
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid glycoconjugation with glycosyl amines†
Mareike A.
Rapp
,
Oliver R.
Baudendistel
,
Ulrich E.
Steiner
 and
Valentin
Wittmann
*
and
Valentin
Wittmann
*
Department of Chemistry and Konstanz Research School Chemical Biology (KoRS-CB), University of Konstanz, Universitätsstraße 10, 78457 Konstanz, Germany. E-mail: mail@valentin-wittmann.de
First published on 29th October 2021
Abstract
Conjugation of unprotected carbohydrates to surfaces or probes by chemoselective ligation reactions is indispensable for the elucidation of their numerous biological functions. In particular, the reaction with oxyamines leading to the formation of carbohydrate oximes which are in equilibrium with cyclic N-glycosides (oxyamine ligation) has an enormous impact in the field. Although highly chemoselective, the reaction is rather slow. Here, we report that the oxyamine ligation is significantly accelerated without the need for a catalyst when starting with glycosyl amines. Reaction rates are increased up to 500-fold compared to the reaction of the reducing carbohydrate. For comparison, aniline-catalyzed oxyamine ligation is only increased 3.8-fold under the same conditions. Glycosyl amines from mono- and oligosaccharides are easily accessible from reducing carbohydrates via the corresponding azides by using Shoda's reagent (2-chloro-1,3-dimethylimidazolinium chloride, DMC) and subsequent reduction. Furthermore, glycosyl amines are readily obtained by enzymatic release from N-glycoproteins making the method suited for glycomic analysis of these glycoconjugates which we demonstrate employing RNase B. Oxyamine ligation of glycosyl amines can be carried out at close to neutral conditions which makes the procedure especially valuable for acid-sensitive oligosaccharides.
Introduction
Carbohydrate components of glycoproteins and glycolipids are massively involved in both physiological and pathological recognition events,1 and the elucidation of their biological functions is a worldwide endeavor.2 Essential for unraveling carbohydrate recognition partners is the conjugation of isolated glycans to surfaces or analytical probes by chemoselective ligation reactions.3 In particular, the oxyamine ligation, i.e., the reaction of the aldehyde function of unprotected (reducing) glycans with oxyamines, has found widespread application in the field3a for example to prepare carbohydrate microarrays,4 nanoparticles,5 or fluorescently labeled carbohydrates.6 The reaction has also been used to prepare glycopeptide mimetics,7 PET labeling probes,8 and carbohydrate polymers.9 Although the ligation is highly chemoselective, it suffers from low reaction rates, especially when carbohydrates with a 2-acetamido substituent, such as GlcNAc derivatives, are reacted.10 In contrast to open-chain aldehydes, the aldehyde function of carbohydrates is masked by formation of a hemiacetal that accounts to more than 99.9% in equilibrium, shielding the carbonyl group from nucleophilic attack of the oxyamine. Oxime formation11 can be catalyzed by aniline and derivatives thereof as pioneered by Jenks12 and further developed by Dawson.13 Later, Jensen reported that the oxyamine ligation of carbohydrates in analogy with oxime formation of open-chain aldehydes is catalyzed by aniline14 and derivatives, such as 1,4-phenylenediamine (PDA).15 However, aniline and especially PDA, that have to be added in high concentrations to be effective, are toxic16 and can cause the formation of N-arylglycosylamine side products. Furthermore, PDA is chemically unstable, leading to deeply colored solutions even after reaction times of only a few hours and to precipitate formation.16 Here, we report that the rate of the oxyamine ligation of carbohydrates can be significantly accelerated without the need of a catalyst by using readily available glycosyl amines. The resulting ligation rates are increased up to 500-fold compared to the reaction of the reducing carbohydrate. For comparison, aniline-catalyzed oxyamine ligation is only increased 3.8-fold under the same conditions and 5-fold in the best case.Results and discussion
Scheme 1 shows possible reaction pathways of the oxyamine ligation of GlcNAc derivatives 1 which represent the reducing end of N-glycans. In the uncatalyzed reaction, the oxyamine reacts with the open-chain aldehyde 2 leading to oxime 3 that exists in equilibrium with N-glycosyloxyamine 4. The catalytic intermediate of the aniline-catalyzed oxyamine ligation14 reaction is glycosyl imine 5. This phenyl imine is more easily protonated than the parent carbonyl compound 2 which facilitates the attack of the oxyamine under weakly acidic conditions. While searching for an alternative to aniline catalysis that would allow fast ligation without a large excess of an external catalyst, we noticed that glycans enzymatically released from N-glycoproteins 8 by use of peptide N-glycosidase F (PNGase F)17 are obtained as free amines 7 that are in equilibrium with imine 6. Imine 6 resembles catalytic intermediate 5 of aniline catalysis. Thus, glycosyl amines 7 might be suitable substrates for rapid oxyamine ligation. It is expected that 6 is much more easily protonated than aryl imine 5, rendering it a more efficient intermediate for oxime formation, potentially also at higher (neutral) pH. The extent of the imine pKa decrease caused by the phenyl group can be estimated by comparing the pKa values of glucosyl amine (5.6) and glucosyl aniline (<1.5).18 Accordingly, we expect the pKa of 6 to be several pH units higher than that of 5.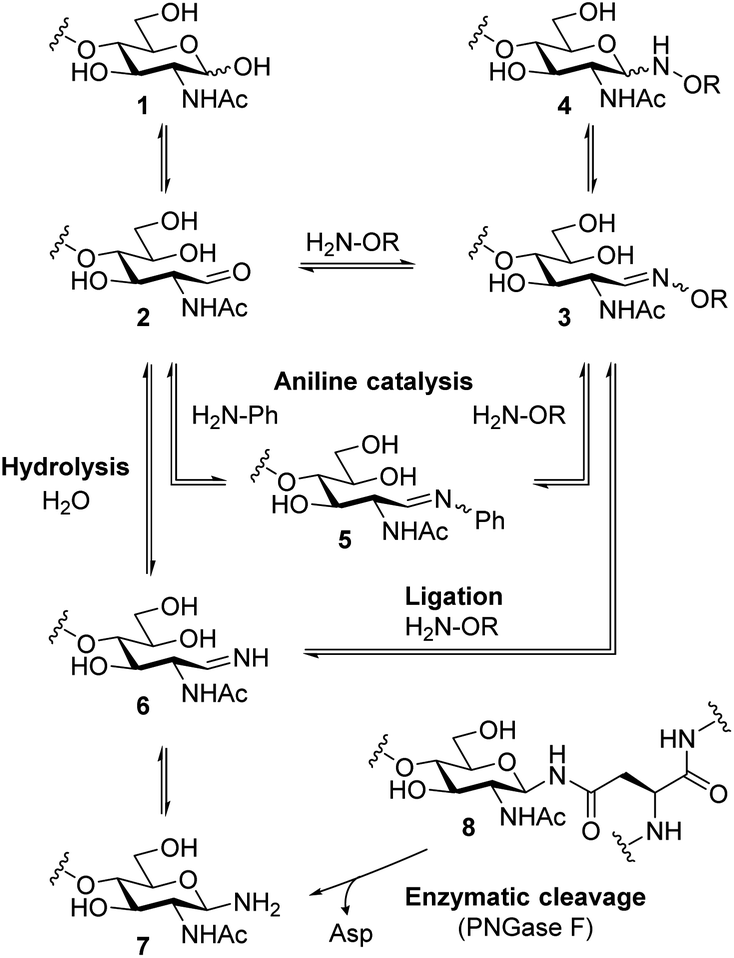 | ||
| Scheme 1 Simplified reaction pathways of oxyamine ligation of reducing oligosaccharides and oligosaccharyl amines. | ||
Conventionally, oligosaccharyl amines 7 released from N-glycoproteins are used for oxyamine formation after hydrolysis to 2 (or 1).3b Whereas hydrolysis is fast under acidic conditions, glycosyl amines 7 are stable enough at pH 8.5 to be acylated.19 Since hydrolysis of glycosyl amines competes with their reaction with oxyamines leading to oximes 3, we first determined hydrolysis rates of the GlcNAc-derived glycosyl amine 10 serving as model compound at different pH values. To prepare the glycosyl amine, GlcNAc was converted to glycosyl azide 9 using Shoda's reagent (2-chloro-1,3-dimethylimidazolinium chloride, DMC)20 followed by catalytic hydrogenation to give 10 in 82% yield (Scheme 2). Glycosyl amine 10 was dissolved in buffer at pH values ranging from 5 to 8 and the formation of hydrolysis product GlcNAc was followed by 1H NMR spectroscopy. Measurement of the pH after the experiments ensured that the pH did not increase due to the release of NH3. Fig. 1 shows the kinetics of the hydrolysis experiments (for more details see ESI†). As expected, the hydrolytic stability of 10 increased with increasing pH. At pH 5, hydrolysis of 90% of imine 6 occurs within 22 min, whereas it takes 2.5 h at pH 6, 13.5 h at pH 7, and 43 h at pH 8.
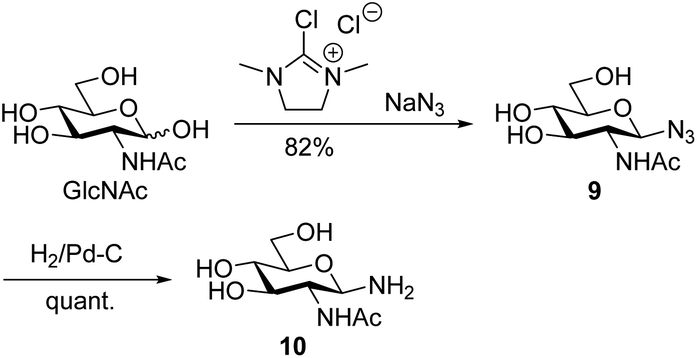 | ||
| Scheme 2 Synthesis of GlcNAcNH210. | ||
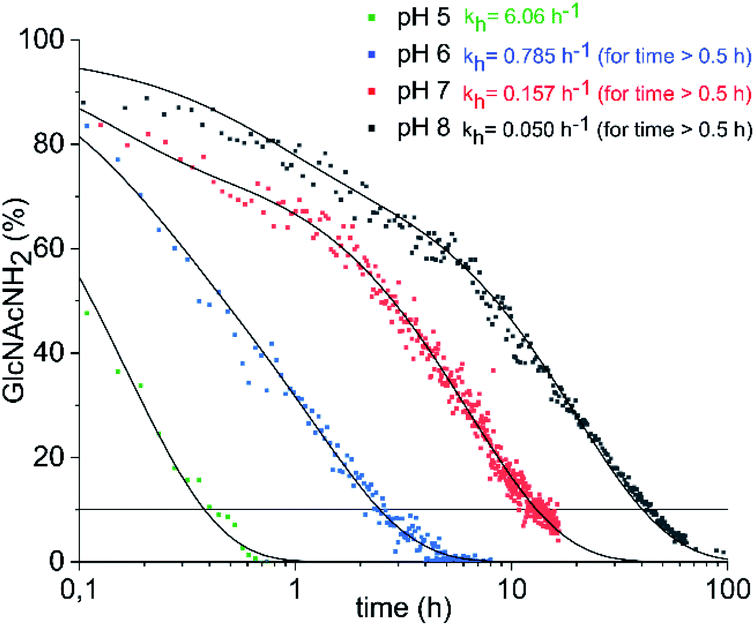 | ||
| Fig. 1 Hydrolysis of GlcNAcNH2 (10) in ammonium acetate-d7 (pH 5–7) and KD2PO4 buffer (pH 8) at 39 °C. The exponential decay constants given refer to the final 60–70% of decay (for details see ESI†). | ||
Next, we studied the oxyamine ligation using glycosyl amine 10 which was freshly prepared from shelf-stable azide 9 for each experiment and ethoxyamine. Again, we followed the reaction by 1H NMR spectroscopy. In contrast to HPLC analysis, NMR spectroscopy allowed us to quantify all products that formed during ligation in real time regardless of their stability during any workup. From the yields of the individual ligation products ((E)- and (Z)-oximes 11, N-glycoside 12) we determined combined yields (CY) in dependence on the reaction time (Fig. 2 and Table 1). For comparison, we also performed the ligation reaction with GlcNAc with and without aniline catalysis (see Fig. S1 and S2† for yields of individual ligation products and sample NMR spectra). Oxyamine ligations are typically carried out at a pH between 4 and 5. This pH represents the best compromise between reaction rate and equilibrium yield.21 Due to the higher pKa value of imine 6 compared to aldehyde form 2, we expected that ligation reactions with glycosyl amine 10 are also efficient at higher, close to neutral pH values, a pH range that is more relevant for reactions of sensitive biomolecules. Accordingly, we carried out the experiments at pH 5 to 8. Fig. 2 shows the combined yield CY of ligation products over time and kinetic fits obtained from a simplified kinetic model with three processes described by pseudo first order kinetics: reversible reaction from glycosyl amine 10 to ligation products (k1, k−1), reversible hydrolysis of 10 (kh, k−h), and reversible product formation from GlcNAc formed by hydrolysis or used as a starting material (k2, k−2) (for complete description see ESI†). In Table 1 combined yields after a reaction time of 1 h and the observed rate constants kobs of product formation are given.
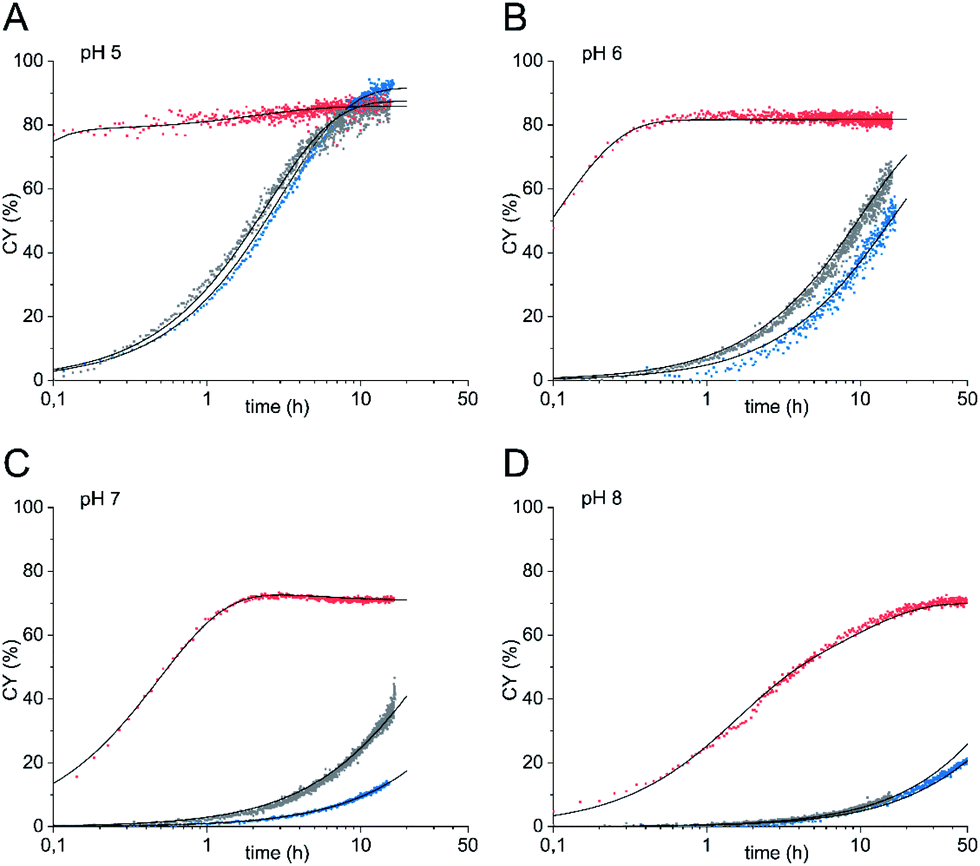 | ||
| Fig. 2 Oxyamine ligation reaction of glycosyl amine 10 (red) and GlcNAc with 100 mM aniline (grey) or without aniline (blue) (36 mM sugar) with 5 equiv. ethoxyamine. (A) pH 5, (B) pH 6, (C) pH 7, (D) pH 8. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | pH | Sugar | Aniline (100 mM) | 36 mM sugar | 5 mM sugar | ||
| CY after 1 h [%] | k obs [h−1] | CY after 1 h [%] | k obs [h−1] | ||||
| 1 | 5 | 10 | — | 81 | 25 | 71 | 10.6 |
| 2 | 5 | GlcNAc | + | 29 | 0.35 | 22 | 0.250 |
| 3 | 5 | GlcNAc | — | 25 | 0.30 | 5 | 0.051 |
| 4 | 6 | 10 | — | 82 | 8.0 | 63 | 5.0 |
| 5 | 6 | GlcNAc | + | 7.6 | 0.080 | 4 | 0.040 |
| 6 | 6 | GlcNAc | — | 4.8 | 0.050 | 1 | 0.012 |
| 7 | 7 | 10 | — | 64 | 1.5 | 37 | 0.55 |
| 8 | 7 | GlcNAc | + | 2.9 | 0.030 | 1 | 0.010 |
| 9 | 7 | GlcNAc | — | 0.9 | 0.010 | 0 | 0.002 |
| 10 | 8 | 10 | — | 25 | 0.37 | 6 | 0.060 |
| 11 | 8 | GlcNAc | + | 0.6 | 0.006 | 0 | 0.00 |
| 12 | 8 | GlcNAc | — | 0.5 | 0.005 | 0 | 0.0007 |

When glycosyl amine 10 was reacted at a concentration of 36 mM with an excess (5 equiv.) of ethoxyamine at pH 5, the ligation was so fast (k1 = 25 h−1), that even at the first data point (taken after 6 min) CY was close to the equilibrium yield and thereafter increased only slightly further. In comparison, the reaction of GlcNAc resulted in a k2 of 0.30 h−1 and CY of only 25% after 1 h. The aniline-catalyzed reaction of GlcNAc was slightly faster under these conditions (k2 = 0.35 h−1, 29% CY after 1 h). At pH 6, the reaction of glycosyl amine 10 reached a k1 of 8.0 h−1 and a plateau of approx. 82% CY after 1 h whereas CY of the reaction of GlcNAc (k2 = 0.050 h−1) was only 4.8% and that of the aniline-catalyzed reaction of GlcNAc (k2 = 0.080 h−1) was 7.6% at that time. At neutral pH, the reaction of glycosyl amine 10 reached a plateau after approx. 2 h (64% CY after 1 h, k1 = 1.5 h−1). The combined yields of the reaction of GlcNAc after 1 h were 0.9% with k2 = 0.010 h−1 (uncatalyzed) and 2.9% with k2 = 0.030 h−1 (aniline-catalyzed). Even at pH 8, the ligation reaction is significant for glycosyl amine 10 (k1 = 0.37 h−1), although the plateau is reached later, whereas both the uncatalyzed and the aniline-catalyzed reaction are too slow to be useful. These experiments clearly demonstrate that the use of glycosyl amines results in very fast oxyamine ligations over a broad pH range. Although hydrolysis and ligation are competing reactions, high ligation yields are obtained with 10 after short reaction times. The optimum of this advantage of the glycosyl amine ligation was found at a pH of 6 (k1/k2 = 160) with not much loss until pH 7 (k1/k2 = 150), which indicates that the distinct optimum should be somewhere between these two pH values.
So far, reactions were carried out at sugar concentrations of 36 mM. To demonstrate that ligations of glycosyl amine 10 can also be carried out at concentrations typically used during biological applications,22 we determined reaction kinetics at sugar concentrations of 5 mM at pH values from 5 to 8 (Fig. 3A–D). In these experiments, we used 10 equiv. of ethoxyamine (corresponding to a concentration of 50 mM) instead of the 5 equiv. employed in the experiments with higher sugar concentration (corresponding to a concentration of 180 mM). The rationale behind the increased number of equivalents was that the ratio between hydrolysis and ligation reaction is dependent on the ratio of the absolute concentrations of water (55.5 M when used as the solvent) and oxyamine. Using only 5 equiv. would have resulted in a too high proportion of hydrolysis of 10 in parallel to the ligation reaction. Again, all reactions with glycosyl amine 10 were significantly faster than the aniline-catalyzed GlcNAc ligation at all pH values investigated. At pH 6, the rate constant k1 of the reaction of 10 is 5.0 h−1 which is 417 times higher than the rate constant of the uncatalyzed reaction of GlcNAc (k2 = 0.012 h−1) and 125 times higher than the rate constant of the aniline-catalyzed GlcNAc ligation (k2 = 0.04 h−1). At this pH, the gain of reaction rate of the ligation of glycosyl amine 10 compared to the aniline-catalyzed reaction of GlcNAc is highest. At pH 7, k1 of the reaction of 10 is 275 times higher than the rate constant k2 of the uncatalyzed reaction of GlcNAc and 55 times higher than the rate constant of the aniline-catalyzed GlcNAc ligation.
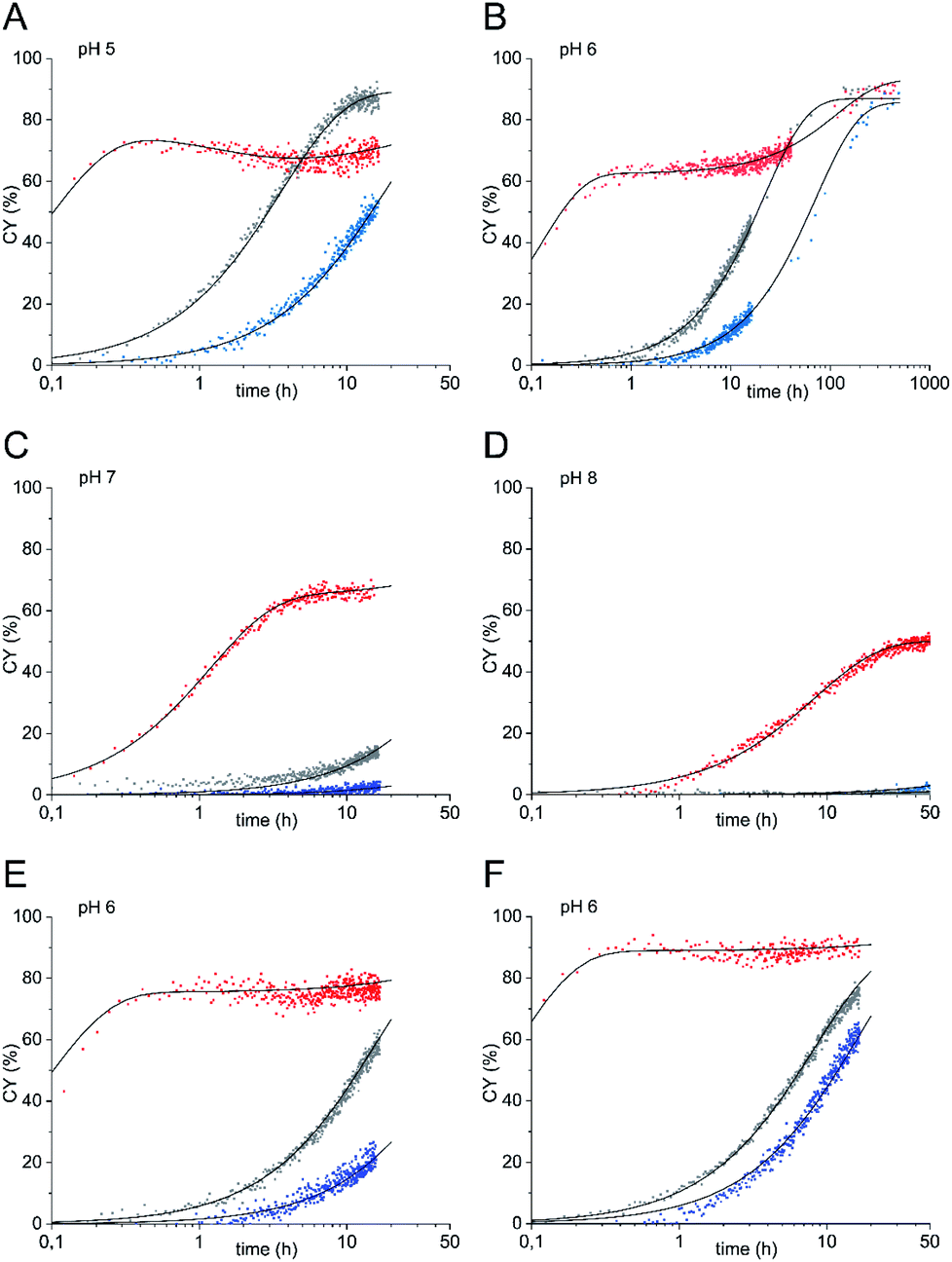 | ||
| Fig. 3 Oxyamine ligation reaction of glycosyl amine 10 (red) and GlcNAc with 100 mM aniline (grey) or without aniline (blue) (5 mM sugar) with 10 equiv. of ethoxyamine at (A) pH 5, (B) pH 6, (C) pH 7, (D) pH 8, and at pH 6 with 20 equiv. (E) and 50 equiv. of ethoxyamine (F). | ||
In the experiments represented by Fig. 3A–D, we observed two stages of the ligation reaction. First, glycosyl amine 10 reacts in a very fast reaction with the alkoxyamine to the ligation products until consumption of 10 partly also by hydrolysis. In the second stage, hydrolysis product GlcNAc undergoes the slower ligation until the equilibrium is reached. The two stages of the reaction are particularly visible at pH 6 (Fig. 3B) where we followed the reaction over a longer reaction time of up to 500 h and partially at pH 7 (Fig. 3C) whereas at pH 8 the second step is too slow to be observed.
A particular case seems to prevail with the amine ligation kinetics at pH 5 shown in Fig. 3A. Here, after reaching a first plateau after about half an hour, the product yield slightly diminishes again and starts the ascent to the final plateau yield only after approx. 5 h. Formally, the pertinent kinetic curve, when drawn over a log(t) scale, as employed in all figures shown here, exhibits three inflection points. Based on a system of linear differential equations for describing the kinetics, such a behavior can be only expected if at least four species are involved. In that case the reservoir of the fourth species X might first be filled from the fraction of product formed in the fast reaction through the amine pathway, thus leading to a decrease of the product before its renewed increase through the slower hydrolysis pathway. The species X might be an intermediate on the way from GlcNAc to oximes 11 or – more likely – a consecutive product to the main oxime ligation product, possibly the aminal formed upon addition of a further molecule of ethoxyamine – a species that remains unrecognized by NMR spectroscopy due to massive signal overlap. The kinetics has been treated based on a reaction scheme entailing both reaction pathways (see ESI† for a detailed discussion). Thereby, a smooth simulation of the observed kinetics is possible.
To test whether the proportion of hydrolysis of glycosyl amine 10 during the first reaction stage can be reduced by using a higher alkoxyamine concentration, we carried out the ligation reactions at pH 6 also with 20 and 50 equiv. of ethoxyamine23 corresponding to concentrations of 100 mM and 250 mM, respectively (Fig. 3E and F). Pleasantly, this procedure increased the yield reached in the first stage of the reaction significantly to a CY of 76% after 1 h (20 equiv.) and even 90% after only 30 min (50 equiv.). For comparison, the aniline-catalyzed and the uncatalyzed reaction of GlcNAc resulted in yields of 5.8% at most at the same points of time for both 20 and 50 equiv. of oxyamine. The highest acceleration of the glycosyl amine ligation over the uncatalyzed reaction of GlcNAc by a factor of 500 is observed with 20 equiv. of ethoxyamine (k1 = 8.0 h−1, k2 = 0.016 h−1).
Next, we verified the applicability of the glycosyl amine ligation to the analysis of glycans released from a glycoprotein by treatment with PNGase F. Ribonuclease B (RNase B) is a commercially available glycoprotein with a single N-glycosylation site at Asn-34 to which glycans of the high-mannose type with 5 to 9 mannose residues (Man5–9GlcNAc2) are attached.24 To release the glycans, we treated RNase B with rapid PNGase F for 10 min at pH 7.5 (Fig. 4A, for experimental details see ESI†). After deglycosylation, the mixture was lyophilized and incubated with oxyamine-Cy3 conjugate 13 (75 mM, for complete formula see ESI†) in ammonium acetate buffer at 39 °C and pH 6 (Fig. 4B). 13 is a multipurpose reagent featuring an aminooxy group for carbohydrate ligation, an additional amino group for the possibility of a further modification (e.g., the preparation of carbohydrate microarrays), and a fluorophore with a permanent charge for detection by fluorescence spectroscopy or mass spectrometry. After 4 h, excess of 13 was scavenged by addition of aldehyde-functionalized TentaGel resin and protein components were removed with a 10 kDa molecular weight cut-off filter. The obtained crude glycoconjugate mixture was subjected to nano-LC-MS/MS analysis which allowed the identification of the glycan composition. As shown in Fig. 4C and S7,† all expected glycan conjugates could be identified which illustrates the efficiency of the new ligation procedure.
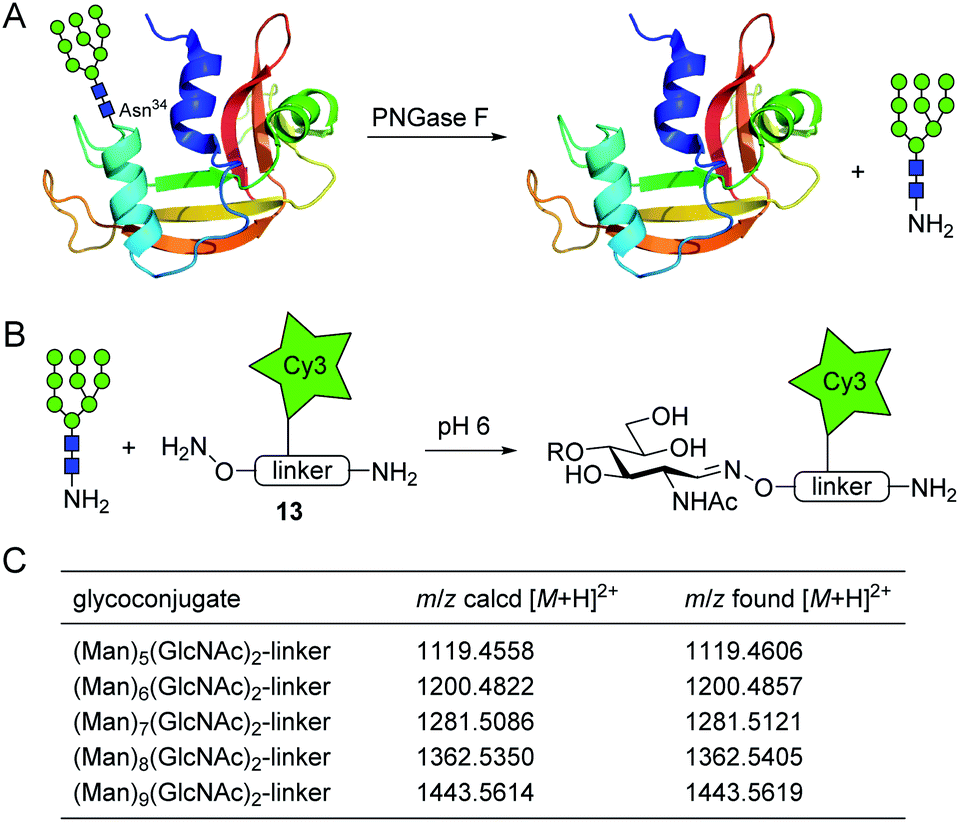 | ||
| Fig. 4 Application of glycosyl amine ligation for glycan analysis of RNase B. (A) Release of N-linked glycans with PNGase F (exemplarily shown for Man9GlcNAc2). (B) Ligation of released glycosyl amines with oxyamine-Cy3 conjugate 13. (C) Results of nano-LC-MS/MS analysis of glycan conjugates. | ||
Conclusions
In summary, we discovered that the oxyamine ligation of unprotected carbohydrates can be significantly accelerated when using glycosyl amines as starting material. Ligation rates are not only faster than the uncatalyzed reaction of reducing sugars but also much faster than their aniline-catalyzed ligation. Glycosyl amines from mono- and oligosaccharides are easily accessible from reducing carbohydrates via the corresponding azides by using Shoda's reagent and subsequent reduction. They are furthermore obtained by PNGase-catalyzed release of N-glycans from glycoproteins facilitating their glycomic analysis which was illustrated by the analysis of N-glycans released from RNase B. The oxyamine ligation of glycosyl amines can be carried out under very mild, close to neutral conditions which is beneficial for the analysis of acid-sensitive glycans, such as sialylated structures. The formation of glycoconjugates using glycosyl amines is expected to have a major impact on numerous applications in the field of glycobiology.Data availability
Related data are contained in the ESI.†Author contributions
Conceptualization – ORB, VW; investigation – MAR, ORB; data curation – MAR, ORB, UES, VW; formal analysis – UES; funding acquisition – VW; supervision – VW; writing, original draft – MAR, VW; writing, review & editing – MAR, ORB, UES, VW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg (33-7533-7-11.9/7/2). We thank Anke Friemel and Ulrich Haunz from the NMR Core Facility of the University of Konstanz for their help in recording reaction kinetics and the Proteomics Center for MS analysis of glycan conjugates.References
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2015-2017 Search PubMed.
- (a) K. K. Palaniappan and C. R. Bertozzi, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS PubMed; (b) L.-X. Wang and B. G. Davis, Chem. Sci., 2013, 4, 3381–3394 RSC.
- (a) K. Villadsen, M. C. Martos-Maldonado, K. J. Jensen and M. B. Thygesen, ChemBioChem, 2017, 18, 574–612 CrossRef CAS PubMed; (b) L. R. Ruhaak, G. Zauner, C. Huhn, C. Bruggink, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2010, 397, 3457–3481 CrossRef CAS PubMed.
- (a) Y. Liu, T. Feizi, M. A. Campanero-Rhodes, R. A. Childs, Y. Zhang, B. Mulloy, P. G. Evans, H. M. I. Osborn, D. Otto, P. R. Crocker and W. Chai, Chem. Biol., 2007, 14, 847–859 CrossRef CAS PubMed; (b) S. Park, M.-R. Lee and I. Shin, Bioconjugate Chem., 2009, 20, 155–162 CrossRef CAS PubMed; (c) S. Park, J. C. Gildersleeve, O. Blixt and I. Shin, Chem. Soc. Rev., 2013, 42, 4310–4326 RSC.
- M. B. Thygesen, J. Sauer and K. J. Jensen, Chem.–Eur. J., 2009, 15, 1649–1660 CrossRef CAS PubMed.
- X.-P. He, Y.-L. Zeng, X.-Y. Tang, N. Li, D.-M. Zhou, G.-R. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 13995–13999 CrossRef CAS PubMed.
- (a) S. E. Cervigni, P. Dumy and M. Mutter, Angew. Chem., Int. Ed. Engl., 1996, 35, 1230–1232 CrossRef CAS; (b) Y. Zhao, S. B. H. Kent and B. T. Chait, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1629–1633 CrossRef CAS PubMed; (c) S. Peluso and B. Imperiali, Tetrahedron Lett., 2001, 42, 2085–2087 CrossRef CAS; (d) D. Specker and V. Wittmann, Top. Curr. Chem., 2007, 267, 65–107 CrossRef CAS.
- S. Dall'Angelo, Q. Zhang, I. N. Fleming, M. Piras, L. F. Schweiger, D. O'Hagan and M. Zanda, Org. Biomol. Chem., 2013, 11, 4551–4558 RSC.
- (a) R. Novoa-Carballal and A. H. E. Müller, Chem. Commun., 2012, 48, 3781–3783 RSC; (b) I. Vikøren Mo, Y. Feng, M. Øksnes Dalheim, A. Solberg, F. L. Aachmann, C. Schatz and B. E. Christensen, Carbohydr. Polym., 2020, 232, 115748 CrossRef PubMed.
- A. Lohse, R. Martins, M. R. Jørgensen and O. Hindsgaul, Angew. Chem., Int. Ed., 2006, 45, 4167–4172 CrossRef CAS PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- W. P. Jencks, Prog. Phys. Org. Chem., 1964, 2, 63–128 CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- M. B. Thygesen, H. Munch, J. Sauer, E. Cló, M. R. Jørgensen, O. Hindsgaul and K. J. Jensen, J. Org. Chem., 2010, 75, 1752–1755 CrossRef CAS PubMed.
- M. Østergaard, N. J. Christensen, C. T. Hjuler, K. J. Jensen and M. B. Thygesen, Bioconjugate Chem., 2018, 29, 1219–1230 CrossRef PubMed.
- L. H. Yuen, N. S. Saxena, H. S. Park, K. Weinberg and E. T. Kool, ACS Chem. Biol., 2016, 11, 2312–2319 CrossRef CAS PubMed.
- A. L. Tarentino, C. M. Gomez and T. H. Plummer, Biochemistry, 1985, 24, 4665–4671 CrossRef CAS PubMed.
- G. Legler, Biochim. Biophys. Acta, 1978, 524, 94–101 CrossRef CAS.
- (a) H. S. Isbell and H. L. Frush, Methods Carbohydr. Chem., 1980, 8, 255–259 CAS; (b) J. R. Rasmussen, J. Davis, J. M. Risley and R. L. Van Etten, J. Am. Chem. Soc., 1992, 114, 1124–1126 CrossRef CAS.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-i. Shoda, Chem. Commun., 2009, 3378–3379 RSC.
- O. R. Baudendistel, D. E. Wieland, M. S. Schmidt and V. Wittmann, Chem.–Eur. J., 2016, 22, 17359–17365 CrossRef CAS PubMed.
- Y. Kawaharada, S. Kelly, M. Wibroe Nielsen, C. T. Hjuler, K. Gysel, A. Muszynski, R. W. Carlson, M. B. Thygesen, N. Sandal, M. H. Asmussen, M. Vinther, S. U. Andersen, L. Krusell, S. Thirup, K. J. Jensen, C. W. Ronson, M. Blaise, S. Radutoiu and J. Stougaard, Nature, 2015, 523, 308–312 CrossRef CAS PubMed.
- Using an excess of ethoxyamine or an aminooxy-functionalized linker does not represent an experimental disadvantage because it can be easily removed after ligation either by evaporation or extraction or with an aldehyde-functionalized scavenger resin as shown below.
- (a) N. Kawasaki, M. Ohta, S. Hyuga, O. Hashimoto and T. Hayakawa, Anal. Biochem., 1999, 269, 297–303 CrossRef CAS PubMed; (b) J. M. Prien, D. J. Ashline, A. J. Lapadula, H. Zhang and V. N. Reinhold, J. Am. Soc. Mass Spectrom., 2009, 20, 539–556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures, kinetic treatment of hydrolysis and ligation reaction, MS data. See DOI: 10.1039/d1sc05008g |
| This journal is © The Royal Society of Chemistry 2021 |