
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRNA-inspired intramolecular transesterification accelerates the hydrolysis of polyethylene-like polyphosphoesters†
Tobias P.
Haider
a,
Oksana
Suraeva
a,
Ingo
Lieberwirth
 a,
Piotr
Paneth
b and
Frederik R.
Wurm
*c
a,
Piotr
Paneth
b and
Frederik R.
Wurm
*c
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: frederik.wurm@utwente.nl
bInternational Center for Research on Innovative Biobased Materials (ICRI-BioM), Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
cSustainable Polymer Chemistry, Department of Molecules and Materials, MESA+ Institute for Nanotechnology, Faculty of Science and Technology, Universiteit Twente, PO Box 217, 7500 AE Enschede, The Netherlands
First published on 23rd November 2021
Abstract
To synthesize new (bio)degradable alternatives to commodity polymers, adapting natural motives can be a promising approach. We present the synthesis and characterization of degradable polyethylene (PE)-like polyphosphoesters, which exhibit increased degradation rates due to an intra-molecular transesterification similar to RNA. An α,ω-diene monomer was synthesized in three steps starting from readily available compounds. By acyclic diene metathesis (ADMET) polymerization, PE-like polymers with molecular weights up to 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 were obtained. Post-polymerization functionalization gave fully saturated and semicrystalline polymers with a precise spacing of 20 CH2 groups between each phosphate group carrying an ethoxy hydroxyl side chain. This side chain was capable of intramolecular transesterification with the main-chain similar to RNA-hydrolysis, mimicking the 2′-OH group of ribose. Thermal properties were characterized by differential scanning calorimetry (DSC (Tmca. 85 °C)) and the crystal structure was investigated by wide-angle X-ray scattering (WAXS). Polymer films immersed in aqueous solutions at different pH values proved an accelerated degradation compared to structurally similar polyphosphoesters without pendant ethoxy hydroxyl groups. Polymer degradation proceeded also in artificial seawater (pH = 8), while the polymer was stable at physiological pH of 7.4. The degradation mechanism followed the intra-molecular “RNA-inspired” transesterification which was detected by NMR spectroscopy as well as by monitoring the hydrolysis of a polymer blend of a polyphosphoester without pendant OH-group and the RNA-inspired polymer, proving selective hydrolysis of the latter. This mechanism has been further supported by the DFT calculations. The “RNA-inspired” degradation of polymers could play an important part in accelerating the hydrolysis of polymers and plastics in natural environments, e.g. seawater.
400 g mol−1 were obtained. Post-polymerization functionalization gave fully saturated and semicrystalline polymers with a precise spacing of 20 CH2 groups between each phosphate group carrying an ethoxy hydroxyl side chain. This side chain was capable of intramolecular transesterification with the main-chain similar to RNA-hydrolysis, mimicking the 2′-OH group of ribose. Thermal properties were characterized by differential scanning calorimetry (DSC (Tmca. 85 °C)) and the crystal structure was investigated by wide-angle X-ray scattering (WAXS). Polymer films immersed in aqueous solutions at different pH values proved an accelerated degradation compared to structurally similar polyphosphoesters without pendant ethoxy hydroxyl groups. Polymer degradation proceeded also in artificial seawater (pH = 8), while the polymer was stable at physiological pH of 7.4. The degradation mechanism followed the intra-molecular “RNA-inspired” transesterification which was detected by NMR spectroscopy as well as by monitoring the hydrolysis of a polymer blend of a polyphosphoester without pendant OH-group and the RNA-inspired polymer, proving selective hydrolysis of the latter. This mechanism has been further supported by the DFT calculations. The “RNA-inspired” degradation of polymers could play an important part in accelerating the hydrolysis of polymers and plastics in natural environments, e.g. seawater.
1. Introduction
Polyethylene (PE) is the most produced commodity plastic, with an annual demand in Europe of more than 15 million tonnes (in 2017).1 Its excellent mechanical properties and high chemical resistance make PE a suitable material for a variety of applications. Yet, a high chemical resistance is accompanied by a low degradability in natural environments. With rising global plastic production and increased plastic waste, new concepts for the end-of-life of polymers are needed. Besides new concepts for recycling, also degradable alternatives to commodity polymers are an important puzzle piece for more sustainable polymeric materials.2To enhance the degradability of PE, one approach utilizes functional groups that are built into the aliphatic polymer backbone and act as breaking points.3 Van der Waals interactions between the hydrocarbon segments in such materials result in semi-crystalline polymers with thermal properties comparable to conventional polyolefins. However, a high degree of crystallinity and hydrophobicity minimizes the degradation rate, as it was shown for the hydrolytic degradation of so-called “long-chain” polyesters.4 Thus, incorporating functional groups that are more prone to hydrolytic degradation is advisable. In this way, our group recently reported acid-labile long-chain polyorthoesters, which resulted in adjustable hydrolysis rates depending on the orthoester structure.5 However, fast hydrolysis of monomers makes the handling difficult and further functionalization of the orthoester groups is a synthetic challenge.
Herein, we present an RNA-inspired degradation unit that was installed into an aliphatic polymer backbone and controlled the backbone scission by an intramolecular transesterification similar to RNA-hydrolysis, similar to recent studies in polylactide.6 Further, the pendant hydroxyl groups allowed the functionalization of the polymers.6
The stability and degradation profiles of natural polymers are perfectly adjusted to their tasks. Deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are polyphosphodiesters that are negatively charged at physiological pH.7 This is important on one hand as the resulting polyanions will not pass through cell membranes.8 On the other hand, those negatively charged polypohosphodiesters are less susceptible to hydrolysis, which is essential for the storage of the genetic code by DNA. In contrast, RNA is constantly reproduced and has lower stability insufficient for ongoing gene expression but is also based on a polyphosphodiester. The rate of spontaneous hydrolysis of RNA is 100 times higher than that of DNA at room temperature, mainly due to the hydroxyl group at the C2 position of ribose, which is missing in DNA.9 Due to the proximity of this hydroxyl group to the polymer backbone, an intramolecular transesterification via a 5-membered cyclic intermediate leads to chain scission (Scheme 1A).8 This RNA-inspired module was installed into PE-like polyphosphoesters (PPE), which led to an accelerated hydrolysis rate compared to analog PE-like PPEs without the pendant ethoxy hydroxyl group.
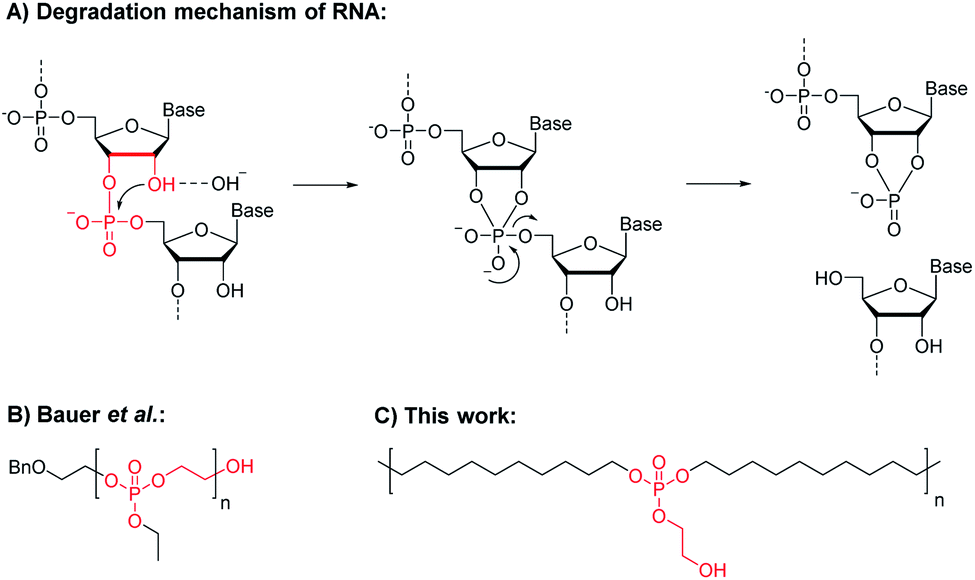 | ||
| Scheme 1 Intramolecular transesterification and chain scission. Top: The hydrolytic degradation mechanism of RNA. Bottom: Synthetic polyphosphoesters with a terminal hydroxyl group in close proximity, i.e. poly(ethyl ethylene phosphate) (reported by Bauer et al. recently),10 which led to a backbiting degradation, and poly-1H presented in this work, carrying the same RNA-analog motif along a hydrophobic long-chain PPE, which is supposed to lead to chain scission by intramolecular transesterification. | ||
Recently, we were able to prove that poly(ethyl ethylene phosphate) with the terminal RNA-inspired ethoxy hydroxyl group degraded mainly via a backbiting mechanism via a 5-membered cyclic intermediate, which was detected by NMR spectroscopy (Scheme 1B).10 When the end-group was blocked, dramatically reduced hydrolysis rates were determined. Herein, we propose this RNA-inspired transesterification as a general chemical module to increase polymer degradation by intramolecular transesterification and to establish seawater-degradable polymers.11 We prepared RNA-inspired polyethylene mimics carrying phosphate groups with pendant ethoxy hydroxyl groups that should be able to accelerate hydrolytic degradation of these hydrophobic polymers (Scheme 1C). Due to its proximity, the pendant hydroxyl group should be capable to undergo transesterification with the phosphoester in the polymer backbone. Acyclic diene metathesis (ADMET) polymerization and subsequent hydrogenation were used to obtain a linear polyphosphate, which was characterized in detail. Further, the hydrolytic degradation of polymer films in water (at different pH values) was studied. We were able to prove that such polymers degraded faster than the analogs without the pendant ethoxy hydroxyl groups. In addition, we proved that the degradation followed the anticipated and by DFT calculations predicted intramolecular degradation pathway by studying hydrolysis in solution and films.
We believe that this general motif of ethoxy hydroxyl phosphate can be used to accelerate the degradation of other polymers as well and that it can be an important strategy for fighting plastic pollution as a safeguard when polymers are littered into the ocean. Moreover, the herein presented OH-functional PPEs further enlarge the family of functional PPEs as the OH-groups could also be used for post-polymerization functionalization. Exemplary, we prepared the first PPE grafted with PLA chains by the grafting-from polymerization of lactide. This approach might further be used to alter the degradability of such complex macromolecular architectures.
2. Results and discussion
Monomer synthesis
Monomer 1 was synthesized in a two-step reaction starting from POCl3 (Fig. 1A). In the first step, 2-(benzyloxy)ethanol was reacted with an excess of POCl3 in the presence of triethylamine as an HCl scavenger to 1a. The excess of POCl3, which prevented the formation of di- or tri-functionalized phosphoesters, was removed at reduced pressure after the esterification and 1a was used directly in the following step. Compound 1a was reacted with 10-undecen-1-ol to give monomer 1 as a viscous oil in high purity in 57% yield (cf. NMR spectra in the ESI†).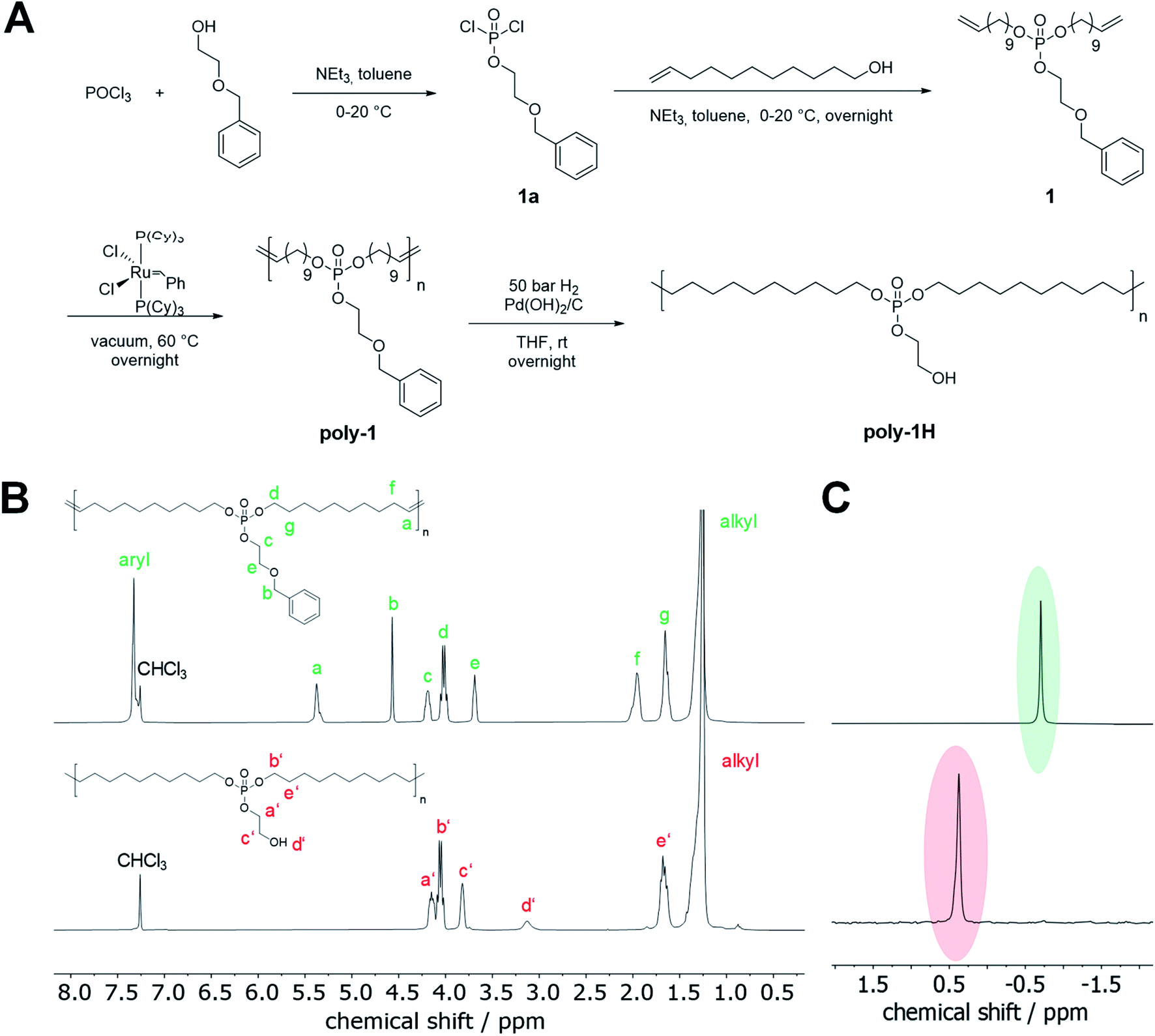 | ||
| Fig. 1 (A) Synthesis of monomer 1, ADMET polymerization and post-polymerization functionalization. (B) 1H NMR (300 MHz at 298 K, in CDCl3) of poly-1 (top) and poly-1H (bottom). (C) 31P NMR (121 MHz at 298 K, in CDCl3) of poly-1 (top) and poly-1H (bottom). | ||
The benzyl ether protection group was chosen for two reasons: First, it does not interfere with the ADMET polymerization process. Second, hydrogenation of the unsaturated ADMET polymer and debenzylation was conducted in a single catalytic hydrogenation step.
ADMET polymerization and hydrogenation
ADMET polymerization is a step-growth polymerization that produces linear polymers by a polycondensation reaction of a diene under the elimination of ethylene.12 High vacuum is usually applied during the polymerization to remove the evolving ethylene and to achieve high molecular weights. ADMET polymerization of 1 was carried out in bulk at 60 °C at high vacuum using 1st generation Grubbs catalyst. Polymers with reproducible molecular weights up to 38400 g mol−1 were obtained (Table 1). Comparing the 1H NMR spectrum of the polymer with the monomer spectrum, the resonances for the terminal double bonds at 5.81 and 4.95 ppm vanished, while a new signal at 5.36 ppm appeared corresponding to the formed internal double bonds (Fig S4† and 1B). Signals at 7.35 and 4.57 ppm indicated that the benzyl ether protection group remained intact during the polymerization process. Moreover, there was no shift in the 31P NMR spectrum detectable (Fig. S6† and 1C). ADMET polymerization produces unsaturated polymers with mixed cis/trans double bonds which hinder the crystallization of the polymer,13 resulting in polymers with low melting points. Poly-1 was obtained as a viscous, amorphous oil at room temperature. To increase the thermal and mechanical properties of the material, Pd-catalyzed hydrogenation was performed to prepare the saturated poly-1-H in quantitative yields. At the same time, the benzyl ether protection group was removed to release the pendant hydroxyl groups (Fig. 1A).
| Entry | Scale [g] | Unsaturated polymers (Poly-1) | Saturated polymers (Poly-1H) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M n [g mol−1] | M w [g mol−1] | M w/Mna | M n [g mol−1] | M w [g mol−1] | M w/Mnb | Deprotectione [%] | T m [°C] | ΔmHc [J g−1] | Crystalinityd [%] | ||
| a Determined by SEC in THF vs. polystyrene standards. Values for unsaturated polymers. b Determined by SEC in THF vs. polystyrene standards. Values for hydrogenated polymers. c Determined by DSC. Values for hydrogenated polymers. d Relative to 100% crystalline PE (ΔHm = −293 J g−1). e Removal of benzyl ether protecting group during hydrogenation step. n.d. = not determined. f As this sample reached only 91% deprotection, the respective data is labeled with poly-1H-co-poly-1Bn in the manuscript and the ESI. | |||||||||||
| 1 | 0.8 | 16700 |
34900 |
2.08 | 15500 |
37200 |
2.40 | 100 | n.d. | n.d. | n.d. |
| 2 | 1.2 | 20300 |
38400 |
1.89 | 7400 | 21000 |
2.84 | 100 | 86 | −102 | 35 |
| 3 | 1.3 | 16000 |
31300 |
1.96 | 11700 |
21000 |
1.79 | 100 | 84 | −95 | 32 |
| 4 | 1.2 | 15600 |
45500 |
2.92 | 24500 |
46600 |
1.90 | 91f | n.d. | n.d. | n.d. |
The benzyl ether group was reluctant to deprotection, as standard reaction conditions for debenzylation using Pd/C as a catalyst led to no or only little removal of the benzyl ether group. Only when Pd(OH)2 on charcoal with 20 wt% Pd(OH)2/C was used, complete removal of benzyl protecting groups was achieved. After hydrogenation, the resonances for the benzyl ether groups at ca. 7.35 ppm disappeared in the 1H NMR spectrum as well as the signals at 5.36 and 1.96 ppm, indicating quantitative hydrogenation (Fig. 1B). Signals attributed to the CH2 groups in the phosphate side chain were slightly shifted and a new, broad resonance at 3.13 ppm was detected, which corresponded to the hydroxyl groups. In the 31P NMR spectrum, a clear and complete shift from −0.70 ppm to 0.36 ppm after hydrogenation was detected (Fig. 1C). SEC data further indicates a decrease in molar mass after hydrogenation due to the change in the hydrodynamic radii of the polymers. The pendant hydroxyl groups were further studied to graft polylactide to the PPE and how they influence the thermal properties and hydrolytic stability (see below). For studies on the degradation mechanism, poly-1Bn was synthesized by selective hydrogenation of the olefins in the polymer backbone but without cleavage of the benzyl ether group. For this approach, a Fischer carbene prepared by the reaction of Grubbs catalyst 1st generation with ethyl vinyl ether was used as a hydrogenation catalyst.14
Solid-state properties
Poly-1H was obtained as a colorless solid material at room temperature. A 3 mm thick and 4 × 4 cm large film of the semi-crystalline polymer was prepared by pressing the polymer melt between two metal plates (Fig. 2A). The film was flexible to some extent, but relatively brittle, presumably, due to the relatively low molecular weight of the polymer (for comparable long-chain polyesters, a brittle-to-ductile transformation for an Mw between 53 × 103 and 78 × 103 g mol−1 was reported15). Its thermal stability was examined by thermal gravimetric analysis (TGA) with an onset degradation temperature (Ton) after 5% degradation at ca. 275 °C, which was similar to other previously prepared long-chain PPEs16 and ca. 150 °C lower compared to Ton of HDPE but well above the melting point (Fig. S24†). The char yield of ca. 19 wt%, however, is higher than for HDPE, as phosphate-containing char was produced during the decomposition, which might add additional flame-retardant properties to these materials and will be a subject of further studies in our group (theoretical phosphate amount: 22 wt%).17 The melting (Tm) and the crystallinity of the polymer were determined by differential scanning calorimetry (DSC). Poly-1H exhibited a melting event at ca. 86 °C and a glass transition temperature of ca. −17 °C (Fig. 2B). In contrast to other PPEs with similar structures but without pedant OH-groups, poly-1H exhibited a relatively high Tm that might be attributed to intermolecular hydrogen bonds of the hydroxyl groups. A structural analog PPE with an ethyl side chain but the same spacer length of 20 CH2 groups exhibited a ca. 35 °C lower melting temperature (Tm of 51 °C).16 The strong effect of hydrogen bonding was shown by Wagener et al. for hydroxyl-functionalized polyethylenes: rather than the length of the polyethylene segment between each hydroxyl-bearing carbon, hydrogen bonding dominated the polymers' thermal behavior.18 In accordance, a long-chain phosphoric acid diester with a spacer length of 20 CH2 groups synthesized by Tee et al. exhibited a melting point of 93 °C due to strong hydrogen bonding between phosphoric acids.19 The crystallinity of poly-1H was calculated by comparing its melting enthalpy ΔHm of −102.0 J g−1 (entry 2) to ΔH of 100% crystalline polyethylene (ΔHm = 293 J g−1)20 to give a value of ca. 35%. This value is significantly higher in comparison to an ADMET pyrophosphate with the same aliphatic spacer of 20 CH2 groups (23%),21 but lower than for comparable long-chain polyesters like e.g. poly(pentadecalactone) with a crystallinity of 64%.22 The carboxylic acid esters have a lower impact on the crystallization of the polyethylene segments in long-chain polyesters and the polymers generally crystallize in an orthorhombic crystal structure like polyethylene.3 In contrast, long-chain PPEs with a PE segment of 20 CH2 groups had been proven to crystallize with the bulky phosphate groups being expelled from the lamellar crystal.23,24 The crystal structure of poly-1H was determined by wide angle X-ray scattering (WAXS) and compared to HDPE (Fig. 2C). While the orthorhombic HDPE has two distinct reflections at 21.4 and 23.8, poly-1H exhibited a single, broader peak at 21.1. The peak pattern of poly-1H suggests a pseudo-hexagonal crystal structure as reported for other PPEs previously.16,23,25 The broadening of the peak indicates a lower crystallinity in comparison to HDPE.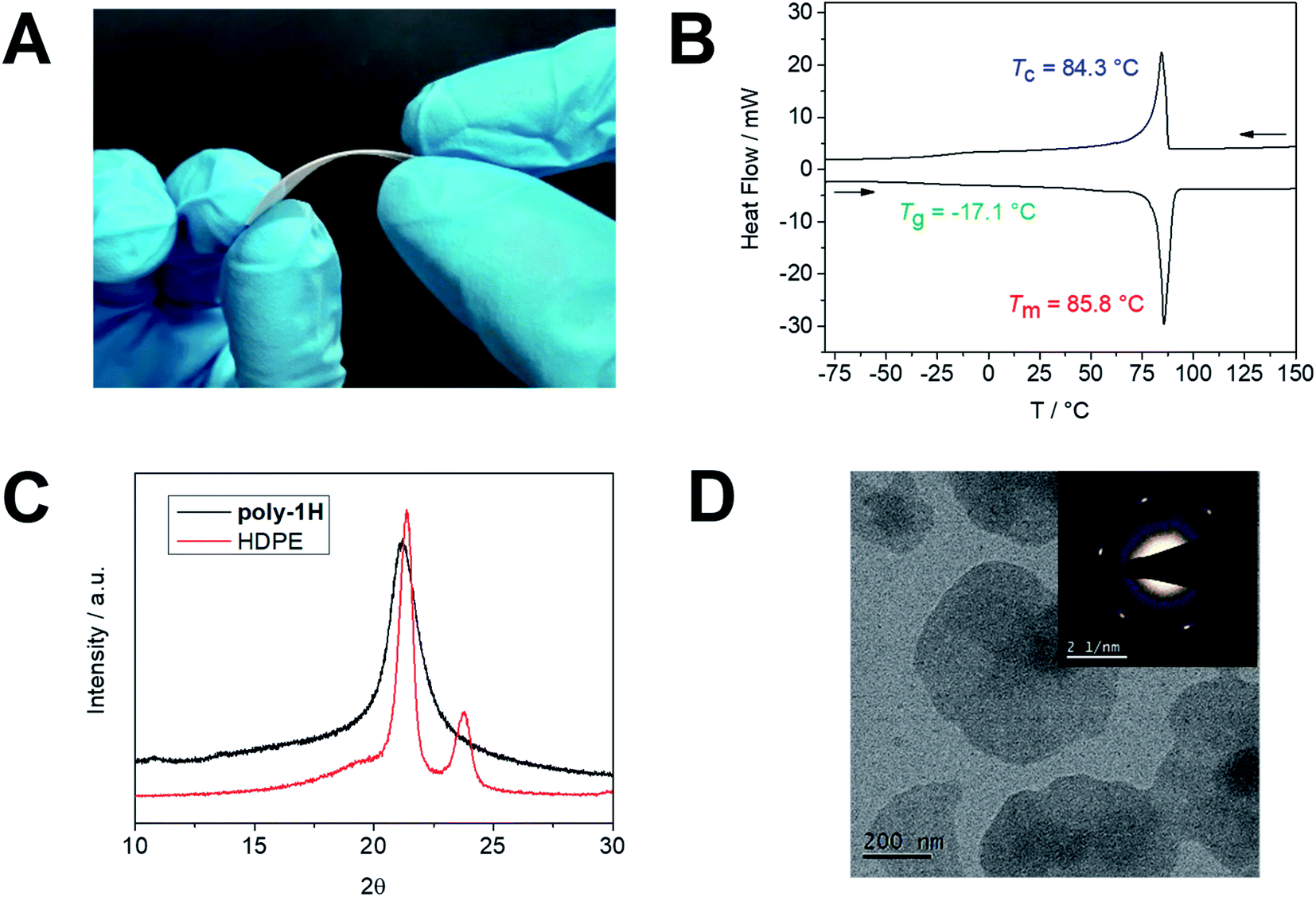 | ||
| Fig. 2 (A) Photograph of poly-1H film. (B) DSC thermogram of poly-1H. (C) XRD diffractogram of poly-1H (black) and HDPE (red). (D) TEM bright-field micrograph and corresponding diffraction pattern (inset) of solution-grown crystals of poly-1H (by cooling a 0.05 mg mL−1 solution in ethyl acetate). | ||
Besides bulk properties, we prepared solution-grown polymer crystal platelets of poly-1H. A 0.05 mg mL−1 solution of poly-1H in ethyl acetate was heated to 70 °C and slowly cooled to room temperature to induce crystallization. The resulting dispersion of anisotropic polymer platelets was drop-cast onto a transmission electron microscopy (TEM) grid and visualized (Fig. 2D). The average lateral sizes of the anisotropic polymer crystal-platelets were ca. 400 nm. Compared to crystallization in bulk from the melt, solution-crystallized polymers have a substantially higher crystallinity and thin anisotropic crystals with a thickness of <10 nm can be prepared.26 The electron diffraction (Fig. 2D inset) correlated with WAXS data and reveals the single-crystal pattern of pseudo-hexagonal polymer crystals.
Hydrolytic degradation
PPEs can be degraded by enzymes or hydrolysis.27 Penczek et al. investigated the hydrolytic degradation of the water-soluble poly(methyl ethylene phosphate).28 While the (slow) acidic degradation occurred by a nucleophilic attack of water at the α-carbon atom in the side chain, the degradation under basic conditions proceeded via nucleophilic attack of a hydroxyl ion at the phosphorus center.28 They reported a trigonal bipyramidal transition state, from which the side-chain or main-chain could be cleaved at the same rate. A similar degradation pattern had been reported by Becker et al. for PPEs prepared by ADMET polymerization.29 Recently, Bauer et al. proved that the ethoxy hydroxy end group in linear and water-soluble poly(ethyl ethylene phosphate) resulted in a predominant degradation via a backbiting mechanism (under basic conditions). The hydroxyl end group attacked the last repeat unit and a 5-membered transition state was formed, which led to the release of a single repeat unit.10 Leong et al. synthesized two different water-soluble polyphosphates with a propylene backbone and an ethylamine and ethyl alcohol side chain, respectively.30,31 They found an increased degradation rate of both water-soluble polymers compared to other PPEs even at physiological pH. These indicative findings led us to the assumption that the RNA-inspired intramolecular transesterification could be general pathway to increase polymer hydrolysis rates by installing ethoxy hydroxyl groups also in hydrophobic and PE-like polymers, which are typically relative recalcitrant to hydrolysis. In contrast to water-soluble polymers, polyethylene-like PPEs are in general remarkably robust to hydrolytic degradation due to increased crystallinity and hydrophobicity. A polyphosphate with an ethoxy side chain and an aliphatic spacer of 20 CH2 groups did not show any degradation in phosphate-buffered saline (PBS) solution (pH 7.1) at 37 °C for 2 months.21 Only at pH > 12 the polymer was degradable within this timeframe. Other PE-mimics like long-chain polyesters showed similar degradation profiles: poly(pentadecalactone) was stable at pH 7.2 for 2 years and showed no decrease in molecular weight.4 Polyethylene itself is remarkably stable towards hydrolysis, as the polymer consists only of non-polar C–C and C–H bonds which do not provide centers for electrophilic or nucleophilic attacks.32 Oxygen-containing groups (mainly carbonyls) can be incidentally present in the polymer chain by air-oxidation during polymer extrusion or abiotic oxidation. However, these defects are very rare, minimizing their influence on the overall degradability of polyethylene. As a result, in an accelerated degradation test of polyethylene microplastic in artificial seawater over eight weeks, only minor changes such as micro-cracks of the pellet's surface or an increased number of oxidized groups along the polymer chain could be observed, but no significant degradation of the polymer chain itself.33 Also, blending polyethylene with degradable polymers like starch does produce a higher surface/volume ratio of the polyethylene matrix, however, does not increase the weight loss of the material in a 20 month-long degradation test in the Baltic Sea.11A conventional long-chain PPE, i.e. without the RNA-inspired motif, degrades by statistical hydrolysis (under basic conditions) of the main chain and pendant chain leading to a (poly)phosphodiester.27 The phosphodiester is relatively stable against a further attack of hydroxyls, due to its negative charge.27 For the RNA-inspired long-chain PPEs, different scenarios are possible (as shown in Scheme 2).
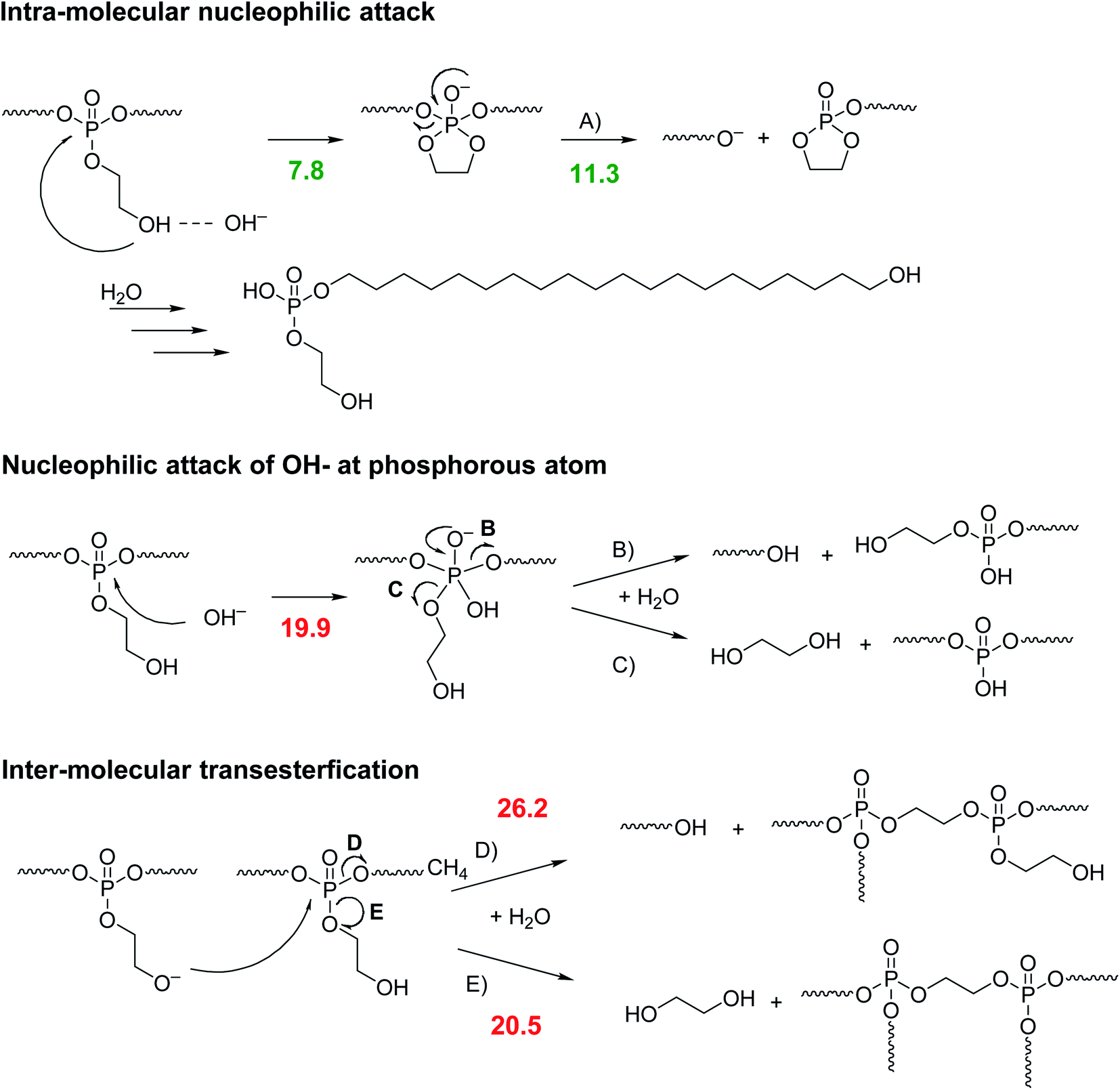 | ||
| Scheme 2 Potential hydrolysis pathways for poly-1H either by intra or inter-molecular transesterification (the numbers indicate the Gibbs free energies for the rate-determining steps in kcal mol−1 for the respective pathway). | ||
The degradation by the envisioned intra-molecular nucleophilic attack of the pendant hydroxyl group (pathway A) via the cyclic intermediate state, which was supported by DFT calculations (see numbers in Scheme 2). This pathway would lead eventually to a single amphiphilic degradation product with surface-active properties. In contrast, if water or hydroxyls would attack the polymer backbone, phosphodiester anions would be generated, too (pathways B and C). However, also the release of ethylene glycol would be detectable. Inter-molecular transesterification (pathways D and E) would also be possible and would lead to the formation of branched or cross-linked species and the release of ethylene glycol (pathway E).
The mechanistic alternatives have been approached by using theoretical tools that proved successful in our previous studies of the reactions mechanisms of organophosphorus compounds.34 Gibbs free energies for the rate-determining steps in kcal mol−1 are listed in Scheme 2. In the case of pathway A, the cyclization step is less energy demanding than the subsequent departure of one part of the polymer chain, which is overall rate-limiting. As in previous calculations, no direct SN2(P) reaction has been observed. Upon hydroxyl attack, an adduct intermediate is formed. This addition step by about 9 kcal mol−1 more energy demanding than the RNA-inspired pathway A and thus mechanisms that comprise the initial nucleophilic attack of the hydroxyl anion can be excluded from consideration. Alternative intermolecular nucleophilic substitution by the deprotonated pendant has been found to require even more energy and with the barrier higher than 26 kcal mol−1 can be safely disregarded.
To elucidate the degradation mechanism experimentally, hydrolytic degradation tests of films prepared from poly-1H were performed at 37 °C at different pH values. Therefore, films of poly-1H with a diameter of ca. 2 cm and a weight of ca. 25 mg each, were coated on glass cover slides and were immersed in NaHCO3/NaOH buffer (pH 11.1) or NaOH solution (pH 13.1), respectively. Samples were taken after one, two, and three weeks. All polymer films were detached from the glass cover slides and disintegrated into small pieces after one week (Fig. 3A). The surface tensions of all solutions decreased in comparison to the initial solutions, indicating the presence of surface-active phosphodiester degradation products (Table S1†). In addition, the pH values of the NaHCO3/NaOH buffer dropped to 10.6, indicating the formation of phosphoric acid derivatives (cf. Table S2†). The residual solids were washed with water and dried at reduced pressure. Samples at pH 13.1 were insoluble in any solvent after degradation, while samples at pH 11.1 were still soluble in CHCl3 and THF but the solubility decreased over time. Thus, only samples at pH 11.1 were analyzed by SEC and NMR spectroscopy. SEC measurements revealed for pH 11 no residual polymer after one week and later. In 1H NMR, new peaks at 3.63 and 1.55 ppm appeared, corresponding to the deprotonated phosphodiester as the main degradation product (Fig. 3B).
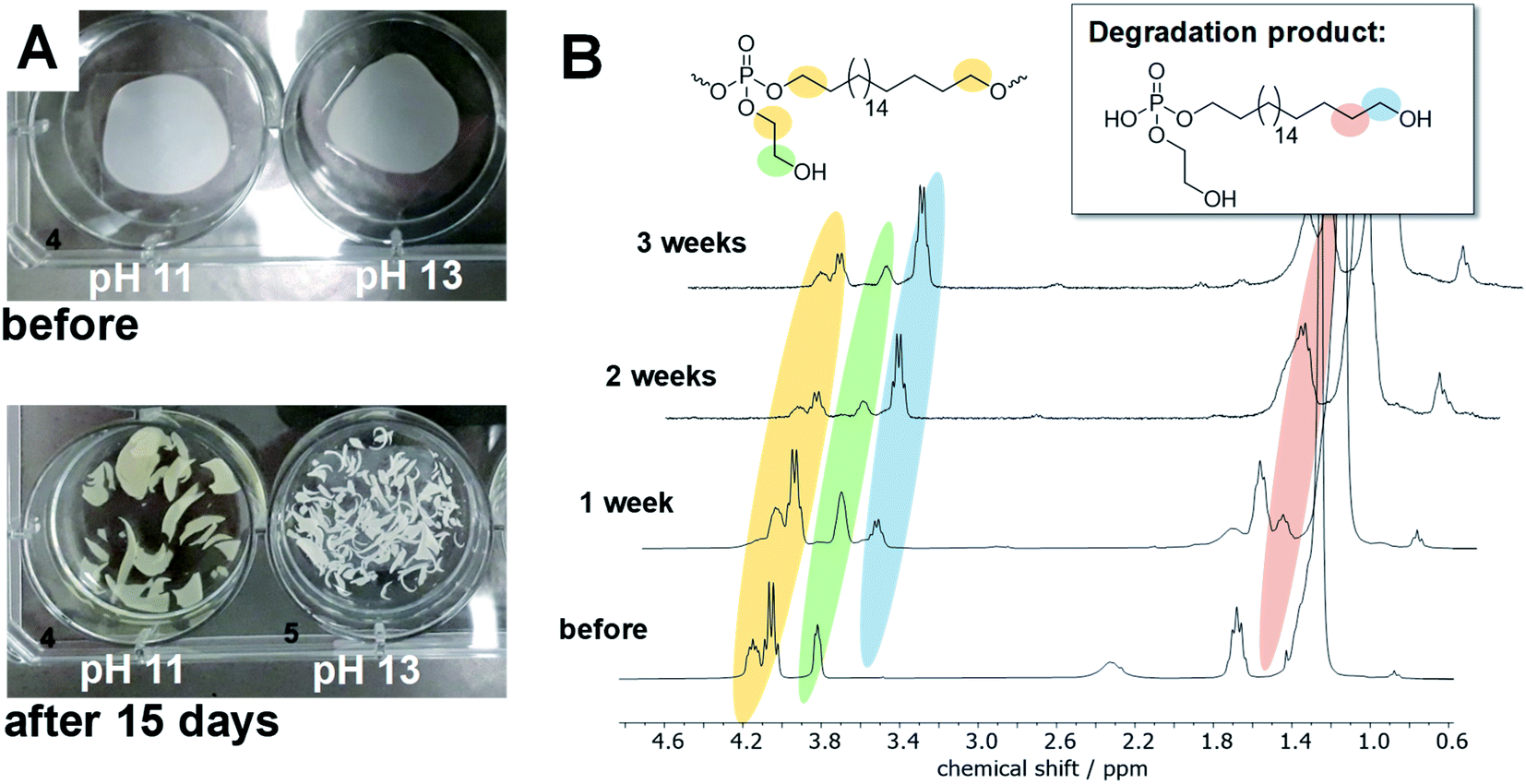 | ||
| Fig. 3 (A) Films of poly-1H before and after 15 days immersing in different buffer solutions at 37 °C. Diameter of films is ca. 2 cm. (B) 1H NMR (300 MHz, CDCl3) overlay of poly-1H after degradation at pH 11.1. | ||
31P NMR showed beside the initial peak at 0.36 ppm additional peaks at 1.91 (phosphodiester), 0.18, and 0.09 ppm. For the hydrolysis of water-soluble poly(ethyl ethylene phosphate) (PEEP), Bauer et al. detected the signal corresponding to the cyclic species at ca. 18 ppm in the 31P NMR while monitoring the degradation in solution.10 As the degradation of poly-1H was conducted in a film and degradation products were studied after dissolving the remaining material, the cyclic intermediate was not detectable as it ring-opens quickly. In contrast, when poly-1H was dissolved in CDCl3 together with a five-fold excess of diazabicycloundecene (DBU) as a strong base, a new resonance in the 31P NMR spectra at ca. 17.5 ppm indicated the presence of the cyclic intermediate state (Fig. 4), which was presumably in equilibrium with the deprotonated phosphotriester and the phosphodiester degradation product.
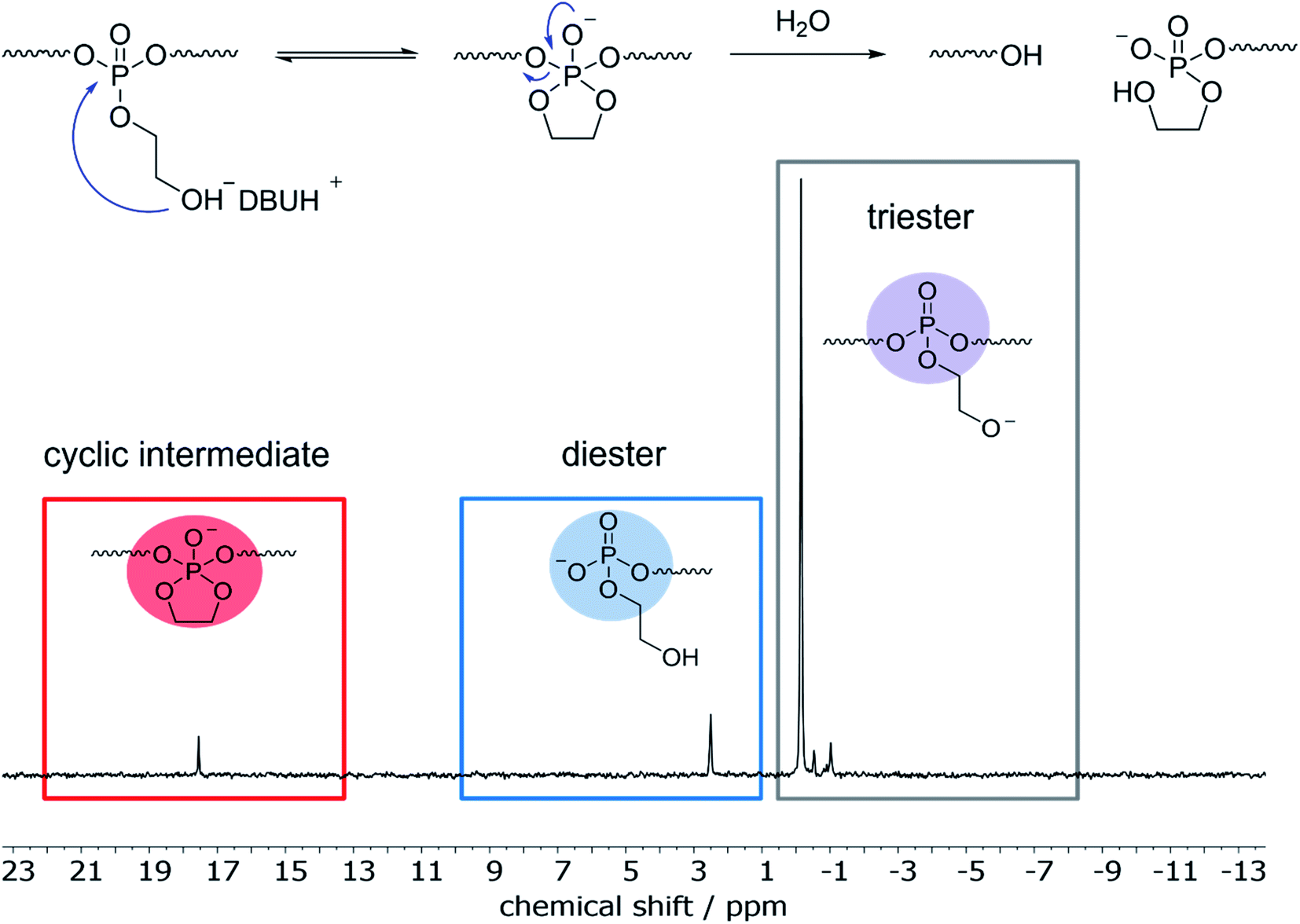 | ||
| Fig. 4 Top: Indication of the major degradation pathway by the intramolecular attack of the pendant OH-groups on the phosphate groups with subsequent main-chain cleavage. Bottom: 31P NMR (121 MHz, CDCl3) spectrum of poly-1H after addition of DBU showing the cyclic intermediate due to the RNA-inspired degradation pathway. | ||
Moreover, no ethylene glycol was detected by 1H NMR when poly-1H was kept as a powder in the open air for several months, which only resulted in partial hydrolysis of the material (Fig. S11†). These findings suggest that the hydrolysis of poly-1H occurred by an intramolecular nucleophilic attack according to pathway A in Scheme 2.
To further prove the degradation mechanism and to exclude degradation via an inter-molecular nucleophilic attack, the basic hydrolysis of a blended film of poly-1H and poly-1Bn was studied by NMR and SEC for two months. As a reference, separate films of both polymers were monitored over the same time frame (Fig. 5). Due to the pendant benzyl groups along poly-1Bn, the polymer could be selectively detected by SEC as it is UV-active, while poly-1H did not exhibit a UV signal at 254 nm absorbance. Since the free hydroxyl group in the pendant side chain is blocked, hydrolytic degradation of poly-1Bnvia an intramolecular nucleophilic attack is excluded. Separate films of poly-1H and poly-1Bn and a blended film of poly-1H/poly-1Bn (50/50 wt%) were cast on glass cover slides and immersed into an aqueous NaOH/NaHCO3 buffer at pH 11.1. After 2 months at 37 °C, the film of poly-1H disintegrated and lost all its mechanical strength (Fig. S27†), while the film prepared from poly-1Bn remained intact. The blended film showed partial disintegration. After washing and drying, the film of poly-1Bn remained fully soluble in organic solvents and did not show signs of degradation by NMR and SEC. The poly-1H/poly-1Bn blend did not entirely dissolve in CHCl3 and THF, the residual poly-1H polymer was completely insoluble. In the 31P NMR spectrum of the polymer blend after degradation, the signal corresponding to poly-1H vanished completely while the signal corresponding to poly-1Bn remained unchanged (Fig. 5A). No additional peaks were visible in the 31P NMR spectrum. So, any inter-molecular transesterification between poly-1H and poly-1Bn can be excluded.
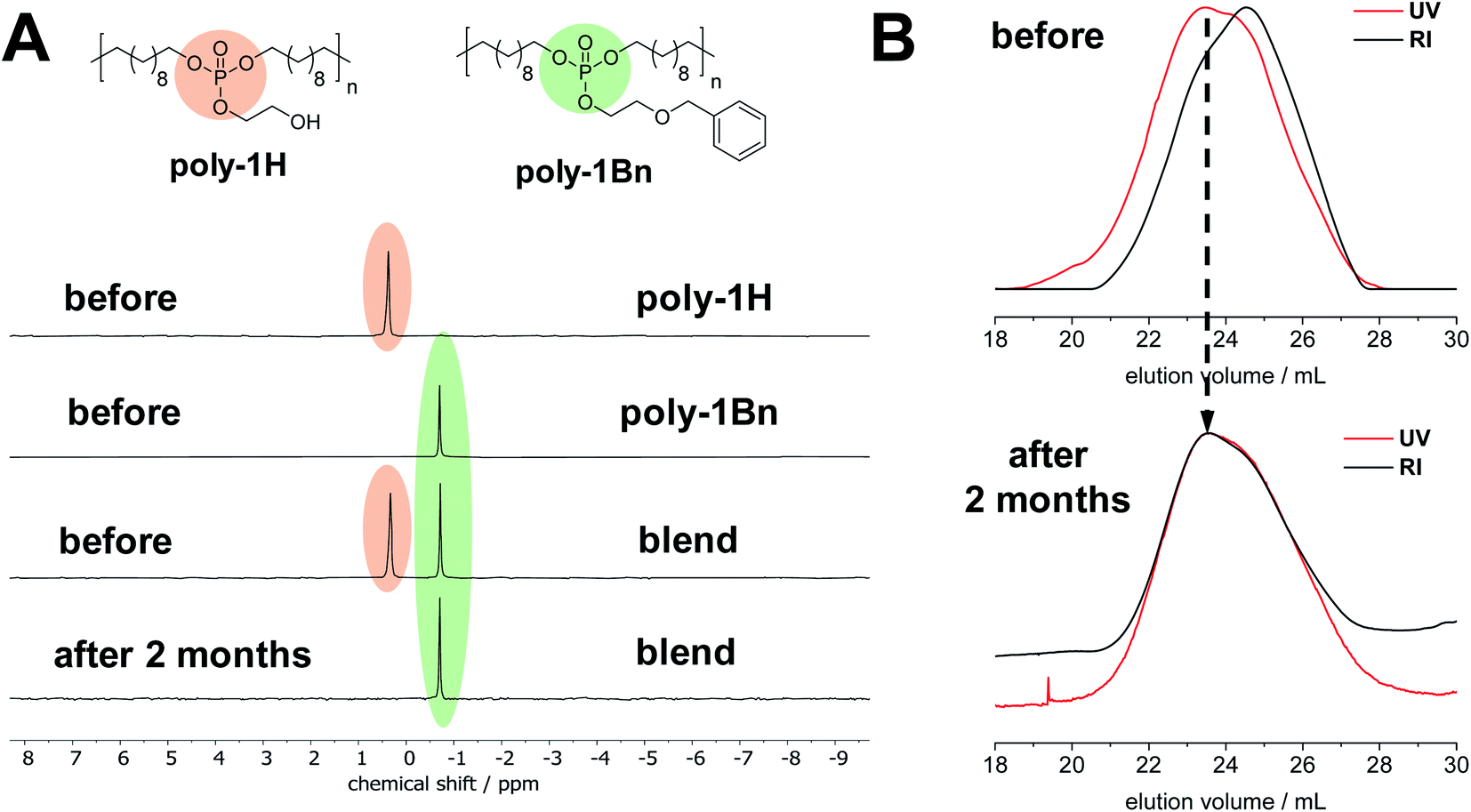 | ||
| Fig. 5 Simultaneous hydrolytic degradation test of poly-1H/poly-1Bn blend at pH 11. (A) Overlay of 31P NMR spectra (121 MHz, CDCl3) of poly-1H/poly-1Bn blend and homopolymers before and after degradation studies. (B) Overlay of SEC traces (UV: red, RI: black) in THF of poly-1H/poly-1Bn blend before (top) and after (bottom) degradation. | ||
Moreover, SEC measurements after degradation revealed a selective hydrolytic degradation of poly-1H (Fig. 5B). In contrast to the SEC elugram of the polymer blend before degradation, the RI and UV trace after degradation overlap with each other. Since only poly-1Bn is UV-active, this indicates the single presence of the poly-1Bn, while poly-1H is fully degraded. The differences in degradation rate at pH 11 theoretically could be explained by a difference in hydrophobicity (with poly-1Bn being more hydrophobic than poly-1H). Yet, all findings combined suggest that the enhanced degradation rate of poly-1H is best explained by its hydrolytic degradation via an intramolecular nucleophilic attack by the deprotonated OH-group in the side chain at the central phosphorus atom similar to the degradation of RNA.
After having elucidated the degradation mechanism at rather drastic conditions at pH 11 and 13, we investigated the degradation behavior of poly-1H at conditions more relevant to natural environments. Therefore, we performed hydrolytic degradation tests at room temperature with polymer films immersed in an aqueous solution of phosphate-buffered saline (PBS) at pH 7.4 and in artificial seawater at pH 8.1. The polymer batch (entry 4) used for these degradation tests was slightly more hydrophobic than the polymer previously used, due to incomplete removal of the benzyl ether protection group during polymer synthesis. The obtained copolymer poly-1H-co-poly1Bn had a remaining benzyl ether content of ca. 9% (cf.1H and 31P NMR in ESI†). Polymer degradation tests were performed for one, two, and four weeks before the samples were isolated and analyzed by NMR and SEC. All polymer films immersed in PBS solutions remained intact during the degradation tests. Polymer films immersed in artificial seawater were partly detached from the glass slide after 4 weeks. After a washing and drying step, these polymer films were further disintegrated (Fig. S28†). SEC measurements revealed no significant change in molecular weight for the PBS samples (Fig. S23†). These findings were supported by 1H NMR (Fig. S14†), as no additional peaks were observed in comparison to the initial copolymer spectrum. In contrast, a signal at −1.06 ppm with low intensity was visible in the 31P NMR spectrum after 4 weeks (Fig. S15†). A signal at the same chemical shift was observed in the 31P NMR spectrum for polymer films after they had been immersed in artificial seawater for four weeks (Fig. S17†). In addition, the 1H NMR spectrum (Fig. S16†) shows two triplets at 3.63 and 1.56 ppm corresponding to the phosphodiester degradation product. Compared to the 1H NMR spectra of degraded polymers films at pH 11 and 13, the intensity of the signals was lower, indicating slower hydrolysis kinetics in seawater. The overlay of the SEC elugrams of poly-1H-co-poly-1Bn degraded in artificial seawater for one, two, and four weeks shows a shift to higher elution volume over time (Fig. 6). The number average molar mass Mn decreased during this period from initially 24500 g mol−1 to 8300 g mol−1 (vs. PS calibration).
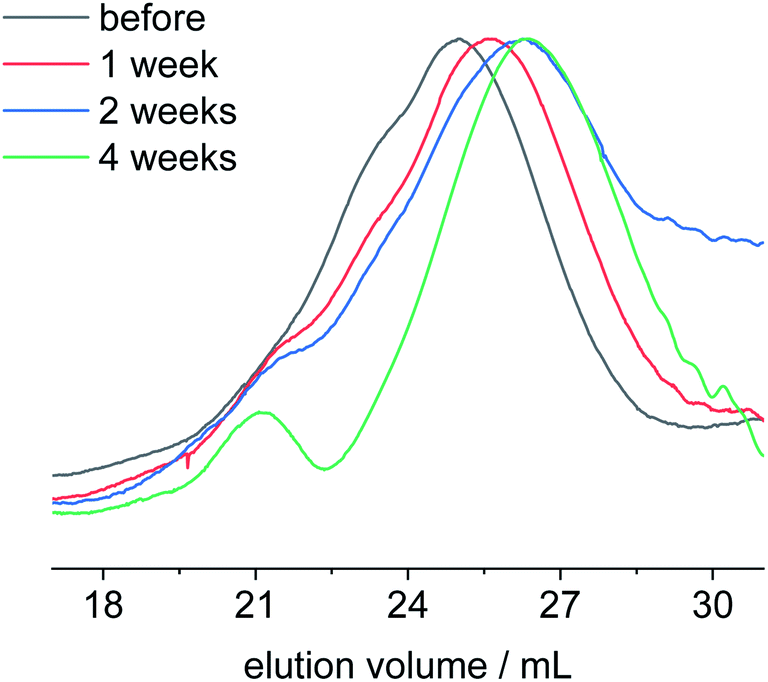 | ||
| Fig. 6 SEC elugrams in THF of poly-1H-co-poly-1Bn before (black) and after degradation in artificial seawater at pH 8.1 for one week (red), two weeks (blue) and four weeks (green). | ||
Overall, poly-1H-co-poly-1Bn seems to be stable at a neutral, physiological pH of 7.4 and room temperature. At an increased pH value, however, the hydrolysis of the material takes place by intramolecular transesterification, while the degradation kinetics are related to the pH value, making it an attractive PE-alternative that degrades in seawater.
Use as macro-initiator for grafting of lactide
Besides increased hydrolysis, the free hydroxyl groups in the phosphate side chain of poly-1H enable further functionalization. To prove the accessibility of the OH-groups, we prepared a PPE, grafted with another biodegradable polymer, namely polylactide. Via a grafting-from approach, poly-1H was used as a macroinitiator for the organocatalyzed anionic ring-opening polymerization (AROP) of lactide (Fig. 7A). Prior, Iwasaki et al. reported the synthesis of a PPE-graft-poly(2-methacryloyloxyethyl phosphorylcholine) by a grafting-from approach via atom transfer radical polymerization (ATRP).35 Furthermore, PPE-graft-poly(ethylene oxide) copolymers were obtained by grafting-through36 and grafting-onto approaches.37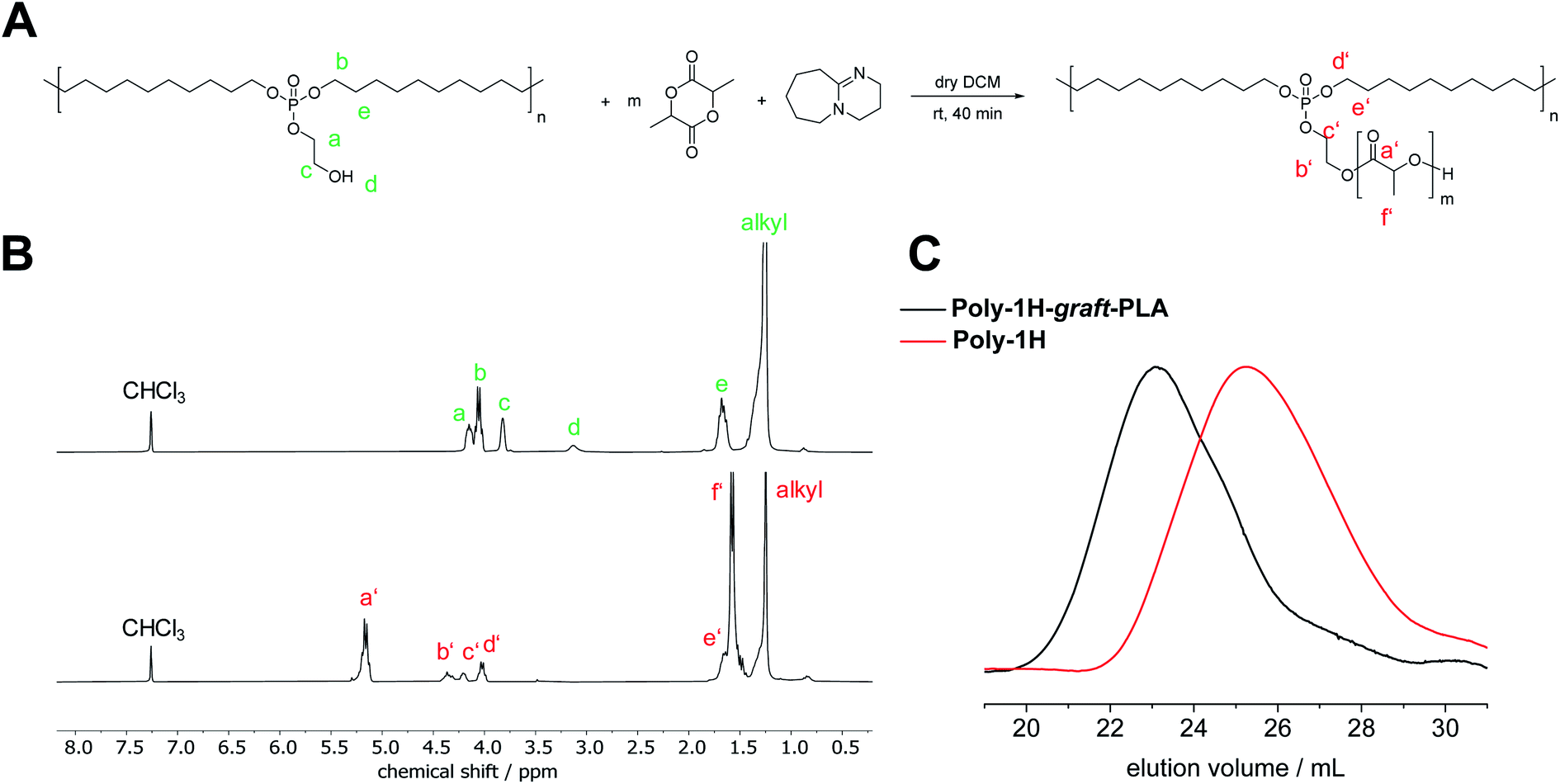 | ||
| Fig. 7 (A) Anionic ring-opening polymerization (AROP) of lactide with poly-1H as a macroinitiator. (B) 1H NMR (300 MHz at 298, in CDCl3) of poly-1H (top) and poly-1H-graft-PLA (bottom). (C) SEC elugrams of poly-1H (red) and poly-1H-graft-PLA (black) in THF. | ||
As a proof of concept, we conducted the polymerization in dry CH2Cl2 at room temperature with used diazabicycloundecene (DBU) as a catalyst. After 40 min, the reaction was terminated with formic acid. Poly-1H-graft-PLA was purified by precipitation into methanol. Poly-1H-graft-PLA was obtained as a solid, colorless powder. Successful grafting was proven by SEC by a shift to lower elution volumes compared to the initial poly-1H, indicating an increase in molar mass (Fig. 7C). Mn increased from 7400 to 32800 g mol−1 and Mw increased from 21000 to 57400 g mol−1 (vs. PS calibration, Table 2).
In the 1H NMR spectrum in CDCl3 (Fig. 7B), the broad at 3.13 ppm peak corresponding to the free hydroxyl group disappeared completely while the signals of the CH2 groups in the phosphate side-chain both shifted to lower field, indicating successful esterification. New resonances for PLA were detected at 5.17 ppm for the CH group and 1.55 ppm for the methyl group of PLA. The degree of polymerization of the grafted PLA was determined by the integration of the PLA–CH peak at 5.17 ppm relative to the –P–O–CH2– peak in the polymer backbone to give a value of 12.
In the 2D Diffusion Ordered Spectroscopy (DOSY) NMR spectrum, all peaks correlating to the PLA side chain and phosphoester backbone have the same diffusion coefficient, proofing that the PLA side chain is covalently bonded to the macroinitiator polymer (Fig. S19†). By DSC, a decrease of Tm by 35 °C to 56 °C was determined (Fig. S25†) which can be explained by the absence of hydrogen bonding after esterification of the hydroxyl groups. A glass transition point (Tg) at 22 °C correlates to the PLA chains. Compared to high molecular weight PLA with a Tg of about 55 to 60 °C, the Tg of oligomeric PLA is significantly lower.38 Since racemic lactide was used for the copolymerization, the PLA chains do not crystallize but stay in an amorphous state. This combination of two degradable polymers in one brush-like copolymer visualizes the potential of poly-1H as a platform for further interesting polymer structures with tunable degradation profiles and mechanical and chemical properties.
3. Conclusions
We prepared RNA-inspired long-chain polyphosphoesters, which were stable in water under neutral conditions (pH = 7.4 for at least 4 weeks) but hydrolyzed under slightly basic conditions (pH = 8.1) as they are found in seawater. An intramolecular transesterification of pendant ethoxy hydroxyl groups are responsible for this increased hydrolysis of these PE-mimics compared to previously reported PPEs that did not carry these pendant groups. The hydrolysis in these RNA-inspired PPEs (poly-1H) followed an intramolecular transesterification, similar to the 2′-OH-group of ribose in RNA, which is responsible for faster hydrolysis of RNA compared to DNA. A cyclic intermediate was predicted by DFT calculations and detected by 31P NMR, indicating intramolecular transesterification. In addition, the basic hydrolysis of a blended film of poly-1H and OH-protected poly-1Bn strengthened the conclusion that the enhanced degradability of poly-1H was caused by hydrolytic degradation via an intramolecular nucleophilic attack. The successful synthesis of a graft copolymer with PLA brushes following a macroinitiator approach showed the potential of poly-1H as a platform polymer for more sophisticated degradable polymer architectures. Future works shall focus on increasing the molecular weight of the polymers to improve the mechanical properties. Overall, the herein presented strategy is a promising example of how to incorporate binding motifs inspired from biopolymers into synthetic polymers to synthesize novel degradable polymers. Intramolecular transesterification hereby can largely increase degradation rates of different polymers.Data availability
Data will be made available on request.Author contributions
T. H. and O. S. performed the experiments and analyses. T. H. and F. R. W. wrote and edited the paper. P. P. analysed the data and conducted calculations. I. L. and F. R. W. conceived the idea and supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the German Federal Ministry for Education and Research (BMBF) for their support of the program “Research for sustainable development (FONA)”, “PlastX – Plastics as a systemic risk for social-ecological supply systems” (grant number: 01UU1603A). The authors thank the Deutsche Forschungsgemeinschaft (WU750/6-2) for funding. The authors thank Michael Steiert (MPIP) for XRD measurements and Christine Rosenauer (MPIP) for GPC. Further support for synthesis by Angelika Manhart (MPIP) is acknowledged. PP acknowledges support from the Foundation for Polish Science (MAB PLUS/2019/11) and allocation of the computer time at the PLGRID, Cyfronet, Kraków.References
- S. Penczek, J. Pretula and K. Kaluzynski, Biomacromolecules, 2005, 6, 547–551 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- F. Stempfle, P. Ortmann and S. Mecking, Chem. Rev., 2016, 116, 4597–4641 CrossRef CAS PubMed.
- I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404–3410 CrossRef CAS PubMed.
- T. Haider, O. Shyshov, O. Suraeva, I. Lieberwirth, M. von Delius and F. R. Wurm, Macromolecules, 2019, 52, 2411–2420 CrossRef CAS PubMed.
- T. Rheinberger, J. Wolfs, A. Paneth, H. Gojzewski, P. Paneth and F. R. Wurm, J. Am. Chem. Soc., 2021, 143(40), 16673–6681 CrossRef CAS PubMed.
- D. Voet and J. Voet, Biochemistry, John Wiley & Sons Inc., Hoboken, 2004 Search PubMed.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed.
- J. Eigner, H. Boedtker and G. Michaels, Biochim. Biophys. Acta, 1961, 51, 165–168 CrossRef CAS.
- K. N. Bauer, L. Liu, M. Wagner, D. Andrienko and F. R. Wurm, Eur. Polym. J., 2018, 108, 286–294 CrossRef CAS.
- G. X. Wang, D. Huang, J. H. Ji, C. Völker and F. R. Wurm, Adv. Sci., 2020, 2001121 Search PubMed.
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, Polym. Int., 2017, 66, 7–12 CrossRef CAS.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- P. Ortmann, F. P. Wimmer and S. Mecking, ACS Macro Lett., 2015, 4, 704–707 CrossRef CAS.
- C. Liu, F. Liu, J. Cai, W. Xie, T. E. Long, S. R. Turner, A. Lyons and R. A. Gross, Biomacromolecules, 2011, 12, 3291–3298 CrossRef CAS PubMed.
- K. N. Bauer, H. T. Tee, I. Lieberwirth and F. R. Wurm, Macromolecules, 2016, 49, 3761–3768 CrossRef CAS.
- J. C. Markwart, A. Battig, L. Zimmermann, M. Wagner, J. Fischer, B. Schartel and F. R. Wurm, ACS Appl. Polym. Mater., 2019, 1, 1118–1128 CrossRef CAS.
- D. Thompson, R. Yamakado and K. B. Wagener, Macromol. Chem. Phys., 2014, 215, 1212–1217 CrossRef CAS.
- H. T. Tee, K. Koynov, T. Reichel and F. R. Wurm, ACS Omega, 2019, 4, 9324–9332 CrossRef CAS PubMed.
- H. Busch, E. Schiebel, A. Sickinger and S. Mecking, Macromolecules, 2017, 50, 7901–7910 CrossRef CAS.
- H. T. Tee, I. Lieberwirth and F. R. Wurm, Macromolecules, 2019, 52, 1166–1172 CrossRef CAS PubMed.
- M. Letizia Focarete, M. Scandola, A. Kumar and R. A. Gross, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 1721–1729 CrossRef.
- Y.-R. Zheng, H. T. Tee, Y. Wei, X.-L. Wu, M. Mezger, S. Yan, K. Landfester, K. Wagener, F. R. Wurm and I. Lieberwirth, Macromolecules, 2016, 49, 1321–1330 CrossRef CAS.
- N. Hasan, K. Busse, T. Haider, F. R. Wurm and J. Kressler, Polymers, 2020, 12, 2408 CrossRef CAS PubMed.
- T. Haider, O. Suraeva, M. L. O'Duill, J. Mars, M. Mezger, I. Lieberwirth and F. R. Wurm, Polym. Chem., 2020, 11, 3404–3415 RSC.
- P. Welch and M. Muthukumar, Phys. Rev. Lett., 2001, 87, 218302 CrossRef CAS PubMed.
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 61–122 CrossRef CAS.
- J. Baran and S. Penczek, Macromolecules, 1995, 28, 5167–5176 CrossRef CAS.
- G. Becker, L.-M. Ackermann, E. Schechtel, M. Klapper, W. Tremel and F. R. Wurm, Biomacromolecules, 2017, 18, 767–777 CrossRef CAS PubMed.
- S.-W. Huang, J. Wang, P.-C. Zhang, H.-Q. Mao, R.-X. Zhuo and K. W. Leong, Biomacromolecules, 2004, 5, 306–311 CrossRef CAS PubMed.
- J. Wang, H.-Q. Mao and K. W. Leong, J. Am. Chem. Soc., 2001, 123, 9480–9481 CrossRef CAS PubMed.
- M. Koutny, J. Lemaire and A. M. Delort, Chemosphere, 2006, 64, 1243–1252 CrossRef CAS PubMed.
- J. P. Da Costa, A. R. Nunes, P. S. M. Santos, A. V. Girão, A. C. Duarte and T. Rocha-Santos, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2018, 53, 866–875 CrossRef CAS PubMed.
- A. Paneth and P. Paneth, J. Mol. Model., 2019, 25, 286 CrossRef PubMed.
- Y. Iwasaki and K. Akiyoshi, Macromolecules, 2004, 37, 7637–7642 CrossRef CAS.
- J.-Z. Du, D.-P. Chen, Y.-C. Wang, C.-S. Xiao, Y.-J. Lu, J. Wang and G.-Z. Zhang, Biomacromolecules, 2006, 7, 1898–1903 CrossRef CAS PubMed.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavía-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- N. Burgos, D. Tolaguera, S. Fiori and A. Jiménez, J. Polym. Environ., 2014, 22, 227–235 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05509g |
| This journal is © The Royal Society of Chemistry 2021 |