
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMediation of metal chelation in cysteine-derived tetramate systems†
Ruirui
Zhang
 a,
Miroslav
Genov
c,
Alexander
Pretsch
c,
Dagmar
Pretsch
c and
Mark G.
Moloney
*ab
a,
Miroslav
Genov
c,
Alexander
Pretsch
c,
Dagmar
Pretsch
c and
Mark G.
Moloney
*ab
aThe Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: mark.moloney@chem.ox.ac.uk
bOxford Suzhou Centre for Advanced Research, Building A, 388 Ruo Shui Road, Suzhou Industrial Park, Jiangsu 215123, P. R. China
cOxford Antibiotic Group, The Oxford Science Park, Magdalen Centre, Oxford OX4 4GA, UK
First published on 2nd December 2021
Abstract
A study of bicyclic tetramates modified with a bulky ester, which leads to steric hindrance of distal chelating atoms as a route for the alteration of metal binding ability is reported. This approach required the development of a direct method for the synthesis of different esters of cysteine from cystine, which then provided access to bicyclic tetramates by Dieckmann cyclisation. Further derivation to ketones and carboxamides by Grignard addition and transamination reactions respectively provided rapid access to a chemical library of tetramates with diverse substitution. Of interest is that bicyclic tetramate ketones and carboxamides showed different tautomeric and metal binding behaviour in solution. Significantly, in both systems, the incorporation of bulky C-5 esters at the bridging position not only reduced metal binding, but also enhanced antibacterial potencies against Gram-positive MRSA bacteria. Those tetramates with antibacterial activity which was not metal dependent showed physiochemical properties of MSA of 559–737 Å2, MW of 427–577 Da, clogP of 1.8–6.1, clogD7.4 of −1.7 to 3.7, PSA of 83–109 Å2 and relative PSA of 12–15% and were generally Lipinski rule compliant. A subset of tetramates exhibited good selectivity towards prokaryotic bacterial cells. Given that the work reported herein is synthesis-led, without the underpinning detailed mechanistic understanding of biological/biochemical mechanism, that the most active compounds occupy a small region of chemical space as defined by MW, clogP, PSA and %PSA is of interest. Overall, the bicyclic tetramate template is a promising structural motif for the development of novel antibacterial drugs, with good anti-MRSA potencies and appropriate drug-like physiochemical properties, coupled with a potential for multi-targeting mechanisms and low eukaryotic cytotoxicity.
Introduction
Tetramates1 exhibit wide ranging antibacterial activity coupled with low levels of toxicity,2–5 but are also well known for their metal chelating ability,6,7 which, it has been suggested recently, should be explicitly considered during any study of their biological effects.8 For example, metal chelates of a 3-acyltetramic acid, melophlin C, with Ga(III), La(III) and Ru(II), all of which were considered to be Fe(III) mimics, were found to be active against Micrococcus luteus (M. luteus).9 Streptolydigin, a tetramate-containing natural product, was reported to inhibit bacterial RNA polymerase (RNAP) but not eukaryotic RNA polymerases, and non-catalytic Mg(II) was found to be essential for its binding.10 However, while some tetramic acids demonstrated increased toxicity as a metal complex,11 in others, toxicity was attenuated by metal chelation.12 Naturally occurring 3-acyltetramates have been reported to chelate a wide variety of metals,6–8,13,14 including most commonly Cu(II),15–17 Mg(II), Ca(II),11 Fe(III) and Ni(II),17 but also Zn(II), Ga(III), La(III), Ru(II),9 Al(III),18 Pd(II),19 Co(II),20 Rh(I),21 Na(I), K(I),22 Cs(I),23 Cd(II),24 Pb(II),25 Ba(II),26 Hg(II),27 and Mn(II).28 X-ray photoelectron spectroscopic (XPS) analysis of some derivatives clearly identified the presence of Ca(II)29 in metal-chelated tetramates and a high abundance of Ca(II), together with Na(I), Mg(II), Fe(III) and Zn(II), was also found using inductively coupled plasma mass spectrometry (ICP-MS).30 The metal chelating properties of tetramates are most immediately evident by post-chromatographic signal broadening in 1H NMR spectra, which can only be removed by acid wash,29–32 and the metal complexation behaviour of tetramates was also found to be both structure and metal-ion dependent.33 We have described earlier that cysteine-derived bicyclic tetramates 1 are excellent metal chelating agents, and established that the C-6, C-8 and/or C-9 carbonyl groups were directly involved in that metal complexation, but that it could be blocked by protection of the enolic system;34 it seemed that modification of the C-5 ester (CO2R1, Fig. 1), along with the concave-convex shape of the bicyclic tetramate system, might be further exploited to alter the metal binding ability of the tetramate system by steric interference distal to the region of metal chelation.33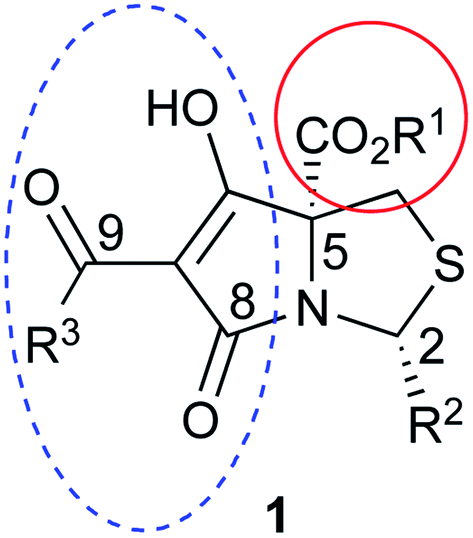 | ||
| Fig. 1 Cysteine-derived tetramates with C-5 ester functionalisation. | ||
Specifically, target compounds were designed with increasing steric bulk at C-5, with R1 varying from methyl to allyl, isopropyl, cyclohexyl, benzyl, nitro-benzyl and bromo-benzyl esters (Fig. 2). Polar pyridyl rings R2 were incorporated at C-2 to increase overall hydrophilicity. Various C-9 side chains R3 such as cyclohexyl, phenyl, phenethyl, styrenyl, and heterocyclic pyridyl and thiazolyl systems in tetramate ketones as well as cyclohexyl and adamantyl amide pendants in tetramate carboxamides were examined (Fig. 2). The cyclohexyl ketone pendant is a simplified mimic of the trans-decalin system found in many tetramate-containing natural products, such as kibdelomycin, altersetin, zopfiellamide A and signermycin B. Phenethyl and styrenyl derivatives were also structurally similar to naturally occurring tetramates with alkyl ketone side chains such as epicoccarine A, streptolydigin and virgineone, as well as the endogenous substrates of undecaprenyl pyrophosphate synthase (UPPS) such as isopentenyl pyrophosphate (IPP) and farnesyl pyrophosphate (FPP); UPPS is a known biological target of tetramates.35–37 Thus, a C-9 ethyl ketone series and a C-9 heterocyclic ketone series was synthesised to compare structural activity relationships, to demonstrate the versatility of the synthetic route for functionalisation.
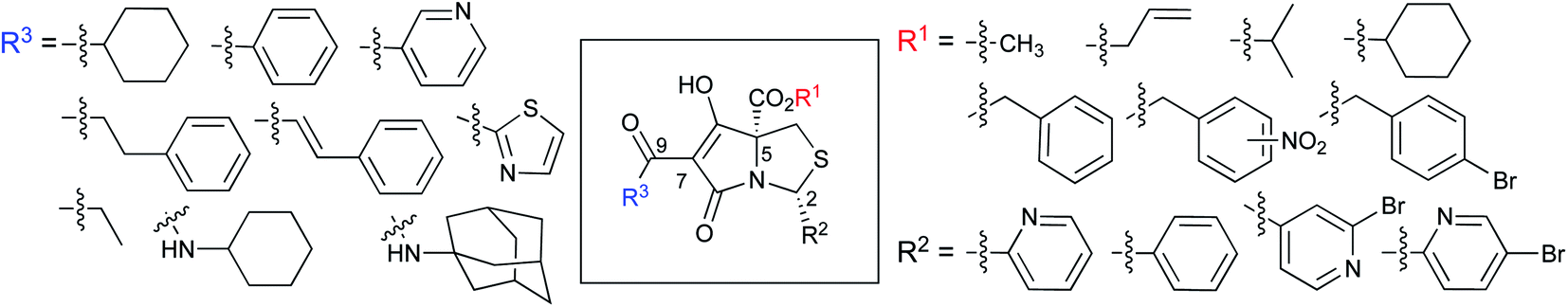 | ||
| Fig. 2 Target library of cysteine-derived tetramates. | ||
Results and discussion
In order to access C-5 ester variants, our initial approach sought to exploit the fact that N-acylthiazolidines 2 with aromatic C-2 substituents (R2 = Ar or HetAr) selectively cyclise from the side chain enolate onto the C-5 methyl ester to form tetramate esters 3 as the major product, leaving no C-5 substituents, but that N-acylthiazolidines (R2 = t-Bu) with a bulkier C-2 t-butyl group cyclise from the enolate of the C-5 methyl ester to form the tetramate core 4; unfortunately, however, the methyl ester of the latter could not be directly transesterified, suggesting unusual stability, probably arising from the significant steric hindrance of the bicyclic system (Scheme 1). | ||
| Scheme 1 Reagents and conditions: (a) KOtBu, THF, reflux, 4 h; (b) R1OH, catalytic p-TsOH monohydrate or DMAP, toluene, reflux. | ||
Alternatively, attempts to adjust the identity of the ester earlier in the synthetic sequence, by for example, direct transesterification of N-acylthiazolidine 2 (R2 = Ph), in fact favoured reaction at the less hindered ethyl ester on the side chain (Scheme 2A); transesterification of the C-5 ester group could not be achieved, with the harsh conditions resulting instead in ring opening and degradation of the N-acylthiazolidine 2 (R2 = Ph). Transesterification of the unprotected thiazolidine, known to be less stable than the N-acylthiazolidine derivative, not surprisingly led to ring opening; only transesterified L-cysteine and the product with alkylation on the thiol group were detected by mass spectrometry (Scheme 2B).
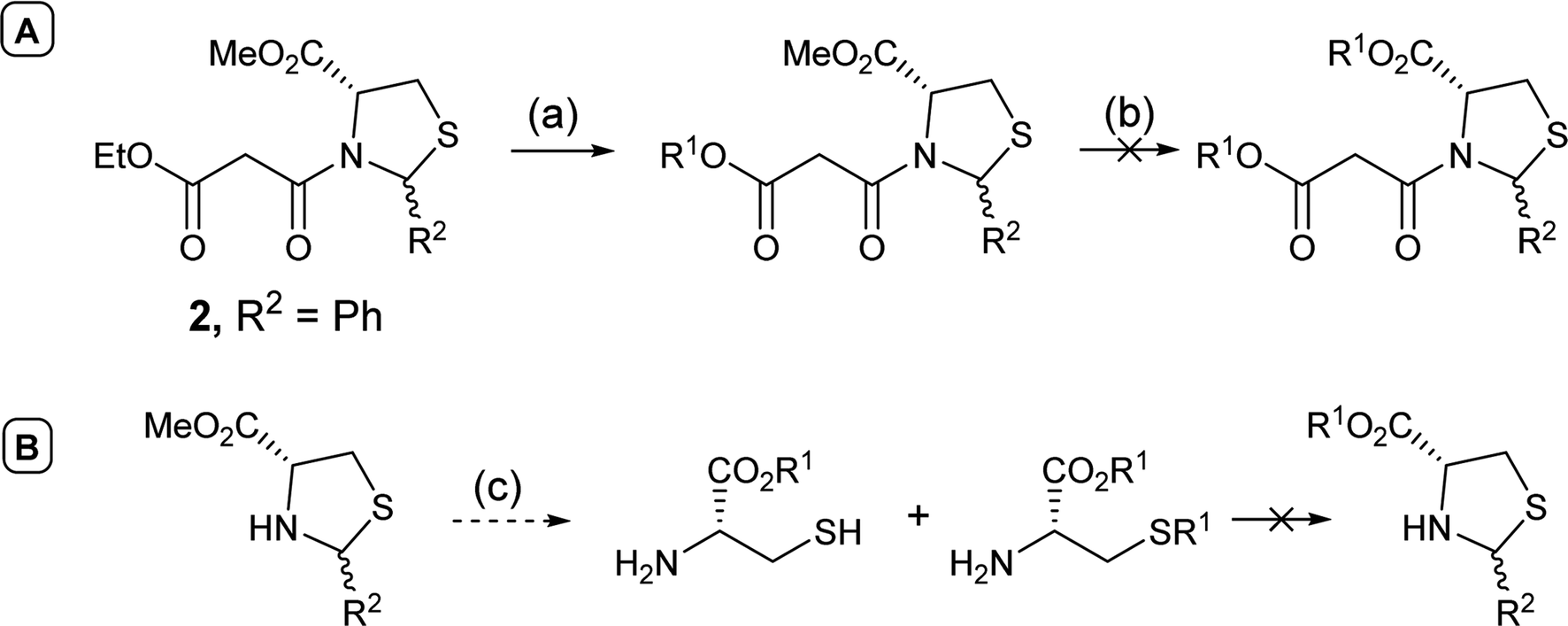 | ||
| Scheme 2 (A) Direct transesterification of N-acylthiazolidine; (B) transesterification of the unprotected thiazolidine. Reagents and conditions: (a) R1OH, R1 = p-methoxybenzyl, catalytic DMAP, toluene, reflux, o.n.; (b) additional equiv. of R1OH, toluene, reflux, o.n.; (c) R1OH, R1 = benzyl or p-methoxybenzyl, catalytic p-TsOH monohydrate or DMAP, toluene, reflux, o.n. | ||
With the failure to directly functionalise both thiazolidines and the derived tetramate core at the C-5 ester group, it became apparent that access to esters of L-cysteine at the outset would be required. The formation of such systems is complicated by the presence of the highly nucleophilic thiol group, and while application of an elegant literature method to prepare esters of L-serine and L-threonine38 using amine protection with ethyl acetoacetate followed by esterification with alkyl halides under basic conditions and subsequent amine deprotection with methanolic HCl indeed gave the desired ester, it was also obtained along with unwanted reaction at the thiol side chain (Scheme 3).
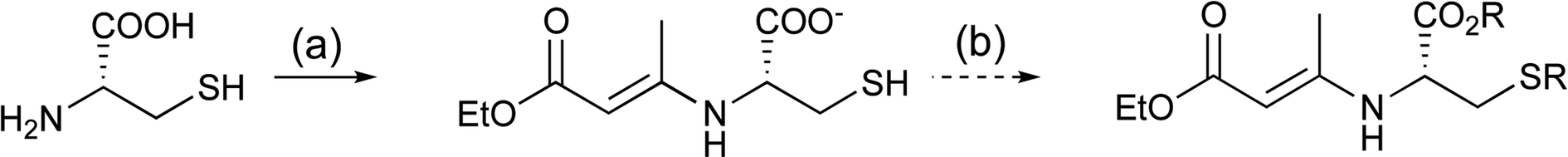 | ||
| Scheme 3 Reagents and conditions: (a) N,N,N,N-tetramethylguanidine, anhydrous DMF, 0 °C for 30 min followed by addition of ethyl acetoacetate, 0 °C to r.t., 18 h; (b) RBr, (R = benzyl), r.t., 24 h. | ||
We therefore focussed on a strategy exploiting the attenuated nucleophilicity of sulphur in the disulphide group of L-cystine 5. Esterification of L-cystine by thionyl chloride and simple alcohols occurred in quantitative yield,39 although reaction with bulkier alcohols such as benzyl alcohol and p-methoxybenzyl alcohol was more limited and gave incomplete reaction, with starting material mostly being recovered. Acid-catalysed esterification with p-TsOH monohydrate in toluene proved to be more reliable,40,41 and was even better using cyclohexane as solvent,42–44 giving diesters of L-cystine 6a–g which precipitated as their bis(p-toluenesulphonate) salts (Scheme 4).45–47 Although widely used as starting materials for synthesising biologically relevant molecules, these systems have generally been incompletely characterised.46–52
 | ||
| Scheme 4 Reagents and conditions: (a) p-TsOH monohydrate, R1OH, cyclohexane, reflux, 5–16 h; (b) DTT, Et3N, DCM, N2, r.t., 24 h; (c) R2CHO, petrol 40/60, reflux, 18 h; (d) mono-ethyl malonate, DCC, DMAP, DCM, 0 °C to r.t., 18 h; (e) N-methoxy-N-methylcarbomoyl chloride 22, dry pyridine, MgBr2, DCM, 0 °C to r.t., 18–24 h; (f) DBU dry THF, r.t., 24 h; (g) R3MgBr (prepared fresh with R3Br, Mg, dibromoethane, dry THF, reflux, 1 h), dry THF, −15 °C, 1 h or o.n.; (h) R3NH2, THF/toluene, reflux, 18 h. | ||
These diesters of L-cystine 6a–g were then reduced at the disulphide bond to produce esters of L-cysteine 7a–g. Reduction by sodium borohydride by the literature method53 also gave unwanted reduction of the ester groups, but fully selective cleavage of the disulphide bond was successfully achieved with dithiothreitol (DTT) under weakly alkaline conditions (Et3N) (Scheme 4) in good yield.45,54–56
With these materials reliably in hand, condensation of L-cysteine esters 7a–g with aromatic aldehydes (R2CHO) gave the corresponding thiazolidines 8–14 as a mixture of cis/trans-2,5 diastereomers (ratio varying from 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 4.5:1) in good yield (Scheme 4 and Table S1, ESI†). The characteristic and highly conserved H-2 and H-5 chemical shifts were consistent with those previously found in C-5 methyl ester thiazolidines,33 both being more downfield in the trans-diastereomers.
:1 to 4.5:1) in good yield (Scheme 4 and Table S1, ESI†). The characteristic and highly conserved H-2 and H-5 chemical shifts were consistent with those previously found in C-5 methyl ester thiazolidines,33 both being more downfield in the trans-diastereomers.
Using DCC/DMAP coupling conditions, thiazolidines 8–14 were converted to the corresponding N-acylthiazolidines 15–21 as a mixture of cis/trans-2,5 diastereomers in high yield (Scheme 4 and Table S2, ESI†). In the NMR spectrum, complexity arose since each diastereomer appeared as a rotameric pair, and H-2 chemical shifts were consistent with that observed previously in N-acylthiazolidines with C-2 aromatic substituents.33 Thus, in the C-2 phenyl series, the H-2 chemical shifts for the major trans rotamers were consistently more downfield; this trend was reversed in the minor rotamers. In the C-2 pyridyl series, the H-2 chemical shifts for major trans rotamers were consistently more upfield, and this trend was preserved in the minor rotamers. In the case of 17i, two diastereomers of the starting thiazolidine 10i could be separated by column chromatography and each was taken for separate N-acylation. The reaction with cis-10i produced a mixture of predominantly cis-malonamide 17i (cis/trans ratio of 4.8:1), while the reaction with trans-10i yielded a mixture of predominantly trans-malonamide 17i (cis/trans ratio of 0.2:1). That these two separate reactions starting from a pure single diastereomer produced a mixture of diastereomers, and that the cis/trans ratio changes from thiazolidines 8–14 to N-acylthiazolidines 15–21, indicated the existence of an equilibrium between the two thiazolidine diastereoisomers of 8–14 under the reaction conditions. It was generally observed that when groups at C-2 and C-5 became bulkier, as in the cases 18–21, the production of the more stable cis-diastereomer was favoured, presumably since in that case, both groups were able to assume pseudodiequatorial positions.
Noteworthy is that when N-acylthiazolidine 18iii (with a bulkier C-5 benzyl ester) was subjected to Dieckmann cyclisation, it cyclised from the more stable side chain enolate to the C-5 ester, giving tetramate ester 3iii as the major product and therefore with loss of the C-5 ester (Scheme 5); the cyclisation started with a 1.9:1 mixture of cis/trans diastereomers, and while the trans diastereomer of 18iii cyclised to form 3iii, the cis diastereomer was recovered partially (degradation was observed). This chemoselectivity in cyclisation has been consistently observed in N-acylthiazolidines with C-2 aryl substituents.32 Since this reaction outcome was the undesired chemoselectivity, to a avoid this, a Weinreb amide was introduced under Sucunza's acylation conditions,57 an approach which had been used previously for the acylation of N-acyloxazolidines or thiazolidines with a C-2 t-butyl group.29 Successful C-acylation on the side chain of the N-acylthiazolidines 15–21 was achieved to give 23–30 (Scheme 4 and Table S3, ESI†), but even after 24 hours, reactions were not complete, with starting malonamides enriched in trans-diastereomers being recovered, even with 1.5 equivalents of the acid chloride 22. Each of products 23–30 was obtained as an approximately equal ratio mixture of C2′′ R/S diastereomers (Table S3, ESI†). Consistent with previous observations,58 the side chain tricarbonyl system existed in the keto-form, indicated by the presence of an H-2′′ singlet in 1H NMR spectra. Exclusive formation of the cis diastereomer was found in all cases except for 27ii. The relative stereochemistry was supported by NOE analysis (Fig. S1, ESI†), while 2D NOESY also confirmed the presence of rotamers in the mixture. In the case of 26i, the predominantly cis mixture of 17i (cis/trans = 4.8:1) produced cis-26i exclusively under the acylation conditions in 68% yield. The reaction with the predominantly trans mixture of 17i (cis/trans = 0.2:1) was less straightforward; thus, unreacted trans starting material 17i was recovered in 42% yield, and product 27a was formed in 38% yield as a mixture of cis/trans (0.4:1) and C2′′ R/S (0.8:1) diastereomers. This result suggested that the cis-malonamides reacted more favourably under the acylation conditions and that there could be interconversion between cis/trans diastereomers to favour the cis-2,5 products. The diastereomers were not separable by flash column chromatography and the mixture was used directly for Dieckmann cyclisation (see below).
 | ||
| Scheme 5 | ||
Using literature conditions with DBU,29 successful Dieckmann cyclisation of 23–30 was achieved generally in good yield, leading to the desired tetramate Weinreb amides 31–39 with different C-5 esters (Scheme 4 and Table S4, ESI†). The presence of basic C-2 pyridyl groups (R2) and hence the corresponding increase in product polarity added to the difficulty of acidic work-up after the reactions, lowering yields significantly. For cyclisation with 24i and 25i, which contained a C-2 unsubstituted pyridyl ring, although the product was detected by mass spectrometry, no product could be extracted; increasing the lipophilicity of groups at C-2 and C-5 avoided this difficulty. Under these Dieckmann cyclisation conditions, the cis diastereomer of tricarbonyl malonamides cyclised from the C-5 enolate onto the side chain carbonyl by placing the C-2 substituents at the less hindered exo-face of the bicyclic ring, producing tetramate derivatives 31–39 with retention of the C-5 ester groups. However, in the reaction with trans-27i, no tetramate product was observed and starting material was recovered, presumably due to the greater bulk of the C-5 ester. Noteworthy was the efficiency and chemoselectivity of the synthetic route, and with bulky C-5 esters, the cis-diastereomer was the only diastereomer that cyclised under the Dieckmann cyclisation, retaining the C-5 ester. The basic conditions with DBU also helpfully favoured exclusively reaction at the ester of the side chain, so that the Weinreb amide remained available for late-stage functionalisation at C-7. Weinreb amides 31–39 were completely enolic, with no C-7 proton observed in 1H NMR spectra, and in the endo-enolic tautomeric pair AB form (Fig. 3 and Scheme 4), as supported by HMBC analysis (Fig. S2, ESI†) and by comparison of carbonyl 13C NMR peaks with earlier reports.29 They were also a single diastereomer at C-2/C-5 with characteristic and consistent H-2 chemical shifts (Table S4, ESI†). The cis relationship between C-5 ester and C-2 aromatic rings was supported by NOE analysis (Fig. S2, ESI†).
 | ||
| Fig. 3 Tautomeric behaviour of tetramate ketones. | ||
In the reactions of 28iv and 29iv, which contained only C-5 nitrobenzyl esters, the product was unexpectedly a 1:1 inseparable mixture of tetramate Weinreb amides with the respective C-5 nitrobenzyl ester along with ethyl ester 37iv (Table S4, ESI†). The latter by-product 37iv would arise from transesterification with ethoxide generated under the basic conditions of the reaction. Similar by-product formation was observed from the crude product of the reaction of 35 and 39 but to a much less extent and desired products could be easily purified with column chromatography. The by-product formation with bulky C-5 esters limited the scope of the process, and made 39 all the more valuable, since the bromo-substituted ester allowed expansion of ester diversity via late-stage functionalisation.
The presence of the C-7 Weinreb amide allowed further functionalisation by introducing either C-7 acyl substituents using Grignard reagents or C-7 amides using transamination. Successful C-7 acylation of Weinreb amides was achieved using aryl, alkyl and alkenyl Grignard reagents. High chemoselectivity at the side chain (Tables 1–3 and Scheme 4), with no reactions at the C-5 esters, was observed, except in the Grignard addition to 38iv which gave some C-5 ketone formation leading to 40 (Scheme 6). The high chemoselectivity of Grignard addition was attributed to both the hindered nature of the bicyclic tetramate system and the stability of a magnesium-chelated intermediate with the Weinreb amide, which also avoided over-addition leading to tertiary alcohols.59 Since it has been reported that this Mg-chelated intermediate was only stable at low temperature, the process required a low-temperature reaction quench.59 In the synthesised tetramate ketones, the cis relative stereochemistry at C-2/C-5 was supported by NOE analysis (Fig. S2, ESI†). With consistent H-2 chemical shifts across the series, cis-2,5 stereochemistry was assumed to be conserved from the starting tetramate Weinreb amides (Tables 1–3).
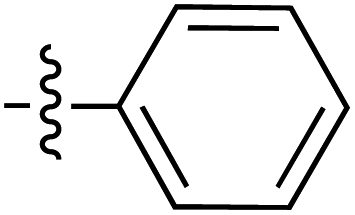
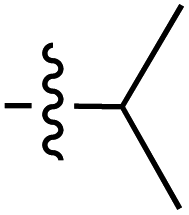
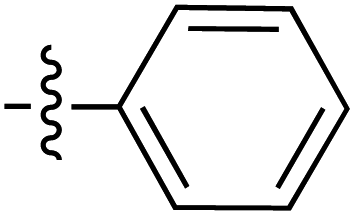
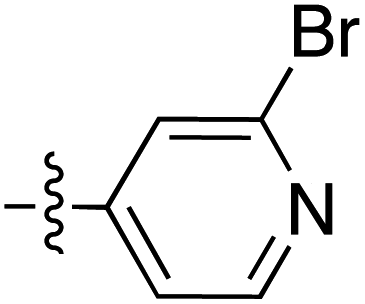
| Grignard R3MgBr | R1 = | R2 = | δ (ppm) | AB/CD | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| H-2 | C-6 | C-8 | C-9 | ||||||
| 41 |

|
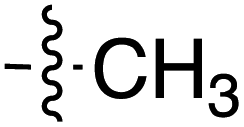
|
|
6.42 | 192.3 | 175.6 | 187.3 | 4.1:1 |
71 |
| 6.47 | — | — | — | ||||||
| 42 |

|
6.42 | 192.3 | 175.8 | 187.3 | 3.7:1 |
45 | ||
| 6.46 | — | — | — | ||||||
| 43 |
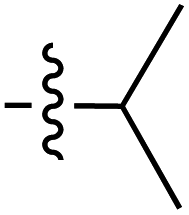
|
6.41 | 192.0 | 175.9 | 187.6 | 4.2:1 |
40 | ||
| 6.46 | — | — | — | ||||||
| 44 |
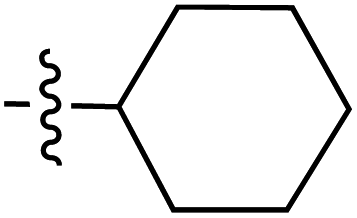
|
6.40 | 191.9 | 175.8 | 187.7 | 4.5:1 |
80 | ||
| 6.44 | — | — | — | ||||||
| 45 |
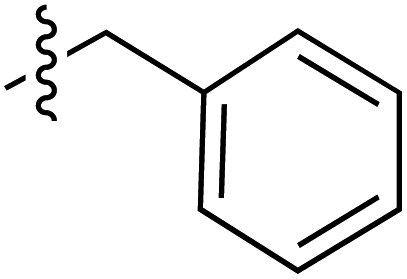
|
6.40 | 192.3 | 175.9 | 187.3 | 4.2:1 |
43 | ||
| 6.44 | — | — | — | ||||||
| 46 |

|
6.40 | 192.5 | 175.9 | 187.3 | 3.3:1 |
42 | ||
| 6.42 | — | — | — | ||||||
| 47 |
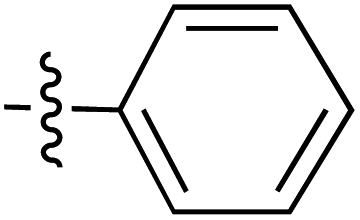
|

|

|
6.49 | 185.9 | 177.7 | 182.9 | CD | 33 |
| 48 |
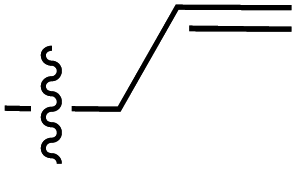
|
6.49 | 189.9 | 177.1 | 181.5 | 0.2:1 |
46 | ||
| 6.40 | 185.9 | 177.9 | 182.9 | ||||||
| 49 |
|
6.49 | 186.2 | 177.9 | 182.6 | CD | 62 | ||
| 50 |

|
6.48 | 186.4 | 177.9 | 182.5 | CD | 53 | ||
| 51 |

|
6.47 | 185.9 | 178.0 | 182.9 | CD | 30 | ||
| Grignard R3MgBr | R1 = | R2 = | δ (ppm) | AB/CD | %Yield | ||||
|---|---|---|---|---|---|---|---|---|---|
| H-2 | C-6 | C-8 | C-9 | ||||||
| 52 |

|
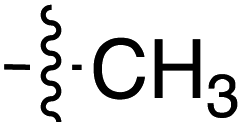
|
|
6.41 | 195.3 | 176.1 | 187.1 | 3.8:1 |
35 |
| 6.44 | — | — | 182.4 | ||||||
| 53 |

|
|
6.28 | 195.9 | 176.4 | 186.3 | 3.3:1 |
33 | |
| 6.30 | — | — | — | ||||||
| 54 |

|
|
6.41 | 195.3 | 176.3 | 187.1 | 4.0:1 |
39 | |
| 6.44 | — | — | — | ||||||
| 55 |

|
|
6.40 | 195.1 | 176.4 | 187.4 | 4.0:1 |
30 | |
| 6.43 | — | — | — | ||||||
| 56 |
|

|
6.39 | 195.0 | 176.4 | 187.6 | 4.3:1 |
57 | |
| 6.42 | — | — | — | ||||||
| 57 |
|
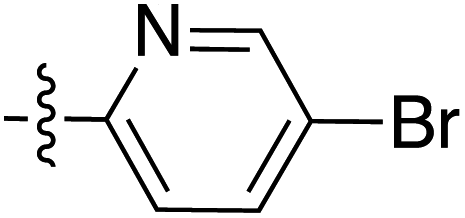
|
6.35 | 195.7 | 176.8 | 187.0 | 3.7:1 |
29 | |
| 6.38 | 194.3 | 172.9 | 186.0 | ||||||
| 58 |
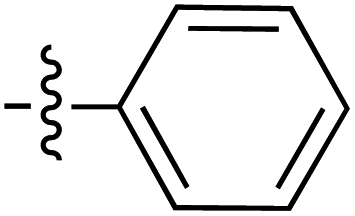
|
6.38 | 195.3 | 176.4 | 187.1 | 4.6:1 |
37 | ||
| 6.42 | — | — | — | ||||||
| 59 |
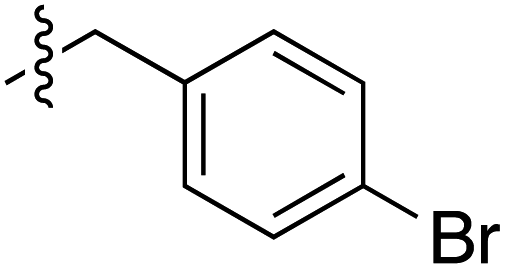
|
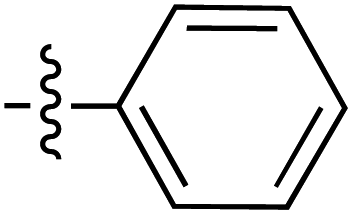
|
6.38 | 195.5 | 176.4 | 186.9 | 5.1:1 |
36 | |
| 6.41 | — | — | — | ||||||
| 60 | Phenethyl |
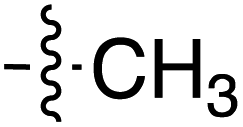
|

|
6.40 | 189.8 | 175.3 | 187.1 | CD | 40 |
| 61 |
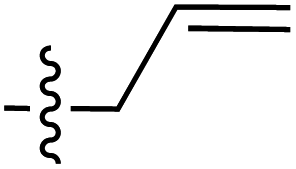
|
6.41 | 191.5 | 174.7 | 188.1 | 0.8:1 |
33 | ||
| 6.40 | 189.8 | 175.5 | 187.2 | ||||||
| 62 |
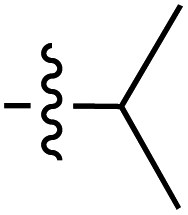
|
6.41 | — | — | — | 0.1:1 |
45 | ||
| 6.40 | 189.5 | 175.6 | 187.5 | ||||||
| 63 |

|
6.42 | — | — | — | 0.2:1 |
73 | ||
| 6.40 | 189.4 | 175.6 | 187.6 | ||||||
| 64 |
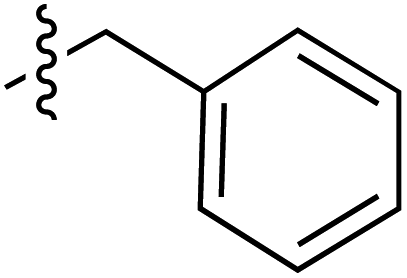
|
6.38 | 189.8 | 175.6 | 187.1 | CD | 40 | ||
| 65 |

|
6.43 | — | — | — | 0.2:1 |
31 | ||
| 6.38 | 190.1 | 175.6 | 187.1 | ||||||
| Grignard R3MgBr | R1 = | R2 = | δ (ppm) | AB/CD | %Yield | ||||
|---|---|---|---|---|---|---|---|---|---|
| H-2 | C-6 | C-8 | C-9 | ||||||
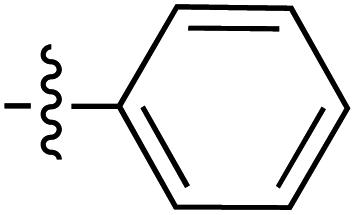
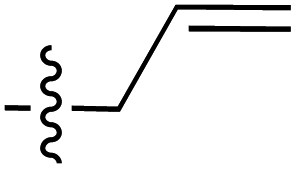
| a The E stereochemistry of the styrenyl ketone pendant was confirmed by the J coupling value of H-1′′ and H-2′′ (consistently at J = 16.0 Hz, indicating the trans relationship between the two protons) on the side chain. | |||||||||
| 66 | Styrenyla |
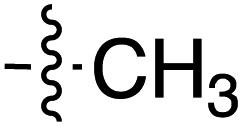
|
|
6.51 | — | — | — | 0.26:1 |
58 |
| 6.46 | 187.5 | 176.0 | 176.2 | ||||||
| 67 |

|

|
6.36 | — | — | — | 0.34:1 |
31 | |
| 6.32 | 186.7 | 176.3 | 176.6 | ||||||
| 68 |
|

|
6.51 | 196.3 | 175.0 | 176.7 | 0.17:1 |
58 | |
| 6.46 | 187.5 | 176.1 | 176.2 | ||||||
| 69 |

|

|
6.50 | 196.7 | — | 176.5 | 0.28:1 |
77 | |
| 6.46 | 187.7 | 176.0 | 176.3 | ||||||
| 70 |

|
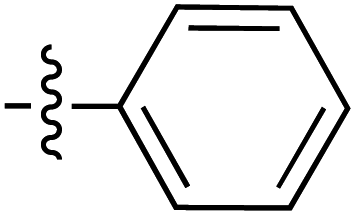
|
6.49 | 196.9 | — | 176.5 | 0.28:1 |
53 | |
| 6.45 | 187.9 | 175.9 | 176.3 | ||||||
| 71 |
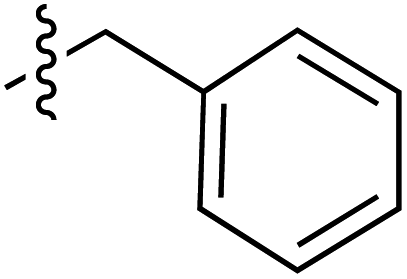
|

|
6.44 | 196.1 | 175.5 | 177.0 | 0.43:1 |
29 | |
| 6.40 | 187.4 | 176.5 | 176.7 | ||||||
| 72 |

|
6.49 | — | — | — | 0.26:1 |
55 | ||
| 6.44 | 187.4 | 176.2 | 176.3 | ||||||
| 73 |

|
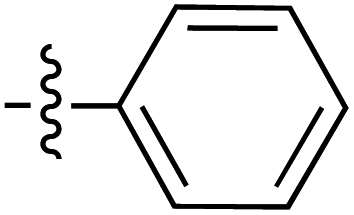
|
6.49 | — | — | — | 0.23:1 |
59 | |
| 6.44 | 187.3 | 176.2 | 176.3 | ||||||
 | ||
| Scheme 6 | ||
Tautomers existed for tetramates 41–73 as evidenced by the presence of two H-2 chemical shifts for each tetramate and the distinctive two sets of carbonyl chemical shifts across the series (Tables 1–3). The assignment of tautomeric forms was assisted by HMBC analysis (Fig. S2, ESI†) allowing assignment of C-6, C-8 and C-9 carbonyl shifts, which indicated that the Z-exo-enolic D as the major or exclusive tautomeric form in the R3 = phenyl, phenethyl and styrenyl series and that the E-exo-enolic form B as the major tautomer in the R3 = ethyl and cyclohexyl series (Fig. 3), was favoured. For the major forms, tetramate ketones with R3 = ethyl and cyclohexyl consistently exhibited more downfield C-6 chemical shifts than the R3 = phenyl, phenethyl and styrenyl series (Tables 1–3), which supported the assignment of the former as tautomeric form B and the latter as form D. The C-6 chemical shifts in tetramate Weinreb amides 31–39 assigned as endo-enolic form A (Table S4, ESI†) were the lowest of C-6 chemical shifts among tetramate ketones. C-8 and C-9 chemical shifts of tautomeric pair BD were very similar, with C-8 being generally more downfield in D because of hydrogen bonding. C-9 Chemical shifts in the R3 = styrenyl series were more upfield than others due to the extended conjugation system of the styrenyl side chain. The trend observed in carbonyl chemical shifts and the assignment of tautomeric forms was consistent to those in previously synthesised tetramate ketones with a C-2 t-butyl group. Besides the nature of the solvents used for NMR analysis,29,31 the ratio of tautomeric forms was also dependent on the nature of the C-9 ketone substituent.
To expand the scope of this process, heterocyclic Grignard reagents such as pyridyl and thiazolyl systems were investigated for Grignard addition. Pyridyl magnesium bromide was prepared with ethyl bromide as an entrainment agent and successful Grignard addition was achieved to give 74 along with 42 (Scheme 7). Although the yield of the Grignard reaction is reported to be increased by raising the ratio of ethyl bromide to 3-bromopyridine to 3:1,60 this proved not to be optimal with tetramate Weinreb amides since full reaction selectivity was lost, and the C-9 ethyl tetramate ketone 42 was obtained as by-product in every reaction.
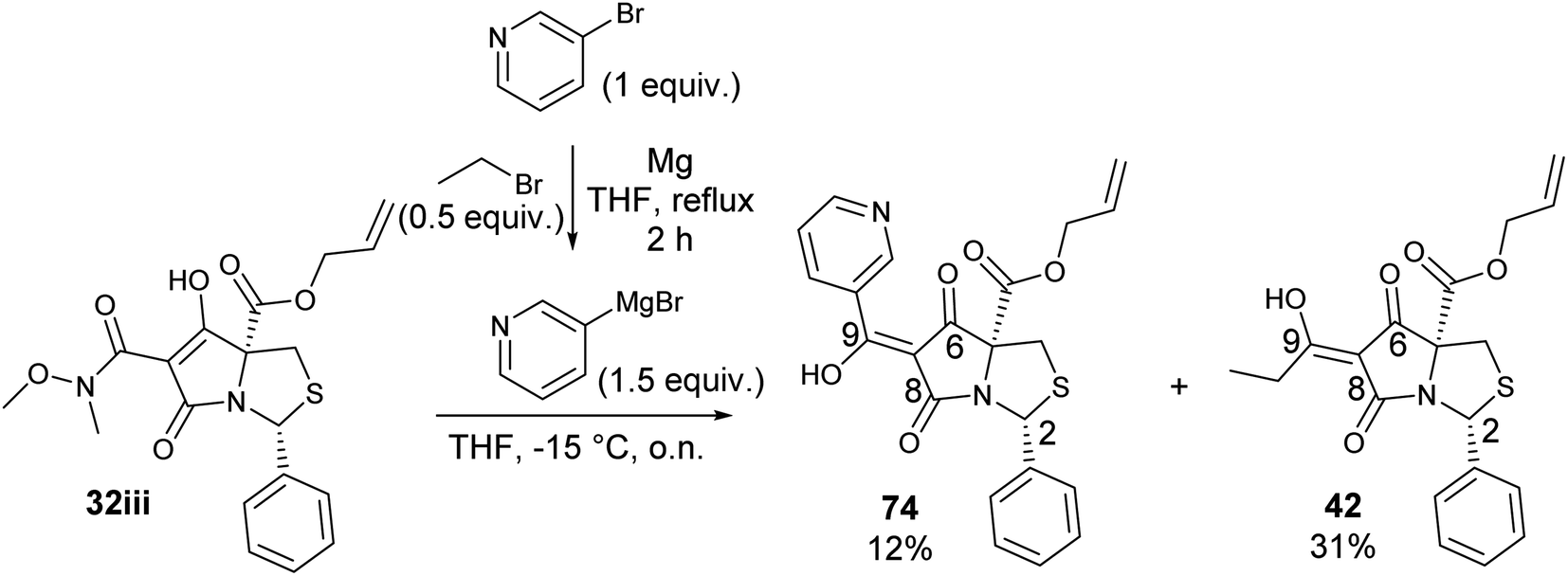 | ||
| Scheme 7 | ||
Another widely used method of preparing organomagnesium reagents is through halogen–metal exchange,61 and this was applied to the 2-bromothiazole at room temperature with iPr2Mg.62–64 Subsequent Grignard addition to Weinreb amide 32iii gave ketone formation but accompanied by hydrolysis and decarboxylation, giving derivative 75 (Scheme 8). Exo-enolic tautomeric form D was found for both tetramate ketones 74 and 75 (Table 4) based on comparison of the carbonyl shifts with the previously synthesised ketone series (Tables 1–3).
 | ||
| Scheme 8 | ||
| Grignard R3MgBr or R3MgiPr | R1 = | R2 = | δ (ppm) | AB/CD | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| H-2 | C-6 | C-8 | C-9 | ||||||
| 74 |

|

|

|
6.47 | 189.6 | 176.2 | 181.6 | CD | 12 |
| 75 |
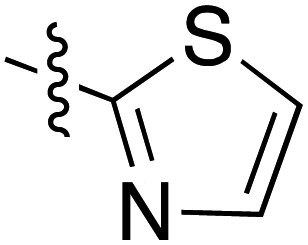
|
— | 6.48 | 189.2 | 173.0 | 177.9 | CD | 23 | |
Along with the C-7 ketones 41–75 described above, C-7 carboxamides 76–94 could also be accessed from tetramate Weinreb amides 31–39 by transamination using the corresponding amines at reflux (Scheme 4). Interestingly, these carboxamides exhibited two distinctive sets of H-2 shifts and carbonyl peaks for each compound, which were of approximately equal ratio (Table 5). The two sets of H-2 peaks were present in 1H NMR spectra of crude material, but did not arise after column chromatography or post-column acid wash. For 76, 2D NOESY ruled out rotameric behaviour as the two sets of peaks did not show rapid dynamic exchange while 1D NOE analysis supported the same cis-relative stereochemistry at C-2 and C-5 (Fig. S2, ESI†). The assignment of carbonyl peaks was assisted by HMBC correlations, which showed that each of the two H-2 shifts correlated with a different C-8 carbonyl (Fig. S2, ESI†). The only plausible explanation for the two sets of chemical shifts from a single reaction was tautomeric behaviour. The range of carbonyl chemical shifts was comparable and the consistent downfield trend in carbonyl peaks in tautomeric pair CD over AB was also similar to that observed with carboxamides. This tautomeric behaviour is also solvent dependent, so that, for example, for 88, in methanol, only one set of chemical shifts was observed in both 1H and 13C NMR spectra (Fig. S3C and D, ESI†), which appeared to be that of an averaged hybrid structure due to equilibration of the tautomeric form; different behaviour was observed in CDCl3 (Fig. S3A and B, ESI†). Similar equilibration was observed in other polar solvents such as CD3CN and THF-d8 but with broader signals, suggesting different rates of the solvent-assisted proton-transfer reactions.
| Amine R3NH2 | R1 = | R2 = | δ (ppm) | AB/CD | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| H-2 | C-6 | C-8 | C-9 | ||||||
| 76 |
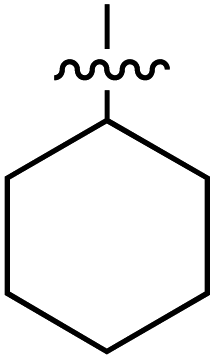
|

|

|
6.27 | 187.1 | 173.2 | 165.8 | 1:1 |
30 |
| 6.31 | 189.0 | 178.4 | 165.9 | ||||||
| 77 |
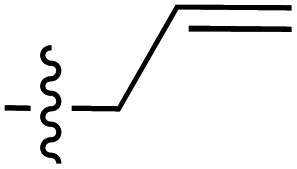
|
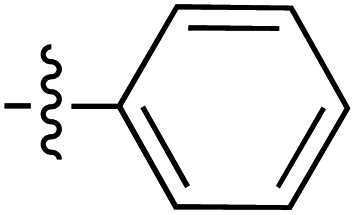
|
6.26 | 187.0 | 173.3 | 165.7 | 0.9:1 |
48 | |
| 6.31 | 188.8 | 178.5 | 165.9 | ||||||
| 78 |

|

|
6.26 | 187.4 | 173.4 | 165.7 | 1.1:1 |
49 | |
| 6.31 | 188.6 | 178.7 | 165.9 | ||||||
| 79 |

|
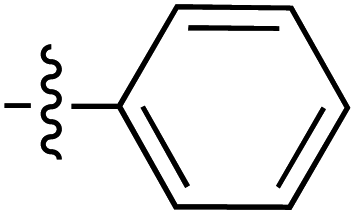
|
6.25 | 187.5 | 173.4 | 165.7 | 1.1:1 |
61 | |
| 6.30 | 188.6 | 178.7 | 165.9 | ||||||
| 80 |

|

|
6.24 | 187.1 | 173.5 | 165.8 | 0.9:1 |
58 | |
| 6.27 | 189.4 | 178.6 | 165.8 | ||||||
| 81 |
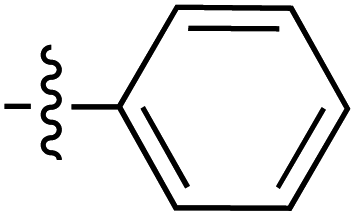
|
6.25 | 187.1 | 173.5 | 165.8 | 0.9:1 |
35 | ||
| 6.29 | 188.9 | 178.7 | 165.9 | ||||||
| 82 |

|

|
6.25 | 186.9 | 173.4 | 165.8 | 0.9:1 |
47 | |
| 6.29 | 189.0 | 178.6 | 165.9 | ||||||
| 83 |
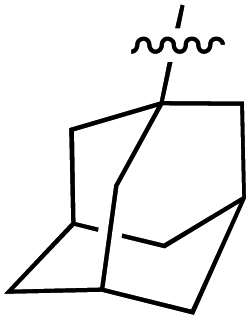
|

|

|
6.26 | 187.0 | 173.4 | 166.7 | 0.9:1 |
56 |
| 6.31 | 189.4 | 178.6 | 166.8 | ||||||
| 84 |

|

|
6.14 | 186.6 | 173.5 | 166.7 | 1:1 |
52 | |
| 6.18 | 189.7 | 178.5 | 166.8 | ||||||
| 85 |
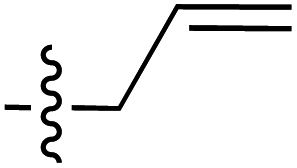
|
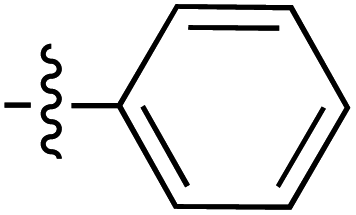
|
6.26 | 187.0 | 172.6 | 166.7 | 1:1 |
46 | |
| 6.31 | 189.3 | 178.8 | 166.8 | ||||||
| 86 |
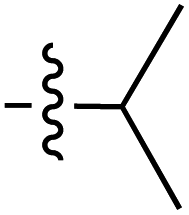
|
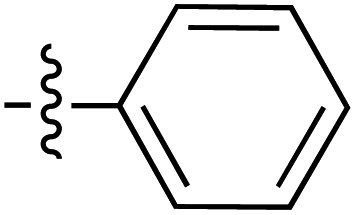
|
6.24 | 187.2 | 173.5 | 166.5 | 1:1 |
44 | |
| 6.30 | 189.0 | 178.9 | 166.6 | ||||||
| 87 |

|

|
6.31 | 187.4 | 173.6 | 166.4 | 1:1 |
33 | |
| 6.35 | 189.5 | 179.0 | 166.7 | ||||||
| 88 |

|
6.25 | 187.3 | 173.5 | 166.4 | 1:1 |
57 | ||
| 6.30 | 189.1 | 178.9 | 166.5 | ||||||
| 89 |
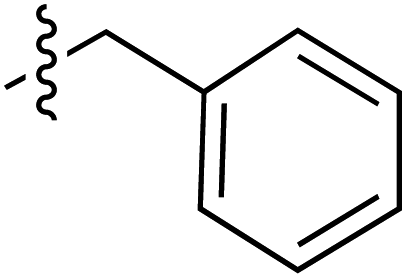
|
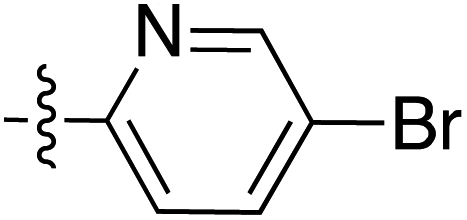
|
6.23 | 187.0 | 173.6 | 166.7 | 0.9:1 |
34 | |
| 6.26 | 189.8 | 178.9 | |||||||
| 90 |
|
6.23 | 187.0 | 173.6 | 166.6 | 1:1 |
67 | ||
| 6.28 | 189.3 | 178.9 | 166.7 | ||||||
| 91 |

|
|
6.34 | 187.1 | 173.8 | 166.7 | 0.8:1 |
52 | |
| 6.36 | 189.1 | 179.0 | |||||||
| 92 |

|

|
6.14 | 186.5 | 173.5 | 166.7 | 1:1 |
43 | |
| 6.18 | 189.7 | 178.5 | 166.7 | ||||||
| 93 |

|
|
6.23 | 187.0 | 173.6 | 166.7 | 0.9:1 |
44 | |
| 6.27 | 189.9 | 178.9 | 166.7 | ||||||
| 94 |
|
6.24 | 186.9 | 173.6 | 166.7 | 0.9:1 |
47 | ||
| 6.29 | 189.4 | 178.8 | 166.8 | ||||||
Metal chelating properties of the synthesised tetramates
C-7 acylated tetramate ketones 41–75 were unsurprisingly metal-chelating, with their chelating abilities influenced by the bulkiness of both C-5 esters and C-9 ketone pendant groups. With the abundance of Mg2+ ions after the Grignard reactions, no meaningful product peaks could be detected in the NMR spectra of crude reaction mixture of 60, for example, prior to acidic work-up (Fig. S4A, ESI†). However, once the crude material was washed with 0.5 M HCl, immediate sharpening of the resulting NMR spectrum was observed (Fig. S4B, ESI†). When subjected to flash column chromatography, the tetramate ketone again bound metal impurities from the silica, giving broad product peaks in the 1H NMR spectrum (Fig. S4C, ESI†). Tetramate ketones were therefore routinely washed with 0.5 M HCl post-column to render them metal-free, which immediately sharpened the signals (Fig. S4D, ESI†). Similar metal chelating behaviours were observed across the C-9 ethyl, phenethyl and styrenyl tetramate ketone series 41–73. With bulkier C-5 esters, such as 65, there was an improvement in the resolution of peaks post-column as compared to tetramates with smaller C-5 methyl esters (Fig. S4E and F, ESI†), supporting the hypothesis that steric hindrance posed by neighbouring C-5 esters reduced metal chelation. C-9 cyclohexyl ketones 52–56 demonstrated a similar broadness of peaks post-column chromatography but before acid wash. Interestingly, for tetramates 58 and 59 with bulkier C-5 benzyl or substituted benzyl esters, sharper peaks were detected on 1H NMR spectroscopic examination of both the crude and post-column samples prior to acid wash (Fig. S5A, C and E, ESI†), consistent with reduced metal binding abilities in these compounds, although similar metal chelation behaviour was nonetheless discernible (Fig. S5C–F, ESI†). Comparison with tetramate ketones with the same C-5 ester also indicated the role played by the C-9 ketone pendant group in reducing metal binding. Thus, the presence of C-2 pyridyl rings in 57 and 71 broadened the post-column product peaks before acid wash, suggesting that the C-2 pyridyl rings could also participate in extended metal chelation (Fig. S6A, ESI†). As previously reported, tetramate ketones with a C-5 methyl ester required 2 M HCl solution to wash out metal ions,29 and the fact that 57 and 71 could be washed free from metal ions with a much weaker 5% citric acid solution to give well-resolved and sharp 1H NMR peaks suggested much reduced metal chelation resulting from the presence of a bulky C-5 ester (Fig. S6B, ESI†).In order to investigate in more detail the types of metal ions potentially involved in chelation, metal-free tetramate ketone 60 was deliberately washed with 1 M aq. Ca(NO3)2 solution (Fig. S7A, ESI†) and aq. 1 M FeSO4 solution (Fig. S7B, ESI†). The extent of peak broadening suggested a higher affinity of metal chelation for Ca2+ than Fe2+. Peak broadening induced by washing metal-free samples with aq. Ca(NO3)2 solution has also been observed previously in tetramate ketones with C-2 t-butyl substituents. This observation in tetramate ketones was in contrast to tetramate esters and carboxamides, which demonstrated a preference for Fe2+ with significant peak broadening (likely also due to paramagnetic behaviour) when washed with the same strength of metal solution.29 This difference again highlighted the structural dependency of metal chelation in tetramate derivatives. In addition, analysis by SFC-MS of a post-column sample of 63 prior to acid wash indicated the presence of m/z peaks that could be attributed to [M + Ca]2+ and [M + Mg]2+ but not [M + Fe]2+ or [M + Fe]3+ (Fig. S8, ESI†). The presence of Ca2+ ions in post-column metal-chelated tetramate ketones was also previously indicated by XPS analysis.29
Tetramate carboxamides 76–94 were also similarly found to be metal-chelating, displaying broadness of product peaks post-column prior to acid wash (Fig. S9A, ESI†). Tetramate carboxamides were also routinely washed with 0.5 M HCl solution to remove metal ions post-column (Fig. S9B, ESI†), although 5% citric acid solution was used instead if pyridyl groups were present. For tetramate carboxamide 91 with a bulky C-5 nitro-benzyl ester, a 5% citric acid wash resulted in a better-resolved and sharp 1H NMR spectrum, while the H-2 and H-5 signals of 95 still remained relatively broad after acidic wash as has been reported previously (Fig. S9C and D, ESI†).33 The relative extent of metal binding with increasing bulkiness of C-5 ester was not discernible by visual comparison of 1H NMR spectra. With an NH linker, the bulkiness of the C-9 amide pendant probably played a less important steric influence than the C-9 ketone pendant in reducing metal chelation.
A primary goal of this work was to modulate metal binding properties of the tetramates, and the concept behind chemical modifications of C-5 esters was to reduce metal chelation through steric influence of the neighbouring C-5 groups. In order to quantify the extent of metal chelation, peak broadening as measured by width-at-half-height (W1/2) values for the H-2 signals in post-column 1H NMR spectra was used (Fig. 4 and Table S5, ESI†). In cases where two sets of H-2 signals were present in the NMR spectrum of a single compound due to tautomeric behaviour, the major peak was taken for calculation. Moreover, since the principle behind chemical modifications of C-5 esters was to reduce metal chelation through steric influence of the neighbouring C-5 groups, their bulk was estimated by calculation of their van der Waals volumes (Å3) and correlated with the post-column H-2 widths at half height (Fig. 4). Besides groups at the C-5 position, the metal chelation behaviour was also complicated by the identity of C-2 and C-9 pendants. Therefore, compounds were grouped into their respective sub-series and the trend of increasing C-5 steric bulkiness analysed separately. Across all series, an increase in the bulkiness of C-5 ester generally correlated with a decrease in peak broadening, suggesting reduced metal-binding ability and hence validating the working principle behind chemical modifications at C-5 esters (Fig. 4). Tetramates with C-2 heterocycles were generally more metal-binding, as heterocycles could also participate in extended metal chelation. The smallest extent of peak broadening among polar heterocycle-containing tetramate carboxamides was observed with 87 with C-2 pyridin-2-yl ring and C-5 cyclohexyl ester (Table S5, ESI†). The best combination for reducing metal chelation was 58 with C-9 cyclohexyl ketone pendant and C-5 benzyl ester, with a post-column H-2 width at 2.4 Hz, which is almost as sharp as non-metal binding tetramates.34
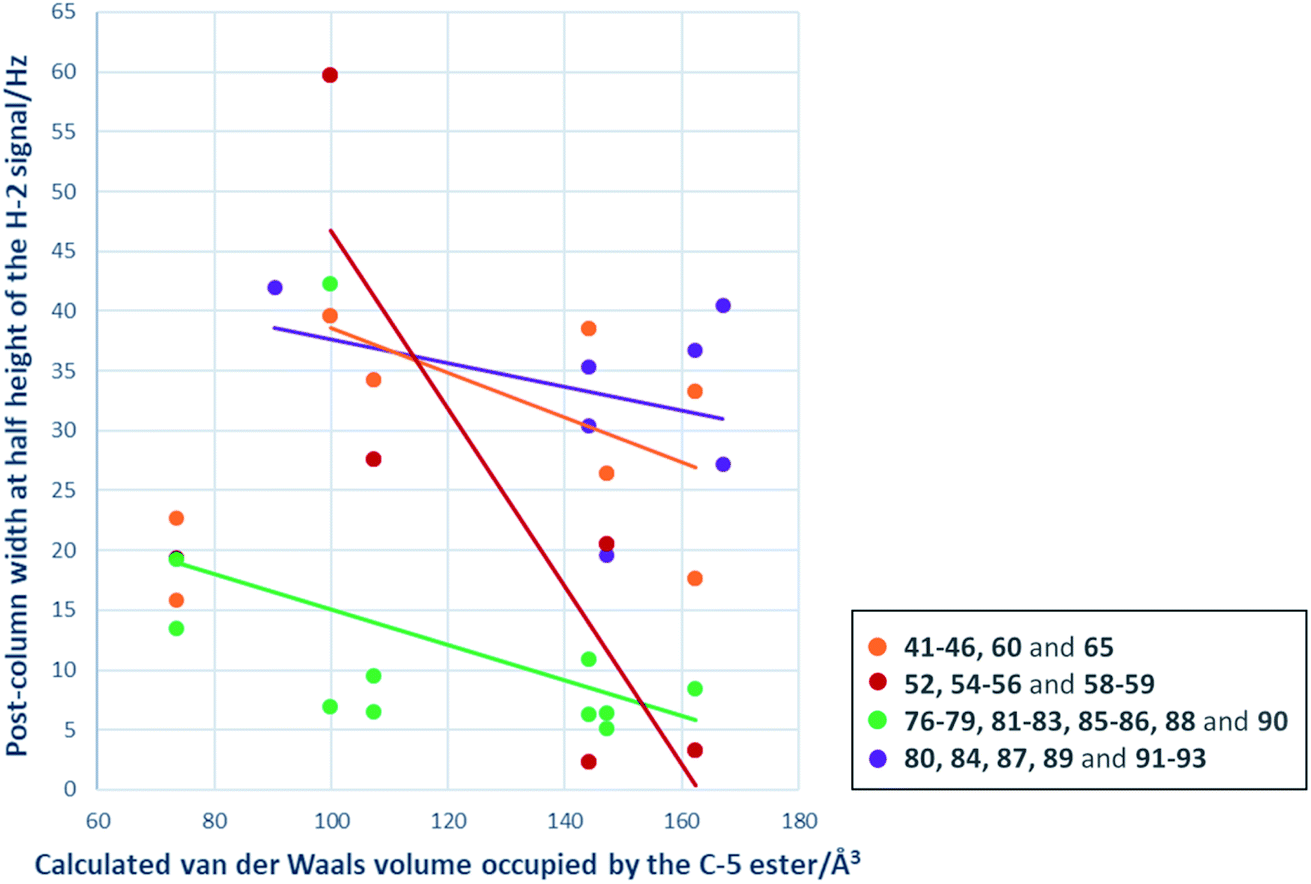 | ||
| Fig. 4 Plot of post-column widths at half height/Hz for H-2 signals against van der Waals volume occupied by the respective C-5 ester. Subseries of compounds: orange – R3 = ethyl or phenethyl, R2 = phenyl; red – R3 = cyclohexyl, R2 = phenyl; green – R3 = N-cyclohexyl or N-(1-adamantyl), R2 = phenyl; purple – R3 = N-cyclohexyl or N-(1-adamantyl), R2 = pyridin-2-yl or bromo-pyridinyl. | ||
Biological evaluation
Tetramate ketones 41–75 and carboxamides 76–94 were tested for their antibacterial activities against Gram-positive MRSA and Streptomyces and Gram-negative E. coli and the most active systems are reported in Table 6. No activity against E. coli was observed. Tetramate ketones 41–51 with C-9 ethyl or phenyl ketone pendants were not active while C-9 cyclohexyl, phenethyl and styrenyl ketone pendants 52–73 were more active against Gram-positive MRSA and Streptomyces (Table S6, ESI†). Tetramate carboxamides 76–94 with C-9 cyclohexyl or adamantyl amide pendants also demonstrated activities against Gram-positive bacteria (Table S6, ESI†). A key initial observation was that modification at C-5 esters did not result in activity loss, with bulkier C-5 esters actually enhancing the antibacterial potency against MRSA.| Cpd No. | MW/Da | clogP | clogD7.4 | PSA/Å2 | MSA/Å2 | %PSA | HBD | HBA | MIC against | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Streptomyces | MRSA | ||||||||||
| μg ml−1 | μg ml−1 | μM | |||||||||
| a MW was calculated by ChemDraw 19.1.1.32; clogP, clogD7.4, PSA, MSA, hydrogen bond donors (HBD) and acceptors (HBA) were calculated with Marvin (20.3.0), 2020, ChemAxon; %PSA was calculated by %PSA/MSA. (−) means tested but not active; n.t. means not tested. MIC values against MRSA that are <8 μg ml−1 are highlighted in red. | |||||||||||
| 54 | 427.52 | 3.56 | 0.88 | 83.91 | 572.86 | 14.6 | 1 | 4 | 3.91 | 7.81 | 18.3 |
| 55 | 429.53 | 3.58 | 0.91 | 83.91 | 602.20 | 13.9 | 1 | 4 | 7.81 | 7.81 | 18.2 |
| 56 | 469.50 | 4.41 | 1.72 | 83.91 | 662.98 | 12.7 | 1 | 4 | 3.91 | 3.91 | 8.33 |
| 58 | 477.58 | 4.60 | 1.84 | 83.91 | 647.55 | 13.0 | 1 | 4 | 3.91 | 3.91 | 8.19 |
| 59 | 556.47 | 5.39 | 2.60 | 83.91 | 668.03 | 12.6 | 1 | 4 | 0.49 | 1.95 | 3.50 |
| 61 | 449.52 | 4.09 | 1.54 | 83.91 | 587.27 | 14.2 | 1 | 4 | n.t. | 0.98 | 2.18 |
| 62 | 451.54 | 4.11 | 1.58 | 83.91 | 616.53 | 13.6 | 1 | 4 | n.t. | 3.91 | 8.66 |
| 63 | 491.60 | 4.94 | 2.39 | 83.91 | 677.79 | 12.4 | 1 | 4 | n.t. | 0.98 | 1.99 |
| 64 | 499.59 | 5.13 | 2.51 | 83.91 | 662.21 | 12.7 | 1 | 4 | n.t. | 3.91 | 7.83 |
| 68 | 447.51 | 4.22 | 1.98 | 83.91 | 559.30 | 15.0 | 1 | 4 | 7.81 | 7.81 | 17.5 |
| 69 | 449.52 | 4.24 | 2.02 | 83.91 | 588.30 | 14.3 | 1 | 4 | 7.81 | 1.95 | 4.33 |
| 70 | 489.59 | 5.07 | 2.83 | 83.91 | 650.24 | 12.9 | 1 | 4 | 1.95 | 1.95 | 3.98 |
| 72 | 497.57 | 5.26 | 2.95 | 83.91 | 634.90 | 13.2 | 1 | 4 | 7.81 | 3.91 | 7.86 |
| 73 | 576.46 | 6.05 | 3.70 | 83.91 | 655.43 | 12.8 | 1 | 4 | n.t. | 0.98 | 1.70 |
| 79 | 484.61 | 2.79 | −0.72 | 95.94 | 684.24 | 14.0 | 2 | 4 | 1.95 | 3.91 | 8.05 |
| 86 | 496.62 | 2.10 | −1.42 | 95.94 | 680.97 | 14.1 | 2 | 4 | 1.95 | 7.81 | 15.7 |
| 87 | 537.68 | 1.89 | −1.66 | 108.83 | 736.12 | 14.8 | 2 | 5 | 7.81 | 7.81 | 14.5 |
| Ave. | 484.26 | 4.20 | 1.51 | 86.79 | 640.40 | 13.6 | 1 | 4 | |||
The drug-likeness of active tetramates was further assessed by calculation of their physiochemical properties, for which high oral availability, aqueous solubility and low protein plasma binding are considered to be important.65 The parameters MW (Da), clogP, clogD7.4, polar surface area (PSA/Å2) and relative PSA (as a % of molecular surface area, MSA/Å2) of the synthesised tetramates were calculated (Table 6). clogP and clogD7.4 predictions used Marvin 20.3.0, 2020, ChemAxon where the pool of fragments with assigned logP values is based on the data set of Viswanadhan et al.66 While an MIC of 16–32 μg ml−1 against tested pathogenic organism is considered a suitable minimum at a preliminary evaluation stage,67 effective antibacterial activity of cysteine-derived tetramates synthesised in this project was defined by an MIC against MRSA of <8 μg ml−1.68 The ranges of physiochemical properties for active tetramate derivatives were MSA of 559–737 Å2, MW of 427–577 Da, clogP of 1.8–6.1, clogD7.4 of −1.7–3.7, PSA of 83–109 Å2 and relative PSA of 12–15%. These physiochemical boundaries are comparable to antibacterial active tetramate carboxamides previously reported (MSA of 530–600 Å2, clogP of 1.2–2.5, clogD7.4 of −1.1–0, PSA of 65–80 Å2 and relative PSA of 11–14%)30,69 and these properties also resemble those of clinically used antibacterial fluoroquinolones (MSA of 380–550 Å2, clogP of 0.6–3.7, clogD7.4 of −2.7–0.5, PSA of 60–120 Å2 and relative PSA of 13–24%).68 The chemical space occupied by the active tetramates was further delineated by plots of clogP, clogD7.4, PSA and relative PSA against MW (Fig. 5). For active tetramates, their clogP and clogD7.4 values broadly correlate with MW while relative PSA changes in the inverse direction to MW (Fig. 5A, B and D). Such trends in structure–activity relationships were previously noted for antibacterial active tetramate carboxamides.69 Most of the active tetramates possess a PSA of 82–84 Å2 (Fig. 5C). The narrow ranges of PSA and relative PSA indicate the small variation in polarity which is required to maintain antibacterial activity against MRSA. Given that the work reported herein is synthesis-led, without the underpinning detailed mechanistic understanding of biological/biochemical mechanism, that the most active compounds occupy a small region of chemical space as defined by MW, clogP, PSA and %PSA (Fig. 5A–D) clearly highlights the benefit and role of chemical library synthesis and detailed evaluation in antibacterial drug discovery.
 | ||
| Fig. 5 Plots of (A) clogP, (B) clogD7.4, (C) PSA/Å2 and (D) %PSA against MW/Da. Data representing the active tetramate are highlighted in red (MIC against MRSA, <8 μg ml−1); tetramates exhibiting mild activity (8 μg ml−1 < MIC against MRSA ≤ 32 μg ml−1) are represented in yellow; and tetramates with no activity are shown in blue. | ||
While some tetramates demonstrated good antibacterial activity against Gram-positive strains of bacteria including MRSA, no activity in Gram-negative E. coli was observed. This observation was nevertheless in accordance with previous observations that Gram-positive and Gram-negative activities have different requirements of physiochemical properties.68 It was previously reported that larger and more lipophilic C-7 acyltetramates with MW > 620 Da, clogP > 3, clogD7.4 > 1, relative PSA <12% were more easily transported by efflux pumps in Gram-negative H. influenza.69 Among tetramates that were tested active against MRSA, while none of their MW exceeds 620 Da and most of their relative PSA were > 12%, the majority of them have clogP > 3 and clogD7.4 > 1, which increases their likelihood of being transported out by efflux pumps in Gram-negative bacteria. The trend of differential requirements between Gram-positive and Gram-negative activities was also noted by O'Shea and Moser where they reported the average physiochemical properties for selective Gram-negative activity as MW = 414, clogP = −0.1, clogD7.4 = −2.8 and relative PSA = 42% and for selective Gram-positive activity as MW = 813, clogP = 2.1, clogD7.4 = −0.2 and relative PSA = 30%.68 Most tetramate-containing natural products exhibit selective activity against Gram-positive bacteria while only a few (such as epicoccarine A, zopfiellamide A and kibdelomycin) have been reported to be active against Gram-negative bacteria and mycobacteria; this has been attributed to their more complex cell wall and membrane structures which impede membrane permeability.70–77 This suggests that tetramate-based derivatives generally occupy a chemical space more intrinsically in accordance with the requirement for Gram-positive activities. The average values of physiochemical properties for the active tetramates are MW = 484, clogP = 4.2, clogD7.4 = 1.5 and relative PSA = 14% (Table 7), which fit better into the chemical space for selective Gram-positive activities as described by O'Shea and Moser.68
| Cpd no. | MIC (μg ml−1) against MRSA | IC50a (μg ml−1) against | MRSA selectivityb | |||
|---|---|---|---|---|---|---|
| HeLa | HEK-239 | CaCo | MDCK | |||
| a IC50 of 62.5–125 μg ml−1 is considered non-toxic; IC50 of 15.6–31.3 μg ml−1 – less toxic and IC50 <8 μg ml−1 – toxic. b The selectivity index for MRSA was calculated as IC50/MIC. | ||||||
| 50 | 31.3 | 31.3 | 31.3 | 31.3 | 62.5 | 1.0–2.0 |
| 60 | 15.6 | 31.3 | 31.3 | 31.3 | 62.5 | 2.0–4.0 |
| 61 | 0.98 | 7.81 | 15.6 | 7.81 | 7.81 | 8.0–16.0 |
| 62 | 3.91 | 15.6 | 31.3 | 15.6 | 31.3 | 4.0–8.0 |
| 63 | 0.98 | 7.81 | 15.6 | 7.81 | 15.6 | 8.0–16.0 |
| 64 | 3.91 | 31.3 | 31.3 | 31.3 | 31.3 | 8.0 |
Interestingly, unlike C-6/C-9 modified tetramates,34 reduced metal chelation ability in the C-5 modified tetramates did not result in a loss of antibacterial activity. Those C-5 modified tetramates that showed some antibacterial activity with MIC values against MRSA ≤ 62.5 μg ml−1 were selected, and their MIC values plotted against post-column peak broadening (W1/2/Hz of the H-2 signal) (Fig. 6). The absence of correlation between activity and peak broadening indicated that among tetramates with C-5 esters, antibacterial activity does not depend on metal chelation, with 56, 58–59, 79 and 86–87 showing reduced metal binding along with good antibacterial activity with MIC values against MRSA ≤ 7.81 μg ml−1.
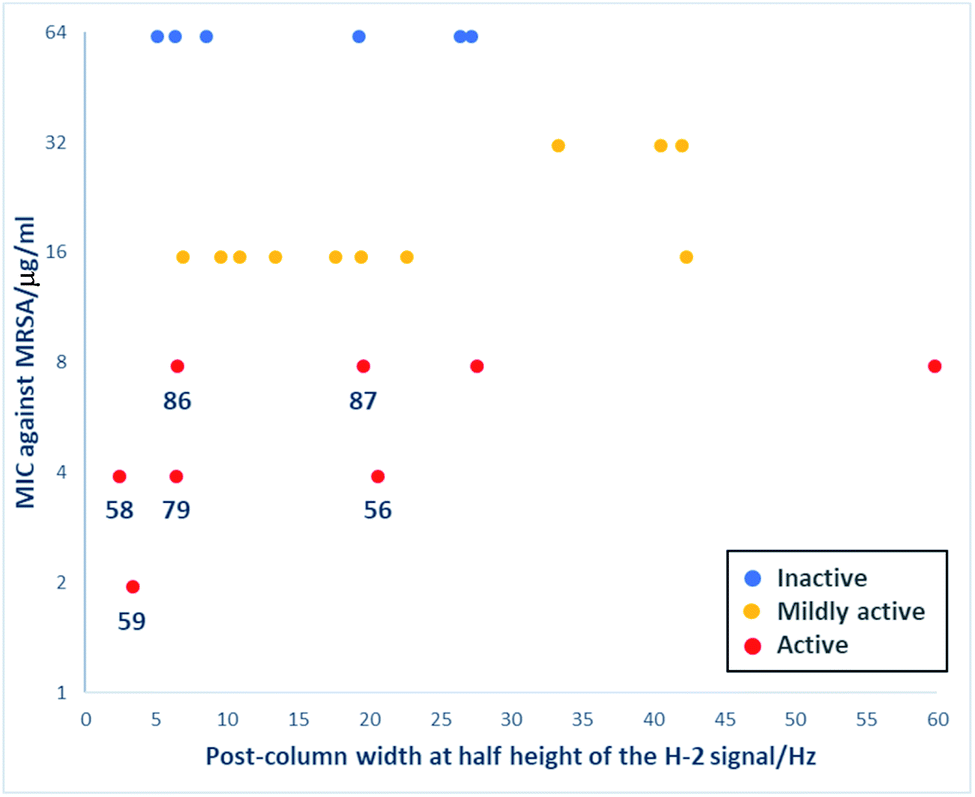 | ||
| Fig. 6 Plot of MIC values against MRSA (μg ml−1) against post-column widths at half height of the H-2 signal (Hz) for C-5 modified tetramates. Data representing the active tetramate are highlighted in red (MIC against MRSA, <8 μg ml−1); tetramates exhibiting mild activity (8 μg ml−1 < MIC against MRSA ≤ 32 μg ml−1) are represented in yellow; and tetramates with no activity are shown in blue. | ||
Tetramate ketones 54, 56, 59 were further tested against MRSA in the presence of metal ions (culture medium containing Fe3+, Ca2+, Mg2+ and Zn2+ in the same concentrations as in blood) and in this case, antibacterial activity was retained, with MIC values of 7.81, 7.81 and 0.98 (μg ml−1) respectively. While there is evidence of reduced metal chelation in these tetramate systems, it clearly does not compromise their antibacterial activity, which is in contrast with our previous attempt to directly block metal complexation.34 However, complete loss of activity in the presence of serum/blood was found, likely due to plasma protein binding due to relatively high lipophilicity (Table 7). This outcome highlights the challenge in antibacterial drug discovery of simultaneous multiparametric optimization, including consideration of at least efficacy, toxicity, membrane permeability and plasma binding.
The selectivity of tetramates 50 and 60–64 to prokaryotic bacteria cells over eukaryotic mammalian cells was analysed using cytotoxicity screens against HeLa, HEK-239, CaCo and MDCK cell lines. The selectivity index for MRSA was calculated as the ratio of IC50 in respective cell lines against the MIC value (IC50/MIC) (Table 7). Tetramates 61–64 demonstrated enhanced selectivity towards MRSA, with 61 and 63 being more active with a MIC value of 0.98 μg ml−1 and selectivity of more than 8-fold. While 62 and 64 were less active, they were also generally less toxic, with selectivity for MRSA as good as 8-fold. The different cytotoxicity towards the tested cell lines also suggested that there could be a viable therapeutic window for these tetramates even at high dose so long as specific tissue toxicities were also observed. It has been proposed that a reduction in toxicity could be achieved by increasing compound complexity with introduction of sp3 hybridised carbon and reduction of aromatic rings to improve specificity of target interaction.78,79 In this regard, the chiral bicyclic tetramate template is an attractive scaffold and among the tetramates tested for cytotoxicity, and tetramate ketones 61 and 63 are particularly promising leads for further optimisation.
Conclusion
The synthesis of cysteine-derived tetramate ketones and carboxamides with various C-5 esters, designed to decrease metal chelation by increasing bulkiness of the groups near the centre of metal binding, is reported. To achieve this goal, a library of L-cysteine esters was synthesised using acid-catalysed esterification of L-cystine in cyclohexane followed by selective reduction of the disulphide bond, with high yield and enantiopurity. This was followed by condensation with aldehydes and N-acylation, and a key step introduced in the synthetic route to incorporate a Weinreb amide to the N-acyl side chain of malonamides under Sucunza's acylation conditions allowed subsequent chemoselective Dieckmann cyclisation with DBU to proceed in the direction that retained the C-5 ester and C-7 Weinreb amide. The resulting key intermediates, tetramate Weinreb amides, allowed further functionalisation to easily access C-7 ketones and carboxamides. The target tetramates were synthesised with a cis relationship between C-2 substituents and C-5 esters, as mixtures of tautomers in non-polar solvent such as CDCl3. Qualitative comparison of post-column 1H NMR spectra suggested reduced metal binding in tetramates with bulkier C-5 esters and C-9 ketone pendants. Interestingly, modifications towards bulkier C-5 esters resulted in an enhancement of antibacterial activity against Gram-positive bacteria including MRSA, which indicated the potential for controlling metal chelation without compromising biological activity at C-5 position of the bicyclic tetramate system. Some synthesised tetramate ketones and carboxamides exhibited promising antibacterial potencies against Gram-positive MRSA with MIC values <8 μg ml−1, which were found to be independent from metal chelation. The antibacterial activity of these tetramates was maintained when tested in solvent media with metal ions of the same concentrations as in blood. Analysis of their physiochemical properties delineated a chemical space occupied by the active tetramates, with MSA of 559–737 Å2, MW of 427–577 Da, clogP of 1.8–6.1, clogD7.4 of −1.7–3.7, PSA of 83–109 Å2 and relative PSA of 12–15%. The structure–activity relationships indicated that clogP and clogD7.4 of the active tetramates change approximately in proportion to MW while relative PSA changes in the inverse direction but there is only a narrow activity window for PSA variations. The active tetramate derivatives are generally Lipinski's rule compliant, which predict for good oral availability. The set of physiochemical properties fit to the chemical space of antibacterial drugs that possess selective Gram-positive activity. A subset of tetramates was tested for their cytotoxicity in mammalian cells and some tetramates exhibit good selectivity towards prokaryotic bacterial cells. Overall, the suitability of the natural product inspired bicyclic tetramate template as a promising structural motif for the development novel antibacterial drugs, with good anti-MRSA potencies and appropriate physiochemical properties, to be drug-like, coupled with a potential for multi-targeting mechanisms and low eukaryotic cytotoxicity, has been demonstrated.Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Author contributions
RRZ conducted chemical synthesis, data collection and analysis, and manuscript preparation; MG, AP and DP conducted bioassays and reviewed the manuscript; MGM supervised the work and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.References
- G. Athanasellis, O. Igglessi-Markopoulou and J. Markopoulos, Bioinorg. Chem. Appl., 2010, 315056 CAS.
- M. Petermichl and R. Schobert, Synlett, 2017, 28, 654–663 CrossRef CAS.
- G. T. Zhang, W. J. Zhang, S. Saha and C. S. Zhang, Curr. Top. Med. Chem., 2016, 16, 1727–1739 CrossRef CAS PubMed.
- R. Schobert and A. Schlenk, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS PubMed.
- B. J. L. Royles, Chem. Rev., 1995, 95, 1981–2001 CrossRef CAS.
- P. Dandawate, S. Padhye, R. Schobert and B. Biersack, Expert Opin. Drug Discovery, 2019, 14, 563–576 CrossRef CAS PubMed.
- A. K. Renfrew, Metallomics, 2014, 6, 1324–1335 CrossRef CAS PubMed.
- M. Zaghouania and B. Nay, Nat. Prod. Rep., 2016, 33, 540–548 RSC.
- B. Biersack, R. Diestel, C. Jagusch, F. Sasse and R. Schobert, J. Inorg. Biochem., 2009, 103, 72–76 CrossRef CAS PubMed.
- S. Zorov, Y. Yuzenkova, V. Nikiforov, K. Severinov and N. Zenkin, Antimicrob. Agents Chemother., 2014, 58, 1420–1424 CrossRef PubMed.
- P. S. Steyn and C. J. Rabie, Phytochemistry, 1976, 15, 1977–1979 CrossRef CAS.
- G. F. Kaufmann, R. Sartorio, S.-H. Lee, C. J. Rogers, M. M. Meijler, J. A. Moss, B. Clapham, A. P. Brogan, T. J. Dickerson and K. D. Janda, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 309–314 CrossRef CAS PubMed.
- D. Matiadis, Catalysts, 2019, 9, 50–76 CrossRef.
- G. De Tommaso, M. M. Salvatore, R. Nicoletti, M. DellaGreca, F. Vinale, A. Bottiglieri, A. Staropoli, F. Salvatore, M. Lorito, M. Iuliano and A. Andolfi, Molecules, 2020, 25, 2147 CrossRef CAS PubMed.
- D. Matiadis, D. Tsironis, V. Stefanou, O. Igglessi-Markopoulou, V. McKee, Y. Sanakis, K. N. Lazarou, A. Chrissanthopoulos, S. N. Yannopoulos and J. M. Markopoulos, Bioinorg. Chem. Appl., 2017, 7895023 Search PubMed.
- A. Dippenaar, C. W. Holzapfel and J. C. A. Boeyens, J. Cryst. Mol. Struct., 1977, 7, 189–197 CrossRef CAS.
- M.-H. Lebrun, P. Duvert, F. Gaudemer, A. Gaudemer, C. Deballon and P. Boucly, J. Inorg. Biochem., 1985, 24, 167–181 CrossRef CAS PubMed.
- Z. Shang, L. Li, B. P. Espósito, A. A. Salim, Z. G. Khalil, M. Quezada, P. V. Bernhardt and R. J. Capon, Org. Biomol. Chem., 2015, 13, 7795–7802 RSC.
- E. Gavrielatos, G. Athanasellis, B. T. Heaton, A. Steiner, J. F. Bickley, O. Igglessi-Markopoulou and J. Markopoulos, Inorg. Chim. Acta, 2003, 351, 21–26 CrossRef CAS.
- E. Gavrielatos, C. Mitsos, G. Athanasellis, B. T. Heaton, A. Steiner, J. F. Bickley, O. Igglessi-Markopoulou and J. Markopoulos, J. Chem. Soc., Dalton Trans., 2001, 639–644 RSC.
- B. T. Heaton, C. Jacob, J. Markopoulos, O. Markopoulou, J. Nähring, C.-K. Skylaris and A. K. Smith, J. Chem. Soc., Dalton Trans., 1996, 1701–1706 RSC.
- G. Lang, A. L. J. Cole, J. W. Blunt and M. H. G. Munro, J. Nat. Prod., 2006, 69, 151–153 CrossRef CAS PubMed.
- T. Rosett, R. H. Sankhala, C. E. Stickings, M. E. U. Taylor and R. Thomas, Biochem. J., 1957, 67, 390–400 CrossRef CAS.
- D. Matiadis, V. Stefanou, D. Tsironis, A. Panagiotopoulou, O. Igglessi-Markopoulou and J. Markopoulos, Arch. Pharm., 2021, e2100305 Search PubMed.
- J. Rouleau, A. Korovitch, C. Lion, M. Hémadi, N.-T. Ha-Duong, J.-M. E. H. Chahine and T. L. Gall, Tetrahedron, 2013, 69, 10842–10848 CrossRef CAS.
- M. Petroliagi, O. Igglessi-Maikopoulou and J. Markopoulos, Heterocycl. Commun., 2000, 6, 157–164 CAS.
- O. Markopoulou, J. Markopoulos and D. Nicholls, J. Inorg. Biochem., 1990, 39, 307–316 CrossRef CAS.
- N. Umetsu, J. Kaji and K. Tamari, Agric. Biol. Chem., 1972, 37, 451–452 CrossRef.
- L. Josa-Culleré, C. Towers, A. L. Thompson and M. G. Moloney, Eur. J. Org. Chem., 2017, 7055–7059 CrossRef.
- T. D. Panduwawala, S. Iqbal, A. L. Thompson, M. Genov, A. Pretsch, D. Pretsch, S. Liu, R. H. Ebright, A. Howells, A. Maxwelld and M. G. Moloney, Org. Biomol. Chem., 2019, 17, 5615–5632 RSC.
- Y.-C. Jeong and M. G. Moloney, J. Org. Chem., 2011, 76, 1342–1354 CrossRef CAS PubMed.
- T. D. Panduwawala, S. Iqbal, R. Tirfoin and M. G. Moloney, Org. Biomol. Chem., 2016, 14, 4464–4478 RSC.
- R. Zhang, X. Li, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, J. Org. Chem., 2020, 85, 12393–12407 CrossRef CAS PubMed.
- R. Zhang, X. Li, M. Genov, A. Pretsch, D. Pretsch and M. G. Moloney, J. Org. Chem., 2021, 86, 12886–12907 CrossRef CAS PubMed.
- L. V. Lee, B. Granda, K. Dean, J. Tao, E. Liu, R. Zhang, S. Peukert, S. Wattanasin, X. Xie, N. S. Ryder, R. Tommasi and G. Deng, Biochemistry, 2010, 49, 5366–5376 CrossRef CAS PubMed.
- S. Peukert, Y. Sun, R. Zhang, B. Hurley, M. Sabio, X. Shen, C. Gray, J. Dzink-Fox, J. Tao, R. Cebula and S. Wattanasin, Bioorg. Med. Chem. Lett., 2008, 18, 1840–1844 CrossRef CAS PubMed.
- W. Sinko, C. s. d. Oliveira, S. Williams, A. V. Wynsberghe, J. D. Durrant, R. Cao, E. Oldfield and J. A. McCammon, Chem. Biol. Drug Des., 2011, 77, 412–420 CrossRef CAS PubMed.
- J. Maclaren, Aust. J. Chem., 1972, 25, 1293–1299 CrossRef CAS.
- R. P. Patel and S. Price, J. Org. Chem., 1965, 30, 3575–3576 CrossRef CAS.
- C. J. Gray, M. Quibell, K.-L. Jiang and N. Baggett, Synthesis, 1991, 1991, 141–146 CrossRef.
- H. Poras, G. Kunesch, J.-C. Barriere, N. Berthaud and A. Andremont, J. Antibiot., 1998, 51, 786–794 CrossRef CAS PubMed.
- C. Bolchi, F. Bavo and M. Pallavicini, Amino Acids, 2017, 49, 965–974 CrossRef CAS PubMed.
- L. Zervas, M. Winitz and J. P. Greenstein, J. Org. Chem., 1957, 22, 1515–1521 CrossRef CAS.
- G. Fölsch, Acta Chem. Scand., 1959, 13, 1407–1421 CrossRef.
- L. Zervas and I. Photaki, J. Am. Chem. Soc., 1962, 84, 3887–3897 CrossRef CAS.
- A. F. Lailey, L. Hill, I. W. Lawston, D. Stanton and D. G. Upshall, Biochem. Pharmacol., 1991, 42, S47–S54 CrossRef CAS PubMed.
- S. Bechaouch, B. Coutin and H. Sekiguchi, Macromol. Rapid Commun., 1994, 15, 125–131 CrossRef CAS.
- M. Hobbs, M. Butterworth, G. Cohen and D. Upshall, Biochem. Pharmacol., 1993, 45, 1605–1612 CrossRef CAS PubMed.
- E. Dauty, J.-S. Remy, T. Blessing and J.-P. Behr, J. Am. Chem. Soc., 2001, 123, 9227–9234 CrossRef CAS PubMed.
- A. Vogel, C. P. Katzka, H. Waldmann, K. Arnold, M. F. Brown and D. Huster, J. Am. Chem. Soc., 2005, 127, 12263–12272 CrossRef CAS PubMed.
- L. N. Poloni, Z. Zhu, N. Garcia-Vázquez, A. C. Yu, D. M. Connors, L. Hu, A. Sahota, M. D. Ward and A. G. Shtukenberg, Cryst. Growth Des., 2017, 17, 2767–2781 CrossRef CAS PubMed.
- B. Deka and R. J. Sarma, ChemistrySelect, 2018, 3, 5786–5791 CrossRef CAS.
- C. R. Stahl and S. Siggia, Anal. Chem., 1957, 29, 154–155 CrossRef CAS.
- J. Butler, S. P. Spielberg and J. D. Schulman, Anal. Biochem., 1976, 75, 674–675 CrossRef CAS.
- P. C. Jocelyn, Methods Enzymol., 1987, 143, 246–256 CAS.
- B. F. Erlanger and R. M. Hall, J. Am. Chem. Soc., 1954, 76, 5781–5782 CrossRef CAS.
- D. Sucunza, D. Dembkowski, S. Neufeind, J. Velder, J. Lex and H.-G. Schmalz, Synlett, 2007, 16, 2569–2573 Search PubMed.
- L. Josa-Culleré, A. Pretsch, D. Pretsch and M. G. Moloney, J. Org. Chem., 2018, 83, 10303–10317 CrossRef PubMed.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- R. P. Kurkjy and E. V. Brown, J. Am. Chem. Soc., 1952, 74, 6260–6262 CrossRef CAS.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- M. Abarbri, J. Thibonnet, L. Bérillon, F. Dehmel, M. Rottländer and P. Knochel, J. Org. Chem., 2000, 65, 4618–4634 CrossRef CAS PubMed.
- W. Strohmeier and F. Seifert, Chem. Ber., 1961, 94, 2356–2357 CrossRef CAS.
- R. E. Dessy and G. S. Handler, J. Am. Chem. Soc., 1958, 80, 5824–5826 CrossRef CAS.
- M. Gajdács, Molecules, 2019, 24, 892 CrossRef PubMed.
- V. N. Viswanadhan, A. K. Ghose, G. R. Revankar and R. K. Robins, J. Chem. Inf. Comput. Sci., 1989, 29, 163–172 CrossRef CAS.
- A. J. O'Neill and I. Chopra, Expert Opin. Invest. Drugs, 2004, 13, 1045–1063 CrossRef PubMed.
- R. O'Shea and H. E. Moser, J. Med. Chem., 2008, 51, 2871–2878 CrossRef PubMed.
- Y.-C. Jeong, M. Anwar, M. G. Moloney, Z. Bikadi and E. Hazai, Chem. Sci., 2013, 4, 1008–1015 RSC.
- M. Dafernera, T. Ankea and O. Sternerb, Tetrahedron, 2002, 58, 7781–7784 CrossRef.
- M. Figueroa, H. Raja, J. O. Falkinham, A. F. Adcock, D. J. Kroll, M. C. Wani, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2013, 76, 1007–1015 CrossRef CAS PubMed.
- V. Hellwig, T. Grothe, A. Mayer-Bartschmid, R. Endermann, F. Geschke, T. Henkel and M. Stadler, J. Antibiot., 2002, 55, 881–892 CrossRef CAS PubMed.
- J. W. Phillips, M. A. Goetz, S. K. Smith, D. L. Zink, J. Polishook, R. Onishi, S. Salowe, J. Wiltsie, J. Allocco, J. Sigmund, K. Dorso, S. Lee, S. Skwish, M. d. l. Cruz, J. Martín, F. Vicente, O. Genilloud, J. Lu, R. E. Painter, K. Young, K. Overbye, R. G. K. Donald and S. B. Singh, Chem. Biol., 2011, 18, 955–965 CrossRef CAS PubMed.
- S. V. Pronin, A. Martinez, K. Kuznedelov, K. Severinov, H. A. Shuman and S. A. Kozmin, J. Am. Chem. Soc., 2011, 133, 12172–12184 CrossRef CAS PubMed.
- S. B. Singh, P. Dayananth, C. J. Balibar, C. G. Garlisi, J. Lu, R. Kishii, M. Takei, Y. Fukuda, S. Ha and K. Younga, Antimicrob. Agents Chemother., 2015, 59, 3474–3481 CrossRef CAS PubMed.
- H. V. K. Wanguna and C. Hertweck, Org. Biomol. Chem., 2007, 5, 1702–1705 RSC.
- T. Watanabe, M. Igarashi, T. Okajima, E. Ishii, H. Kino, M. Hatano, R. Sawa, M. Umekita, T. Kimura, S. Okamoto, Y. Eguchi, Y. Akamatsu and R. Utsumi, Antimicrob. Agents Chemother., 2012, 56, 3657–3663 CrossRef CAS PubMed.
- F. Lovering, MedChemComm, 2013, 4, 515–519 RSC.
- T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05542a |
| This journal is © The Royal Society of Chemistry 2021 |