DOI:
10.1039/D1SC05370A
(Edge Article)
Chem. Sci., 2021,
12, 15339-15346
Intelligent demethylase-driven DNAzyme sensor for highly reliable metal-ion imaging in living cells†
Received
29th September 2021
, Accepted 28th October 2021
First published on 29th October 2021
Abstract
The accurate intracellular imaging of metal ions requires an exquisite site-specific activation of metal-ion sensors, for which the pervasive epigenetic regulation strategy can serve as an ideal alternative thanks to its orthogonal control feature and endogenous cell/tissue-specific expression pattern. Herein, a simple yet versatile demethylation strategy was proposed for on-site repairing-to-activating the metal-ion-targeting DNAzyme and for achieving the accurate site-specific imaging of metal ions in live cells. This endogenous epigenetic demethylation-regulating DNAzyme system was prepared by modifying the DNAzyme with an m6A methylation group that incapacitates the DNAzyme probe, thus eliminating possible off-site signal leakage, while the cell-specific demethylase-mediated removal of methylation modification could efficiently restore the initial catalytic DNAzyme for sensing metal ions, thus allowing a high-contrast bioimaging in live cells. This epigenetic repair-to-activate DNAzyme strategy may facilitate the robust visualization of disease-specific biomarkers for in-depth exploration of their biological functions.
Introduction
Molecular probes are crucial tools for the visualization of specific molecular and biological processes in live cells.1–3 Accordingly, live-cell imaging probes have been rapidly developed by targeting tumor-relevant RNAs,2,4–6 proteins,7–9 specific metal ions,10–12etc. In particular, these metal-ion-imaging fluorescent probes occupy a significant space for the in-depth exploration of their related cell biology. Metal ions play pivotal roles in various biological systems, and their aberrant homeostasis is correlated with a variety of human diseases.13,14 To explore the vital role of metal ions in biological processes, probes that enable the accurate imaging of metal ions in living cells are of great promise. Numerous fluorescent probes have thus been developed for the intracellular imaging of metal ions, and these include small organic molecules15–17 and fluorescent proteins.18–22 However, these small-molecule probes are only suitable for a limited range of metal-ion targets and might bring undesired biotoxicity to live cells. Meanwhile, although these genetically engineered fluorescent protein probes are encoded with high biocompatibility, they require tedious biosynthesis procedures and post-synthetic modifications. It is thus highly desirable to develop a simple yet versatile fluorescent probe for the intracellular imaging of metal ions. Toward this goal, DNAzymes have aroused wide interest due to their specific requirement for metal-ion cofactors that contribute to the high catalytic activities of DNAzymes.23–26 Accordingly, a series of DNAzyme-powered fluorescence sensors have been developed for imaging metal ions, such as Zn2+,27,28 Mg2+,29 Pb2+,30 and Na+.31 Despite these rapid progresses, the extensive biosensing application of DNAzymes in live cells remains a challenge due to the annoying signal leakage (off-target activation), originating from their invariably catalytic ON state, which can non-specifically react with the surrounding metal ions. There is thus an urgent need to develop a stimuli-responsive DNAzyme sensor that can only be specifically activated in target cells and stay inert prior to reaching the desired destination. This site-specific DNAzyme regulation strategy is especially appealing for realizing the precise and high-contrast intracellular bioimaging applications because the annoying off-site signal leakage could be substantially eliminated by the inactive transit configuration of the DNAzyme sensor while the desired on-site signal gain could be retained by the full restoration of the DNAzyme biocatalysis.
Currently, continuous efforts have been devoted to promoting the responsiveness of DNAzyme to light32 and temperature33 stimuli. For example, DNAzyme can be modified with a photolabile group to prohibit its biocatalytic activity until the chemical group is removed by photodissociation.32 However, the activation efficiency is inevitably constrained by the limited light penetration depth, meanwhile, the high dosages of laser radiation may cause an adverse effect on living systems. Also, temperature stimulation can be introduced by integrating a heat-sensitive glyoxal molecule, which can react with nucleobases for cloaking the DNAzyme configuration, and can then be thermo-released for restoring the catalytic activity of DNAzyme.33 Besides heat stimulation, reactive oxygen species (ROS) stimuli have also been introduced for regulating the activity of DNAzyme by integrating ROS-sensitive chemical modifications on DNAzyme.34 Despite these exciting achievements, these DNAzyme activation strategies have been mainly achieved to date by external stimuli, while the endogenous stimuli toolbox has been rarely explored. Recently, endonuclease enzymes have been used to stimulate DNAzyme-mediated metal-ion imaging in living cells.29,35 Here, the catalytic activity of DNAzyme is initially caged by an integrated endonuclease-recognizing duplex segment, which can then be specifically removed by the corresponding endonuclease for liberating the DNAzyme strand via a sophisticated structural reconstitution procedure. However, this endonuclease-stimulation strategy is constrained by the inevitable activity loss and the tardy response of DNAzyme for those grafted functional duplexes, which could, at least partially, impair the integrity of DNAzyme structures and the DNAzyme regulation might then proceed via an indirect structural rearrangement pathway. Also, these complicated DNAzyme-grafting and structural reconstitution procedures need to be carefully tuned to obtain a high-contrast stimuli-responsive DNAzyme system that could initially guarantee efficient DNAzyme blockage and subsequently achieve the most efficient DNAzyme biocatalysis after endogenous stimulation. Therefore, it is highly desirable to expand the endogenous stimuli toolbox considering that the exploration of endogenous stimuli-responsive DNAzyme systems is still limited. In particular, such simpler and more direct endogenous enzyme-guided nucleobase-caging/de-caging strategies are highly appealing for elaborately regulating the DNAzyme activity in live cells.
Herein, we report a simple yet versatile endogenous demethylase (DMase)-driven DNAzyme (called DMADz hereafter) for imaging metal ions in living cells by introducing an exquisitely engineered multifunctional DNAzyme that was site-specifically modified by an N6-methyladenine (m6A) group (Fig. 1). The as-integrated m6A modification could substantially deactivate the DNAzyme for eliminating the undesired off-site activation with signal leakage, and could then be specifically removed by intracellular m6A demethylase,36–38e.g., FTO (with abnormal expression in many cancer cells rather than normal cells),39,40 for restoring the original DNAzyme structure with high catalytic activity in the specific DMase-containing cellular environment. This intracellular DMase-activated DNAzyme could then mediate the successive cleavage of the substrate with a multiple turnover rate for achieving site-specific metal-ion sensing in live cells. This endogenous DMase-activated DNAzyme system is more attractive for intracellular imaging based on the following advantages: first, this m6A chemical modification can effectively cloak the catalytic activity of DNAzyme and can then be obliterated for recovering the high catalytic activity of DNAzyme via the DMase-mediated m6A removal procedure, thus achieving high-contrast intracellular metal-ion imaging. Second, the endogenous stimuli-responsive DNAzyme strategy guarantees a more reliable intracellular DNAzyme-sensing system by avoiding undesired off-site activation events prior to its entrance into live cells, and allows on-site activation in target cells. Lastly, the endogenous DMase-responsive DNAzyme can only be activated in DMase-overexpressed cancer cells while maintaining an inactive state in DMase-deficient normal cells, making the DMase-guided DNAzyme system more appealing for realizing amplified metal-ion imaging in specific cancer cells. Considering the simple yet versatile DNAzyme regulation and the extensive variety of DNAzyme-recognizing targets, our direct and successive DNAzyme regulation strategy may expand the availability of endogenous stimulated DNAzyme systems for monitoring other crucial biomarkers in the complex biological environment with a high-contrast imaging ability.
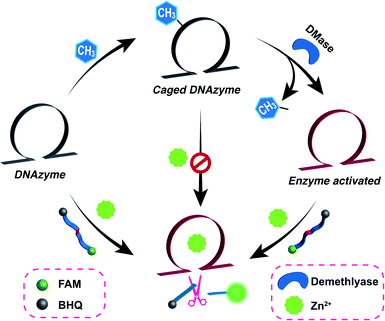 |
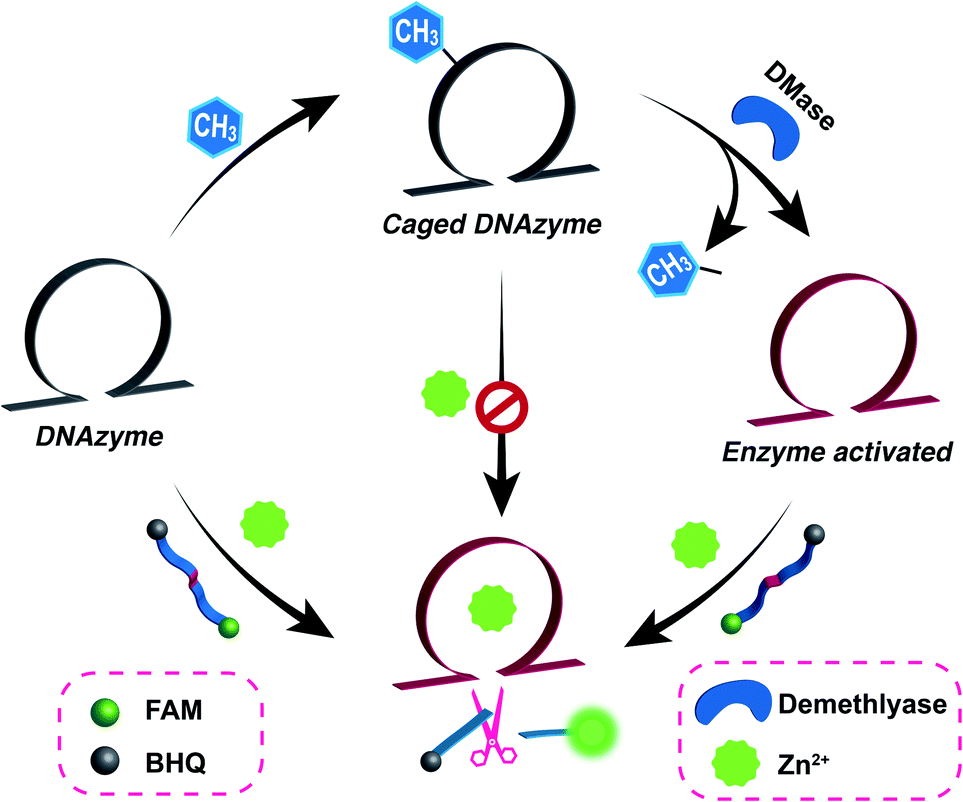
| | Fig. 1 Schematic illustration of the versatile demethylase (DMase)-guided DNAzyme-mediated intracellular metal-ion-imaging system, where the DNAzyme configuration was initially cloaked by the endogenous DMase-recognizing m6A groups. | |
Results and discussion
Validation of the DMase-guided DNAzyme for metal-ion sensing
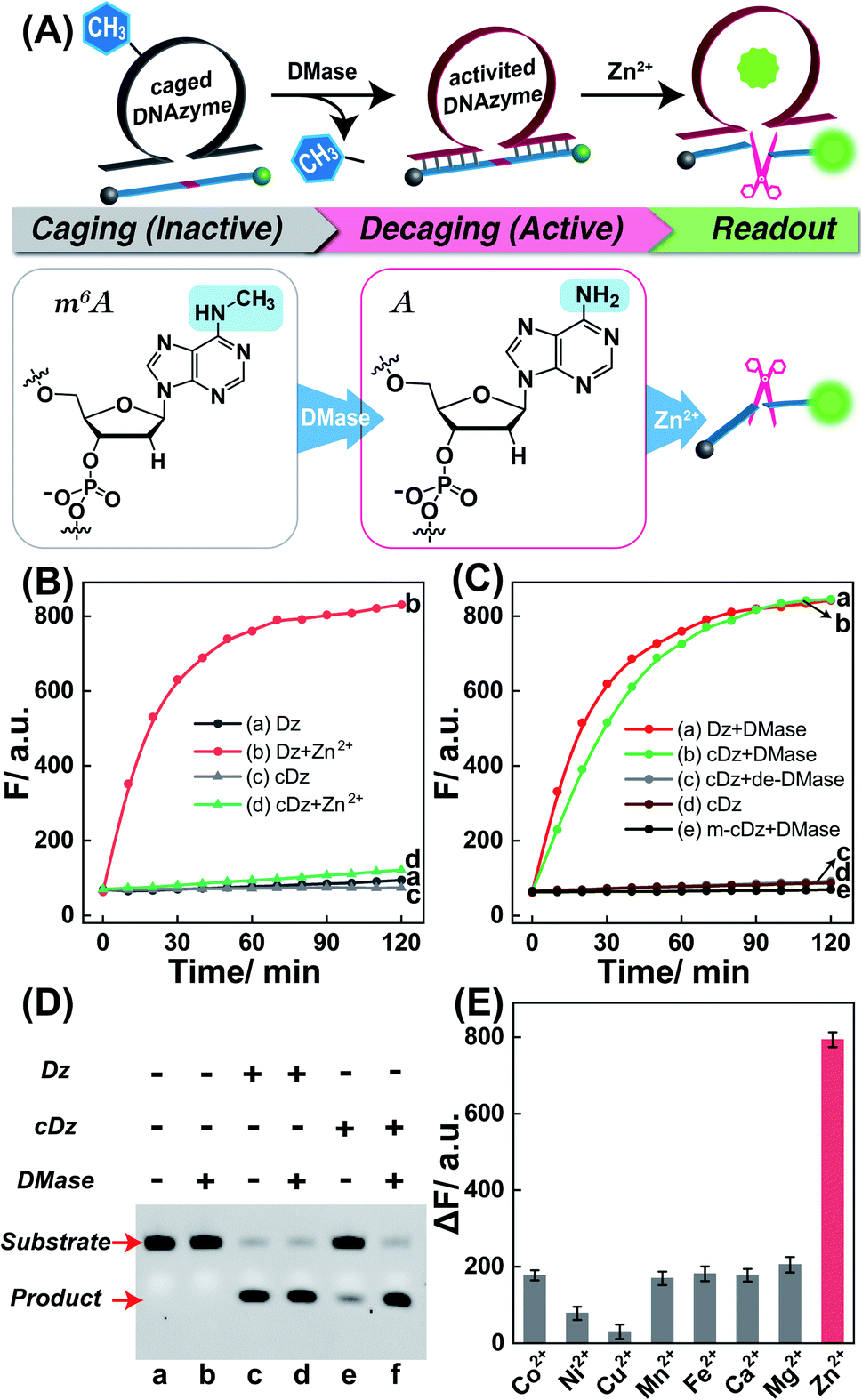
Zn2+ homeostasis is closely related to extracellular or intracellular signal regulation and transduction, and the fluctuation of Zn2+ is associated with many pathological processes.41–43 Therefore, increasing attention has been devoted to the exploration of versatile molecule probes for selectively imaging cellular Zn2+ ions. As a proof of concept, the 8–17 DNAzyme was chosen as a model system. As shown in Fig. 2A, the DMADz system consisted of a Zn2+-targeting DNAzyme strand and the corresponding substrate strand. Here the key catalytic site of DNAzyme was meticulously integrated with a specific DMase-recognizing m6A modification for effectively inhibiting DNAzyme-mediated substrate cleavage. The substrate strand contained an adenosine ribonucleotide (rA) at the cleavage site and was also functionalized at its 5′- and 3′-end with a fluorophore (FAM) and quencher (BHQ), respectively. Initially, the reporter substrate stayed in a fluorescence-quenched state based on the fluorescence resonance energy transfer (FRET) between FAM and BHQ. This m6A-caging strategy can guarantee an inactive DNAzyme configuration before its entrance into the destination cells, while it can also enable specifically restoring the catalytic activity of DNAzyme in an enzyme-activated manner. In the presence of endogenous demethylase (DMase), the m6A modification could be efficiently removed to recover the initial configuration of DNAzyme with a highly efficient catalytic activity. With Zn2+ cofactors, the DMase-liberated DNAzyme could then successively cleave its substrate with multiple turnover rates to generate an obviously amplified fluorescence readout signal for sensing Zn2+ ions. The feasibility of our DMADz system (m6A-caged DNAzyme strand and substrate stand) was first examined in an ideal buffer solution. The detailed nucleotide sequences are listed in Table S1.† In the presence of Zn2+ ions, the intact DNAzyme (Dz, without m6A modification) revealed a dramatically increased fluorescence signal (curve b, Fig. 2B), while the m6A-caged DNAzyme (cDz) showed a much weaker fluorescence change (curve d, Fig. 2B), demonstrating that the DMADz system was efficiently prohibited by the m6A-caging strategy. Then, the highly effective FTO DMase-guided demethylation of the DMADz system was explored for sensing target Zn2+ ions (Fig. 2C). The DMase-treated DMADz displayed a rapid increase in fluorescence in response to Zn2+, quickly reaching a plateau at 1.5 h (curve b, Fig. 2C), which was comparable with that of the intact DNAzyme (curve a, Fig. 2C). This indicated that DMase could effectively remove the m6A modification from the m6A-caged DNAzyme strand for restoring the initial DNAzyme catalytic activity. Meanwhile, heat-deactivated DMase was also introduced to treat this DMADz system to verify the DMase-specific activation of the m6A-caged DNAzyme. As expected, the deactivated DMase-treated m6A-caged DNAzyme could not recover the intrinsic catalytic activity of DNAzyme as revealed by the slight fluorescence change (curve c, Fig. 2C), which was comparable with that of intact m6A-caged DNAzyme (curve d, Fig. 2C). Further, the essential role of DNAzyme biocatalysis was explored by introducing a mutant m6A-caged DNAzyme (m-cDz) control, in which the guanine nucleobase of the DNAzyme catalytic core was substituted with a thymine nucleobase. Under this circumstance, the m-cDz control could not restore the catalytic activity of DNAzyme, even after the DMase-mediated demethylation process. Unsurprisingly, the DMase-treated m-cDz control failed to activate the DNAzyme, as presented by the invariable fluorescence change (curve e, Fig. 2C). The corresponding fluorescence spectra of these different systems are shown in Fig. S1,† and were consistent with the kinetics curves (Fig. 2B and C). Polyacrylamide gel electrophoresis (PAGE) was further used to verify the feasibility of our DMADz system (Fig. 2D), and showed that the DMase had no influence on the DNAzyme substrate (bands a and b, Fig. 2D) as well as on the DNAzyme biocatalysis (bands c and d, Fig. 2D). The activity of DNAzyme could be sufficiently caged by the m6A group as revealed by the intact DNAzyme substrate without cleavage (band e, Fig. 2D), and could then be fully restored to cleave its substrate after the efficient DMase-mediated removal of m6A modification (band f, Fig. 2D). Thus, the feasibility of this DMase-driven DNAzyme strategy was confirmed for Zn2+ detection. The DMase-activated DMADz system showed satisfactory selectivity for Zn2+ detection against other metal ions and possible interference analytes (Fig. 2E and S2†), indicating its favorable application potential in the complex biological environment.
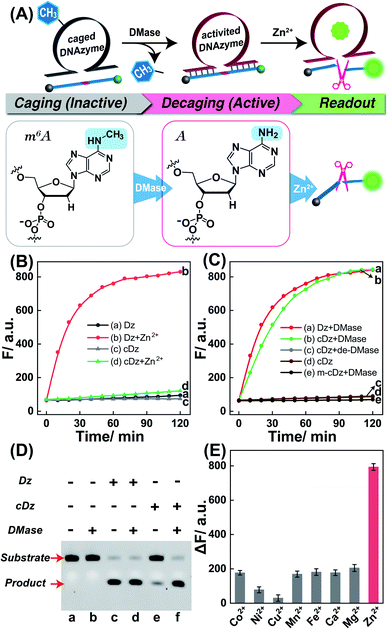 |
| | Fig. 2 (A) Scheme of the on-site activation of the metal-ion-sensing DNAzyme via the endogenous demethylase (DMase)-mediated demethylation reaction. The detailed variation of adenine nucleotide treated with DMase is also presented. (B) Time-dependent fluorescence changes of the intact DNAzyme or m6A-modified DNAzyme for sensing Zn2+ ions (100 μM): (a) Dz, (b) Dz + Zn2+, (c) cDz, (d) cDz + Zn2+, (λex = 480 nm; λem = 520 nm). The system was composed of 50 nM intact DNAzyme or 50 nM m6A-caged DNAzyme and 200 nM substrate. (C) Time-dependent fluorescence changes of (a) intact Dz, (b) DMase-treated cDz, (c) the heat-deactivated DMase-treated cDz, (d) intact cDz, and (e) DMase-treated mutant cDz. Here DMase denotes FTO demethylase. (D) PAGE analysis of the DMase-activated DNAzyme for cleaving its substrate. The “+” and “−” represent the presence and absence of the relevant component, respectively. (E) Specificity of the DMase-driven DNAzyme sensor against Zn2+ and other metal ions. The system was treated with 100 μM Zn2+, or 100 μM Co2+, or 100 μM Ni2+, or 100 μM Cu2+, or 100 μM Mn2+, or 100 μM Fe2+, or 1 mM Ca2+, or 1 mM Mg2+. Error bars were acquired from n = 3 experiments. | |
Exploration of the Zn2+-sensing performance of the DMase-driven DNAzyme system
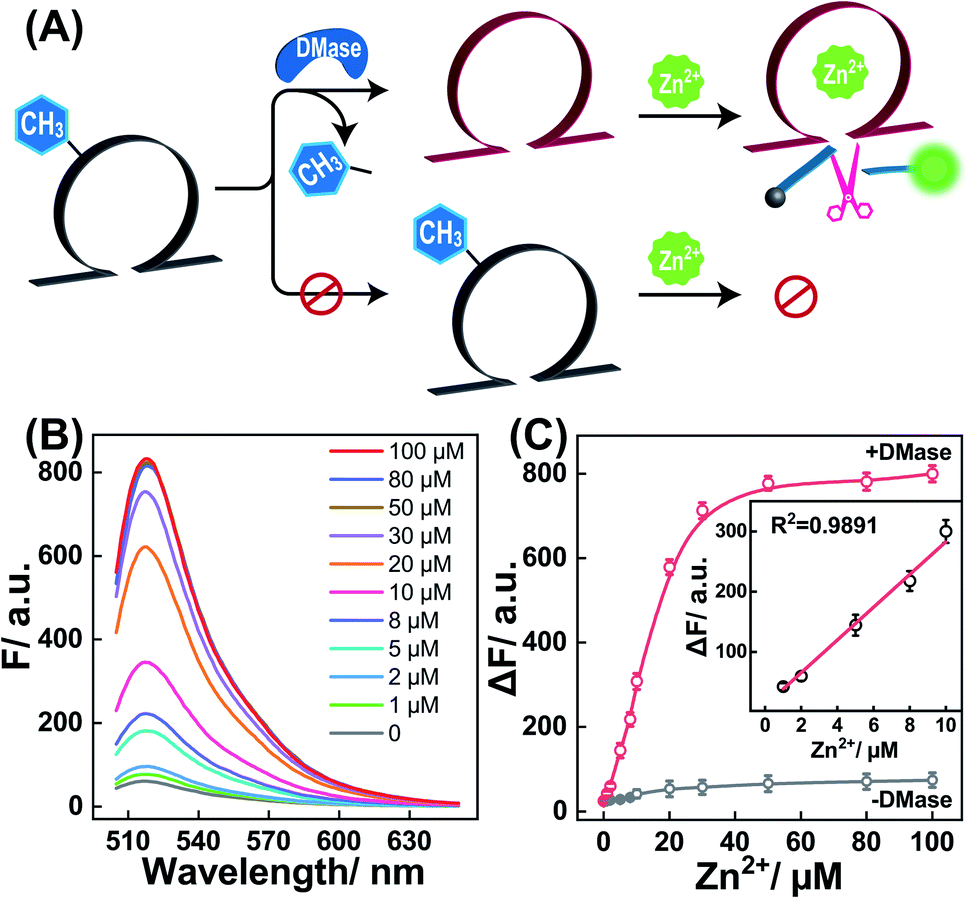
Having proven the DMase-guided DNAzyme strategy, the DMADz system was then optimized to achieve a rational sensing performance for Zn2+ ions. The surrounding temperature was first investigated for guaranteeing the efficient DNAzyme biocatalysis, and the Zn2+-targeting DMADz system revealed a higher signal-to-background ratio at 37 °C (Fig. S3†), which was thus chosen as the optimized temperature for the subsequent experiments. Then the dosage and duration of DMase were explored to achieve an optimal demethylation efficiency for guaranteeing efficient Zn2+-sensing (Fig. S4†). The fluorescence signal gradually intensified with an increasing concentration of DMase (from 5 nM to 400 nM), reaching a saturated value at 250 nM DMase. Accordingly, 250 nM was chosen as the optimized DMase dosage for the subsequent experiments. Moreover, DMase exhibited an efficient demethylation execution within 15 min incubation. For guaranteeing an efficient demethylation extent, 1 h of enzymatic reaction was chosen as the optimized demethylation duration. Under these optimized reaction conditions, the Zn2+-sensing performance of our DMADz system was investigated. Here the DMADz system was activated, only through the DMase-mediated demethylation process, for liberating DNAzyme to catalyze the Zn2+-mediated successive substrate cleavage. However, without DMase, the catalytic activity of m6A-caged DNAzyme was substantially prohibited, thus the DMADz system stayed in a catalytically inactive state even in the presence of Zn2+ ions (Fig. 3A). Therefore, the initially caged DNAzyme stayed inert until it reached the destination site for achieving on-site activation, leading to high-contrast metal-ion sensing with minimal off-target signal leakage. The fluorescence intensity of our DMADz system was enhanced with the increasing concentration of the Zn2+ target (Fig. 3B). Also, the detection limit of Zn2+ was acquired as 0.4 μM from the calibration curve (Fig. 3C), which was comparable with the current Zn2+-analysis methods (Table S2†). Nevertheless, without DMase treatment, a dramatically lower fluorescence response was observed for the m6A-caged DNAzyme system (Fig. 3C), indicating the indispensable role of DMase in the site-specific activation of DNAzyme for efficient and reliable Zn2+-ion sensing.
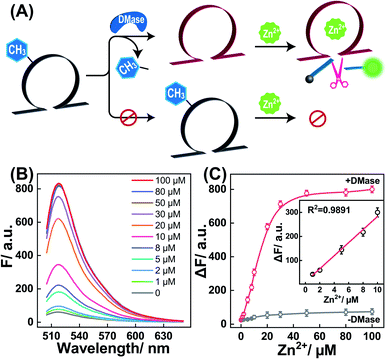 |
| | Fig. 3 (A) Schematic diagram of the DMase-guided selection of DNAzyme system for sensing Zn2+ ions. (B) Fluorescence spectra of the DMase-activated DNAzyme system for sensing Zn2+ with different concentrations (from 0 to 100 μM). Here DMase represents FTO demethylase. (C) Fluorescence intensity changes of the intact or DMase-treated caged-DNAzyme system for analyzing different concentrations of Zn2+. Inset: calibration curve of the DMase-treated caged-DNAzyme system. Error bars were acquired from n = 3 experiments. | |
Specificity and stability of the DMase-driven DNAzyme system
The satisfactory sensing performance of the DMADz platform inspired us to further explore its in-depth utilization for the intracellular imaging of Zn2+ ions in living cells. To achieve this goal, the DMase-responsive specificity and stability of our platform were investigated to prevent undesired signal leakage prior to its delivery into live cells. Fig. S5A† shows that a high fluorescence signal was obtained from the specific DMase-treated DNAzyme system, while little fluorescence enhancement was obtained from the other endogenous small molecules even with high abundance, including hydrogen peroxide (H2O2), adenosine triphosphate (ATP), glutathione (GSH), and glucose. In addition, the caged-DNAzyme system could only be activated by the DMase-mediated demethylation process, while other proteins, such as bovine serum albumin (BSA), dam methyltransferase (Dam), polynucleotide kinase (PNK), CpG methyltransferase (M. SssI), and alkaline phosphatase (ALP), failed to activate the caged-DNAzyme system (Fig. S5B†). The satisfactory stimuli-responsiveness of the DMADz system was thus clearly demonstrated. Furthermore, the stability of the DMADz system was explored in cell lysates (Fig. S6A†), which showed that the caged-DNAzyme could exist stably in cell lysates without apparent degradation. Meanwhile, the DMase-treated caged-DNAzyme could effectively catalyze the successive cleavage of the substrate in the cell lysates (Fig. S6B†). These above results convincingly prove that the DMADz system possessed excellent DMase-specific responsiveness against other control proteins, and also possessed an additional high anti-interference capacity in the cell lysate environment, thus paving the way for intracellular Zn2+ imaging in a complex biological environment by employing this DMase-activatable DNAzyme platform.
Feasibility demonstration of the endogenous DMase-driven DNAzyme-amplified Zn2+-imaging platform in live cells
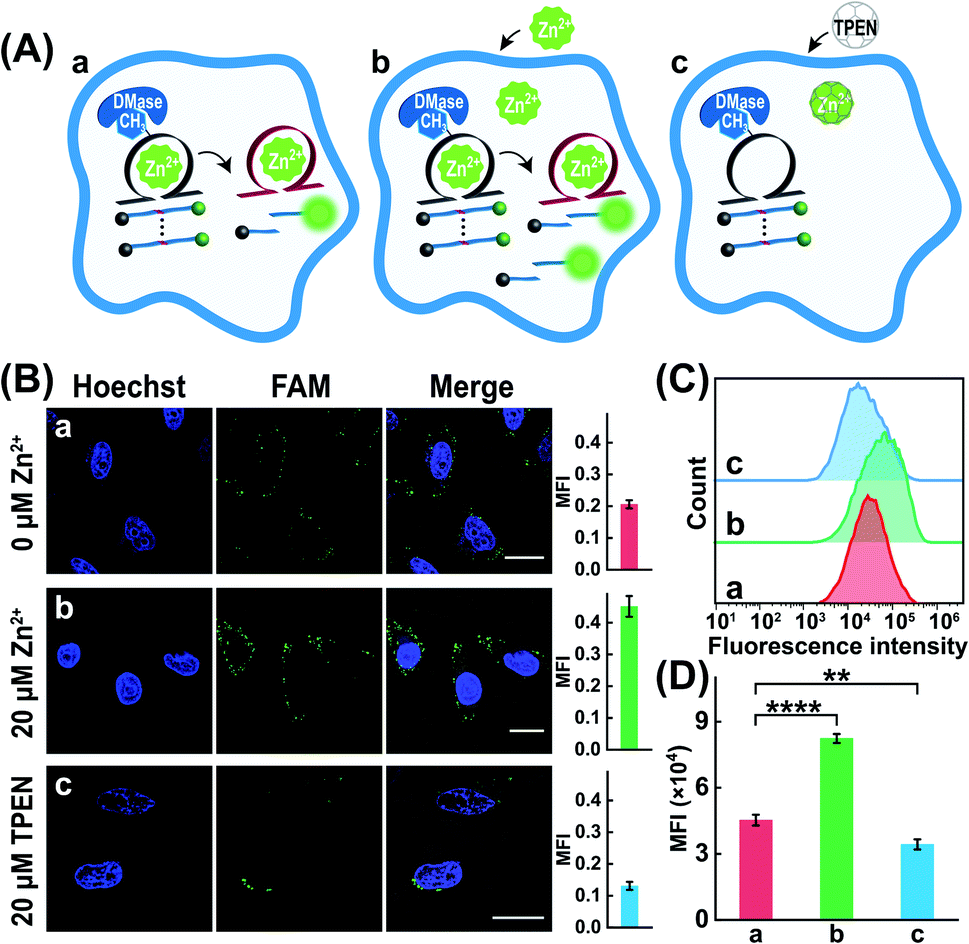
The characteristic DMase-overexpressed MCF-7 cells were first used as a model system to probe the feasibility of the DMADz-mediated intracellular imaging of Zn2+ ions. Here the DMADz system was delivered into the cytoplasm of live cells by a commercial lipofectamine 3000 transfection reagent. The appropriate duration of cellular incubation with the DMADz system was initially explored by flow cytometric analysis (Fig. S7†). This analysis showed that the fluorescence signal continuously increased with prolonging the incubation duration, reaching to a plateau value after 4 h, which was thus chosen as the optimized incubation time for the following experiments. Then confocal laser scanning microscopy (CLSM) was used to explore intracellular fluorescence Zn2+-imaging performance of the DMADz system in MCF-7 cells (Fig. 4A). As anticipated, the endogenous Zn2+ could motivate the generation of high fluorescence originating from the endogenous DMase (FTO or ALKBH5)-activated DNAzyme that catalyzes the efficient cleavage of substrate (sample a, Fig. 4B), while the additional supplementary Zn2+ ions could contribute to the generation of a higher fluorescence readout (sample b, Fig. 4B). Moreover, the lowly concentrated Zn2+ ions have little influence on the activity of DMase (Fig. S8†), indicating the negligible interference of Zn2+ ions on cellular FTO DMase. Accordingly, an extra 20 μM Zn2+ was supplemented to investigate the more accurate and reliable DNAzyme biocatalysis event. To ensure the Zn2+-DNAzyme-mediated fluorescence generation, the N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN, a cell membrane permeable reagent to chelate intracellular Zn2+) was introduced to cloak the endogenous Zn2+. As expected, the TPEN-pretreated MCF-7 cells presented a weak fluorescence intensity (sample c, Fig. 4B) as compared with the intact MCF-7 cells (sample a, Fig. 4B), indicating that the fluorescence readout was certainly obtained from Zn2+-involved DNAzyme biocatalysis. Furthermore, a complementary flow cytometric analysis was used to verify the above CLSM demonstrations (Fig. 4C and D). As anticipated, the Zn2+-upregulated MCF-7 cells (with an extra pretreatment of Zn2+ ions) contributed an intensified fluorescence signal, while the Zn2+-downregulated MCF-7 cells (with an additional TPEN-pretreatment) presented a weak fluorescence output as compared to the cells without any extra pretreatment, which was consistent with that of CLSM results. All these results demonstrated that our DMADz system was indeed favorable for reliable intracellular imaging of Zn2+ ions in live cells.
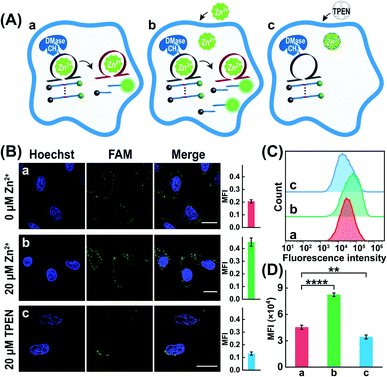 |
| | Fig. 4 (A) Schematic diagram of the DMADz-incubated MCF-7 cells with different treatments: (a) intact DMADz, (b) pretreated with 20 μM Zn2+ + intact DMADz, (c) pretreated with 20 μM TPEN + intact DMADz. (B) CLSM images of the DMADz-incubated MCF-7 cells with different treatments. The corresponding statistical histogram of mean fluorescence intensity was shown aside. (C) Flow cytometry analysis of these differently treated MCF-7 cells. (D) Mean fluorescence intensity of the flow cytometry results as shown in (C). ****p < 0.0001 (one-way ANOVA followed by Tukey's multiple comparisons test). Error bars were acquired from n = 3 experiments. Scale bar = 20 μm. | |
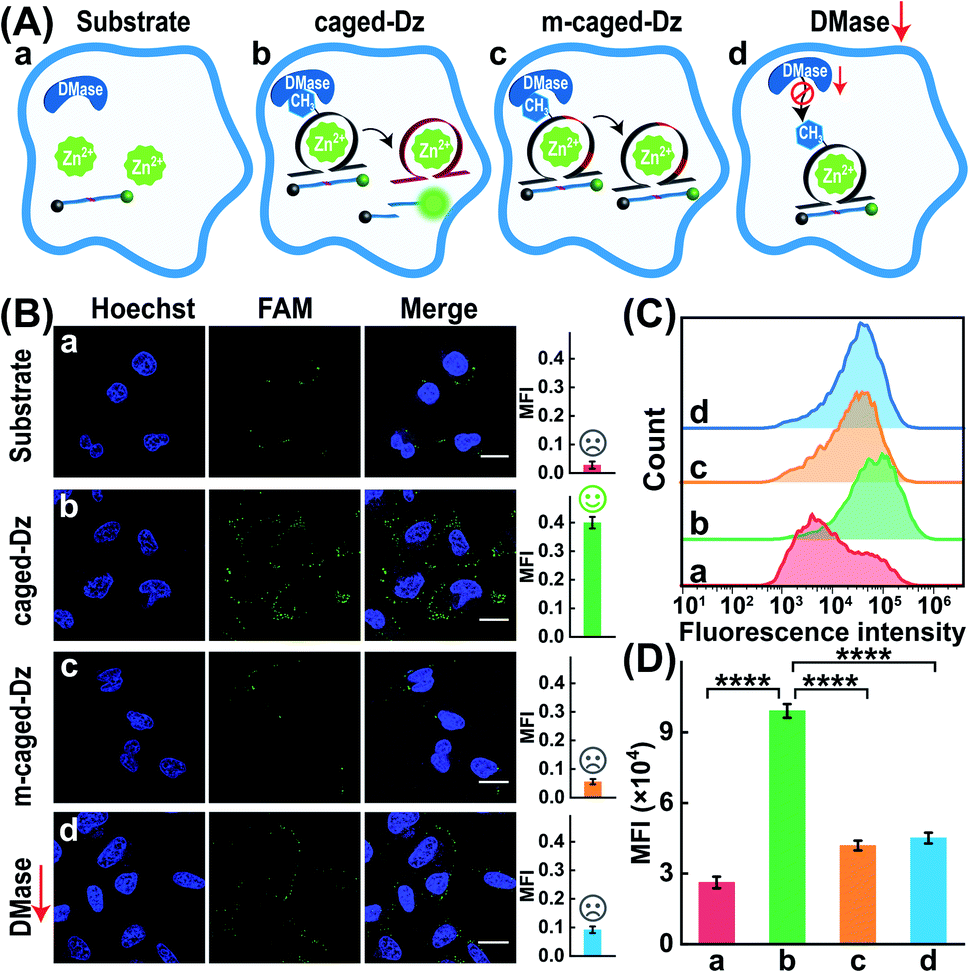
Furthermore, the robustness of the DMADz-mediated Zn2+-imaging system was investigated in live cells (Fig. 5A). Nearly no fluorescence signal was observed for the mere substrate-transfected MCF-7 cells (sample a, Fig. 5B), while an obviously higher fluorescence signal was observed in the intact DMADz-transfected MCF-7 cells (sample b, Fig. 5B), indicating that the integrity of our DMADz system was crucial for motivating the amplified fluorescence readout signal. However, a substantially lower fluorescence signal was obtained from the m6A-caged mutant DNAzyme-integrated DMADz system (sample c, Fig. 5B). This is reasonable for the DMase-liberated mutant DNAzyme, encoded with a catalytically inactive DNAzyme catalytic core, could not cleave the corresponding substrate, demonstrating the indispensable role of the activated DNAzyme biocatalysis for live cells imaging application. Furthermore, to prove the import role of the DMase-mediated demethylation reaction, the MCF-7 cells were pretreated with FTO-silencing siRNA to downregulate the intracellular expression of FTO. Unsurprisingly, a much lower fluorescence signal was obtained in these FTO-downregulated MCF-7 cells (sample d, Fig. 5B) as compared with the intact MCF-7 cells (sample b, Fig. 5B), demonstrating the essential role of endogenous DMase-involved demethylation–activation reaction for the DNAzyme-amplified intracellular imaging application. Moreover, considering the higher demethylation activity of FTO than ALKBH5,44,45 and the nucleus distribution of ALKBH5 (FTO distributed in both cytoplasm and nucleus46,47 while ALKBH5 mainly distributed in nucleus45), we speculated that FTO played an indispensable role for the reliable Zn2+-imaging experiments. The CLSM results were further proved by the complementary flow cytometry analysis (Fig. 5C and D). An obviously increased fluorescence was observed in the intact DMADz-treated MCF-7 cells (sample b), while a substantially weaker fluorescence was revealed for the mere substrate-transfected cells (sample a), the m6A-caged mutant DNAzyme-incubated cells (sample c), as well as the intact DMADz-treated DMase-silenced MCF-7 cells (sample d), which was consistent with that of CLSM demonstration (Fig. 5B). The feasibility and robustness of endogenous DMase-activated DNAzyme platform were thus demonstrated for realizing the efficient intracellular imaging of Zn2+ ions in live cells.
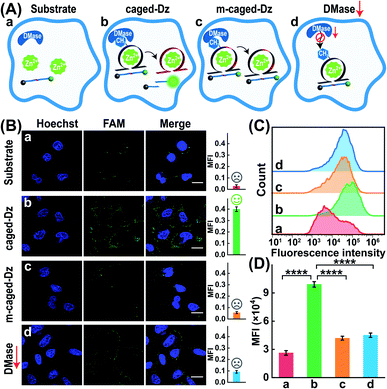 |
| | Fig. 5 Demonstration of the endogenous DMase-driven DNAzyme system for imaging Zn2+ ions in live cells. (A) Schematic diagram of MCF-7 cells underwent with different treatments: (a) only substrate, (b) intact DMADz, (c) m6A-caged mutant DNAzyme and its substrate, (d) siRNA-pretreated cells with intact DMADz. (B) CLSM characterization of the DMase-activated DNAzyme platform for intracellular imaging of Zn2+ ions. The corresponding statistical histogram of mean fluorescence intensity was shown aside. (C) Flow cytometry analysis of these differently treated MCF-7 cells. (D) Mean fluorescence intensity of the flow cytometry results as shown in (C). ****p < 0.0001 (one-way ANOVA followed by Tukey's multiple comparisons test). Scale bar = 20 μm. | |
Cell-selective imaging of Zn2+ ions by the DMase-driven DNAzyme system
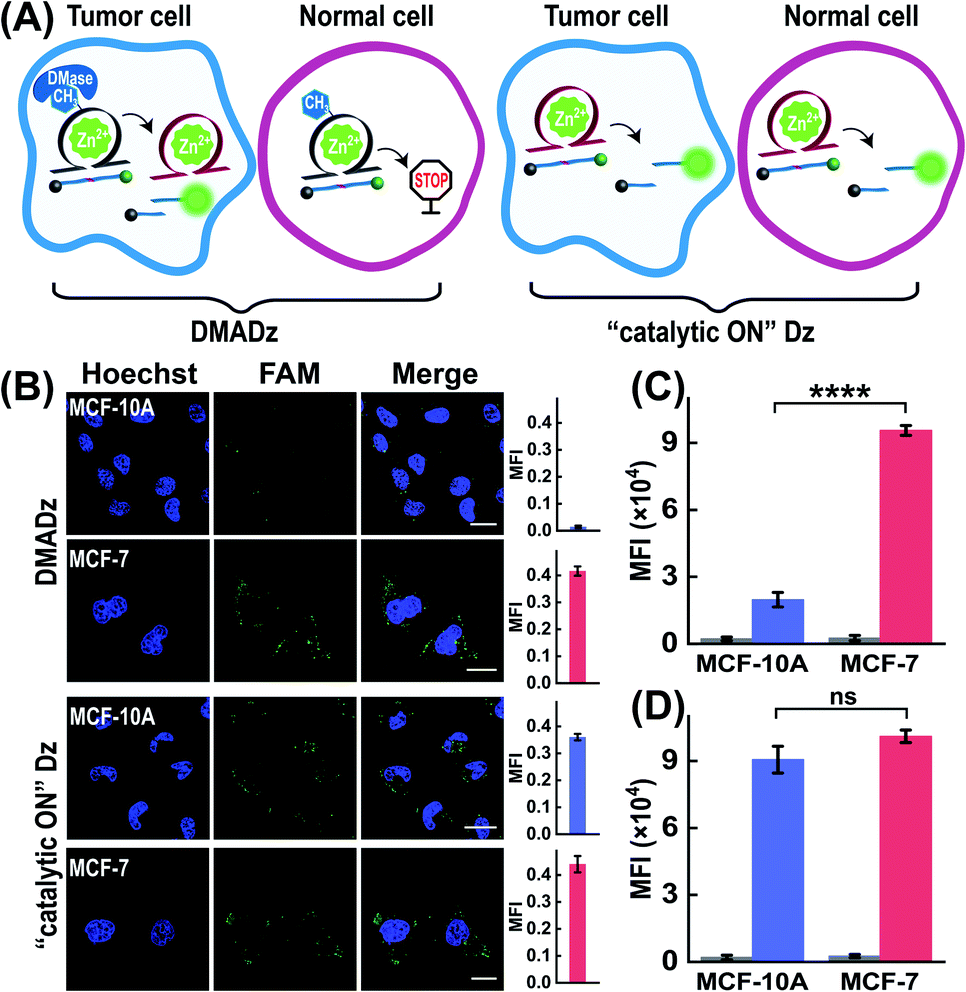
Considering that the overexpressed DMase was always observed in cancer cells but not in normal cells, the DMADz system might be especially suitable for realizing cell-selective Zn2+ imaging. To investigate the cell-selective Zn2+ imaging capacity of the DMADz system, more different cells were chosen for investigating the Zn2+-imaging performance of our DMADz system (Fig. 6A), including MCF-10A cells (healthy breast cells with low DMase expression39 and Zn2+ ions content48,49), and MCF-7 cells (with a comparably higher DMase expression39 and Zn2+-ion content than MCF-10A cells48,49). Fig. 6B shows that the DMADz-treated MCF-7 cells displayed a dramatically increased fluorescence readout with the addition of Zn2+ ions. In contrast, nearly no fluorescence signal was observed in the DMADz-treated MCF-10A cells with the addition of Zn2+ ions. This suggested that the lowly expressed DMase could not liberate the m6A-caged DNAzyme for biocatalytic transduction in MCF-10A cells. Clearly, our DMADz system could be successfully activated by the endogenously and highly expressed DMase in cancer cells, but stayed inactive in normal cells due to the deficient expression of DMase therein. Meanwhile, the always “catalytic ON” Dz with no DMase responsiveness was employed as the control to further demonstrate the cell-selectivity of our DMADz platform. As expected, the “catalytic ON” Dz-treated MCF-10A cells and MCF-7 cells all presented high fluorescence signals, indicating that the DMase-guaranteed repair-to-activate feature of the DMADz system was vital for its cell-specific bioimaging capacity. A complementary flow cytometry analysis was further executed to verify the above CLSM characterizations, where the fluorescence signal in DMADz-treated MCF-7 cells was found to be distinctly higher than that in MCF-10A cells (Fig. 6C and S9A†), while nearly no obvious fluorescence difference was observed in the “catalytic ON” Dz-treated cells (Fig. 6D and S9B†). As expected, the results from the flow cytometry analysis were consistent with that of CLSM demonstration, further confirming the favorable cell-selective Zn2+-imaging utilization of our DMADz system. Moreover, to further expand the applicability of the DMADz system, L02 cells (human normal liver) and HepG2 cells (human hepatocellular carcinoma) were applied for exploring its site-specific Zn2+-imaging ability. As illustrated in Fig. S10A,† the DMADz-treated HepG2 cells (with a relatively high expression of DMase50,51) presented a stronger fluorescence signal with the addition of Zn2+, while the DMADz-treated L02 cells (with a comparably low expression of DMase50,51) showed a weaker fluorescence signal with the addition of Zn2+. These results demonstrated that the DMADz could be specifically activated by endogenous DMase in cancer cells but still remained silent in normal cells. By comparison, both L02 cells and HepG2 cells showed a high fluorescence readout when they were treated with the always “catalytic ON” Dz system, further confirming that the endogenous DMase-activated DMADz strategy was indispensable for its cell-specific biosensing behavior. Meanwhile, flow cytometry analysis was used to verify the above CLSM characterizations. Unsurprisingly, the results from the flow cytometry analysis were consistent with the CLSM results (Fig. S10B and C†), further confirming that the DMADz system was suitable for realizing cell-selective Zn2+ imaging and could be used for various types of cells according to their different DMase expressions.
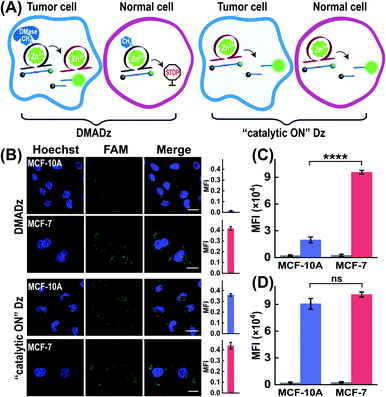 |
| | Fig. 6 Demonstration of the cell-specific imaging of intracellular Zn2+ ions by our DMase-activated DNAzyme platform. (A) Schematic diagram of different cells treated with the DMADz system or “catalytic ON” Dz system. (B) CLSM images of these different cells treated with the DMADz system or “catalytic ON” Dz system. The corresponding statistical histogram of the mean fluorescence intensity are shown at the side. (C) Mean fluorescence intensity of flow cytometry results of different cells treated with the DMADz system. (D) Mean fluorescence intensity of the flow cytometry results of different cells treated with the “catalytic ON” Dz system. ****p < 0.0001 (t tests), ns, no significance. Scale bar = 20 μm. | |
Conclusion
In conclusion, we demonstrated a simple yet versatile epigenetic regulation strategy for endogenously and bioorthogonally activating the catalytic activity of DNAzyme by integrating a demethylase-recognizing m6A site into the catalytic core of DNAzyme, thus achieving the ultimate high-contrast intracellular imaging of metal ions in live cells. The as-integrated m6A modification group could substantially prohibit the activity of DNAzyme for eliminating off-site activation (signal leakage) induced by the extracellular metal ions during the delivery process, while the m6A-incapacitated DNAzyme could be simultaneously repaired and activated by cell-specific endogenous demethylase for accurately sensing metal ions in live cells. Thus, through the endogenous DNA-repairing demethylase enzyme, this on-site epigenetic DNAzyme regulatory manner consequently achieved high-contrast intracellular bioimaging. Based on a systematic investigation, this exquisite stimuli-responsive DNAzyme system achieved the cell-selective imaging of metal ions due to the different expressions of demethylase between normal cells and cancer cells. More importantly, this epigenetically repaired-to-activated DNAzyme strategy might be extended for site-specifically detecting and imaging other crucial disease-relevant biomarkers in living systems due to the highly programmable DNAzyme structure and the extensive variety of DNAzyme-recognizing targets, thus contributing to our deciphering of the underlying biological functions of specific disease-related biomarkers.
Data availability
All experimental data is available in the ESI.†
Author contributions
F. Wang and C. Hong conceived the study and designed the experiment. C. Hong performed the main experiments and analyzed the data. Q. Wang, Y. Chen, Y. Gao, and J. Shang assisted with cell experiments and modeling construction. All authors participated the writing of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by National Natural Science Foundation of China (No. 21874103 and 22074112) and the Central Funds Guiding the Local Science and Technology Development of Shenzhen (2021Szvup101).
Notes and references
- Y. Zhao, K. Wei, F. Kong, X. Gao, K. Xu and B. Tang, Anal. Chem., 2019, 91, 1368–1374 CrossRef CAS PubMed.
- H. Wu, T.-T. Chen, X.-N. Wang, Y. Ke and J.-H. Jiang, Chem. Sci., 2020, 11, 62–69 RSC.
- F. Li, H. Zhang, Z. Wang, X. Li, X.-F. Li and X. C. Le, J. Am. Chem. Soc., 2013, 135, 2443–2446 CrossRef CAS PubMed.
- J. Wei, X. Gong, Q. Wang, M. Pan, X. Liu, J. Liu, F. Xia and F. Wang, Chem. Sci., 2018, 9, 52–61 RSC.
- R. Deng, K. Zhang, Y. Sun, X. Ren and J. Li, Chem. Sci., 2017, 8, 3668–3675 RSC.
- R. Deng, K. Zhang, L. Wang, X. Ren, Y. Sun and J. Li, Chem, 2018, 4, 1373–1386 CAS.
- K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 13386–13395 CrossRef CAS PubMed.
- L. Francés-Soriano, M. Leino, M. C. Dos Santos, D. Kovacs, K. E. Borbas, O. Söderberg and N. Hildebrandt, Anal. Chem., 2021, 93, 1842–1850 CrossRef PubMed.
- F. Li, Y. Lin and X. C. Le, Anal. Chem., 2013, 85, 10835–10841 CrossRef CAS PubMed.
- J. M. Goldberg, F. Wang, C. D. Sessler, N. W. Vogler, D. Y. Zhang, W. H. Loucks, T. Tzounopoulos and S. J. Lippard, J. Am. Chem. Soc., 2018, 140, 2020–2023 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- V. Juvekar, S. J. Park, J. Yoon and H. M. Kim, Coord. Chem. Rev., 2021, 427, 213574 CrossRef CAS.
- H. Kozlowski, A. Janicka-Klos, J. Brasun, E. Gaggelli, D. Valensin and G. Valensin, Coord. Chem. Rev., 2009, 253, 2665–2685 CrossRef CAS.
- N. Nelson, EMBO J., 1999, 18, 4361–4371 CrossRef CAS PubMed.
- P. Ravichandiran, S. A. Subramaniyan, A. P. Bella, P. M. Johnson, A. R. Kim, K. S. Shim and D. J. Yoo, Anal. Chem., 2019, 91, 10095–10101 CrossRef CAS.
- L. Marbella, B. Serli-Mitasev and P. Basu, Angew. Chem., Int. Ed., 2009, 48, 3996–3998 CrossRef CAS PubMed.
- F. Qian, C. Zhang, Y. Zhang, W. He, X. Gao, P. Hu and Z. Guo, J. Am. Chem. Soc., 2009, 131, 1460–1468 CrossRef CAS.
- J. G. Park, Y. Qin, D. F. Galati and A. E. Palmer, ACS Chem. Biol., 2012, 7, 1636–1640 CrossRef CAS PubMed.
- Z.-j. Chen and H.-w. Ai, Anal. Chem., 2016, 88, 9029–9036 CrossRef CAS PubMed.
- S. V. Wegner, H. Arslan, M. Sunbul, J. Yin and C. He, J. Am. Chem. Soc., 2010, 132, 2567–2569 CrossRef CAS PubMed.
- A. Miyawaki, J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura and R. Y. Tsien, Nature, 1997, 388, 882–887 CrossRef CAS.
- L. Zarowny, A. Aggarwal, V. M. S. Rutten, I. Kolb, R. Patel, H.-Y. Huang, Y.-F. Chang, T. Phan, R. Kanyo, M. B. Ahrens, W. T. Allison, K. Podgorski and R. E. Campbell, ACS Sens., 2020, 5, 1959–1968 CrossRef CAS PubMed.
- Y. Li and R. R. Breaker, Curr. Opin. Struct. Biol., 1999, 9, 315–323 CrossRef CAS PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS.
- P.-J. J. Huang, D. de Rochambeau, H. F. Sleiman and J. Liu, Angew. Chem., Int. Ed., 2020, 59, 3573–3577 CrossRef CAS PubMed.
- W. J. Moon, Y. Yang and J. Liu, ChemBioChem, 2021, 22, 779–789 CrossRef CAS PubMed.
- C. Yang, X. Yin, S.-Y. Huan, L. Chen, X.-X. Hu, M.-Y. Xiong, K. Chen and X.-B. Zhang, Anal. Chem., 2018, 90, 3118–3123 CrossRef CAS PubMed.
- M. Xiong, Z. Yang, R. J. Lake, J. Li, S. Hong, H. Fan, X.-B. Zhang and Y. Lu, Angew. Chem., Int. Ed., 2020, 59, 1891–1896 CrossRef CAS PubMed.
- Y. Lin, Z. Yang, R. J. Lake, C. Zheng and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 17061–17067 CrossRef CAS PubMed.
- W. Zhou, W. Liang, D. Li, R. Yuan and Y. Xiang, Biosens. Bioelectron., 2016, 85, 573–579 CrossRef CAS PubMed.
- Z. Wu, H. Fan, N. S. R. Satyavolu, W. Wang, R. Lake, J.-H. Jiang and Y. Lu, Angew. Chem., Int. Ed., 2017, 56, 8721–8725 CrossRef CAS PubMed.
- K. Hwang, P. Wu, T. Kim, L. Lei, S. Tian, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 13798–13802 CrossRef CAS PubMed.
- S. D. Knutson, A. A. Sanford, C. S. Swenson, M. M. Korn, B. A. Manuel and J. M. Heemstra, J. Am. Chem. Soc., 2020, 142, 17766–17781 CrossRef CAS PubMed.
- L. Xiao, C. Gu and Y. Xiang, Angew. Chem., Int. Ed., 2019, 58, 14167–14172 CrossRef CAS PubMed.
- D. Yi, J. Zhao and L. Li, Angew. Chem., Int. Ed., 2021, 60, 6827 CrossRef CAS.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y.-G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- X. Wang, X. Yi, Z. Huang, J. He, Z. Wu, X. Chu and J.-H. Jiang, Angew. Chem., Int. Ed., 2021, 60, 19889–19896 CrossRef CAS PubMed.
- B. Wang, L.-q. Cao, W. Chiuman, Y.-f. Li and Z. Xi, Biochemistry, 2010, 49, 7553–7562 CrossRef CAS PubMed.
- Y. Niu, Z.-y. Lin, A. Wan, H.-l. Chen, H. Liang, L. Sun, Y. Wang, X. Li, X.-f. Xiong, B. Wei, X.-b. Wu and G.-h. Wan, Mol. Cancer, 2019, 18, 46 CrossRef.
- Q. Wang, K. Tan, H. Wang, J. Shang, Y. Wan, X. Liu, X. Weng and F. Wang, J. Am. Chem. Soc., 2021, 143, 6895–6904 CrossRef CAS.
- Y. Chen, Y. Bai, Z. Han, W. He and Z. Guo, Chem. Soc. Rev., 2015, 44, 4517–4546 RSC.
- N. Z. Gammoh and L. Rink, Nutrients, 2017, 9, 624 CrossRef PubMed.
- C. J. Frederickson, J.-Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS PubMed.
- S. Zou, J. D. W. Toh, K. H. Q. Wong, Y.-G. Gao, W. Hong and E. C. Y. Woon, Sci. Rep., 2016, 6, 25677 CrossRef CAS PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C.-M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W.-L. Wang, S.-H. Song, Z. Lu, R. P. G. Bosmans, Q. Dai, Y.-J. Hao, X. Yang, W.-M. Zhao, W.-M. Tong, X.-J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y.-G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed.
- A. Aas, P. Isakson, C. Bindesbøll, E. A. Alemu, A. Klungland and A. Simonsen, PLoS One, 2017, 12, e0168182 CrossRef PubMed.
- P. Gulati, E. Avezov, M. Ma, R. Antrobus, P. Lehner, S. O’Rahilly and G. S. H. Yeo, Biosci. Rep., 2014, 34, e00144 CrossRef PubMed.
- M.-R. Cui, X.-L. Li, J.-J. Xu and H.-Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 13005–13012 CrossRef CAS PubMed.
- Y. Yao, N. Li, X. Zhang, J. Ong’achwa Machuki, D. Yang, Y. Yu, J. Li, D. Tang, J. Tian and F. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 13991–14003 CrossRef CAS PubMed.
- A. Cheong, J. J. A. Low, A. Lim, P. M. Yen and E. C. Y. Woon, Chem. Sci., 2018, 9, 7174–7185 RSC.
- J. Li, L. Zhu, Y. Shi, J. Liu, L. Lin and X. Chen, Am. J. Transl. Res., 2019, 11, 6084 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, DNA sequences, DMADz optimizations, fluorescence demonstrations, intracellular imaging and flow cytometric analysis. See DOI: 10.1039/d1sc05370a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiaoqing
Liu
a,
Xiaoqing
Liu