
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly pure miktoarm spherical nucleic acids: preparation and usage as a scaffold for abiotic intracellular catalysis†
Bohan
Zhang
a,
Silei
Bai
a,
Xiangyu
Chao
a,
Tong
Wu
a,
Zhiyong
Chen
a,
Zehong
Cheng
a,
Yue
Xiao
b,
Ke
Zhang
*bc and
Yugang
Bai
 *a
*a
aState Key Laboratory of Chem-/Bio-Sensing and Chemometrics, Hunan Provincial Key Laboratory of Biomacromolecular Chemical Biology, College of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, China. E-mail: baiyugang@hnu.edu.cn
bSchool of Chemistry and Chemical Engineering, Zhengzhou University, Zhengzhou, Henan 450001, China
cDepartment of Chemistry and Chemical Biology, Northeastern University, Boston, MA 02115, USA
First published on 5th November 2021
Abstract
We present a fullerene-based strategy that allows the synthesis of molecularly pure miktoarm spherical nucleic acids (SNAs) with diverse structures, which, with post-functionalization, could serve as efficient scaffolds for intracellular catalysis. The SNA structure promotes cell permeability, nucleic acid stability, and catalytic efficiency, making the platform ideal for in cellulo reactions. Consequently, the tris(triazole)-bearing miktoarm SNA was able to effectively mediate intracellular copper-catalyzed alkyne–azide cycloaddition at nanomolar level of copper, and facilitate the same reaction in live zebrafish.
Introduction
Intracellular catalysis has received increasing attention in recent years, as it provides a facile and efficient approach to directly governing biochemical processes or to generating molecules of interest in cells.1–3 To obtain enhanced functions and to achieve usefulness for catalysis in cells, it would be desirable to expand the scope of “catalyzable” reactions using these intracellular catalysts, so that more abiotic transformations could be realized. However, most metal complexes with catalytic properties show decreased activity or even complete deactivation under biologically relevant conditions because of the poisoning of coordinating biomolecules in such environments. A widely adopted solution for this problem is to introduce artificial scaffolds, such as single-chain nanoparticles (SCNPs),4–9 dense-shell nanoparticles (DSNPs),10 proteins,11,12 and inorganic nanomaterials, for those catalytic metal complexes.13–16 These intracellular catalytic strategies have been successful in bringing more catalytic species into cells; but accompanying problems such as cytotoxicity, low cell uptake, difficult metal-scaffold assembly, and limited protective effect for the metal centers are still frequently hindering their applications.Spherical nucleic acids (SNAs) are polyvalent nanoparticles with a dense shell of oriented oligonucleotides.17,18 Although SNAs were initially prepared from gold nanoparticles (AuNPs),19 they can be made using a variety of inorganic and organic materials as cores, including but not limited to various inorganic nanoparticles,20–23 liposomes,24 macromolecules,25 proteins26,27 and metal–organic frameworks (MOFs).28 SNAs have attractive and unique properties that are core-independent, such as high biocompatibility, increased binding affinities for complementary sequences, resistance to nuclease, enhanced cellular uptake and lowered immune responses.29 These unique properties have made SNAs versatile tools, serving as in vitro biomacromolecule probes, self-delivering gene regulation materials, chemotherapy agents, and immune system modulators.27,30–35 Particularly, recently reported strategies for preparing molecularly pure SNAs have provided even more tunability and controllability in the SNA structure and function,36 making them more suitable for applications that require tailor-made functions.
Thus, considering their core–shell architecture, high biocompatibility, cell permeability and in vivo stability, we propose that SNAs with appropriate functionalization can serve as excellent scaffolds for catalytic metal complexes, providing a stable, biocompatible and protective shell, all of which are of significance for conducting catalysis in cells. SNAs also have more definite molecular structures, which is of importance in appropriate recognition and feature design for artificially constructed catalytic nanostructures.37 Herein, we report a fullerene-based strategy for the synthesis of molecularly well-defined, miktoarm SNA scaffolds that allow the incorporation of catalytic sites, so that well-defined and modular biocatalytic systems could be created and evaluated.
Results and discussion
We previously reported the synthesis of a hexakis fullerene core using pristine C60 and malonic esters, forming 6-arm or 12-arm cores with Th symmetry.36 Due to the chemical nature of the coupling between C60 and malonates, fullerene as a core provides a more controllable and convenient approach to introduce different moieties in a single step than dendritic or nanoparticle cores, allowing simplified preparation of molecularly pure miktoarm SNAs (Fig. 1a). Thus, we devised a facile approach to generate asymmetric malonic esters using the ring–opening reaction of Meldrum's acid (2,2-dimethyl-1,3-dioxane-4,6-dione) followed by another esterification step. Using this method, various functionalized alcohols were utilized to yield a series of asymmetric malonates, which were then coupled to the C60 core in the presence of 1,5-diaza-bicyclo[4.3.0]non-5-ene (DBU), CBr4, and o-dichlorobenzene (DCB) to yield fullerene cores with two types of conjugation sites (6 for each type). The synthesized cores are summarized in Table 1. They were purified through chromatography and precipitation and were characterized by nuclear magnetic resonance (NMR, see Fig. S1–S8 in the ESI†), high-performance liquid chromatography (HPLC, Fig. S11 in the ESI†), and matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectrometry (Fig. S12 in the ESI†). It should be noted that because the two types of arms are tethered on the vertices of cyclopropane rings on C60 and are thus pointing at fixed directions, there are numerous stereoisomers for each difunctional core. However, these cores are not differentiated by common spectroscopic or chromatographic means and can thus be effectively treated as Th-symmetric. The two conjugation sites were orthogonal in functionalization, allowing simultaneous or step-wise unmasking (Fig. S11 and S12 in the ESI†) or conjugation (Fig. 1b–d and S14 in the ESI†) of different functional moieties. For example, Core-AzAk could be easily derivatized using DBCO-NH2 (DBCO = dibenzocyclooctyne) through copper-free alkyne–azide cycloaddition to form a hexakis-triazole adduct (Core-NH2Ak), and this new core could again react with an azide, 3-azido-7-hydroxycoumarin, through copper-catalyzed alkyne–azide cycloaddition (CuAAC) to achieve biofunctionalization (Fig. S14 in ESI†). Such a derivatization study well supported the structural integrity of the core and the availability of established functional groups on the core.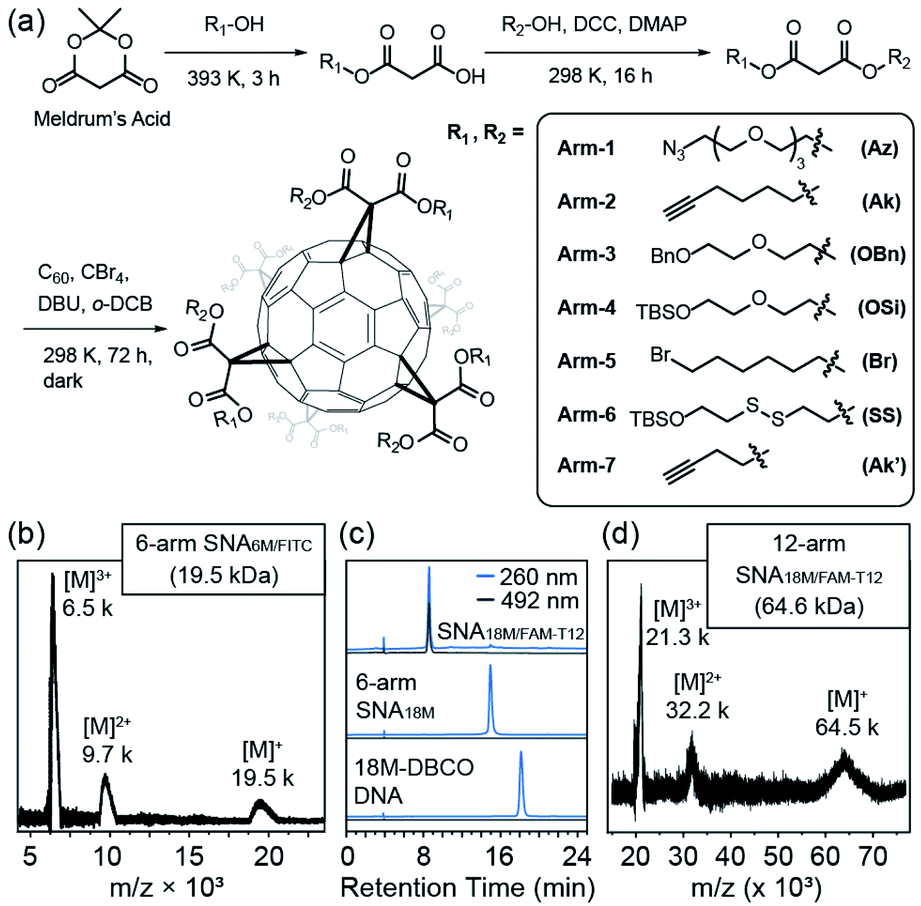 | ||
| Fig. 1 (a) Synthetic route of the asymmetric malonic esters, the miktoarm fullerene core, and the molecularly pure miktoarm SNAs. (b) MALDI-TOF analysis of the 6-arm SNA, SNA6M/FITC. Series of multiple-charged SNA ions were clearly identifiable. (c) HPLC curve overlay of 18M-DBCO DNA, 6-arm SNA18M, and miktoarm SNA18M/FAM-T12. (d) MALDI-TOF analysis of SNA18M/FAM-T12. | ||
| Name | Malonate arms | Theoretical m/z | Conjugation sites provided after unmasking | ||
|---|---|---|---|---|---|
| Type 1 | Type 2 | Type 1 | Type 2 | ||
| Core-AzAk | Arm-1 | Arm-2 | 3020 | –N3 | –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
| Core-AzOBn | Arm-1 | Arm-3 | 3609 | –N3 | –OH |
| Core-AzOSi | Arm-1 | Arm-4 | 3754 | –N3 | –OH |
| Core-SSAk′ | Arm-6 | Arm-7 | 3146 | –SH/–OH | –CCH |
To validate the applicability of such dual-functional cores in the preparation of miktoarm SNAs, we attempted to use Core-AzAk as the starting material for SNA preparation. A short DNA strand, DBCO-6M, was first tried in the synthesis, and a pure 6-arm SNA (SNA6M) was successfully obtained after copper-free “Click” conjugation and HPLC purification (Fig. S15, S18, and S19 in the ESI†). The remaining alkyne groups could be used to introduce 6 fluorophores by performing CuAAC with FITC-N3 (Fig. S15 in the ESI†). Despite the difficulty resulting from the large size, MALDI-TOF mass spectrometric analysis38,39 revealed ion series matching the different multi-charged species from the expected SNA6M/FITC structure (theoretical Mn = 19.5 kDa, Fig. 1b and Fig. S15, S18, and S19 in the ESI†). Importantly, the ratio of DNA strands versus FITC was quantified to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 on the resulting SNA6M/FITC using UV-vis analysis (Fig. S20 in the ESI†). We then moved to longer DNA strands, using 18M-DBCO for the conjugation with Core-AzAk, and obtained another 6-arm SNA with longer arms after a 72 h reaction. Similarly, the resulting SNA has 6 terminal alkyne groups as conjugation sites, and thus another DNA strand, FAM-T12-N3 (Fig. S16 in the ESI†), could be introduced using CuAAC. HPLC analysis showed that the conjugation was highly efficient, with the miktoarm SNA18M/FAM-T12 as the major product, which was easily isolated using reverse-phase HPLC with an acetonitrile/water gradient (Fig. 1c, d and Fig. S18, S19 in the ESI†). Similarly, by establishing a calibration curve using FAM-T12-N3, the ratio of 18M and FAM-T12 arms was determined to be 1:1 (Fig. S20 in the ESI†). These results showed that this strategy, based on dual-functional C60 cores, could indeed afford miktoarm SNAs efficiently with excellent purity.
:1 on the resulting SNA6M/FITC using UV-vis analysis (Fig. S20 in the ESI†). We then moved to longer DNA strands, using 18M-DBCO for the conjugation with Core-AzAk, and obtained another 6-arm SNA with longer arms after a 72 h reaction. Similarly, the resulting SNA has 6 terminal alkyne groups as conjugation sites, and thus another DNA strand, FAM-T12-N3 (Fig. S16 in the ESI†), could be introduced using CuAAC. HPLC analysis showed that the conjugation was highly efficient, with the miktoarm SNA18M/FAM-T12 as the major product, which was easily isolated using reverse-phase HPLC with an acetonitrile/water gradient (Fig. 1c, d and Fig. S18, S19 in the ESI†). Similarly, by establishing a calibration curve using FAM-T12-N3, the ratio of 18M and FAM-T12 arms was determined to be 1:1 (Fig. S20 in the ESI†). These results showed that this strategy, based on dual-functional C60 cores, could indeed afford miktoarm SNAs efficiently with excellent purity.
To show the applicability of such a miktoarm SNA in intracellular catalysis, we synthesized azido-tris(triazole) (azido-TTA) as the ligand for CuAAC and conjugated it to a 6-arm SNA to yield SNA19M/TTA (Fig. 2a–c and Fig. S18 and S19†, a SNA prepared from Core-AzAk and DBCO-19M). Such TTA-type structures are known to be excellent ligands for CuAAC reactions.40,41 We also prepared a linear TTA-DNA conjugate (TTA-19M, Fig. S17 and S19 in the ESI†) using the same DNA sequence as the one in the TTA-SNA synthesis, and this linear conjugate was used as a control to verify if the SNA architecture could provide a beneficial effect in the catalytic process. In our previous study, we observed that an 8-arm molecular SNA with a T8 polyoctahedral silsesquioxanes (POSS) core and a 12-arm SNA with a C60 core both showed improved cell uptake efficiency compared to free DNA strands, with the 12-arm SNA performing better than the 8-arm counterpart.36 However, as such the POSS-SNA already has a low surface DNA intensity; further decrease in the number of arms may lead to a loss of SNA properties, such as enhanced cell uptake and resistance to nucleases. Indeed, our flow cytometry study results revealed that the 6-arm SNA19M-FAM showed very limited improvement in cellular uptake efficiency compared to the free linear DNA (Fig. 2d). Interestingly, however, the results also showed that the non-DNA arms in the miktoarm SNA could help restore the SNA properties. With the TTA ligands attached to the fullerene core, the resulting SNA19M-FAM/TTA with 6 DNA arms and 6 TTA-alkyl arms showed much higher cell permeability compared to the linear DNA, DBCO-19M-FAM, and 6-arm SNA19M-FAM, even outperforming the 12-arm SNA18M/FAM-T12. Such restoration of SNA properties was possibly the result of steric hindrance created by the bulky TTA moieties located near the fullerene core, which forced the nucleic acids to adopt a more extended and oriented conformation similar to prototypical SNAs. An alternative interpretation involves the hydrophobicity of the TTA head that directly leads to better cell permeability. TTA ligand's ability in improving DNA cell uptake was also seen for linear TTA-19M-FAM, as it also showed higher cell permeability than DBCO-19M-FAM. For the improvement on the linear DNA, in addition to the increased hydrophobicity that may have played a role, the self-assembly of TTA-DNA into SNA-like aggregates may be vital for the enhanced cell uptake, as suggested by dynamic light scattering analyses (Fig. S22 in the ESI†).
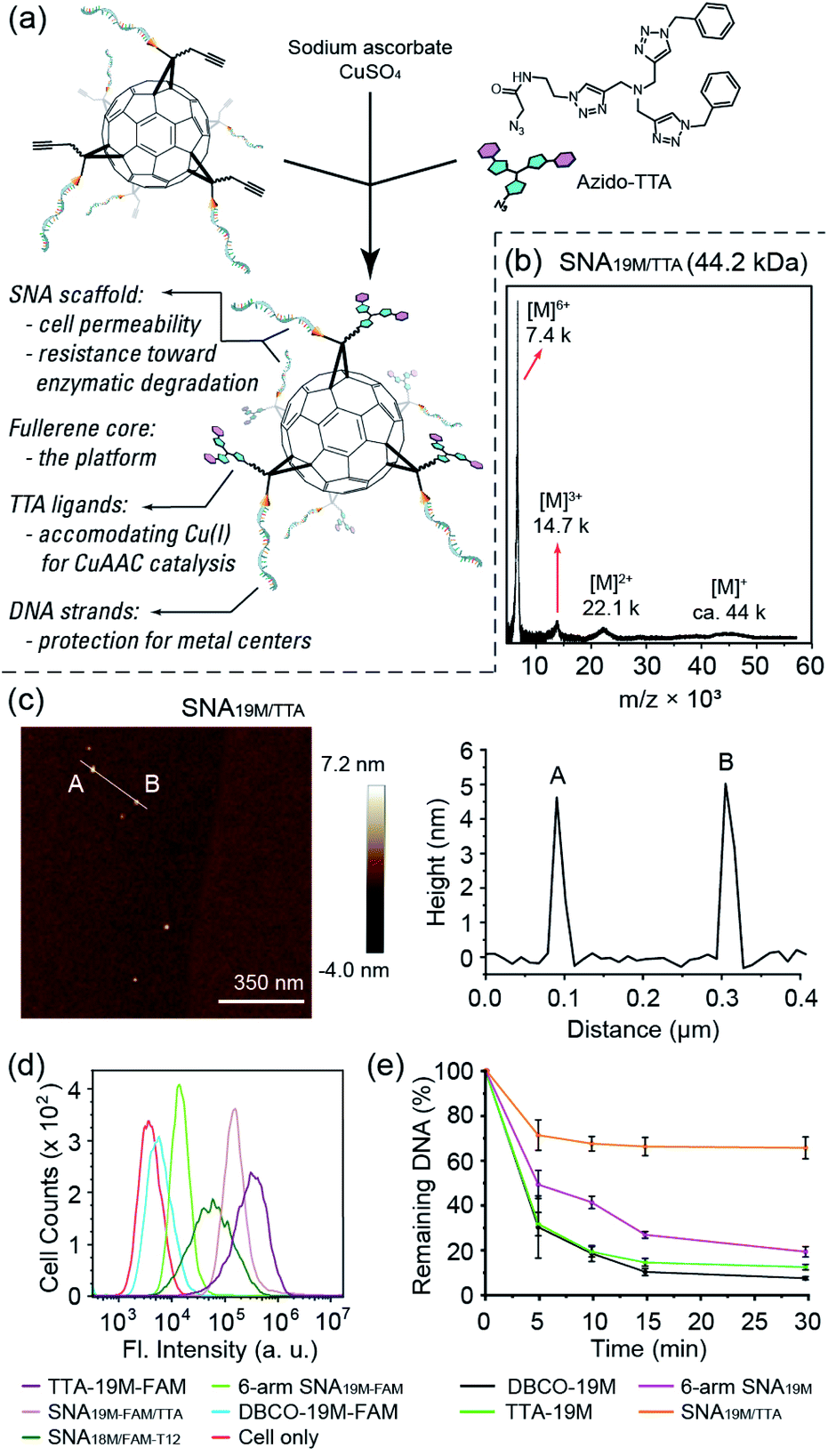 | ||
| Fig. 2 (a) Illustration of the synthesis of a SNA-based “Click” catalyst. (b) MALDI-TOF characterization of miktoarm SNA19M/TTA. (c) AFM image and analysis of SNA19M/TTA. Series of multiple-charged SNA ions were clearly identifiable. (d) Flow cytometry study showed that SNA19M/TTA could enter cells efficiently. (e) The 6 + 6 miktoarm SNA19M/TTA showed good resistance toward DNase according to HPLC analysis. | ||
The miktoarm TTA-SNA also showed improved resistance toward enzymatic degradation. By treating the miktoarm SNA19M/TTA, 6-arm SNA19M, linear TTA-19M and DBCO-19M with DNase I, and monitoring the amount of intact starting material using HPLC, we observed the degradation kinetics of these species. Fig. 2e shows the species' percentages of degradation over time, and very clearly, miktoarm SNA19M/TTA exhibited the highest stability toward the DNase. The linear TTA-19M showed similar degradation kinetics compared to DBCO-19M, indicating that the TTA ligand itself had no stabilization effect. Thus, it is very likely that the TTA arms, despite not being DNA strands in nature, could help restore the unique properties of SNA. Again, these additional arms may force the DNA strands to be more “extended” via the steric effect, as if the strands were in a prototypical SNA with higher surface DNA density.
With the above validation of the necessary properties of miktoarm SNA19M/TTA, we went on to verify if this TTA-bearing SNA had good catalytic capability, which is important for its performance in cells in addition to stability and cell permeability. We used the fluorogenic “Click” reaction between azidocoumarin and ethynylanisole to evaluate the catalyst efficiency in solutions (Fig. 3a). In PBS, commercial catalyst tris(benzyltriazolyl-methyl)amine40 (TBTA, a TTA analog) showed the best performance as the copper ligand, followed by SNA19M/TTA and TTA-19M (Fig. 3b), which was not surprising because the attached DNA chains were likely to bring steric hindrance. As part of the design, such steric effect also protects the catalytic centers, which could be observed when CuAAC reactions were performed in a more complicated environment, i.e., DMEM supplemented with 10% FBS. Under this more biologically relevant condition, the trend was reversed, and SNA19M/TTA became the best scaffold (Fig. 3c). TBTA, in sharp contrast, was almost fully quenched by the media, consistent with previous studies, because amino acids and proteins could all fetch Cu(I) from Cu-TBTA and deactivate the catalyst. The second-order rate constant measured for SNA19M/TTA-mediated CuAAC was approximately 9.1 M−1 s−1 in DMEM containing 10% FBS in the first 30 min, which was remarkable for catalysis performed with such a low catalyst concentration (1.7 μM of SNA19M/TTA, containing 10 μM of CuI) in a complex medium.
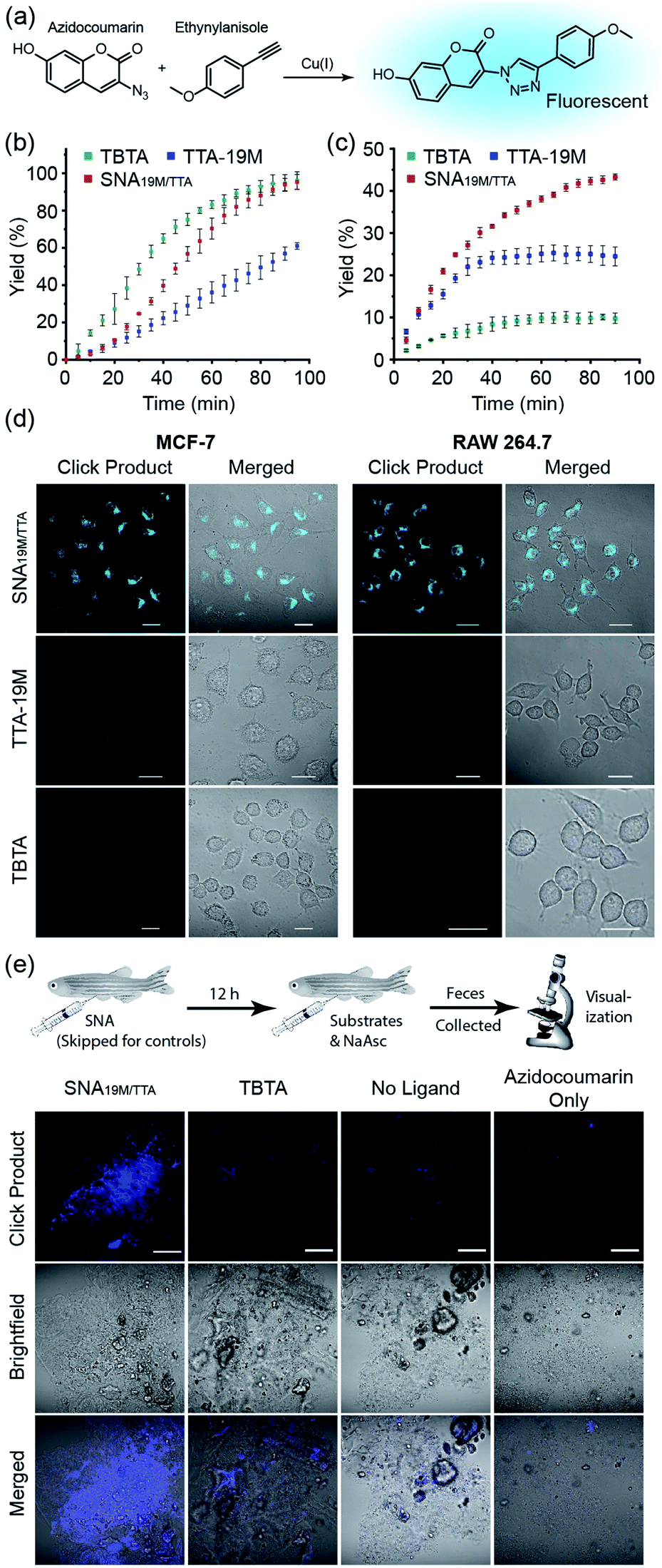 | ||
| Fig. 3 (a) The fluorogenic reaction used to evaluate catalytic efficiencies. (b and c) Catalytic efficiency comparison of copper-loaded miktoarm SNA19M/TTA, TTA-19M, and TBTA in (b) PBS and (c) DMEM containing 10% FBS. [Cu] = [Tris(triazole)] = 10 μM, [Substrate]0 = 20 μM, [NaAsc] = 1 mM. (d) Comparison of the in cellulo activities of the copper-loaded miktoarm SNA19M/TTA, TTA-19M, and TBTA. [Cu] = [Tris(triazole)] = 0.5 μM. Scale bar = 20 μm. (e) Strong fluorescence observed in feces indicating successful CuAAC catalysis by SNA19M/TTA in live zebrafish. Scale bar = 40 μm. | ||
Encouraged by the excellent stability, cell permeability and catalytic efficiency of SNA19M/TTA, we continued to evaluate the catalyst's performance in cells, using TTA-19M and TBTA for comparison. In detail, MCF-7 or RAW 264.7 cells were incubated with CuSO4 and a ligand for 6 h, and the cells were washed with PBS three times. Substrates and ascorbate were then added, and the cells were washed again with PBS after a 2 h incubation. Confocal microscopic imaging on all the cell samples revealed dominating superiority of the SNA-based catalyst, which effectively mediated the intracellular CuAAC reactions in both cell lines and generated a significant amount of fluorescent product, whereas the other two showed no hint of reaction at the same copper and TTA concentration (Fig. 3d and S23 in the ESI†). These results were exciting, as it had reduced the copper concentration required for successful intracellular CuAAC by at least one order of magnitude (to 0.5 μM) compared to previous reports.5,10 In addition, as SNA scaffolds are known for their excellent biocompatibility, this new intracellular CuAAC catalyst is undoubtably the one with the least impact on host cells among all those reported, with no observable cytotoxicity at 4 μM (Fig. S25†). Although fullerenes and other carbon nanomaterials are reported to be cytotoxic,42 it could be seen that the toxicity was largely removed with the cyclopropanation reaction and the resulting DNA strands shielding the fullerene core.
With successful catalysis in live cells by the SNA catalyst at low concentrations, we continued to examine if the catalysis could work in animal models. Zebrafish was chosen as the model, and the fish were divided into four groups (Fig. 3e). For the experiment group, SNA19M/TTA was first intraperitoneally injected, followed by an intraperitoneal injection of a solution containing azidocoumarin, ethynylanisole, sodium ascorbate and CuSO4 in 12 h. For the control groups, (1) TBTA-CuSO4 plus sodium ascorbate and substrates, (2) CuSO4 plus ascorbate and substrates, and (3) azidocoumarin only, was intraperitoneally injected in 12 h, with injected amounts kept consistent with the experiment group. The fish were transferred to fresh water, and their feces at day 2 were collected for visualization under a fluorescence confocal microscope. It was reported that aromatic azide could be metabolically reduced to the corresponding amine in zebrafish;43 but we observed only very weak fluorescence for fish feces from the azidocoumain control group. The other two control groups showed very limited catalysis, revealing weak fluorescence in feces samples, with TBTA-Cu performing slightly better. In sharp contrast, the Cu-SNA19M/TTA system showed significantly improved CuAAC catalysis and gave strong coumarin fluorescence in the feces samples (Fig. 3e and S24 in the ESI†). Importantly, the SNA19M/TTA system showed no toxicity to the fish at its working concentration (Fig. S26 in ESI†). These results served as solid evidence that the SNA could work as a reliable scaffold for abiotic metal catalysis in living systems, as long as suitable ligands were placed into the scaffold to capture metal ions of interest and to promote catalysis.
From a simple comparison of this miktoarm SNA catalyst with the ones reported before, one can easily see that its performance in PBS was mediocre, outperformed significantly by single-chain metal–organic nanoparticle systems,5–8 dense-shell nanoparticle catalysts,10 and dendritic systems.44,45 Thus, its superior performance in cellulo and in vivo can be attributed to its stability in complex environments where coordinating molecules are present, and this is well supported by the kinetic experiment performed in DMEM-FBS. The origin of such good resistance toward hostile environments for the SNA system is an intriguing topic for future studies. A possible reason is that the polyanionic DNA chains around the TTA ligand have provided a weak, polyvalent coordinating environment that traps escaping copper ions by taking advantage of the phosphates' high local concentration, meanwhile hindering the entry of other anionic, coordinating molecules in the environment through charge repulsion and steric stabilization effects.46
Conclusions
In conclusion, we have devised a strategy that allows the synthesis of various di-functional fullerene cores, which in turn provide a platform for the construction of functional miktoarm SNAs bearing different DNA strands or other functionalities as arms. These molecularly pure, well-defined SNAs can be loaded with moieties of interest in a highly definite manner, and we show that they offer great opportunities for conducting intracellular CuAAC catalysis with a low impact on the host, because of the low catalyst concentration required and the high biocompatibility of the SNA scaffolds. Apart from CuAAC, there are many other useful abiotic reactions requiring different metal species in diverse coordination spheres. The SNA construct here is simply serving as a model system to show the possibility of utilizing SNAs as macromolecular scaffolds that can accommodate such catalytic moieties in a modular manner. The DNA arms, on the other hand, offer a programmable approach to control the SNA catalysts' biocompatibility, targeting and recognition capability, and the potential of on-demand assembly into functional nanostructures. Considering the versatility of this synthetic strategy, as well as the tailor-made functions achievable from the SNA scaffold and the metal centres, this study may open new avenues for SNAs toward wide application in intracellular catalyst development.Data availability
All the experimental details and additional data associated with this research can be found in the ESI† of this article.Author contributions
Bohan Zhang: investigation, formal analysis, data curation, and validation, SileiBai: investigation, formal analysis, data curation, and validation, Xiangyu Chao: investigation, and validation, Tong Wu: investigation and formal analysis, Zhiyong Chen: investigation, Zehong Cheng: investigation, Yue Xiao: investigation, Ke Zhang: conceptualization, formal analysis, resources, project administration, funding acquisition, supervision, and writing – review & editing, Yugang Bai: conceptualization, formal analysis, resources, project administration, funding acquisition, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by China’s Fundamental Research Funds for Central Universities, and the funding from the National Natural Science Foundation of China (No. 21877033, and 92163127; Y. B.).Notes and references
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- Y. Liu and Y. Bai, ACS Appl. Bio Mater., 2020, 3, 4717–4746 CrossRef CAS.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- J. Chen, E. S. Garcia and S. C. Zimmerman, Acc. Chem. Res., 2020, 53, 1244–1256 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- J. Chen, J. Wang, Y. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS PubMed.
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 9693–9700 CrossRef CAS PubMed.
- J. Chen, K. Li, J. S. L. Shon and S. C. Zimmerman, J. Am. Chem. Soc., 2020, 142, 4565–4569 CrossRef CAS PubMed.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- Q. Lu, S. Bai, Z. Chen, N. Zheng, X. Feng and Y. Bai, ACS Mater. Lett., 2020, 2, 89–94 CrossRef CAS.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- S. Chordia, S. Narasimhan, A. Lucini Paioni, M. Baldus and G. Roelfes, Angew. Chem., Int. Ed., 2021, 60, 5913–5920 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- F. Wang, Y. Zhang, Z. Du, J. Ren and X. Qu, Nat. Commun., 2018, 9, 1209 CrossRef PubMed.
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS PubMed.
- Z. Gao, Y. Li, Z. Liu, Y. Zhang, F. Chen, P. An, W. Lu, J. Hu, C. You, J. Xu, X. Zhang and B. Sun, Nano Lett., 2021, 21, 3401–3409 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- S. C. P. Williams, Proc. Natl. Acad. Sci., 2013, 110, 13231 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- J.-S. Lee, A. K. R. Lytton-Jean, S. J. Hurst and C. A. Mirkin, Nano Lett., 2007, 7, 2112–2115 CrossRef CAS PubMed.
- J. I. Cutler, D. Zheng, X. Xu, D. A. Giljohann and C. A. Mirkin, Nano Lett., 2010, 10, 1477–1480 CrossRef CAS PubMed.
- K. L. Young, A. W. Scott, L. Hao, S. E. Mirkin, G. Liu and C. A. Mirkin, Nano Lett., 2012, 12, 3867–3871 CrossRef CAS PubMed.
- C. Zhang, R. J. Macfarlane, K. L. Young, C. H. J. Choi, L. Hao, E. Auyeung, G. Liu, X. Zhou and C. A. Mirkin, Nat. Mater., 2013, 12, 741–746 CrossRef CAS PubMed.
- R. J. Banga, N. Chernyak, S. P. Narayan, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 9866–9869 CrossRef CAS PubMed.
- R. J. Banga, B. Meckes, S. P. Narayan, A. J. Sprangers, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 4278–4281 CrossRef CAS PubMed.
- J. D. Brodin, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci., 2015, 112, 4564 CrossRef CAS PubMed.
- D. Samanta, S. B. Ebrahimi, C. D. Kusmierz, H. F. Cheng and C. A. Mirkin, J. Am. Chem. Soc., 2020, 142, 13350–13355 CrossRef CAS PubMed.
- S. Wang, C. M. McGuirk, M. B. Ross, S. Wang, P. Chen, H. Xing, Y. Liu and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 9827–9830 CrossRef CAS PubMed.
- J. L. Rouge, T. L. Sita, L. Hao, F. M. Kouri, W. E. Briley, A. H. Stegh and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 10528–10531 CrossRef CAS PubMed.
- D. S. Seferos, D. A. Giljohann, H. D. Hill, A. E. Prigodich and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 15477–15479 CrossRef CAS PubMed.
- T. L. Halo, K. M. McMahon, N. L. Angeloni, Y. Xu, W. Wang, A. B. Chinen, D. Malin, E. Strekalova, V. L. Cryns, C. Cheng, C. A. Mirkin and C. S. Thaxton, Proc. Natl. Acad. Sci., 2014, 111, 17104 CrossRef CAS PubMed.
- J. D. Brodin, A. J. Sprangers, J. R. McMillan and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 14838–14841 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, N. Chernyak, C. C. Mader, S. Nallagatla, R. S. Kang, L. Hao, D. A. Walker, T. L. Halo, T. J. Merkel, C. H. Rische, S. Anantatmula, M. Burkhart, C. A. Mirkin and S. M. Gryaznov, Proc. Natl. Acad. Sci., 2015, 112, 3892 CrossRef CAS PubMed.
- A. J. Sprangers, L. Hao, R. J. Banga and C. A. Mirkin, Small, 2017, 13, 1602753 CrossRef PubMed.
- X. Tan, X. Lu, F. Jia, X. Liu, Y. Sun, J. K. Logan and K. Zhang, J. Am. Chem. Soc., 2016, 138, 10834–10837 CrossRef CAS PubMed.
- H. Li, B. Zhang, X. Lu, X. Tan, F. Jia, Y. Xiao, Z. Cheng, Y. Li, D. O. Silva, H. S. Schrekker, K. Zhang and C. A. Mirkin, Proc. Natl. Acad. Sci., 2018, 115, 4340 CrossRef CAS PubMed.
- S. Scott, H. Zhao, A. Dey and T. B. Gunnoe, ACS Catal., 2020, 10, 14315–14317 CrossRef CAS.
- H. F. Cheng, S. Wang and C. A. Mirkin, J. Am. Chem. Soc., 2021, 143, 1752–1757 CrossRef CAS PubMed.
- M. E. Distler, M. H. Teplensky, K. E. Bujold, C. D. Kusmierz, M. Evangelopoulos and C. A. Mirkin, J. Am. Chem. Soc., 2021, 143, 13513–13518 CrossRef CAS PubMed.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- P. S. Donnelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459–2461 RSC.
- Z. Peng, X. Liu, W. Zhang, Z. Zeng, Z. Liu, C. Zhang, Y. Liu, B. Shao, Q. Liang, W. Tang and X. Yuan, Environ. Int., 2020, 134, 105298 CrossRef CAS PubMed.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Köster, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS PubMed.
- D. Astruc, L. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630–640 CrossRef CAS PubMed.
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04833c |
| This journal is © The Royal Society of Chemistry 2021 |