanol dehydrogenation to formaldehyde (FA) is coupled with FA condensation with methanol to yield DMM, hydrogen and water. Thermodynamic analysis confirms the general feasibility of this route and also manifests the vital importance of catalyst selectivity. Therein, close proximity of the catalyst's metallic Cu species and acidic sites is crucial. Noticeably, DMM selectivity of the catalyst only evolves within the first 13 hours of operation rising from 5.8 to 77.2%. A maximum DMM selectivity of 89.2 or 80.3% could be reached for 0.4 and 0.7 wt% Cu on Hβ(836) zeolite with 1.9 or 3.6% methanol conversion, respectively. Comprehensive characterizations emphasize adaptation of Cu species and Hβ zeolite under reaction conditions resulting in the decisive weakened dehydrogenation and condensation ability for high DMM selectivity. Process simulations confirm superior exergy efficiency compared to state-of-the-art technologies for DMM production already with the herein developed catalyst and highlights the high potential of further innovations for technical implementation.
Introduction
With the fast popularization of vehicles in the last several decades, the transportation sector has become the largest oil consumer accounting for more than 60% of the global oil consumption.1,2 Diesel- and gasoline-powered vehicles account for the major share (75%) of this tremendous fuel demand.3 Compared with gasoline cars, diesel engines consume less fuel and accordingly emit less CO2. They play a major role in long distance transportation with 94% of freight transportation relying on diesel engines and will continue to govern the transport sector, e.g., in heavy duty applications such as trucks, construction machinery and shipping. However, in recent years, a number of major European cities have declared to ban diesel cars, because of increasing concerns regarding negative impacts on human health and environment caused by the high emissions of soot and NOx during diesel combustion.4 Advanced exhaust after-treatment systems including the selective catalytic reduction system and particulate filter,5 enable to significantly reduce soot and NOx emissions.6 In an effort to reduce the CO2 footprint of combustion engines, numerous efforts are being made to make synthetic fuels, such as Fischer–Tropsch diesel, available from CO2 and renewable hydrogen feedstocks.7,8Recently, DMM (OME1) and its longer homologues (oxymethylene ethers, OME>1), featuring high oxygen content, no C–C bonds in their structures, and high cetane numbers, are considered as a novel category of synthetic fuels to be blended into or directly replace diesel fuel.9–11 Engine studies confirmed their significant potential as fuel additives for reducing soot and NOx emissions. More specifically, an addition of 35 vol% of DMM in standard diesel fuel reduces soot formation by 90% together with significant NOx suppression (from 0.4 to 0.1 g kW−1 h−1).12–14 Interestingly, pollutant reduction of diesel blends with DMM versus OME3–5 are essentially identical.15 For full fuel substitution, the physicochemical properties of OME3–5 are beneficial. However, in a blend with diesel, the higher energy efficiency of DMM production compared to longer chain OMEs presents a clear advantage.16–18 Available synthetic routes to DMM primarily rely on oxidative reaction pathways, wherein methanol is either first oxidized to formaldehyde (FA) in the presence of oxygen followed by an acetalization of methanol with FA to yield DMM in two steps, or methanol is directly oxidized to DMM in one step (Scheme 1a and b). Both routes have been intensively studied with excellent DMM selectivities and methanol conversions reported in literature.16,19 Nevertheless, the established routes exhibit a common deficiency in hydrogen management, an important factor for a sustainable process. Comprehensive process analysis of DMM production emphasized a significant exergy loss caused by formaldehyde formation and the major influence of hydrogen demand on overall energy requirement and global warming potential.13,16,18,20 Targeting optimized hydrogen management via reductive reaction pathways, the one-pot transformation of methanol, CO2 and H2 to DMM catalyzed by a homogeneous ruthenium or cobalt complex and a Lewis acid co-catalyst in a liquid-phase batch reaction at 80 bar pressure has been demonstrated.21–23 Mechanistically, CO2 is reduced to formic acid yielding methyl formate (MF) in the presence of methanol followed by reduction to the methoxymethanol hemiacetal and fast acetalization to DMM. For supported Cu and Ru nanocatalysts, researchers reported the co-production of DMM in H2 production from methanol in the liquid phase.24,25 Despite these advances, a continuous gas-phase process via a non-oxidative synthesis route coupling methanol dehydrogenation and acetalization to yield DMM, H2 and water appears to be an intriguing alternative for technical implementation (Scheme 1c). While this pathway has, to the best of our knowledge, not been intensively studied yet, the released H2 can be potentially used for the upstream hydrogenation of CO2 to methanol optimizing the important hydrogen management. Besides, the elimination of O2 also improves process safety for a wide range of methanol concentration.
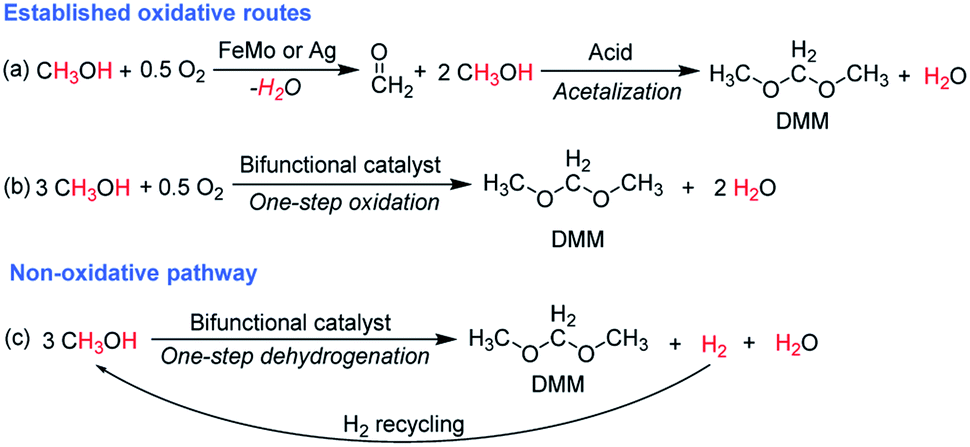 | ||
Scheme 1 Catalytic synthesis of DMM via different routes.16 (a) Industrial route for DMM production either by methanol partial oxidation according to the Johnson Matthey FORMOX Process (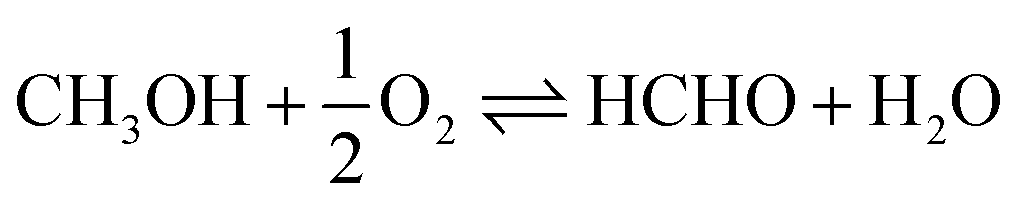 ; ΔrH° = −159 kJ mol−1) or methanol dehydrogenation coupled with hydrogen oxidation to water referred to as BASF process (CH3OH ⇌ HCHO + H2; ΔrH° = +92.4 kJ mol−1 coupled with ; ΔrH° = −159 kJ mol−1) or methanol dehydrogenation coupled with hydrogen oxidation to water referred to as BASF process (CH3OH ⇌ HCHO + H2; ΔrH° = +92.4 kJ mol−1 coupled with 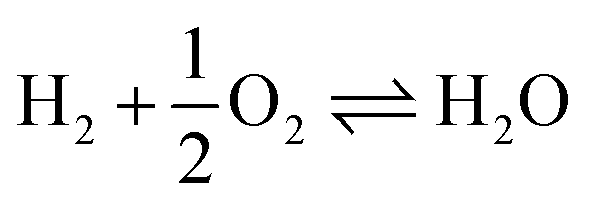 ; ΔrH° = −238.5 kJ mol−1); (b) one-pot oxidation of methanol to DMM; (c) new route via coupling methanol dehydrogenation and acetalization in one step. ; ΔrH° = −238.5 kJ mol−1); (b) one-pot oxidation of methanol to DMM; (c) new route via coupling methanol dehydrogenation and acetalization in one step. | ||
The major challenge arises with the required high reaction temperatures to overcome the thermodynamic limitation of methanol dehydrogenation in the gas-phase and at the same time suppress side reactions. Catalytic functionalities required for DMM formation include metal sites to catalyze dehydrogenation and acid sites to allow acetalization. However, these catalytic functions also facilitate undesirable methanol dehydration to dimethyl ether (DME), methanol dehydrogenation to MF as well as the potential formation of hydrocarbons from methanol. Accordingly, a highly selective catalyst is indispensable, capable of tailoring the complicated reaction network toward the formation of DMM and of minimizing the undesired side product formation, especially towards MF and DME, respectively.
Here, we present the non-oxidative synthesis of DMM in a continuous gas-phase fixed-bed reaction by integrating methanol dehydrogenation to FA with the subsequent acetalization reaction in one reactor. We demonstrate the successful design of a bifunctional catalyst, composed of Cu nanoparticles dispersed on Hβ zeolite, enabling the selective production of DMM (Scheme 1c). A maximum DMM selectivity of 89.2 or 80.3% is reached for 0.4 and 0.7 wt% Cu on Hβ(836) zeolite with 1.9 or 3.6% methanol conversion, respectively. Maximizing DMM yield, 0.7 wt% Cu/Hβ with a high SiO2/Al2O3 ratio (836) facilitates a high DMM selectivity of 77.2% at a single-pass methanol conversion of 4.5% at 220 °C under steady-state operation. Product distribution is determined by the synergistic interplay of Cu and zeolite Hβ, which can be regulated by tuning SiO2/Al2O3 ratio, Cu loading, zeolite type, and the proximity of these two active sites. Interestingly, 0.7 wt% Cu/Hβ shows an induction period featuring a remarkable rise in DMM selectivity from 5.8 to 77.2% with time on stream. The reaction mechanism and the underlying reasons for excellent DMM selectivity are explored by spectroscopic and microscopic characterizations. Process assessment emphasizes the high potential of the presented transformation route with superior exergy efficiency compared to state-of-the-art technologies.
Results and discussion
Thermodynamic considerations
At low temperatures (≤250 °C), methanol dehydrogenation to formaldehyde usually occurs with negligible methanol conversion due to thermodynamic equilibrium limitations, resulting from the highly positive reaction enthalpy and Gibbs reaction energy (Table 1, reaction (1)). Acetalization of FA and methanol is thermodynamically feasible at such low temperatures (Table 1, reaction (2)). Coupling methanol dehydrogenation with acetalization can drive the dehydrogenation of methanol to FA and thermodynamically allow the formation of DMM, as indicated by the notably decreased enthalpy and Gibbs energy (Table 1, reaction (3)). Moreover, our thermodynamic calculations of methanol dehydrogenation to DMM show that with the reaction temperature increasing from 100 to 600 °C, the equilibrium conversion of methanol increases monotonically from 5.5 to 15.6% (ESI Fig. S1†). However, DMM formation via reaction (3) is only possible with a highly selective catalyst that suppresses reactions (4) and (6) very effectively. In fact, if both the desired reaction (3) and the undesired reactions (4) and (6) reach thermodynamic equilibrium, formation of DMM is negligible irrespective of temperature (Fig. 1, top). Only a highly selective catalyst allows effective DMM formation (Fig. 1, bottom).| Reaction | ΔrHθ (298 K, g) (kJ mol−1) | ΔrGθ (298 K, g) (kJ mol−1) |
|---|---|---|
(1) CH3OH(g) ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CH2O(g) + H2(g) CH2O(g) + H2(g) |
+92.4 | +59.7 |
| (2) CH2O(g) + 2CH3OH(g) DMM(g) + H2O(g) |
−79.4 | −27.8 |
| (3) 3CH3OH(g) DMM(g) + H2O(g) + H2(g) |
+12.8 | +31.9 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Side reactions | ||
| (4) 2CH3OH(g) DME(g) + H2O(g) |
−24.1 | −16.8 |
| (5) CH3OH(g) CO(g) + 2H2(g) |
+90.5 | +25.2 |
| (6) 2CH3OH(g) MF(g) + 2H2(g) |
+46.5 | +26.5 |
| (7) CO2(g) + H2(g) CO(g) + H2O(g) |
+41.2 | +67.6 |
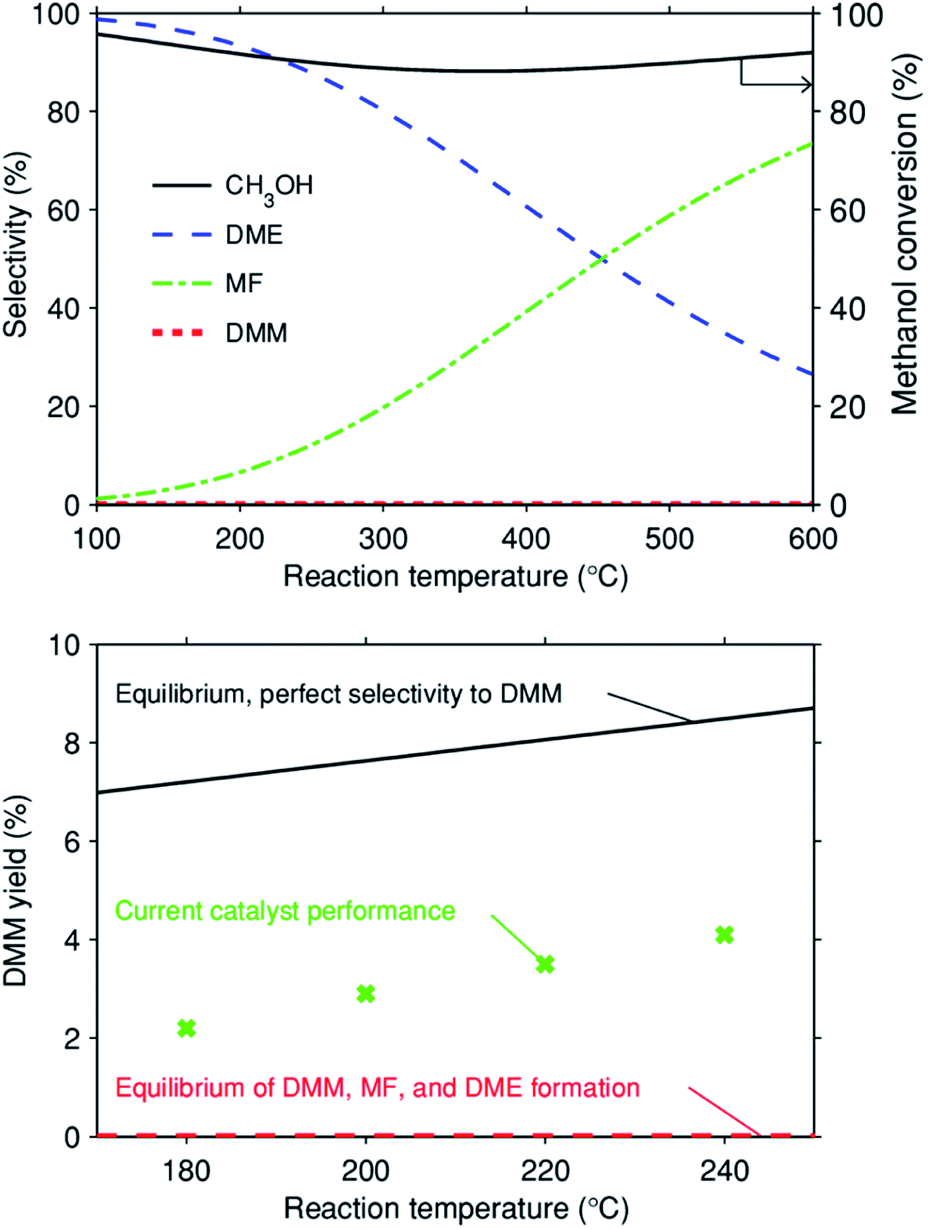 | ||
| Fig. 1 (Top) Equilibrium methanol conversion and selectivity versus reaction temperature assuming three gas-phase reactions: 3CH3OH DMM + H2O + H2, 2CH3OH DME + H2O, and 2CH3OH MF + 2H2 (representing a catalyst that does not suppress any of these three reactions such that all go to equilibrium). Equilibrium was calculated in AspenPlus with the REquil reactor model assuming an ideal gas phase under atmospheric pressure and using the DIPPR-107 heat capacity model4 and pure species properties from the AspenPlus Property Databank APV88 PURE32; (bottom) DMM yield versus reaction temperature for three different scenarios: Equilibrium of the desired reaction 3CH3OH = DMM + H2O + H2 only (cf. ESI Fig. S1†); simultaneous equilibrium of DMM, MF, and DME formation and the experimental results for the 0.7 wt% Cu/Hβ(836) catalyst at GHSV = 14549 mL h−1 gcat.−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 19.4 mL min−1. The reported yield equals the methanol conversion times selectivity to DMM. | ||
Catalyst design
To integrate methanol dehydrogenation and the acetalization of FA with methanol in one reactor, a catalyst possessing active sites for both reaction steps is required. Although a number of transition metal catalysts have been applied for methanol dehydrogenation,26 Cu as the dehydrogenation component appears advantageous considering its low cost, weak adsorption of FA,27,28 and excellent activity in low-temperature methanol dehydrogenation to MF.29,30 The further dehydrogenation of FA intermediate to CO and H2, usually occurring on precious metals, can be suppressed, as a result of the weak adsorption of FA on Cu, thus facilitating subsequent acetalization of FA with methanol. It has been widely demonstrated that zeolites are able to catalyze the acetalization of FA with methanol efficiently.16 In addition, the tuned acidic properties, commercial availability, and porous framework render zeolites the preferred candidate as the acidic component. We therefore envision that the rational combination of Cu with a zeolite into a bifunctional catalyst enables the selective production of DMM via methanol dehydrogenation. Copper was supported on different zeolites by incipient wetness impregnation to assess the role of zeolite structure and module. Three types of zeolites were selected including Hβ (BEA topology), HY (FAU topology), and HZSM-5 (MFI topology), all of which possess three dimensional interconnected channels with 10- or 12-membered ring pores, facilitating the diffusion of reactants/products and the dispersion of Cu species. The catalysts are referred to as xCu/zeolite(y), where x and y denote the measured Cu weight loading and SiO2/Al2O3 ratio (ESI Tables S1 and S2†), respectively.Fig. 2 illustrates the catalytic performances of such bifunctional Cu catalysts with a Cu loading of 7 wt%. All catalysts enable a methanol conversion of around 3%, whereas the type of zeolite significantly influences product selectivity. This effect is very distinct for HZSM-5(358) and Hβ(397), zeolites with similar SiO2/Al2O3 ratio. In the presence of HZSM-5(358), DME presents the major product (90.4% selectivity) with only 8.4% selectivity toward DMM. In contrast, DMM selectivity reaches 67.7% for Cu supported on Hβ(397) and HY(110). Shape-selectivity appears unlikely considering the small kinetic diameters of both DMM and DME.31 Instead, different acidity of these zeolites is inferred to be the origin of selectivity changes. Indeed, with the SiO2/Al2O3 ratio rising from 165 to 836 for Cu/Hβ, DMM selectivity markedly increased from 40.8 to 74.7% with DME selectivity sharply decreasing from 53.6 to 1.7% (Fig. 2). A low Al content appears to facilitate the formation of DMM by suppressing undesired methanol dehydration to DME. Interestingly, MF was produced as the major product without the formation of DME and DMM over silicalite-1, a pure silica zeolite possessing MFI structure as ZSM-5 (ESI Fig. S2†). Compared with the product distribution over ZSM-5(358), we deduce that the presence of Al is indispensable for the formation of DMM at least for MFI type zeolites, and only a small amount of Al is sufficient to reach high DMM selectivity.
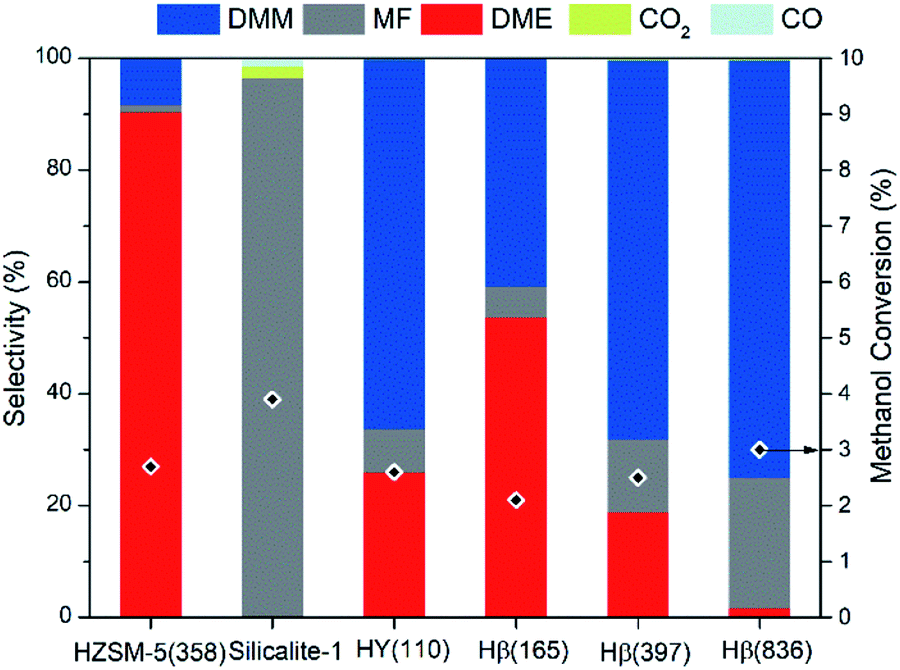 | ||
| Fig. 2 Zeolite screening for Cu/zeolite catalyzed methanol dehydrogenation and acetalization to DMM. Reaction conditions: 1 atm, GHSV = 14549 mL h−1 gcat.−1, 0.1 g of Cu/zeolite catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 19.4 mL min−1, same Cu loading (∼7 wt%) for all catalysts, all zeolites were examined at 180 °C except for HY(110) and silicalite-1 at 200 °C. | ||
To understand the strong dependence of DMM selectivity on SiO2/Al2O3 ratio and zeolite type, temperature programmed ammonia desorption (NH3-TPD) was applied to probe the acidic properties of the zeolite support materials. NH3-TPD profiles indicate that all zeolites, except Hβ(836), possess two broad desorption signals at low temperature (below 350 °C) and high temperature (above 350 °C) (ESI Fig. S3†), corresponding to weak and strong acidic sites, respectively. No obvious desorption signal was detected over silicalite 1 presumably owing to the extremely weak acidity of silanol groups, which were found to be inactive for DMM and DME synthesis, thus leading to the exclusive formation of MF over Cu/silicalite 1. The NH3-TPD curve of ZSM-5(358) exhibited a broad desorption peak centered at 531 °C, indicating the presence of very strong acidic sites. Accordingly, Cu/ZSM-5(358) led to high DME selectivity and only minor formation of DMM. Recently, Grünert et al. reported high DMM selectivity of weak acidic sites in gas-phase acetalization of methanol and formaldehyde.32 Despite a major peak centered at 121 °C corresponding to weak acidic sites and an overall high acid site concentration of 249.5 μmol g−1, Cu/HY(110) only led to medium DMM selectivity. In contrast, Hβ(836) zeolite, showing superior DMM selectivity when coupling with Cu, possesses only weak acid sites with a concentration as low as 36.5 μmol g−1. DMM selectivity seems to depend significantly on zeolite acidity with optimum selectivity achieved at a low concentration of weak acidic sites. It is noteworthy that decreasing acid strength and number of acid sites affect methanol dehydration to DME more significantly than DMM synthesis, in line with the conclusion drawn in the direct oxidation of methanol to DMM.33
Cu loading
With Hβ(836) as support material possessing ideal acidity, we turn to optimize the dehydrogenation function of Cu aiming at further improving DMM selectivity. Hence, a series of Cu/Hβ(836) catalysts with varying Cu loadings were synthesized and examined (Fig. 3). As the Cu loading increased from 0.4 to 7.4 wt%, DMM selectivity decreased monotonically from a maximum of 89.2 to 71%, whereas methanol conversion first underwent a steep increase from 1.9 to 3.6% and then slightly increased further to 3.9%. This suggests that methanol conversion is more likely determined by the dehydrogenation component Cu rather than the acidic component, which showed negligible effects on methanol conversion as mentioned above. Rising DMM selectivity was accompanied by a pronounced decrease in MF selectivity, an indicator of dehydrogenation ability. This reveals that a moderate dehydrogenation ability realized by lowering the Cu loading matched well with the weak acidity of Hβ(836) enabling high DMM selectivity. An optimum Cu loading of 0.7 wt% was chosen since it can facilitate high DMM selectivity (80.3%) without compromising methanol conversion. The aggregation of Cu species became more significant to form large Cu clusters with high Cu loading, as evidenced by high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) (ESI Fig. S4†). At low Cu loading, both 0.4 wt% and 0.7 wt% Cu/Hβ(836) catalysts present a homogeneous distribution of Cu nanoparticles with diameters below 10 nm, in contrast to the coexistence of small and large Cu clusters up to hundreds of nanometers in 3.7 wt% and 7.4 wt% Cu/Hβ (836).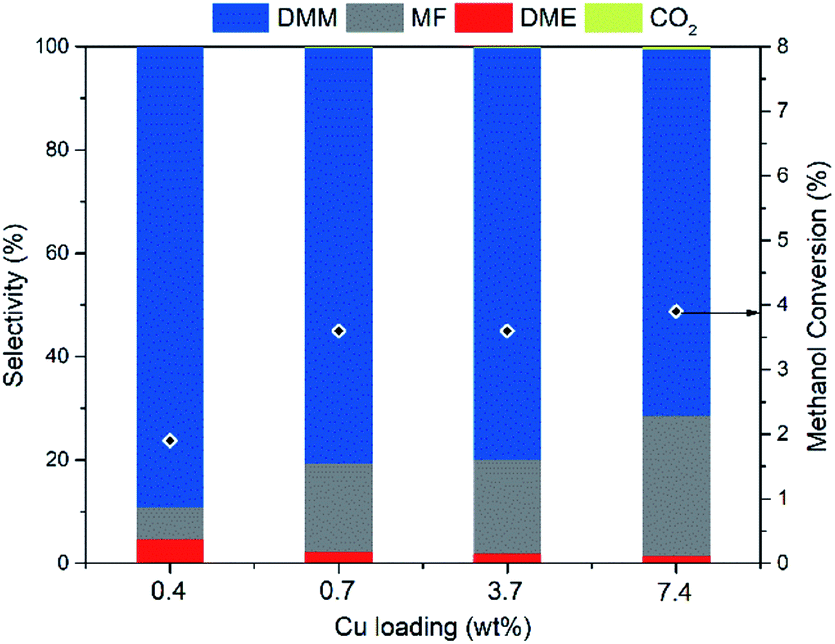 | ||
| Fig. 3 Impact of Cu loading on methanol dehydrogenation to DMM in the presence of Cu/Hβ(836). Reaction conditions: 200 °C, 1 atm, GHSV = 14549 mL h−1 gcat.−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 19.4 mL min−1. | ||
Reaction conditions
The influence of reaction parameters including reaction temperature and gas hourly space velocity (GHSV) was investigated for the optimal catalyst 0.7 wt% Cu/Hβ(836). Given the unfavorable thermodynamics of acetalization as well as the enhanced formation of side products like hydrocarbons, MF, and CO at high temperatures, low reaction temperatures (≤240 °C) were applied in order to achieve high DMM selectivity. Fig. 4 (top) illustrates increasing methanol conversion up to 6.1% with the reaction temperature rising from 180 to 240 °C. In contrast, the product distribution remained almost unchanged with a DMM selectivity of around 80% for a reaction temperature below 220 °C, but further increasing the reaction temperature resulted in a drop of DMM selectivity from 77.2 to 66.2% because of the increased formation of FA, an important intermediate in DMM synthesis. This may be related to the mismatch reaction kinetics, i.e., high reaction temperatures accelerate the dehydrogenation of methanol to FA which cannot be consumed by the following acetalization to yield DMM. The achieved DMM yield in this temperature range is between 30–50% of the equilibrium yield with a hypothetical catalyst with perfect selectivity to DMM (cf.Fig. 1, bottom). At the same time, the achieved yield is by a factor of 80 to 500 larger than the equilibrium DMM yield if MF and DME formation go to equilibrium as well.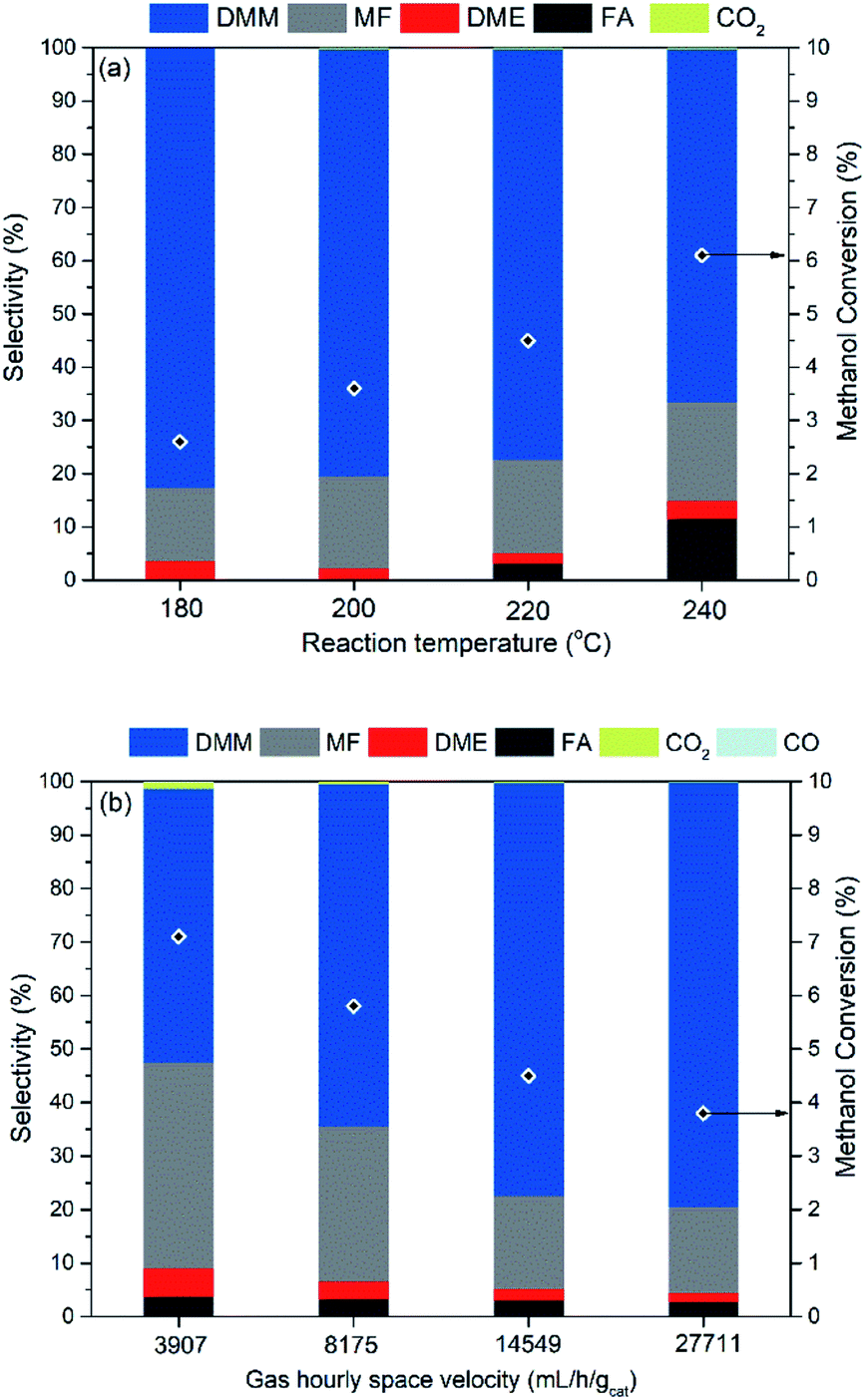 | ||
| Fig. 4 Catalytic performances of 0.7 wt% Cu/Hβ(836) in methanol dehydrogenation to DMM. (Top) Effect of reaction temperature. Reaction conditions: 1 atm, GHSV = 14549 mL h−1 gcat.−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 19.4 mL min; (bottom) effect of gas hourly space velocity. Reaction conditions: 220 °C, 1 atm, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v). | ||
As the space velocity increased from 3907 to 14549 mL h−1 gcat.−1, DMM selectivity significantly improved from 51.2 to 77.2% with a linear decrease in both methanol conversion and MF selectivity (Fig. 4, bottom). Further increasing the space velocity to 27711 mL h−1 gcat.−1 did not change product distribution but led to a slight decrease in methanol conversion. Low space velocity favors the production of undesired products mainly MF, and also CO2 and CO (1.3% selectivity), probably produced from methanol dehydrogenation (reaction (6) in Table 1), steam reforming (CH3OH + H2O CO2 + 3H2) and reverse water–gas shift reaction (reaction (7) in Table 1), respectively. One can conclude that a short contact time between catalyst and methanol is beneficial to obtain high DMM selectivity. Hence, the optimal reaction conditions were fixed at a space velocity of 14549 mL h−1 gcat.−1 and a mild reaction temperature of 220 °C, under which DMM selectivity reached 77.2% at 4.5% methanol conversion, corresponding to a space time yield of up to 14.5 gDMM gCu−1 h−1.
As a result of relatively low methanol conversion, the optimized reaction performance is not comparable to the established two-step route (a) and one-pot route (b) in Scheme 1. By employing a coupled reaction-distillation process, full methanol conversion with high DMM selectivity (99%) can be achieved for route (a).16 A DMM selectivity of 99% at a methanol conversion of 51% represents the state-of-the-art performance for the one-pot route (b) using a VTiSi catalyst.34 However, in terms of DMM formation rate, i.e., the produced DMM normalized by the overall active metal atoms (e.g., Cu and V) on catalysts, the dehydrogenation route over 0.7 wt% Cu/Hβ shows nearly four times higher activity compared to VTiSi-catalyzed one-pot route (b) (12.1 vs. 3.3 h−1).16
Effect of proximity of catalytic functionalities
To understand and clarify the bifunctional nature of Cu/Hβ(836) in methanol dehydrogenation to DMM, we studied the influence of proximity between Cu and Hβ(836) on product distribution by different spatial distributions of these functionalities within the fixed bed (Fig. 5). For comparison, these two components were also examined separately for methanol dehydrogenation. With Hβ(836) as the sole catalyst, DME forms as major product with DMM selectivity as low as 3.8%, agreeing with the acidic nature of Hβ(836) (Fig. 5b). Considering the absence of acidic sites on SiO2, SiO2-supported Cu (2.4 wt% Cu/SiO2) was used to study the catalytic performance of the Cu component. Cu/SiO2 exhibited a very high MF selectivity of up to 94.1% at 37.3% methanol conversion without the formation of DMM and DME (Fig. 5a), consistent with the typical catalytic performance of Cu-based catalysts in methanol dehydrogenation.30 The prevailing formation of MF is related to the exclusive presence of dehydrogenation functions on Cu/SiO2. Similarly, the application of a dual-bed configuration with Hβ(836) placed below Cu/SiO2 separated by a layer of quartz wool also provided a high MF selectivity and no DMM was detected (Fig. 5c). Furthermore, we combined Cu/SiO2 and Hβ(836) by mechanical mixing and grinding for high spatial proximity of these two components (Fig. 5d and e), both resulting in improved DMM selectivity of around 28%. An even closer proximity was realized by dispersing Cu nanoparticles on Hβ(836), typically used in this contribution, which remarkably boosted DMM selectivity up to 71% (Fig. 5f).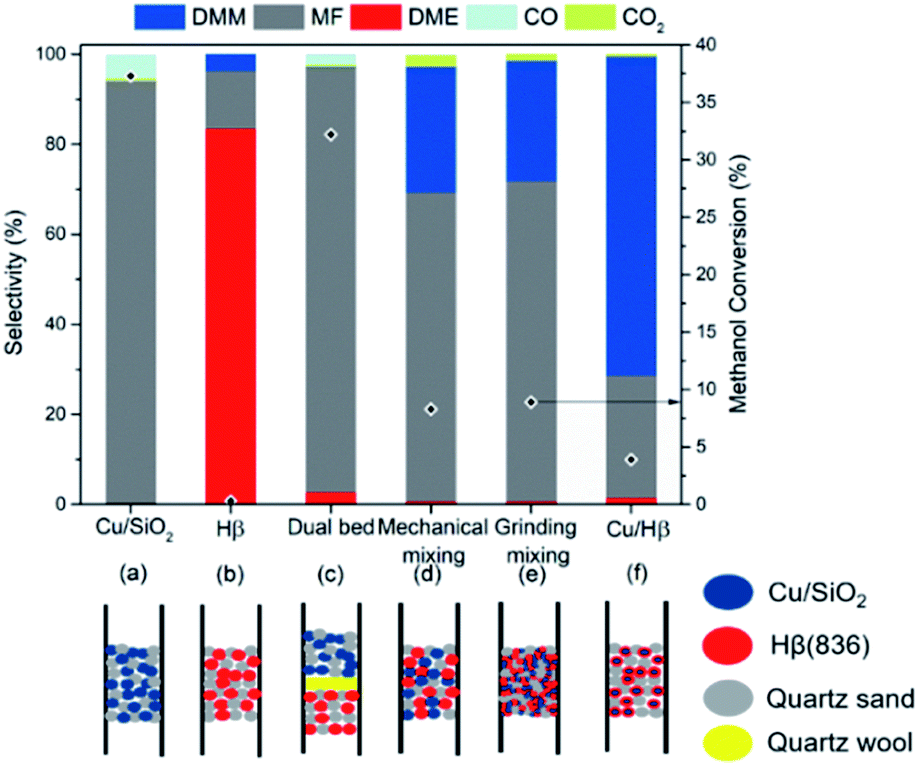 | ||
| Fig. 5 Effect of proximity between the dehydrogenation component Cu and the acidic component Hβ(836) on catalytic behaviors. (a) SiO2-supported Cu catalyst (2.6 wt% Cu, 0.1 g). (b) Hβ(836) (0.1 g). (c) Dual-bed configuration with Hβ(836) (0.05 g) packed below 2.6 wt% Cu/SiO2 (0.05 g) separated by a layer of quartz wool. (d) Mechanical mixing of 2.6 wt% Cu/SiO2 (0.05 g) with Hβ(836) (0.05 g). (e) 2.6 wt% Cu/SiO2 (0.05 g) and Hβ(836) (0.05 g) were mixed by hand grinding in an agate mortar for 10 min. (f) 7.4 wt% Cu/Hβ(836) (0.1 g). Reaction conditions: 200 °C, 1 atm, GHSV = 14549 mL h−1 gcat.−1, 0.9 g of SiC applied for each integration manner, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 19.4 mL min−1. | ||
These results clearly prove that the successful coupling of methanol dehydrogenation to FA with the following acetalization with an excellent DMM selectivity requires a tailored proximity and interaction of Cu and Hβ(836). According to the reported proximity effects in syngas and CO2 transformation,35,36 we speculated that such a tight-contact configuration is advantageous for the quick diffusion of reactive intermediates such as FA and hemiacetal, to the adjacent acidic sites for acetalization, kinetically accelerating the overall reaction to selectively yield DMM.
Time dependent evolution of DMM selectivity
The catalytic stability of Cu/Hβ(836) was investigated by following the evolution of methanol conversion and product distribution with time on stream (Fig. 6). Surprisingly, we observed a dramatic increase in DMM selectivity from 5.8 to 77.2% within the first 13 h on stream accompanied by a sharp decline in MF and DME selectivities. In addition, Cu/Hβ(836) rapidly deactivated indicated by the fast decrease of methanol conversion within the first 8 h on stream. After 20 h on stream at 220 °C, steady-state performance was reached with methanol conversion leveling off at around 4.5% and unchanged product distribution dominated by DMM. Similar evolution trends were also observed for other Hβ-supported Cu catalysts (ESI Fig. S5†). The significantly enhanced DMM selectivity at the expense of MF and DME production suggests the dynamic evolution of catalytically active sites toward weakened dehydrogenation activity and acidity with time on stream. Both are favorable for high DMM selectivity, consistent with the above findings in zeolite screening and variation of Cu loading.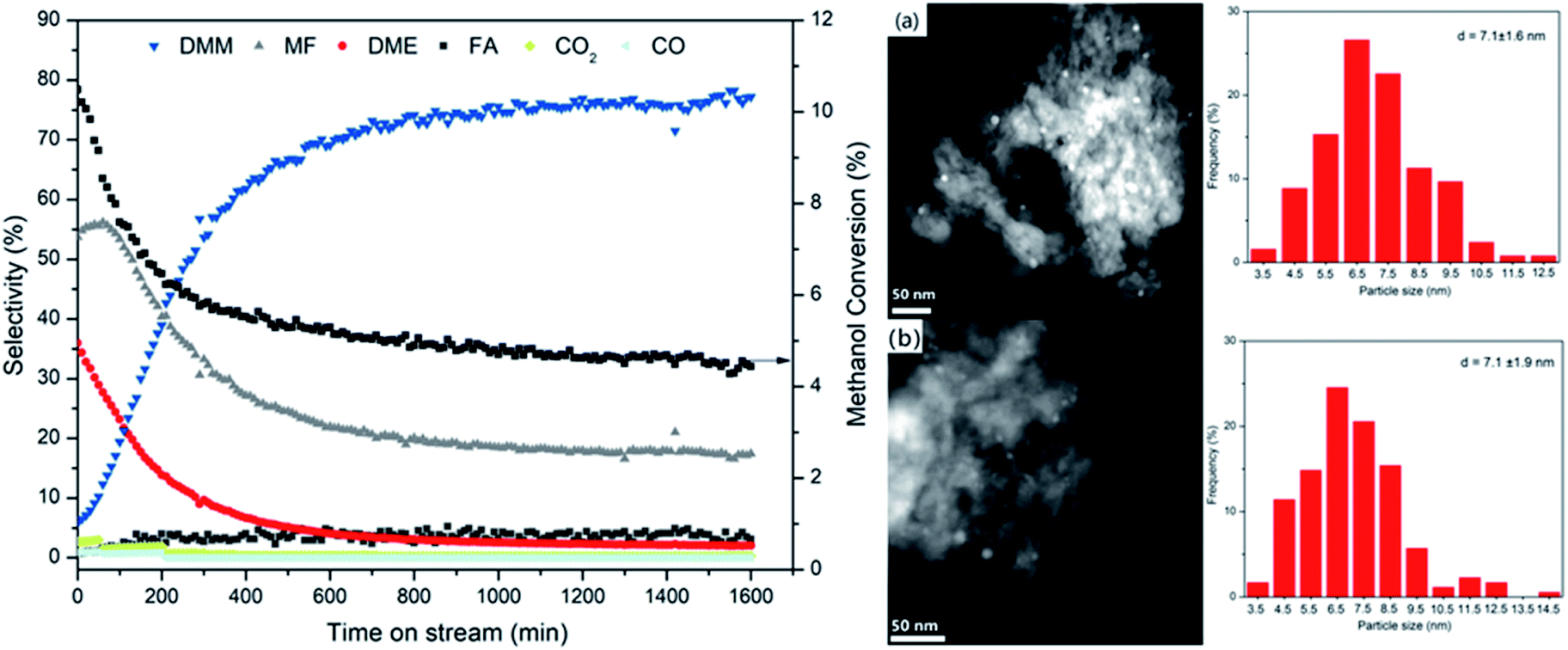 | ||
| Fig. 6 (Left) Stability test for methanol dehydrogenation to DMM in the presence of 0.7 wt% Cu/Hβ(836). Reaction conditions: 220 °C, 1 atm, GHSV = 14549 mL h−1 gcat.−1, 0.1 g of 0.7 wt% Cu/Hβ diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 19.4 mL min−1. (Right) Structural characterization by HAADF-STEM of reduced (a) and spent (b) 0.7 wt% Cu/Hβ(836) catalysts and the corresponding particle size distribution by counting more than 120 particles (the lengths of both scale bars = 50 nm). | ||
Characterization of catalytic function
To unravel the underlying reason for the weakened dehydrogenation ability and acidity with time on stream, a reaction over 0.7 wt% Cu/Hβ(836) without SiC dilution was carried out for 1600 min under otherwise identical conditions. The spent catalyst was characterized by a number of techniques including N2-physisorption, N2O chemisorption, thermogravimetry connected with mass spectrometry (TG-MS), NH3-TPD, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), HADDF-STEM, solid-state magic angle spinning nuclear magnetic resonance (SS MAS NMR), attenuated total reflection infrared spectroscopy (ATR-IR). As shown in Table 2 and ESI Fig. S6,† the reduced and spent catalysts as well as zeolite Hβ(836) exhibited a similar type I isotherm with a hysteresis loop indicating the coexistence of microspores and mesopores likely originating from inter-crystalline voids of zeolite Hβ(836). The introduction of Cu into Hβ(836) led to a decrease in both specific surface area and micropore volume potentially due to partial blockage of the micropore system by Cu nanoparticles. The spent catalyst possessed an even smaller micropore volume than the reduced catalyst (Table 2), indicating partial blockage of the micropore system after reaction likely caused by the aggregation of Cu particles. N2O chemisorption showed a decreased Cu surface atom concentration on the spent catalyst suggesting Cu particle agglomeration. Nevertheless, the same average Cu particle size of around 7.1 nm was observed on both fresh and spent catalysts without significant aggregation observed by HAADF-STEM (Fig. 6 right, a and b). Hence it is reasonable to deduce the presence of ultrasmall Cu clusters inside zeolite channels apart from the visible Cu nanoparticles on the external surface. It was reported that these unstable small Cu clusters tend to aggregate to form big particles following an Ostwald ripening mechanism.37 Nevertheless, the confinement effect of zeolite channels only allows moderate aggregation, which might explain the slight decrease in Cu surface atom concentration and micropore volume. X-ray diffraction patterns (ESI Fig. S7†) confirmed an identical crystal structure of the spent catalyst with regard to diffraction intensity and position as the reduced catalyst, corresponding to the typical BEA topology. Compared to intense reflexes of metallic Cu in the reduced catalyst with high Cu loading (7.4 wt% Cu/Hβ(836)), the absence of metallic Cu reflexes on the fresh and spent catalysts along with N2O chemisorption and STEM confirmed well dispersed Cu species without significant aggregation after long-term reaction. SS MAS 29Si NMR spectra of the reduced and spent catalysts displayed the same resonances with two intense signals (110.8 and 114.1 ppm) corresponding to framework tetrahedral Si (Q4, Si(OSi)4) and a weak shoulder (103.2 ppm) assigned to Q4 Si species (AlSi(OSi)3 or OHSi(OSi)3) (ESI Fig. S8†).38 These findings imply a preserved framework structure of the spent Cu/Hβ(836) catalyst, and moderate agglomeration of Cu particle. The latter is suggested to cause catalyst deactivation and reduce the dehydrogenation function.| Sample | S BET (m2 g−1) | Micropore volumeb (ml g−1) | Cu surface area based on N2Oc (μmol gcat.−1) | Cu particle size based on STEM (nm) |
|---|---|---|---|---|
| a Specific surface area determined by N2 adsorption using BET model, p/p0 = 0.05–0.2. b Determined by t-plot method. c Determined by the consumption of N2O assuming a N2O/Cu stoichiometric ratio of 1/2. | ||||
| Hβ (836) | 584 | 0.19 | — | — |
| Reduced 0.7 wt% Cu/Hβ (836) | 462 | 0.16 | 36 | 7.1 ± 1.6 |
| Spent 0.7 wt% Cu/Hβ (836) | 488 | 0.13 | 29 | 7.1 ± 1.9 |
Carbon-containing organic species accumulating on the spent catalyst investigated by TG-MS and ATR-IR present another possible origin of deactivation and selectivity change. However, only a slight mass loss (around 2.5 wt%) at low temperature appeared, attributed to water desorption (ESI Fig. S9†). The ATR-IR spectrum confirmed bands characteristic of the framework vibrations of Hβ(836) but no additional bands corresponding to C–H vibrations of carbon-containing species (ESI Fig. S10†). Hence, carbon-containing organic molecules appear not to contribute to catalyst deactivation and selectivity changes.
The oxidation states of Cu in the reduced and spent catalyst were studied by XPS. To avoid contacting with air, all samples were prepared, transferred and operated under inert atmosphere. For comparison, the calcined catalyst was also measured, displaying two peaks with binding energies of 933.8 (Cu 2p3/2) and 953.6 eV (Cu 2p1/2), assigned to Cu2+ species (ESI Fig. S11†). For the reduced and spent catalysts, the Cu 2p3/2 and Cu 2p1/2 peaks shifted to lower binding energies at around 932.5 and 952.6 eV, respectively, ascribed to Cu0 or Cu+ species, indicating the absence of Cu2+ on the reduced and spent catalysts. By Cu 2p XPS, conclusive assignment regarding the presence of Cu0 and Cu+ species are difficult, since both hold the same binding energy. LMM Auger spectra are widely used to differentiate Cu0 and Cu+ species. Unfortunately, Cu LMM Auger peaks of Cu/Hβ(836) did not possess sufficient intensity due to the low Cu loading (0.7 wt%). Yang et al. proved metallic Cu rather than Cu+ species to be the catalytically active centers in Cu-based catalysts for methanol dehydrogenation to FA and MF.30 Accordingly, we suggest the presence of Cu0 species on reduced Cu/Hβ(836). As experimental evidences of the Cu oxidation states evolving during reaction, the catalytic performances of the spent catalyst regenerated under N2 and H2 were compared. After regeneration of the spent catalyst with N2, we observed a strong decline of methanol conversion (2.2%) together with an initially high DME selectivity of 65.6% with a very low MF selectivity (5.3%) (Fig. 7 left). In contrast, methanol conversion remained at around 5.8% with a MF selectivity of 20.6% and a DMM selectivity of about 30% over the spent catalyst regenerated under H2 (Fig. 7 right). In both cases, DMM selectivity evolved again accompanied by a decline in methanol conversion as well as DME, and for H2 regeneration also MF selectivity. Both the higher initial MF selectivity and methanol conversion after H2 regeneration compared with N2 activation emphasize partial recovery of the dehydrogenation function. This is highly likely due to reduction of oxidized Cu species on the spent catalysts to form Cu0 species, active for methanol dehydrogenation as mentioned above. Taking together the catalyst regeneration results, XPS analysis and literature reports,39 we propose that Cu0 and Cu+ species coexist on the reduced and spent catalysts, and the possible evolution of Cu0 to Cu+ species during reaction account for catalyst deactivation and the weakened dehydrogenation function. Considering the appreciable amount of water formed both by DMM and also DME formation, an influence on catalyst properties is likely to occur,40 including potentially hydration/oxidation of Cu species and degradation of Al species in a steam environment.41,42
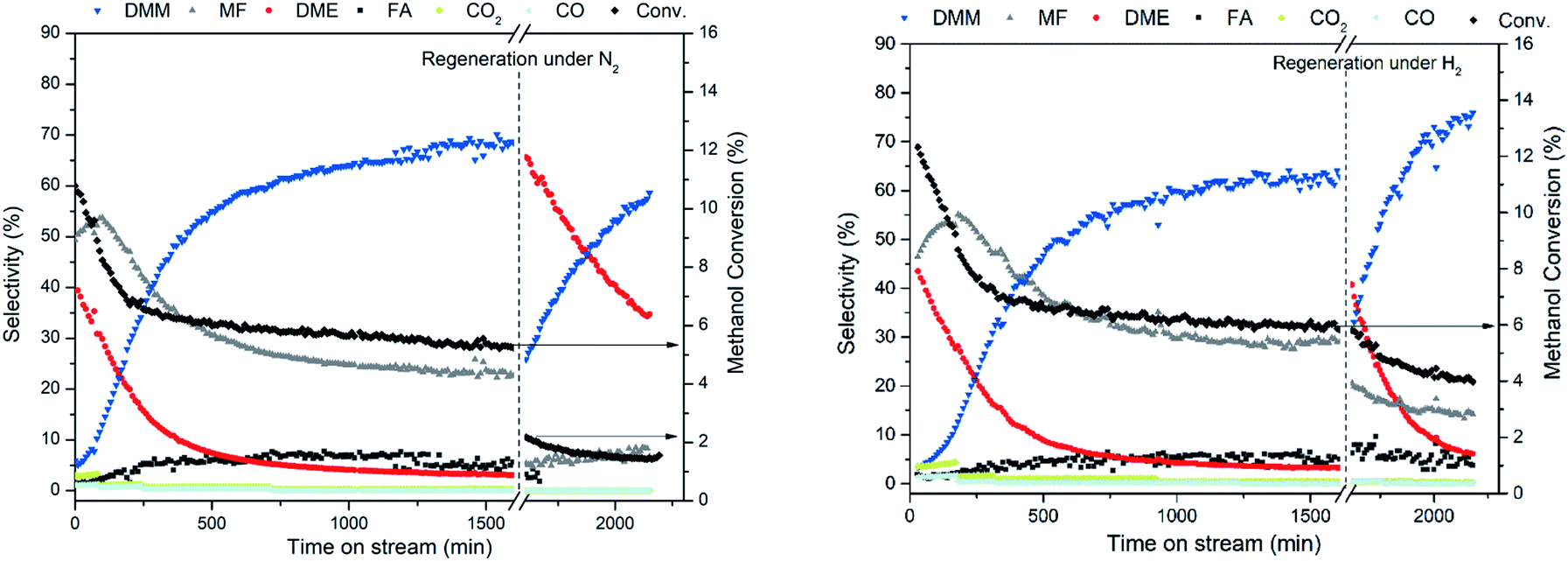 | ||
| Fig. 7 The stability test for methanol dehydrogenation to DMM in the presence of 0.7 wt% Cu/Hβ(836) (left) regenerated under N2 flow at 450 °C for 2 h; (right) regenerated under H2 flow at 450 °C for 2 h. Reaction conditions: 220 °C, 1 atm, GHSV = 8175 mL h−1 gcat.−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24 (v/v), N2 flow rate = 11.2 mL min−1. | ||
Reaction pathway
To gain mechanistic insights regarding reaction intermediates, in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) was applied to detect the surface species formed on Cu/Hβ(836). Upon exposing the reduced catalyst to a methanol-saturated N2 gas stream, two sets of bands were observed at around 1000 and 2900 cm−1, respectively (ESI Fig. S12†). Bands at 1008, 1032, 1056, 2843, and 2981 cm−1 are all assigned to gaseous methanol molecules,30 evidenced by the disappearance of these signals after a N2 purge. The remaining bands at 2858, 2957, and 2922 cm−1 are assigned to methoxy groups on SiO2 and Cu sites,30,43–45 respectively, implying the occurrence of dissociative adsorption of methanol on Cu/Hβ(836). After flushing with N2 for 1 h, two new bands appeared at 1724 and 2995 cm−1, assigned to FA and methoxy groups on SiO2,30,45 respectively. Characteristic bands of MF and DMM were not observed probably due to their low concentrations, as indicated by the very low peak intensity of FA, an intermediate for the production of DMM and MF.Besides, the formation of H2 was investigated by isotope-labeling experiments. H2 was indeed co-produced during reaction supported by the H2 signal in gas chromatography (ESI Fig. S13†). We propose that H2 is released by the combination of H species from O–H bond cleavage of methanol (methanol dissociation) and C–H bond cleavage of methoxy species. Accordingly, the use of isotope-labeled methanol (CD3OH) as substrate would lead to the formation of HD. Two intense signals with m/z of 2 and 3 corresponding to H2 and HD, respectively, appeared in the MS spectra of gas products (ESI Fig. S14†), supporting our hypothesis. The presence of H2 is speculated to be caused by the hydrogen–deuterium exchange between HD and H species potentially from the OH groups of the zeolite or Cu-hydride species generated during reduction.46–48 These spectral findings and labeling experiments reveal that methanol dehydrogenation to DMM was initiated by the dissociative adsorption of methanol on Cu/Hβ(836) to form methoxy species, which are further converted to FA by C–H bond breakage, in line with the proposed elementary steps and intermediates for Cu-catalyzed methanol dehydrogenation.
Process performance
To estimate the potential of the new route from a process point of view, we developed a process for DMM production from H2 and CO2 as potentially renewable raw materials. The process (ESI Fig. S15†) consists of an intermediate methanol production step (i.e., CO2 hydrogenation49) and the subsequent DMM production step (i.e., methanol dehydrogenation). For CO2 hydrogenation, we use the kinetic model of Van-Dal & Bouallou49 for a commercial Cu/ZnO/Al2O3 catalyst. For methanol dehydrogenation, we consider two cases. In the base case, representing the process performance with the developed catalyst in its present form, we use actual experimental results (see Fig. 4 at temperature T = 200 °C). In the idealized case, we consider equilibrium methanol conversion at moderately elevated temperature of 300 °C (i.e., 10%, see ESI Fig. S1†) and a selectivity of 99% towards DMM. This represents a reasonable limit of process performance at mild reaction conditions. For each case, we calculate the exergy efficiency of the overall process as described in ESI.† Due to the early stage of development of the new route, a detailed economic evaluation and a fair comparison to state-of-the-art DMM production exceed the scope of this work. Instead, we focus on changes in H2 consumption and exergy efficiency as a surrogate for the economic and environmental performance of the proposed process compared to state-of-the-art DMM production.The raw material and energy demands per unit of DMM produced via the proposed process (ESI Fig. S15†) are listed in ESI Table S3.† The exergy efficiency of the proposed process is 76% for the base case, and thus 3 percentage points higher than the efficiency of state-of-the-art technologies for DMM production, i.e., via aqueous formaldehyde solution as an intermediate.18 Due to the relatively low conversion of methanol (i.e., 3.6%) high recycle streams are necessary, which result in a high heat demand for separation (i.e., 27 MJ kgDMM−1 at 100 °C). Considerable amounts of MF (i.e., 0.3 kg kgDMM−1) along with small amounts of DME are produced as side products. If this side product stream is not considered as a product in itself but instead burned for energy recovery, the exergy efficiency drops to 65%. This shows that either material use of the side product or further improvements in selectivity are required to exploit the full potential of the route. In the idealized case, the exergy efficiency reaches 85%, both because of the absence of side products and because of the increased methanol conversion, which causes a decrease in heat demand by more than 70%. The H2 consumption (0.223 kg kgDMM−1) is up to 15.8% lower than in state-of-the-art DMM production.18 Considering the high share of H2 cost in renewable DMM production (>70%),50 H2 recycling represents an effective measure for cost reduction. This demonstrates the large potential of methanol dehydrogenation for industrial applications despite the rather low equilibrium limit on methanol conversion (cf. ESI Fig. S1†).
Conclusions
In summary, we have described a novel non-oxidative approach for the continuous production of DMM directly from methanol in the gas phase. In this new route, methanol dehydrogenation to FA and the acetalization of FA with methanol to DMM were successfully integrated by a tailored bifunctional Cu/Hβ catalyst, where metallic Cu sites are mainly responsible for methanol dehydrogenation and Hβ zeolite can provide acidic sites for the acetalization of FA with methanol to DMM. Zeolites with a low amount of weak acidic sites as well as moderate dehydrogenation ability of Cu were demonstrated to be of crucial importance for excellent DMM selectivity. By varying zeolite type, Cu loading, and the spatial distance between Cu and Hβ zeolite, the reaction network could be tailored toward the selective production of DMM with a selectivity as high as 80% at a methanol conversion of 3.6% at 200 °C. Equilibrium calculations show that such DMM yields are only possible if the catalyst is very effective in suppressing side reactions to DME and MF. The stability test disclosed a sharp rise in DMM selectivity over Cu/Hβ with time on stream caused by the oxidation and aggregation of Cu species and the possible degradation of Al species during reaction. From a process point of view, the presented approach enables improved exergy efficiency over state-of-the-art processes for DMM production already with the current state of the catalysts.This study provides a promising route for the efficient synthesis of DMM with a potential benefit of reusing the coproduced H2. For a potential application of this route in the future, further studies are required to improve catalyst stability based on a well-understood mechanism picture of the dynamic changes of Cu and zeolites under reaction conditions using advanced characterization methods. Likewise, more efforts are needed to enhance DMM yield. In terms of thermodynamic equilibrium, further improvements of the DMM yield are possible, but only to a limited extend. Such improvements could enable exergy efficiency as high as 85% for producing DMM from H2 and CO2. To achieve even higher yields and thus potentially even higher efficiencies, the formed H2 could be removed in situ in a membrane reactor to shift the reaction equilibrium toward DMM.
Experimental
Experimental details and characterization methods are provided in the ESI.†Author contributions
The project was led by Regina Palkovits. Ruiyan Sun and Regina Palkovits conceived and designed the research. The experimental work including catalyst preparations, catalytic performance tests, GC analysis, data analysis, IR measurements, N2 physisorption, NH3-TPD, and N2O chemisorption were performed by Ruiyan Sun, assisted by Chalachew Mebrahtu. XPS was conducted and analyzed by Jan Philipp Hofmann. The flow reactor system was designed and built up by Ruiyan Sun and Christian Gierlich. Solid State NMR was carried out and analysed by Peter Hausoul. Process simulation and thermodynamic calculations were supervised by Alexander Mitsos and performed by Dominik Bongartz and Jannik Burre. Ruiyan Sun and Regina Palkovits wrote the manuscript, and all authors contributed to the discussions and revisions of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding of the German Federal Ministry of Education and Research (BMBF) within the Kopernikus Project P2X: Flexible use of renewable resources exploration, validation and implementation (FKZ 03SFK2A) and the project NaMoSyn (Nachhaltige Mobilität durch synthetische Kraftstoffe, FKZ 03SF0566P0).Notes and references
- G. T. Kalghatgi, Int. J. Eng. Res., 2014, 15, 383–398 CrossRef CAS.
- P. Gadonneix, A. Sambo, L. Tie'nan, A. R. Choudhury, J. Teyssen, J. A. V. Lleras, A. A. Naqi, K. Meyers, H. C. Shin and M.-J. Nadeau, Global Transport Scenarios 2050, World Energy Council, 2011 Search PubMed.
- Energy Information Administration (US), and Government Publications Office, International Energy Outlook 2016, with Projections to 2040,Government Printing Office, 2016 Search PubMed.
- World Health Organization, International Agency for Research on Cancer Press Release 213, Diesel Engine Exhaust Carcinogenic, http://press.Iarc.fr/pr213_E.pdf, 2012 Search PubMed.
- T. V. Johnson, Int. J. Eng. Res., 2009, 10, 275–285 CrossRef CAS.
- J. Simböck, M. Ghiasi, S. Schönebaum, U. Simon, F. M. F. de Groot and R. Palkovits, Nat. Commun., 2020, 11, 652 CrossRef.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- J. Burger, M. Siegert, E. Ströfer and H. Hasse, Fuel, 2010, 89, 3315–3319 CrossRef CAS.
- B. Lumpp, D. Rothe, C. Pastötter, R. Lämmermann and E. Jacob, MTZ worldwide eMagazine, 2011, vol. 72, pp. 34–38 Search PubMed.
- L. Lautenschütz, D. Oestreich, P. Seidenspinner, U. Arnold, E. Dinjus and J. Sauer, Fuel, 2016, 173, 129–137 CrossRef.
- A. Omari, B. Heuser and S. Pischinger, Fuel, 2017, 209, 232–237 CrossRef CAS.
- S. Deutz, D. Bongartz, B. Heuser, A. Katelhon, L. Schulze Langenhorst, A. Omari, M. Walters, J. Klankermayer, W. Leitner, A. Mitsos, S. Pischinger and A. Bardow, Energy Environ. Sci., 2018, 11, 331–343 RSC.
- H. Liu, Z. Wang, J. Wang, X. He, Y. Zheng, Q. Tang and J. Wang, Energy, 2015, 88, 793–800 CrossRef CAS.
- A. Omari, B. Heuser, S. Pischinger and C. Rüdinger, Appl. Energy, 2019, 239, 1242–1249 CrossRef CAS.
- R. Sun, I. Delidovich and R. Palkovits, ACS Catal., 2019, 9, 1298–1318 CrossRef CAS.
- J. Burre, D. Bongartz and A. Mitsos, Ind. Eng. Chem. Res., 2019, 58, 5567–5578 CrossRef CAS.
- D. Bongartz, J. Burre and A. Mitsos, Ind. Eng. Chem. Res., 2019, 58, 4881–4889 CrossRef CAS.
- K.-a. Thavornprasert, M. Capron, L. Jalowiecki-Duhamel and F. Dumeignil, Catal. Sci. Technol., 2016, 6, 958–970 RSC.
- L. Lautenschütz, D. Oestreich, P. Haltenort, U. Arnold, E. Dinjus and J. Sauer, Fuel Process. Technol., 2017, 165, 27–33 CrossRef.
- M. Siebert, M. Seibicke, A. F. Siegle, S. Kräh and O. Trapp, J. Am. Chem. Soc., 2019, 141, 334–341 CrossRef CAS PubMed.
- J. Klankermayer, Angew. Chem., Int. Ed., 2017, 56, 10854–10857 CrossRef PubMed.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed.
- N. Meng, T. Yamakawa and S. Shinoda, React. Kinet. Catal. Lett., 1996, 58, 341–348 CrossRef CAS.
- W. Liubi, L. Bolong and Z. Chen, ChemCatChem, 2018, 10, 1140–1147 CrossRef.
- N. Usachev, I. Krukovskii and S. Kanaev, Pet. Chem., 2004, 44, 379–394 Search PubMed.
- N. Iwasa and N. Takezawa, Top. Catal., 2003, 22, 215–224 CrossRef CAS.
- N. Takezawa and N. Iwasa, Catal. Today, 1997, 36, 45–56 CrossRef CAS.
- D.-J. Yuan, A. M. Hengne, Y. Saih and K.-W. Huang, ACS Omega, 2019, 4, 1854–1860 CrossRef CAS PubMed.
- H. Yang, Y. Chen, X. Cui, G. Wang, Y. Cen, T. Deng, W. Yan, J. Gao, S. Zhu, U. Olsbye, J. Wang and W. Fan, Angew. Chem., Int. Ed., 2018, 57, 1836–1840 CrossRef CAS PubMed.
- W. H. Fu, X. M. Liang, H. Zhang, Y. M. Wang and M. Y. He, Chem. Commun., 2015, 51, 1449–1452 RSC.
- A. Grünert, P. Losch, C. Ochoa-Hernández, W. Schmidt and F. Schüth, Green Chem., 2018, 20, 4719–4728 RSC.
- H. Liu, N. Bayat and E. Iglesia, Angew. Chem., Int. Ed., 2003, 42, 5072–5075 CrossRef CAS PubMed.
- Z. Fan, H. Guo, K. Fang and Y. Sun, RSC Adv., 2015, 5, 24795–24802 RSC.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- R. van den Berg, T. E. Parmentier, C. F. Elkjær, C. J. Gommes, J. Sehested, S. Helveg, P. E. de Jongh and K. P. de Jong, ACS Catal., 2015, 5, 4439–4448 CrossRef CAS.
- J. Xu, Q. Wang, S. Li and F. Deng, Solid-State NMR in Zeolite Catalysis, Springer Singapore, Singapore, 2019, pp. 93–132 Search PubMed.
- K. Ma, Y. Tian, Z.-J. Zhao, Q. Cheng, T. Ding, J. Zhang, L. Zheng, Z. Jiang, T. Abe, N. Tsubaki, J. Gong and X. Li, Chem. Sci., 2019, 10, 2578–2584 RSC.
- A. W. Petrov, D. Ferri, F. Krumeich, M. Nachtegaal, J. A. van Bokhoven and O. Kröcher, Nat. Commun., 2018, 9, 2545 CrossRef PubMed.
- O. Ilinich, W. Ruettinger, X. Liu and R. Farrauto, J. Catal., 2007, 247, 112–118 CrossRef CAS.
- C. Montassier, J. M. Dumas, P. Granger and J. Barbier, Appl. Catal., A, 1995, 121, 231–244 CrossRef CAS.
- I. A. Fisher and A. T. Bell, J. Catal., 1999, 184, 357–376 CrossRef CAS.
- G. J. Millar, C. H. Rochester and K. C. Waugh, J. Chem. Soc., Faraday Trans., 1991, 87, 2795–2804 RSC.
- D. B. Clarke, D. K. Lee, M. J. Sandoval and A. T. Bell, J. Catal., 1994, 150, 81–93 CrossRef CAS.
- R. J. Mikovksy, M. Boudart and H. S. Taylor, J. Am. Chem. Soc., 1954, 76, 3814–3819 CrossRef.
- N. W. Cant, S. P. Tonner, D. L. Trimm and M. S. Wainwright, J. Catal., 1985, 91, 197–207 CrossRef CAS.
- K.-D. Jung and A. T. Bell, J. Catal., 2000, 193, 207–223 CrossRef CAS.
- É. S. Van-Dal and C. Bouallou, J. Cleaner Prod., 2013, 57, 38–45 CrossRef.
- S. Schemme, J. L. Breuer, M. Köller, S. Meschede, F. Walman, R. C. Samsun, R. Peters and D. Stolten, Int. J. Hydrogen Energy, 2020, 45, 5395–5414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst synthesis, characterization methods, catalytic test, Fig. S1–S15, and Tables S1–S4. See DOI: 10.1039/d0se01314e |
| This journal is © The Royal Society of Chemistry 2021 |