
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-field transition-induced C–S bond formation from nickelacycles†
Jeongcheol
Shin‡
 ,
Jiseon
Lee‡
,
Jong-Min
Suh
and
Kiyoung
Park
*
,
Jiseon
Lee‡
,
Jong-Min
Suh
and
Kiyoung
Park
*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: kiyoung.park@kaist.ac.kr
First published on 10th November 2021
Abstract
Photoexcitation is one of the acknowledged methods to activate Ni-based cross-coupling reactions, but factors that govern the photoactivity of organonickel complexes have not yet been established. Here we report the excited-state cross-coupling activities of Ni(II) metallacycle compounds, which display ∼104 times enhancement for the C–S bond-forming reductive elimination reaction upon Ni-centered ligand-field transitions. The effects of excitation energy and ancillary ligands on photoactivity have been investigated with 17 different nickelacycle species in combination with four corresponding acyclic complexes. Spectroscopic and computational electronic structural characterizations reveal that, regardless of coordinated species, d–d transitions can induce Ni–C bond homolysis, and that the reactivity of the resulting Ni(I) species determines the products of the overall reaction. The photoactivity mechanism established in this study provides general insights into the excited-state chemistry of organonickel(II) complexes.
Introduction
Ni catalysts for cross-coupling reactions are economical and environment-friendly and show complementary reactivities to traditional Pd catalysts.1 However, in conventional Ni-catalyzed cross-coupling reactions such as Negishi, Suzuki, and Kumada reactions, bond-forming reductive elimination (RE) reactions from the NiII state involve endergonic thermodynamics and thus exhibit slow kinetics.2 To promote this process, several strategies have been practiced. The chemical3 or photochemical2c,4 oxidation of the NiII species to the high-valent states can increase the RE activity. Alternatively, electronic excitation of the NiII species by energy transfer from photocatalysts or direct light irradiation can facilitate the RE reactions. This approach has been successfully implemented for diverse cross-coupling catalyses that include the C–C,5 C–N,6 C–O,6a,7 and C–S6a bond-forming RE reactions.Despite the increasing examples of excited-state Ni chemistries, no consensus has been reached on mechanisms, the understanding of which, though, is necessary for developing highly tunable and efficient photoreactions. For photoexcited Ni-mediated C–O cross-coupling reactions between aryl halide and carboxylic acid,7a several mechanisms have been suggested.8 The transient absorption spectroscopic (TAS) studies of catalytically-relevant NiII(2,2′-bipyridine; bpy)(aryl)Cl and NiII(bpy)(aryl)(acetate) complexes have revealed that Ni-to-bpy charge transfer (CT) transitions lead to a long-lived excited state (τ = 4 ns).8a,c In combination with transient infrared spectroscopy, Doyle et al. has defined this state to be a triplet d–d excited state, which is proposed to induce Ni–C bond homolysis in NiII(bpy)(aryl)Cl on the basis of radical trapping experiment.8b Alternatively, MacMillan et al. has proposed that the long-lived state of NiII(bpy)(aryl)(acetate) can induce the C–O bond-forming RE reaction directly.8c Regarding this system, Chen et al.'s multireference ab initio study has suggested that triplet Ni-to-bpy CT excited states, rather than d–d excited states, are the most viable excited states that can lead to the direct RE reaction.8d Meanwhile, Hadt et al.'s CASSCF study has provided new insights that a repulsive triplet carbon-to-Ni CT excited state can offer a spontaneous pathway for Ni–C bond homolysis.8e
In this study, the excited-state chemistry of three types of NiII metallacycle compounds, with cycloneophyl (–C6H4–o–C(CH3)2CH2–; CC hereinafter) and its oxa- (–OC6H4–o–C(CH3)2CH2–; OC) and thia- (–SC6H4–o–C(CH3)2CH2–; SC) derivative ligands (Scheme 1), have been investigated with five different ancillary ligands and seven different excitation energies. Nickelacycles and their heteroatom derivatives are key intermediates of various cyclization reactions between unsaturated compounds such as alkenes, alkynes, aldehydes, and imines.1a,9 The C–C,9b,10 C–N,9d,11 C–O,9c,12 and C–S9c,13 bonds can be generated from NiII metallacycle compounds, the RE reaction of which often limit catalytic turnover rates.11f,14 Their excited-state reactivity has not been explored, while their electronic structural similarity to other NiII complexes involved in photocatalysis suggests the possibility of light-induced reactions. To elucidate the electronic structures of the nickelacycles, electronic absorption (Abs), magnetic circular dichroism (MCD), and resonance Raman (rR) spectroscopies have been utilized in combination with density functional theory (DFT) computations. The effects of various excitation energies and ancillary ligands on photo-induced RE activity have been explored to specify RE-inducing excited states. The reaction coordinates of the excited-state RE reactions have also been established, providing insights into the factors that determine the RE activity of excited states.
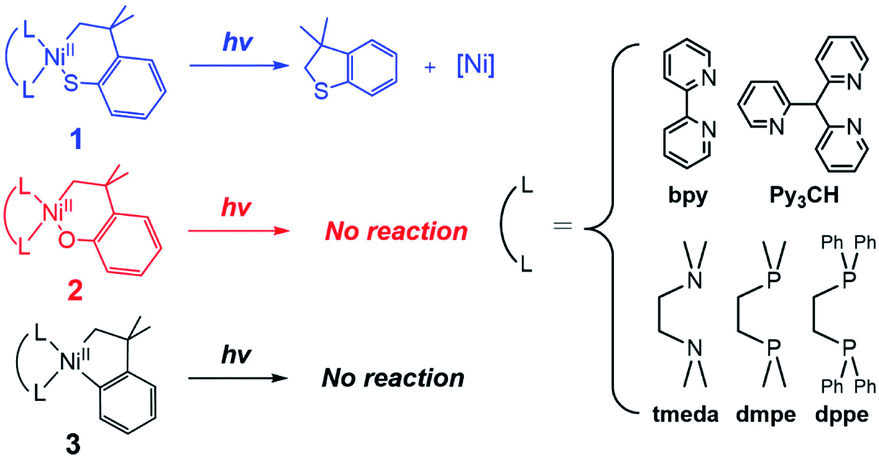 | ||
| Scheme 1 Excited-state RE reactions of nickelacycles. | ||
Results and analysis
C–S bond-forming photoactivity as a function of excitation energy
Given that the bpy ligand had been proposed to play a major role in the photo-induced C–O coupling of the NiII(bpy)(aryl)(acetate) complex, bpy-bound nickelacycles, NiII(bpy)(SC) (1bpy), NiII(bpy)(OC) (2bpy), and NiII(bpy)(CC) (3bpy), were first chosen for testing photoactivities (Fig. 1). These complexes are stable at 20 °C under dark conditions (Table S1†). Upon irradiation with 390 nm 52 W LED light, 1bpy underwent quantitative C–S bond formation, yielding 99% conversion to 2,3-dihydro-3,3-dimethyl-benzo[b]thiophene within 3 hours (Fig. S1†). This photoactivity (k390 ∼ 3.6 × 10−2 min−1) indicates ∼104 times acceleration for the RE reaction, compared to the dark reaction (kdark ∼ 1.5 (±1.0) × 10−6 min−1 predicted for 20 °C by extrapolating Eyring plots from higher temperature data, Fig. S2†). The 390 nm LED-induced RE reaction follows the 1st-order kinetics, implying that the RE reaction is not induced by the generation of NiIII complex via intermolecular electron transfer. Contrarily, no change was observed for 2bpy and 3bpy even after 1 day irradiation (Table S6†). This specific photoactivity of 1bpy was unexpected, given the similarity in the Abs spectra of the three species (Fig. 1c).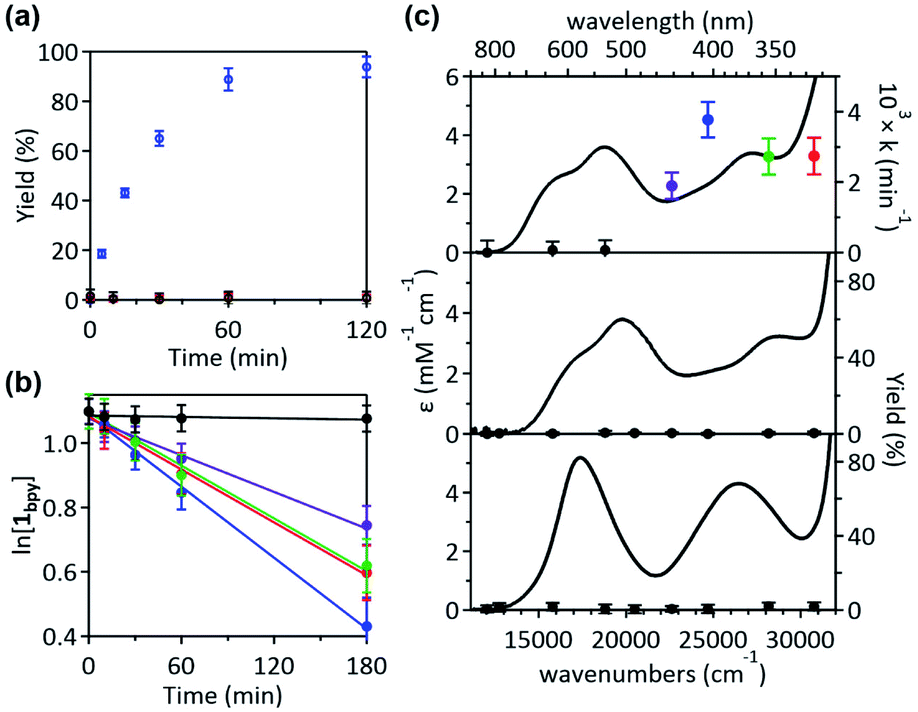 | ||
| Fig. 1 (a) 390 nm LED light-induced formation of RE products from 1bpy (blue), 2bpy (red), and 3bpy (black), monitored by 1H NMR spectroscopy. (b) Plots of the natural logarithm of the 1bpy concentration versus irradiation time, obtained with the 325 (red), 355 (green), 405 (blue), 442 (purple), and 532 (black) nm laser lines with the normalized power of 50 mW. (c) Abs spectra of 1bpy (top), 2bpy (middle), and 3bpy (bottom), overlaid with the 1st-order rate constants of the C–S coupling reaction and the C–O and C–C coupling yields obtained after 8 hours irradiation. | ||
To identify the excited states that lead to the C–S coupling reaction, seven different laser lines, 830, 633, 532, 442, 405, 355, and 325 nm, were examined. With the excitation energy higher than 500 nm, the 1st-order kinetics were observed with rate constants dependent on the laser power (Fig. 1b, S3 & S4†). Contrarily, with the excitation energy lower than 500 nm, the C–S coupling yield was negligible, although the Abs features maxed at 533 and 367 nm display comparable intensities (Fig. 1c & Table S2†). The fastest C–S bond formation was observed with the 405 nm excitation (k405 = 4.2 × 10−3 min−1). This excitation-energy dependence implies that the photo-induced C–S coupling does not follow Kasha's rule.15 Consistent with the 390 nm LED experiment, none of the laser lines could activate 2bpy and 3bpy despite the great similarity of their electronic structures (Fig. S5,†vide infra).
Electronic structure determination
The visible-range Abs and MCD spectra of 1bpy are deconvoluted into five electronic transitions (Fig. 2). The Abs feature at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–22000 cm−1 with negligible RE activities contains bands 1 and 2, while bands 3–5 are associated with the RE-inducing Abs feature at 22000–30500 cm−1. These five transitions display extinction coefficients of ∼4000 M−1 cm−1, indicative of metal-to-ligand charge transfer (MLCT) transitions with ε > 1000 M−1 cm−1 rather than d–d transitions with ε ∼ 50–500 M−1 cm−1.16 The rR spectra of 1bpy support the Ni-to-bpy CT character, exhibiting enhanced intensities for bpy-based vibrations with laser excitation to bands 1 and 2 (Fig. S6 & S7†).17
000–22000 cm−1 with negligible RE activities contains bands 1 and 2, while bands 3–5 are associated with the RE-inducing Abs feature at 22000–30500 cm−1. These five transitions display extinction coefficients of ∼4000 M−1 cm−1, indicative of metal-to-ligand charge transfer (MLCT) transitions with ε > 1000 M−1 cm−1 rather than d–d transitions with ε ∼ 50–500 M−1 cm−1.16 The rR spectra of 1bpy support the Ni-to-bpy CT character, exhibiting enhanced intensities for bpy-based vibrations with laser excitation to bands 1 and 2 (Fig. S6 & S7†).17
 | ||
| Fig. 2 (a) 5 T MCD (top), Abs (middle), and TDDFT-simulated Abs (bottom) spectra of 1bpy. The deconvoluted Gaussian bands and their sum spectra are shown in blue solid and black dashed lines, respectively. (b) MO diagram of 1bpy and frontier MOs relevant to the electronic transitions in the visible region. | ||
This assignment is further confirmed by time-dependent density functional theory (TDDFT) computations, which were validated by the reproducibility of the 1bpy ∼ 3bpy Abs spectra (Fig. S10†). The simulated spectrum obtained with the LC-ωPBE functional18 (ω = 0.15) and the 6-311g(d,p) basis set19 shows that bands 1–5 can be associated with charge transfer transitions from the Ni 3dyz and 3dxz-based molecular orbitals (MOs) to the bpy π*-based MOs (Table S3†). This similar Ni-to-bpy CT character of the five bands contrasts with the distinct photoactivities of bands 3–5 relative to bands 1 and 2. To explain the differential photoactivities, transitions with even small oscillation strengths were thoroughly analyzed, revealing that Ni-centered d–d transitions contribute to the energy range of bands 3–5, but not that of bands 1 and 2. Notably, the Abs and MCD spectral features of 2bpy and 3bpy can also be attributed to Ni-to-bpy π* CT transitions similarly to 1bpy, based on the rR enhancement of bpy-based vibrations and TDDFT-simulated Abs spectra (Fig. S6–S9 and Tables S4, S5†).
Ancillary ligand variations to elucidate photoactive excited states
To unmask the ligand-field transitions from the MLCT-dominant Abs bands, the bpy ligand was replaced with other ancillary ligands. The less-conjugated pyridinyl ligand, tris(2-pyridinyl)methane (Py3CH), was introduced with anticipation of upshifting the MLCT transitions in the spectrum, as the pyridinyl π* system has higher energy than the bpy π* system. However, at 390 nm, 1Py3CH still displays a similar Abs intensity and the comparable photo-induced RE activity as 1bpy (Fig. 3). With the 1,2-bis(diphenylphosphino)ethane (dppe) ligand, 1dppe also displays unexpected Ni-to-dppe phenyl π* transitions in the visible region. Detailed electronic spectral assignments are given in ESI (Fig. S14, S15, and Tables S7, S8†).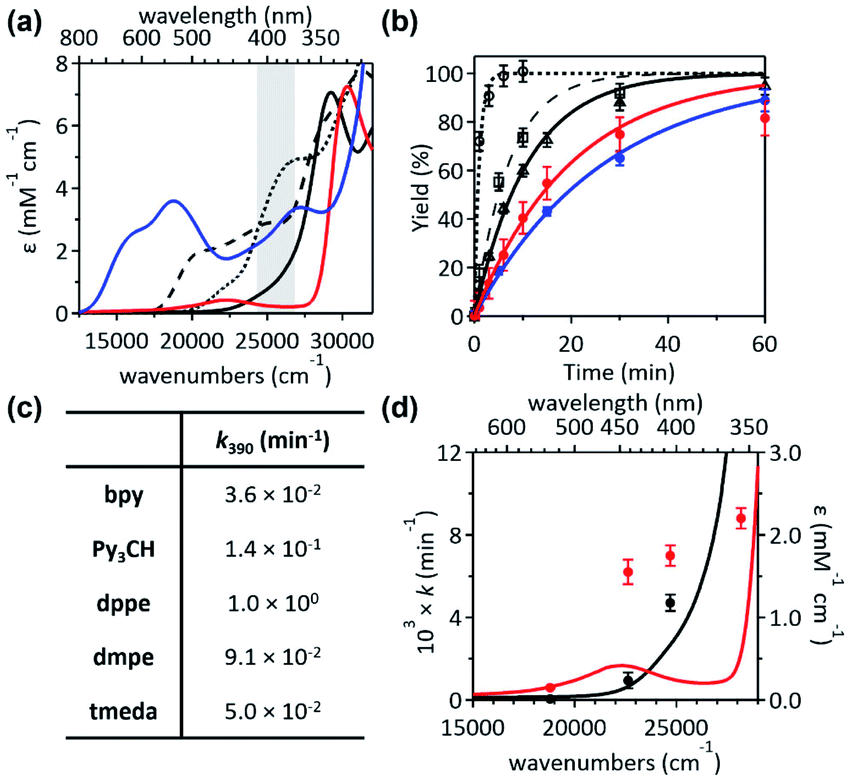 | ||
Fig. 3 (a) Abs spectra and (b) 1H-NMR monitored C–S coupled product formation as a function of 390 nm LED irradiation time of 1bpy (blue solid,  ), 1Py3CH (black dashed, ), 1Py3CH (black dashed,  ), 1dppe (black dotted, ), 1dppe (black dotted,  ), 1dmpe (black solid, ), 1dmpe (black solid,  ), and 1tmeda (red solid, ), and 1tmeda (red solid,  ). The energy window of light emitted from the 390 nm LED lamp is highlighted in gray. (c) 390 nm LED-induced RE rate constants. (d) Excitation energy-dependent C–S coupling rate constants of 1tmeda (red) and 1dmpe (black), overlaid with their Abs spectra. Detailed kinetic analyses are given in Fig. S11–S13 and Table S6.† ). The energy window of light emitted from the 390 nm LED lamp is highlighted in gray. (c) 390 nm LED-induced RE rate constants. (d) Excitation energy-dependent C–S coupling rate constants of 1tmeda (red) and 1dmpe (black), overlaid with their Abs spectra. Detailed kinetic analyses are given in Fig. S11–S13 and Table S6.† | ||
Alternatively, with the aliphatic amine ligand, N,N,N′,N′-tetramethylethylenediamine (tmeda), which does not contain low-lying ligand-based unoccupied MOs, the NiII(tmeda)(SC) complex, 1tmeda, exhibits a significantly reduced Abs intensity at the energy below 350 nm (Fig. 3a). Notably, 1tmeda displays an almost identical photoactivity as 1bpy (k390 = 5.0 × 10−2 min−1 for 1tmeda and 3.6 × 10−2 min−1 for 1bpy), despite the 13 times reduced absorptivity of 1tmeda relative to 1bpy (ε390 = 213 M−1 cm−1 for 1tmeda and 2775 M−1 cm−1 for 1bpy). With the aliphatic phosphine ligand, 1,2-bis(dimethylphosphino)ethane (dmpe), 1dmpe also exhibits low-intensity Abs at 380–450 nm, and undergoes the RE reaction with the similar photoactivity as 1tmeda (Fig. 3d).
To elucidate the low-intensity Abs features in the visible range of 1tmeda, its Abs and MCD spectra were deconvoluted and compared to the TDDFT-calculated electronic transitions (Fig. 4). With the aliphatic ancillary ligand, the frontier MOs (FMOs) of 1tmeda are all Ni 3d-based, and thus only the d–d transitions contribute to the visible energy region (Table S9†). The spectra of 1dmpe could also be assigned similarly (Fig. S16 and Table S10†). These electronic structural characterizations reveal that the ligand-field transitions alone can drive the C–S bond-forming RE reaction. Depending on the ancillary ligands, Abs intensities may vary due to contributions from the MLCT transitions. However, the photoactivities of the five 1 complexes are still comparable, as the d–d excited states of the five species are similarly effective for the RE reaction. The results above do not exclude the possibility of high-energy MLCT transitions, such as those in bands 3–5 of 1bpy, being effective in inducing the RE reaction. Nevertheless, the roles of the ancillary ligands appear to be minor, as d–d transitions suffice for performing the photoreactions of 1's.
 | ||
| Fig. 4 (a) 5 T MCD (top), Abs (middle), and TDDFT-simulated Abs (bottom) spectra of 1tmeda. The deconvoluted Gaussian bands and their sum spectra are shown in colored solid and black dashed lines, respectively. (b) MO diagram of 1tmeda and frontier MOs relevant to the electronic transitions in the visible region. | ||
It should also be noted that, regardless of the ancillary ligands, complexes 2 and 3 remain photo-inactive, although their Abs and MCD spectra are essentially identical to those of corresponding complexes 1 (Fig. S17–S20†). These distinctive photoactivities despite the similar electronic structures imply that the RE-inducing photoactivity would not be solely determined by the initial excited states at the reactant state. Thus, to explore variations in the excited states along the RE reaction coordinate, the excited-state potential energy surfaces (PESs) of 1tmeda, 2tmeda, and 3tmeda have been plotted by performing TDDFT computations on the ground-state PESs (Fig. 5a & S26†).
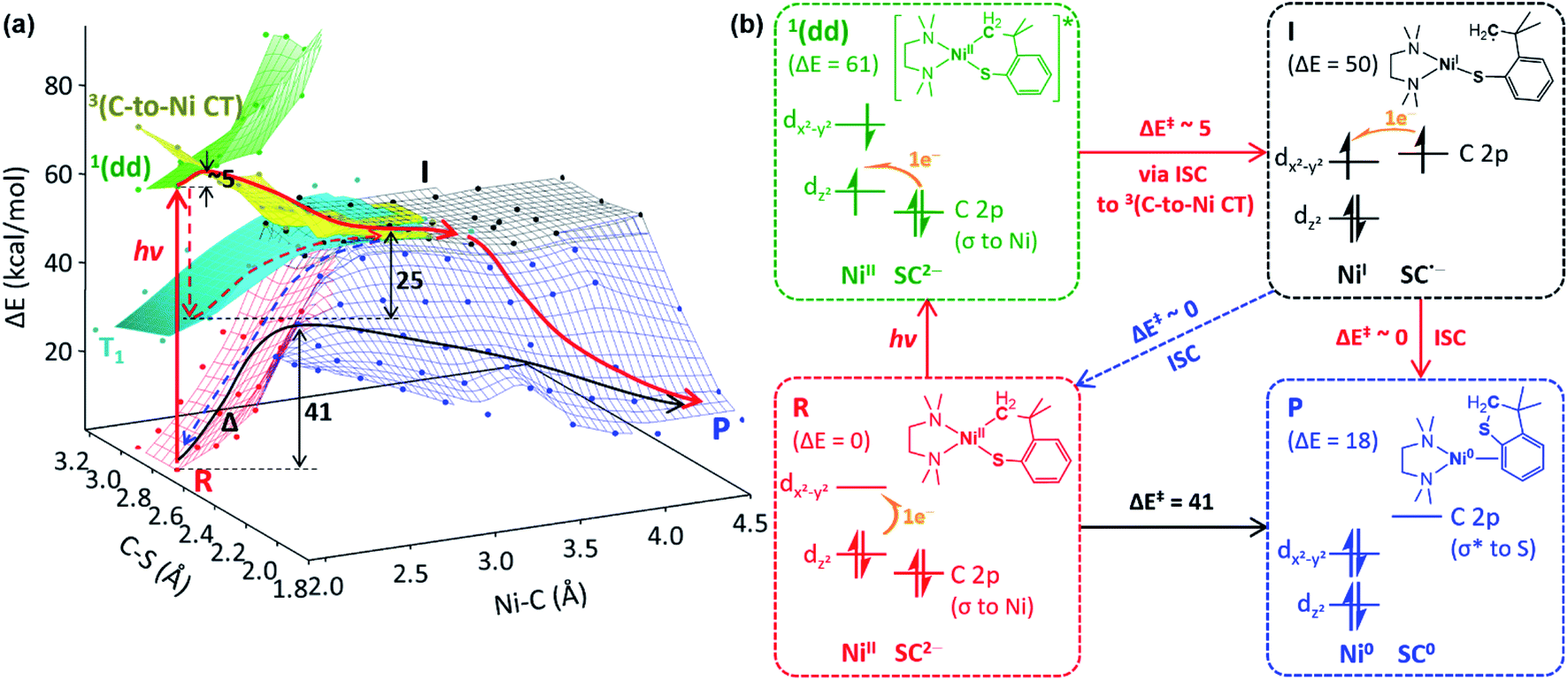 | ||
| Fig. 5 (a) The ground- and excited-state PESs of 1tmeda, colored according to the electronic configurations of the ground state (red) and the 1(dd) (green), 3(C-to-Ni CT) (yellow), and T1 (cyan) excited states of the NiII(SC2−) reactant (R), the NiI(SC˙−) intermediate (I, black), and the Ni0 + SC0 product (P, blue). A plausible photo-induced RE pathway from the 1(dd) state and the dark RE pathway are drawn with red and black solid arrows, respectively. Alternative paths are noted with dashed arrows. (b) Electronic configuration and potential energy (in kcal mol−1) changes over the reactions presented in (a). | ||
Excited-state potential energy surfaces
To search for the reaction coordinate of the C–S bond-forming RE reaction, the PES of 1tmeda has been plotted along the Ni–C and C–S bond lengths. At the ground state, the RE reaction from the NiII(SC2−) reactant (R, red PES in Fig. 5a) to the Ni0 + SC0 product (P, blue PES in Fig. 5a) involves one elementary step with two electrons transferred from the SC ligand to the NiII center. This step is endergonic (ΔG; ΔH; ΔE ∼18, 18, 19 kcal mol−1) with a high kinetic barrier of ΔG‡; ΔH‡; ΔE‡ ∼39, 39, 41 kcal mol−1, consistent with the poor RE activity under dark conditions (Fig. S23†). Alternatively, at the T1 state (cyan PES in Fig. 5a), which is accessible via intersystem crossing (ISC) following photoexcitation, the RE reaction should involve two one-electron transfer steps, as the T1 state contains two singly occupied MOs with the electronic configuration of (dz2)1(dx2–y2)1 (Fig. S24†). The 1st electron transfer occurs from the C-based HOMO to the Ni 3dz2-based spin-down LUMO, leading to the Ni–C bond homolysis and forming the NiI(SC˙−) intermediate (I, black PES in Fig. 5a). I contains two radical sites at the NiI center and the C moiety apart by ∼3.5 Å, far enough to render the singlet and triplet states degenerate. Subsequently, to form the C–S bond in a spin-allowed fashion, the 2nd electron transfer should occur to the Ni 4s-based MO for triplet I, while for singlet I, the lower-energy Ni 3dx2–y2-based MO can be an acceptor. Thus, the former process involves less favorable thermodynamics than the latter one, indicating that the triplet intermediate generated from photoexcitation should undergo ISC prior to the C–S bond formation (Fig. S25†).To form intermediate I from the ligand-field excited states, TDDFT-calculated PESs propose two plausible pathways, one via1(dd) → T1 ISC and the other via1(dd) → 3(C-to-Ni CT) ISC (red dashed vs. solid pathways in Fig. 5a). The former pathway is parallel to the previously suggested mechanism based on the TAS study of NiII(bpy)(aryl)Cl.8b This process is calculated to be uphill by ΔG‡ ∼ 22.5 kcal mol−1 for 1tmeda (cyan PES → black PES in Fig. 5a) and 26.0 for 1bpy (Table S13†). Hadt et al. has shown that DFT calculations tend to underestimate the corresponding barrier.8e Moreover, the excitation energy dependence of the photoreaction of 1bpy proposes that the T1-to-intermediate I conversion is unlikely. Alternatively, in the vicinity of the 1(dd) state (green PES in Fig. 5a), a repulsive PES is calculated, associated with the triplet C ligand-to-Ni CT transition (yellow PES in Fig. 5a). From the 1(dd) state, this 3(C-to-Ni CT) state can be accessed with a minor elongation of the Ni–C bond by ∼0.1 Å and a small barrier of ∼5 kcal mol−1. Since the 3(C-to-Ni CT) state is equivalent to intermediate I in terms of the NiI(SC˙−) electronic configuration (Fig. 5b), once ISC from the 1(dd) state to the 3(C-to-Ni CT) state occurs, the formation of I and subsequently P can proceed in a downhill fashion with no major barriers (red solid path in Fig. 5a). The presence of a similar repulsive PES was predicted in the previous multireference computational study of the NiII(bpy)(Ar) complexes, for which, though, the Ni-to-bpy CT excited state was proposed to be the initial state.8e From the Ni–C bond dissociated intermediate I, R and P are equally accessible with no discernible barriers. This explains a small quantum yield of 6.8 × 10−3 estimated relative to ferrioxalate actinometry (Fig. S27†).
Ni–C bond homolysis induced by ligand-field excitations
To confirm the presence of intermediate I for 1, radical trapping experiments with α-phenyl-N-tertiary-butylnitrone (PBN) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) were attempted but unsuccessful. Nevertheless, this result is understandable, because the subsequent reactions of intermediate I are predicted to have no barriers. Another factor that would limit the lifetime of intermediate I is the chelate effect of the SC ligand, which assists the intramolecular reaction of the carbon radical. Thus, an analogous acyclic complex, NiII(bpy)(Me)(SPh) (4bpy), has been explored. The Abs and MCD spectra of 4bpy are essentially identical to those of 1bpy, as the same type of electronic transitions, Ni-to-bpy CT and d–d transitions, contribute to the spectra (Fig. 6b & S28†). Upon 390 nm LED irradiation, unlike 1bpy that forms the C–S coupled product homogeneously, 4bpy yields ethane majorly (Fig. 6a). These products indicate the photo-induced generation of methyl radicals; i.e., the photoexcitation of 4bpy affects the Ni–C bond homolysis, which could be further confirmed by the radical-trap EPR signal (Fig. S29†). For both 1bpy and 4bpy, the repulsive 3(C-to-Ni CT) PES's are calculated, suggesting that the Ni–C bond homolysis can be photo-induced for both complexes (Fig. S31†). Subsequent to this step, while 1bpy can undergo the intramolecular C–S bond formation with a minor barrier, the dissociated methyl radical of 4bpy kinetically and thermodynamically prefers the ethane formation to the C–S coupled thioanisole generation (Fig. S32†). | ||
| Fig. 6 (a) 390 nm LED-induced reactions of 1bpy (top) and 4bpy (bottom). For 4bpy, the yield is given per methyl group. (b) 5 T MCD (top) and Abs (bottom) spectra of 1bpy (red) and 4bpy (black), with the 405 nm (blue) and 532 nm (green) laser excitation energies shaded. (c) Logarithmic plots for the 1H NMR-monitored decays of 1bpy (top) and 4bpy (bottom) as a function of 405 nm (blue) and 532 nm (green) laser irradiation time. The linear regression fitting gives k532 = 2 × 10−4 min−1 and k405 = 4.2 × 10−3 min−1 for 1bpy and k532 = 3.5 × 10−3 min−1 and k405 = 1.2 × 10−1 min−1 for 4bpy. | ||
The excitation-energy-dependent photoactivity of 4bpy is also equivalent to that of 1bpy, supporting that, regardless of whether the system is cyclic or acyclic, the photo-induced initial step is identical. Fig. 1 shows that 1bpy decays rapidly with excitation to bands 3–5, while excitation to bands 1–2 displays negligible effects. Likewise, a faster decay of 4bpy was observed with the 405 nm irradiation aiming band 3 than with the 532 nm irradiation associated with band 2 (Fig. S30†). Similar rate-constant ratios, k405/k532, were obtained for 1bpy (40) and 4bpy (34), indicating the equivalence of the photo-induced step (Fig. 6c). The excitation energy dependence of the Ni–C bond homolysis supports the significance of the dissociative 3(C-to-Ni CT) PES, which is more accessible from the higher-energy d–d excited states than the lower-energy Ni-to-bpy CT states. The effectiveness of ligand-field excitations on the Ni–C bond homolysis was further confirmed with NiII(dmpe)(Me)(SPh) (4dmpe), which has only d–d transitions in the visible region and exhibits a comparable photoactivity (k405 ∼ 3.7 × 10−2 min−1, Fig. S33†).
NiI intermediates determine the overall photo-reactivity
The above mechanistic studies of 1 and 4 indicate that during the photo-induced Ni–C bond homolysis, the sulfur moiety of the SC ligand does not play a specific role. This finding leaves a question of why 2 and 3 are photo-inactive despite the similar electronic structures. To test whether the Ni–C bond homolysis can still be induced in the absence of the sulfenyl group, the photo-reactivities of NiII(dmpe)(Me)(OPh) (5dmpe) and NiII(dmpe)Me2 (6dmpe) have been compared to 4dmpe. Upon 390 nm LED irradiation which can induce d–d transitions (Fig. S34†), all three complexes produce a sizable amount of ethane (Fig. 7a). This result reveals that the Ni–C bond homolysis can be photo-induced regardless of the ligands, being not unique to the sulfur-ligated systems. This conclusion is further supported by aliphatic nickelacycles, NiII(bpy)(CH2CH2CH2CH2) (7bpy) and NiII(bpy)(OCH2CH2CH2CH2) (8bpy). Under 390 nm irradiation, these systems generate β-hydride elimination products, indicating that the Ni–C bond homolysis can be photo-induced regardless of cyclic or acyclic compounds (Table S15†). These results imply that 2 and 3 could also undergo the photo-induced Ni–C bond homolysis. Consistently, the TDDFT-calculated excited state PESs of 2 and 3 with the tmeda and dmpe ligands predict the presence of the repulsive 3(C-to-Ni CT) states nearby the 1(dd) states (Fig. S26 & S35†).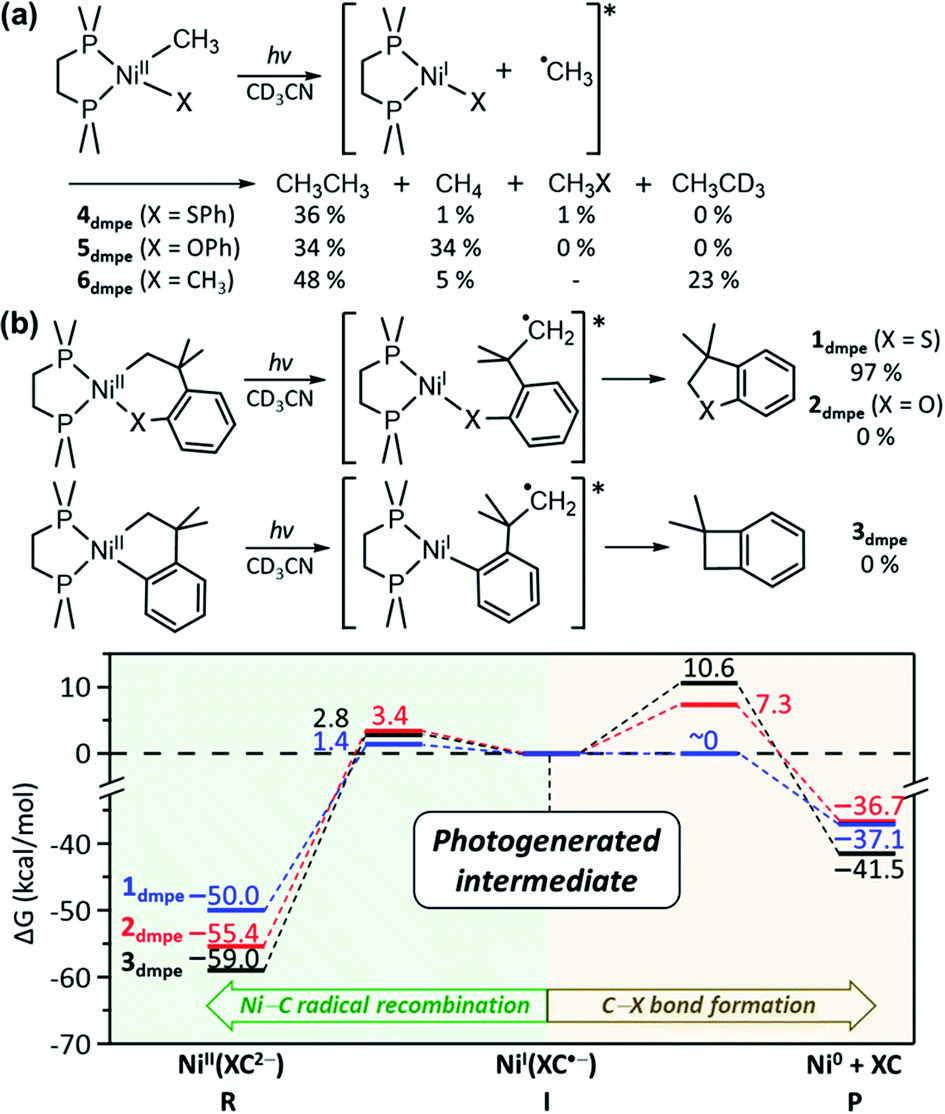 | ||
| Fig. 7 (a) Photoreactions of 4dmpe, 5dmpe, and 6dmpe. The final yields are given with respect to the initial concentrations of the methyl groups. (b) Energy profiles for the reactions of the NiI intermediates generated upon Ni–C bond homolysis of 1dmpe (blue), 2dmpe (red), and 3dmpe (black). | ||
The fact that only 1 generates the RE product while all 1–3 can undergo the Ni–C bond homolysis upon ligand-field excitations proposes that the reactivities of the NiI intermediates can be variable depending on the substrate. In Fig. 5a, the PESs of 1tmeda show that intermediate I can undergo either the C–S bond formation that produces the RE product or the Ni–C radical recombination that leads back to the reactant. The similar pathways are also calculated for 1dmpe ∼ 3dmpe (Fig. S36–S38†). The C–S bond formation from the NiI intermediate of 1dmpe is almost barrierless (Fig. 7b). However, for 2dmpe and 3dmpe, the C–C and C–O bond formations from the NiI intermediates involve 7–11 kcal mol−1 higher kinetic barriers than the Ni–C radical recombination. Therefore, overall only 1dmpe displays the photo-induced RE activity, while 2dmpe and 3dmpe are seemingly photo-inactive. The same tendency is calculated for 1tmeda ∼ 3tmeda and 1bpy (Fig. S39–S43†).
Discussions
We have shown that visible-light photoexcitation can enhance the C–S coupling activity of 1 by four orders of magnitude. To understand its mechanism, the effects of diverse ancillary ligands have been explored, along with electronic structural characterizations by Abs and MCD spectroscopies and TDDFT computations. These analyses reveal that, without bpy-associated transitions, d–d excitations alone can drive the C–S bond-forming RE reaction. The excitation-energy-dependent photoactivity of 1bpy demonstrates that the photo-induced C–S bond formation does not follow Kasha's rule, emphasizing the significance of ISC to the dissociative 3(C-to-Ni CT) state that leads to the Ni–C bond homolysis. This reactivity parallels the C–C coupling photoactivity of high-valent nickelacycles, which display the maximum RE rate upon C-to-Ni CT transitions.20 This excitation-energy-dependent photochemistry is not unprecedented for transition metal complexes. The Cu–Cl bond homolysis of a chlorobis(phenanthroline)CuII complex can be induced by Cl-to-Cu CT transition (427 or 470 nm) but not by d–d transitions (785 or 800 nm).21 Mn2(CO)10 undergoes the Mn–Mn bond homolysis with 350 nm light, while 248 and 193 nm light causes CO loss.22 These examples emphasize the significance of electronic structure characterizations in utilizing excited-state chemistry.The ligand-field transition-induced Ni–C bond homolysis is found to be general for organonickel(II) complexes 1–8, regardless of ancillary ligands and neighboring coordinated species. However, the final product of the photoreactions can be variable, depending on the reactivities of the NiI intermediates and carbon radicals obtained from the photo-induced Ni–C bond homolysis. The DFT-computed reaction coordinates reveal that the NiI intermediates of 1 can undergo the barrierless C–S bond formation, while those of 2–3, acyclic compounds 4–6, and aliphatic nickelacycles 7–8 prefer the recombination of the Ni–C bond, ethane formation, and β-hydride elimination, respectively. This mechanistic elucidation provides a general basis for understanding and designing the excited-state chemistry of organonickel(II) complexes, proposing that to control their photochemistry, the reactivity of intermediates resulting from Ni–C bond homolysis should be concerned. Considering the entropy-driven dissociation of organic radicals in acyclic compounds, this study suggests that nickelacycles can be a good platform for selective photochemistry as shown in the visible-light-induced C–S bond formation of 1.
Data availability
The NMR, Abs, MCD, and rR spectra of Ni complexes and DFT-computation results including Cartesian coordinates can be found in ESI.†Author contributions
J. Shin and J. Lee performed all syntheses, kinetic investigations, and spectroscopic and computational characterizations. J. Suh performed ESI-MS analyses. K. Park supervised research activities. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Samsung Science and Technology Foundation (SSTF-BA1801-05). We thank Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science, for allowing to use computational and rR spectroscopic resources.Notes and references
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (c) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A. M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (d) V. B. Phapale and D. J. Cardenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC; (e) F. S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- (a) R. Y. Han and G. L. Hillhouse, J. Am. Chem. Soc., 1997, 119, 8135–8136 CrossRef CAS; (b) S. A. Macgregor, G. W. Neave and C. Smith, Faraday Discuss., 2003, 124, 111–127 RSC; (c) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330–334 CrossRef CAS PubMed; (d) M. Busch, M. D. Wodrich and C. Corminboeuf, ACS Catal., 2017, 7, 5643–5653 CrossRef CAS; (e) P. M. MacQueen, J. P. Tassone, C. Diaz and M. Stradiotto, J. Am. Chem. Soc., 2018, 140, 5023–5027 CrossRef CAS PubMed.
- (a) K. M. Koo, G. L. Hillhouse and A. L. Rheingold, Organometallics, 1995, 14, 456–460 CrossRef CAS; (b) R. Han and G. L. Hillhouse, J. Am. Chem. Soc., 1998, 120, 7657–7658 CrossRef CAS; (c) N. Nebra, Molecules, 2020, 25, 2015 CrossRef CAS PubMed; (d) F. L. Vaillant, E. J. Reijerse, M. Leutzsch and J. Cornella, J. Am. Chem. Soc., 2020, 142, 19540–19550 CrossRef PubMed.
- (a) O. S. Wenger, Chem.–Eur. J., 2021, 27, 2270–2278 CrossRef CAS PubMed; (b) Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (c) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (d) J. Xuan, T. T. Zeng, J. R. Chen, L. Q. Lu and W. J. Xiao, Chem.–Eur. J., 2015, 21, 4962–4965 CrossRef CAS PubMed; (e) M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016, 138, 1760–1763 CrossRef CAS PubMed; (f) I. Abdiaj, A. Fontana, M. V. Gomez, A. de la Hoz and J. Alcázar, Angew. Chem., Int. Ed., 2018, 57, 8473–8477 CrossRef CAS PubMed; (g) R. Sun, Y. Z. Qin and D. G. Nocera, Angew. Chem., Int. Ed., 2020, 59, 9527–9533 CrossRef CAS PubMed.
- (a) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed; (b) L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed; (c) D. L. Zhu, R. J. Xu, Q. Wu, H. Y. Li, J. P. Lang and H. X. Li, J. Org. Chem., 2020, 85, 9201–9212 CrossRef CAS PubMed.
- (a) R. A. Escobar and J. W. Johannes, Chem.–Eur. J., 2020, 26, 5168–5173 CrossRef CAS PubMed; (b) C. H. Lim, M. Kudisch, B. Liu and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 7667–7673 CrossRef CAS PubMed; (c) M. Kudisch, C. H. Lim, P. Thordarson and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 19479–19486 CrossRef CAS PubMed; (d) T. Kim, S. J. McCarver, C. Lee and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 3488–3492 CrossRef CAS PubMed.
- (a) E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–384 CrossRef CAS PubMed; (b) L. Yang, H. H. Lu, C. H. Lai, G. Li, W. Zhang, R. Cao, F. Y. Liu, C. Wang, J. L. Xiao and D. Xue, Angew. Chem., Int. Ed., 2020, 59, 12714–12719 CrossRef CAS PubMed; (c) D. L. Zhu, H. X. Li, Z. M. Xu, H. Y. Li, D. J. Young and J. P. Lang, Org. Chem. Front., 2019, 6, 2353–2359 RSC.
- (a) B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed; (b) S. I. Ting, S. Garakyaraghi, C. M. Taliaferro, B. J. Shields, G. D. Scholes, F. N. Castellano and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 5800–5810 CrossRef CAS PubMed; (c) L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555–4559 CrossRef CAS PubMed; (d) P. C. Ma, S. H. Wang and H. Chen, ACS Catal., 2020, 10, 1–6 CrossRef CAS; (e) D. A. Cagan, G. D. Stroscio, A. Q. Cusumano and R. G. Hadt, J. Phys. Chem. A, 2020, 124, 9915–9922 CrossRef CAS PubMed.
- (a) G. Wilke, Angew. Chem., Int. Ed., 1988, 27, 185–206 CrossRef; (b) J. Montgomery, Acc. Chem. Res., 2000, 33, 467–473 CrossRef CAS PubMed; (c) T. Kurahashi and S. Matsubara, Acc. Chem. Res., 2015, 48, 1703–1716 CrossRef CAS PubMed; (d) M. Ohashi, Y. Hoshimoto and S. Ogoshi, Dalton Trans., 2015, 44, 12060–12073 RSC.
- (a) A. Nishimura, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2012, 134, 15692–15695 CrossRef CAS PubMed; (b) H. Horie, T. Kurahashi and S. Matsubara, Chem. Commun., 2012, 48, 3866–3868 RSC; (c) S. Ogoshi, A. Nishimura and M. Ohashi, Org. Lett., 2010, 12, 3450–3452 CrossRef CAS PubMed; (d) Y. K. Ni and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 11162–11163 CrossRef CAS PubMed; (e) T. Kawashima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2018, 140, 17423–17427 CrossRef CAS PubMed; (f) L. Liu and J. Montgomery, J. Am. Chem. Soc., 2006, 128, 5348–5349 CrossRef CAS PubMed.
- (a) S. Ogoshi, H. Ikeda and H. Kurosawa, Angew. Chem., Int. Ed., 2007, 46, 4930–4932 CrossRef PubMed; (b) Y. Kajita, S. Matsubara and T. Kurahashi, J. Am. Chem. Soc., 2008, 130, 6058–6059 CrossRef CAS PubMed; (c) M. Ohashi, I. Takeda, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 18018–18021 CrossRef CAS PubMed; (d) Y. Yoshida, T. Kurahashi and S. Matsubara, Chem. Lett., 2011, 40, 1140–1142 CrossRef CAS; (e) T. Shiba, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 13636–13639 CrossRef CAS PubMed; (f) Y. Hoshimoto, T. Ohata, M. Ohashi and S. Ogoshi, Chem.–Eur. J., 2014, 20, 4105–4110 CrossRef CAS PubMed.
- (a) Y. Kajita, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2008, 130, 17226–17227 CrossRef CAS PubMed; (b) I. Koyama, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2009, 131, 1350–1351 CrossRef CAS PubMed; (c) A. Ooguri, K. Nakai, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2009, 131, 13194–13195 CrossRef CAS PubMed; (d) S. Sako, T. Kurahashi and S. Matsubara, Chem. Commun., 2011, 47, 6150–6152 RSC.
- (a) T. Inami, Y. Baba, T. Kurahashi and S. Matsubara, Org. Lett., 2011, 13, 1912–1915 CrossRef CAS PubMed; (b) T. Inami, T. Takahashi, T. Kurahashi and S. Matsubara, J. Am. Chem. Soc., 2019, 141, 12541–12544 CrossRef CAS PubMed.
- L. Adak, W. C. Chan and N. Yoshikai, Chem.–Asian J., 2011, 6, 359–362 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, 1984 Search PubMed.
- A. Basu, H. D. Gafney and T. C. Strekas, Inorg. Chem., 1982, 21, 2231–2235 CrossRef CAS.
- (a) O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed; (b) O. A. Vydrov, J. Heyd, A. V. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106 CrossRef PubMed; (c) S. Ragot and P. J. Becker, J. Chem. Phys., 2006, 125, 154109 CrossRef PubMed.
- (a) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (c) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- J. Shin, S. Gwon, S. Kim, J. Lee and K. Park, J. Am. Chem. Soc., 2020, 142, 4173–4183 CrossRef CAS PubMed.
- R. Fayad, S. Engl, E. O. Danilov, C. E. Hauke, O. Reiser and F. N. Castellano, J. Phys. Chem. Lett., 2020, 11, 5345–5349 CrossRef CAS PubMed.
- (a) D. A. Prinslow and V. Vaida, J. Am. Chem. Soc., 1987, 109, 5097–5100 CrossRef CAS; (b) A. Rosa, G. Ricciardi, E. J. Baerends and D. J. Stufkens, Inorg. Chem., 1996, 35, 2886–2897 CrossRef CAS; (c) C. Daniel, Coord. Chem. Rev., 2003, 238, 143–166 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods and materials, details of photoreactions, NMR, Abs, resonance Raman and MCD spectra of Ni complexes, and DFT-computed results. See DOI: 10.1039/d1sc05113j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |