
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical–anion coupling through reagent design: hydroxylation of aryl halides†
Andrew J.
Greener‡
 ,
Patrycja
Ubysz‡
,
Will
Owens-Ward
,
George
Smith
,
Ivan
Ocaña
,
Adrian C.
Whitwood
,
Victor
Chechik
and
Michael J.
James
*
,
Patrycja
Ubysz‡
,
Will
Owens-Ward
,
George
Smith
,
Ivan
Ocaña
,
Adrian C.
Whitwood
,
Victor
Chechik
and
Michael J.
James
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: michael.james@york.ac.uk
First published on 22nd October 2021
Abstract
The design and development of an oxime-based hydroxylation reagent, which can chemoselectively convert aryl halides (X = F, Cl, Br, I) into phenols under operationally simple, transition-metal-free conditions is described. Key to the success of this approach was the identification of a reducing oxime anion which can interact and couple with open-shell aryl radicals. Experimental and computational studies support the proposed radical-nucleophilic substitution chain mechanism.
Introduction
Arene hydroxylation reactions are powerful enabling synthetic methods which are routinely used in the preparation of high-value pharmaceuticals, agrochemicals, polymers and natural products.1 Many different synthetic approaches have been developed to form aryl C(sp2)–OH bonds,2 but in terms of cost, operational simplicity and toxicity, nucleophilic aromatic substitution (SNAr)3 represents one of the most attractive and frequently used methods.4 However, the broad application and selectivity of this approach is limited by the high basicity and low nucleophilicity of the hydroxide anion. Hydroxide surrogates have been developed to improve these aspects, but their reactivity is still mostly limited to aryl fluorides or chlorides bearing strong electron-withdrawing groups in either the ortho or para positions.5 The development of more general, transition-metal-free6 substitution reactions for arene hydroxylation is therefore a topic of significant importance with wide-reaching synthetic potential.It has long been known that aryl halides that are not activated with strong electron-withdrawing groups can be substituted with a variety of different nucleophiles through the radical-nucleophilic substitution (SRN1) chain mechanism.7 However, hydroxide anions do not participate in SRN1 mechanisms since such processes are driven by electron transfer (ET) and hydroxide anions are poor electron donors. Consequently, the activation barrier for radical–anion coupling is insurmountably high. This is a general problem with oxygen nucleophiles as, to the best of our knowledge, there is no known oxygen-based anion which can engage in intermolecular coupling with aryl radicals to form new C(sp2)–O bonds.7b,8 Our efforts in solving this limitation are outlined herein. In particular, we rationalised that oxime anions could not only be electronically tuned to initiate and favour an SRN1 process, but also serve as hydroxide surrogates. Indeed, based on literature precedent with perfluoroalkyl iodides,9 it was envisaged that oxime anions 1 may readily form charge-transfer complexes10 (CTCs, 2) with aryl halides 3, which could be activated under mild conditions to promote the formation of aryl radical intermediates 4 (Scheme 1a). Radical–anion coupling could then be rendered kinetically favourable by employing a sufficiently reducing oxime anion (Scheme 1b). In addition, it was anticipated that the oxime π-system could also alleviate the need for the aromatic coupling partner to accommodate the unpaired electron in this coupling process (e.g.5vs.6), and therefore enable coupling with a broader range of substrates. Finally, ET from the coupled radical anions 6 to the aryl halides 3 could propagate a radical chain and afford O-aryl oxime intermediates 7 (Scheme 1c), which as demonstrated by Fier and Maloney11 can readily fragment under basic conditions to afford phenols 8.
 | ||
| Scheme 1 Reagent and reaction design: (a) initiation; (b) radical–anion coupling; (c) reactivity of O-aryl oximes. | ||
In this paper, using the design rationale set out in Scheme 1, we report the development of an easily handled oxime-based nucleophile which can selectively substitute an array of electronically diverse arenes bearing every common halide (F, Cl, Br, I) to form phenols under operationally simple, transition-metal-free conditions. The proposed SRN1 chain mechanism is supported by experimental and DFT computational studies.
Results and discussion
Our studies commenced by reacting aryl bromide 3aBr with a range of electronically diverse oximes (9a–d are representative) using KOt-Bu in anhydrous DMSO (0.2 M) at 30 °C for 16 h under nitrogen (Table 1, entries 1–4). In all cases, we observed the formation of phenol 8a in modest to excellent yield, with electron-rich pyrrole-based oxime 9d proving optimal (75%, entry 4). The compatibility of oxime 9d with different bases was also demonstrated (KOH and Cs2CO3), but phenol 8a was obtained in diminished yields (entries 5 and 6). Notably, strongly coloured solutions were observed in every reaction, which can indicate the formation of CTCs. To investigate this possibility further, the reaction using oxime 9d was irradiated with blue LEDs (λmax = 455 nm) for 1 h, which gave phenol 8a in 65% yield instead of 38% yield in the dark or 44% yield when exposed to ambient light from the laboratory (entries 7–9). However, under these photochemical conditions the yield of 8a was partially diminished by the formation of the hydrodehalogenated byproduct 10, which suggested that aryl radicals may be potential intermediates in this reaction. Indeed, reactivity was significantly inhibited by the addition of galvinoxyl or DPPH (1 equiv.) as electron accepting radical scavengers, which reduced the yield of phenol 8a to ≤10% (entries 10 and 11). The addition of TEMPO had a relatively small effect on the yield of phenol 8a (entry 12, no trapped product was detected by high-resolution mass spectrometry but consumption of TEMPO was observed by EPR spectroscopy). However, it should be noted that the coupling of nitroxyl radicals with aryl radicals is known to be relatively slow in polar solvents.12|
|
|||||
|---|---|---|---|---|---|
| Entrya | Oxime | Temp./hv | Base | Time | Yieldb 8a/% |
| a Reactions performed with 0.1 mmol of aryl bromide 3aBr and 0.2 mmol oxime 9a–d with the stated base (0.2 mmol) in DMSO (0.5 mL) under nitrogen. b Determined by 1H NMR spectroscopy against an internal standard (dibromomethane). c Under irradiation with 18 W blue LEDs (λmax = 450 nm) and fan cooling. d Reaction performed in the dark. e Reaction performed in the presence of galvinoxyl (1 equiv.). f Reaction performed in the presence of DPPH (1 equiv.). g Reaction performed in the presence of TEMPO (2 equiv.). | |||||
| 1 | 9a | 30 °C | KOt-Bu | 16 h | 52 |
| 2 | 9b | 30 °C | KOt-Bu | 16 h | 45 |
| 3 | 9c | 30 °C | KOt-Bu | 16 h | 38 |
| 4 | 9d | 30 °C | KOt-Bu | 16 h | 75 |
| 5 | 9d | 30 °C | KOH | 16 h | 38 |
| 6 | 9d | 30 °C | Cs2CO3 | 16 h | 25 |
| 7c | 9d | 450 nm | KOt-Bu | 1 h | 65 |
| 8d | 9d | 30 °C | KOt-Bu | 1 h | 38 |
| 9 | 9d | 30 °C | KOt-Bu | 1 h | 44 |
| 10e | 9d | 30 °C | KOt-Bu | 16 h | 10 |
| 11f | 9d | 30 °C | KOt-Bu | 16 h | <5 |
| 12g | 9d | 30 °C | KOt-Bu | 16 h | 49 |

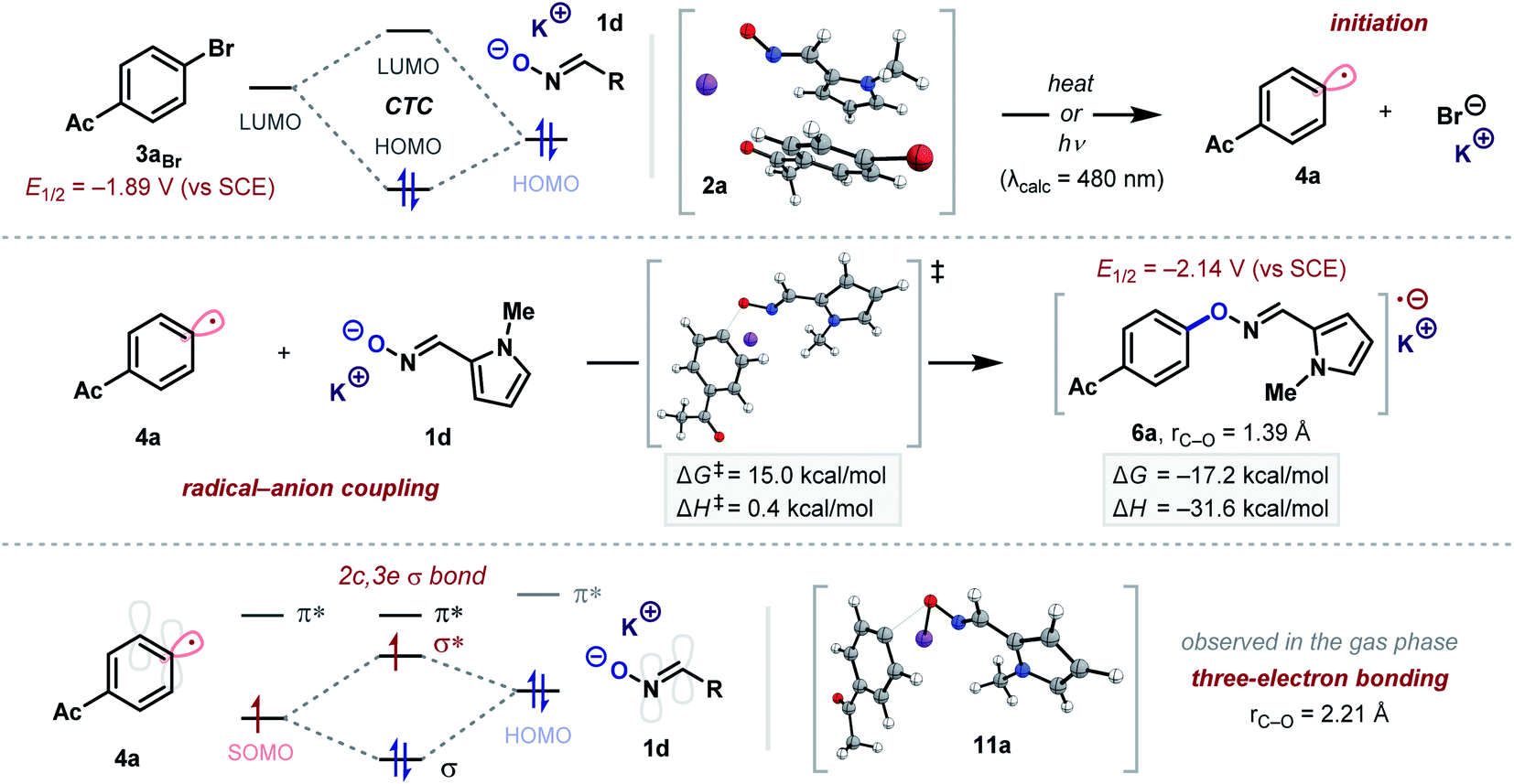
The acceleration of this reaction by light, its inhibition by galvinoxyl and DPPH, and the detection of hydrodehalogenated product 10 all strongly indicated that a radical chain mechanism consistent with an SRN1 reaction was in operation. UV/vis spectroscopic analysis of the reaction mixture and computational studies both supported the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CTC 2a (formed between anion 1d and aryl bromide 3aBr), which may be activated with light or heat10c,d to promote the formation of aryl radical 4a (Scheme 2). The envisaged coupling of 4a with oxime anion 1d was also theoretically explored by DFT computational analysis.13 These studies suggest that radical–anion coupling is exergonic (ΔG = −17.2 kcal mol−1) and there is only a modest activation barrier for radical–anion coupling (ΔG‡ = 15.0 kcal mol−1), which is almost entirely entropic in nature (ΔH‡ = 0.4 kcal mol−1). Considering this, any attractive interaction between the oxime anion and aryl radical could dramatically accelerate the rate of coupling. Indeed, we observed the formation of a weak two-centre three-electron (2c, 3e) σ bonded species 11a in the gas phase.14 In addition, when accounting for concentration effects, the large excess of the oxime anion relative to the radical–anion product will likely lower the activation barrier by ∼4 kcal mol−1 (see the ESI† for details). The calculated redox potential of the coupled radical anion 6a (E1/2 = −2.14 vs. SCE) indicates that propagation of a radical chain by ET to aryl bromide 3aBr (E1/2 = −1.89 vs. SCE)15 would also be exergonic. The resultant neutral O-aryl oxime could then fragment under the basic reaction conditions to afford the observed phenol product. A polar SNAr pathway was considered unlikely to proceed at 30 °C due to the significant activation barrier calculated for the addition of the oxime anion (ΔG‡ = 32.4 kcal mol−1).
:1 CTC 2a (formed between anion 1d and aryl bromide 3aBr), which may be activated with light or heat10c,d to promote the formation of aryl radical 4a (Scheme 2). The envisaged coupling of 4a with oxime anion 1d was also theoretically explored by DFT computational analysis.13 These studies suggest that radical–anion coupling is exergonic (ΔG = −17.2 kcal mol−1) and there is only a modest activation barrier for radical–anion coupling (ΔG‡ = 15.0 kcal mol−1), which is almost entirely entropic in nature (ΔH‡ = 0.4 kcal mol−1). Considering this, any attractive interaction between the oxime anion and aryl radical could dramatically accelerate the rate of coupling. Indeed, we observed the formation of a weak two-centre three-electron (2c, 3e) σ bonded species 11a in the gas phase.14 In addition, when accounting for concentration effects, the large excess of the oxime anion relative to the radical–anion product will likely lower the activation barrier by ∼4 kcal mol−1 (see the ESI† for details). The calculated redox potential of the coupled radical anion 6a (E1/2 = −2.14 vs. SCE) indicates that propagation of a radical chain by ET to aryl bromide 3aBr (E1/2 = −1.89 vs. SCE)15 would also be exergonic. The resultant neutral O-aryl oxime could then fragment under the basic reaction conditions to afford the observed phenol product. A polar SNAr pathway was considered unlikely to proceed at 30 °C due to the significant activation barrier calculated for the addition of the oxime anion (ΔG‡ = 32.4 kcal mol−1).
 | ||
| Scheme 2 Calculations and orbital illustrations to support the proposed radical–anion coupling mechanism. | ||
Importantly, oxime reagent 9d is an easily handled white solid that is prepared on a gram-scale simply by condensing commercial aldehyde 12 with hydroxylamine in the presence of Na2CO3 (Scheme 3). To showcase the utility of designed reagent oxime 9d, the scope of this new arene hydroxylation reaction was fully explored (Table 2). We first sought to determine if halides other than bromine could be substituted by examining a variety of para- and ortho-substituted aromatic carbonyl derivatives (3a–e). Pleasingly, these derivatives could all be converted into the corresponding phenols in good to excellent yields, which demonstrates the compatibility of this reagent with every common halide nucleofuge. However, of the meta-substituted carbonyl derivatives, only fluoride 3fF could be efficiently substituted and that was at elevated temperature (60 °C), which may be due to a switch to a complementary polar SNAr mechanism. Benzonitrile and sulfone derivatives (3g–j) were also examined and the same reactivity pattern was observed: para-substituted derivatives (3g, i) reacted smoothly at 30 °C, whilst the meta-isomers (3h, j) required prolonged reaction times or heating at 60 °C. This reactivity pattern may directly correspond to the rate of radical–anion fragmentation, which is typically ortho > para > meta for aryl halides.7b More strongly electronically activated trifluoromethyl- and nitro-substituted aryl halides (3k–n) were all hydroxylated in typically excellent yields at 30 °C. Relatively unactivated 1-naphthyl and 4-biphenyl halides (3o, p) could also be substituted to afford the desired phenols in modest to excellent yields, although they generally required more forcing reaction conditions (100 °C) and the use of NaOt-Bu as the base. These harsher conditions may be required to overcome higher activation barriers associated with polar pathways (SNAr or benzyne16) or challenging ET initiation events (e.g. from the oxime anion to the arene). However, the ortho-fluorine substituent of dihalogenated biphenyl 3qF could be easily and selectively substituted at 30 °C to afford the phenol in 78% yield. This remarkable reactivity may be due to the sterics of the phenyl ring forcing the fluorine atom to bend out of plane, which could facilitate either a SNAr mechanistic switch or accelerate the rate of radical anion C–F bond fragmentation.17,18 The ortho-fluorine substituent of dihalogenated acetophenone 3rF was also selectively substituted under these reaction conditions. Next, heteroaryl halides were studied (3s–v), and pleasingly activated pyridine 3sBr could be hydroxylated in excellent yield at 30 °C. Unactivated bromo quinolines 3t, u could also be substituted to afford the corresponding phenols in 44–73% yield. Interestingly, as previously observed for dihalogenated arenes, the fluorine atom of pyridine 3vF could also be selectively substituted. Finally, the wider synthetic utility of oxime reagent 9d was demonstrated through the functionalization of aryl halide containing drugs; pleasingly, fenofibrate 3wCl, iloperidone 3xF, etoricoxib 3yCl and blonanserin 3zF were all successfully hydroxylated (47–83% yield).
 | ||
| Scheme 3 Oxime synthesis. | ||


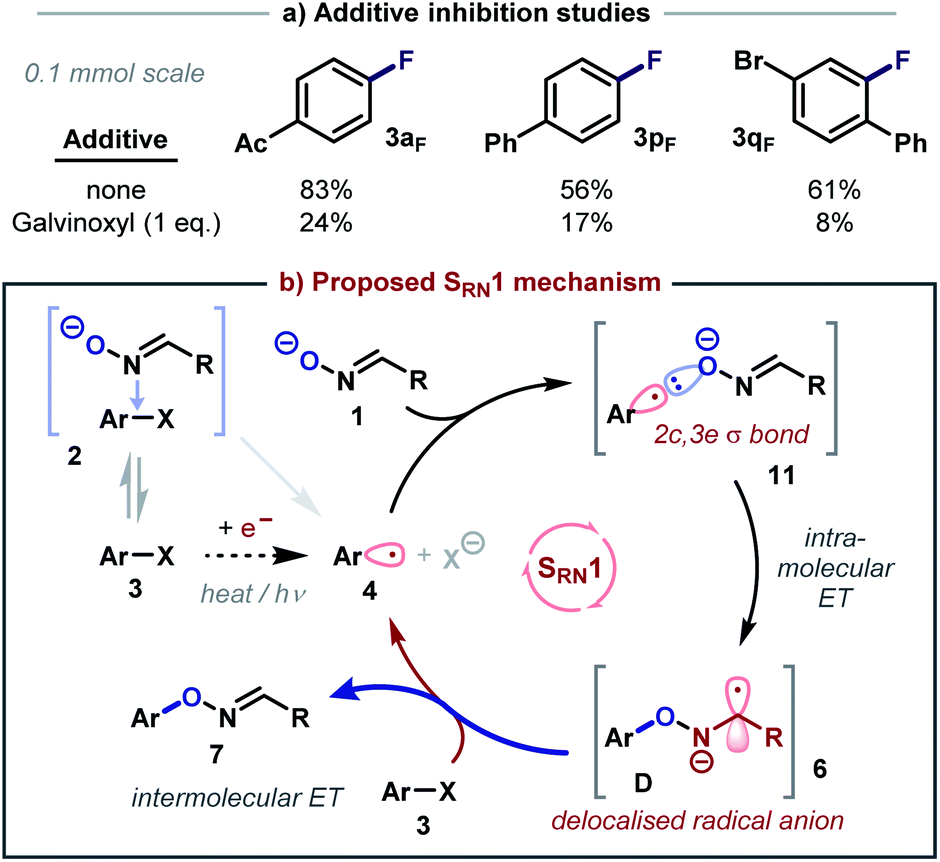
Intrigued by the reactivity and selectivity of some of the aryl fluorides, which could in theory also be substituted via a polar SNAr pathway, their reactions were also studied in the presence of galvinoxyl (Scheme 4a). Interestingly, clear inhibition was observed for every example, which indicates that these reactions are at least partially radical in nature. Alternatively, it is possible that galvinoxyl may disrupt CTC formation, which can theoretically facilitate both polar19 and open-shell reactivity. In this regard, it should also be noted that the formation of strongly coloured reaction mixtures was observed for almost every substrate described in Table 2, which suggests that CTC formation with oxime reagent 9d could be a general process.
 | ||
| Scheme 4 (a) Additional additive inhibition studies; (b) proposed SRN1 mechanism. | ||
Thus, considering these results and our previous observations, it is reasonable to assume that many of the substitution reactions described herein likely proceed via an open-shell mechanism. We therefore propose that an electron-catalysed7c SRN1 chain is initiated by either the formation and activation of a CTC, or a slow thermal (concerted) dissociative ET20 from an anionic electron donor21 (e.g. the oxime anion 1) to the aryl halide 3 (Scheme 4b). The resultant aryl radical 4 can then interact with an oxime anion 1 to form a weakly interacting cluster that may be viewed as a 2c, 3e σ bonded species 11.22 As this bond shortens, a delocalised radical anion 6 (and a standard 2c, 2e bond) is then formed by intramolecular ET from species 11 into a nearby π* orbital (on either the oxime or the aryl ring). Radical anion 6 then reduces another equivalent of 3 through intermolecular ET to regenerate aryl radical 4 and release the coupled product 7, which fragments in situ to afford the observed phenol product.23 However, the contribution of a polar SNAr pathway for some substrates cannot be completely excluded.
Conclusions
In summary, we have reported the design and development of a new oxime-based hydroxylation reagent, which can be used to chemoselectively convert aryl halides into phenols under remarkably simple, transition-metal-free conditions. These reactions are proposed to primarily proceed via the unprecedented intermolecular coupling of an oxygen-based anion with aryl radicals to form new C(sp2)–O bonds. We believe that the synthetic utility of this reagent is likely enhanced by its ability to substitute nucleofuges through complementary polar pathways. It is hoped that these findings will facilitate the rational design of other such anionic reagents and enable new unconventional retrosynthetic strategies to be realised.Data availability
Experimental procedures, characterisation data, computational details, and copies of 1H, 13C and 19F NMR spectra for all compounds featured in this manuscript are provided in the ESI.†Author contributions
Conceptualisation, supervision and writing – M. J. J.; investigation and methodology – A. J. G., P. U., W. O. W., G. S., I. O., A. C. W., V. C., M. J. J.; all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Wild Fund, the University of York, the Leverhulme Trust (for an Early Career Fellowship, ECF-2019-135, M. J. J.) and the Royal Society (Research Grant RGS\R1\201268) for financial support. We would also like to thank Prof. Peter O'Brien and Dr William P. Unsworth for helpful discussions.Notes and references
- Z. Rappoport, The Chemistry of Phenols, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003, ISBN: 978-0-471-49737-0 Search PubMed.
- For selected examples of different arene hydroxylation strategies, see: (a) K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 CrossRef CAS PubMed; (b) A. G. Sergeev, T. Schulz, C. Torborg, A. Spannenberg, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 7595–7599 CrossRef CAS PubMed; (c) A. Tlili, N. Xia, F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 8725–8728 CrossRef CAS PubMed; (d) S. Bracegirdle and E. A. Anderson, Chem. Commun., 2010, 46, 3454 RSC; (e) C. Zhu, R. Wang and J. R. Falck, Org. Lett., 2012, 14, 3494–3497 CrossRef CAS PubMed; (f) K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368–5371 CrossRef CAS PubMed; (g) S. Xia, L. Gan, K. Wang, Z. Li and D. Ma, J. Am. Chem. Soc., 2016, 138, 13493–13496 CrossRef CAS PubMed; (h) J. Börgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026–16031 CrossRef PubMed; (i) L. Yang, Z. Huang, G. Li, W. Zhang, R. Cao, C. Wang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2018, 57, 1968–1972 CrossRef CAS PubMed; (j) R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed; (k) Y. M. Cai, Y. T. Xu, X. Zhang, W. X. Gao, X. B. Huang, Y. B. Zhou, M. C. Liu and H. Y. Wu, Org. Lett., 2019, 21, 8479–8484 CrossRef CAS PubMed.
- (a) J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273–412 CrossRef CAS; (b) F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013, ISBN: 9783527656141 CrossRef; (c) S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 16368–16388 CrossRef CAS PubMed.
- (a) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (b) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- For related hydroxide surrogate substitution reactions, see: (a) A. P. Krapcho and D. Waterhouse, Synth. Commun., 1998, 28, 3415–3422 CrossRef CAS; (b) J. F. Rogers and D. M. Green, Tetrahedron Lett., 2002, 43, 3585–3587 CrossRef CAS; (c) J. I. Levin and M. T. Du, Synth. Commun., 2002, 32, 1401–1406 CrossRef CAS; (d) P. S. Fier and K. M. Maloney, Org. Lett., 2016, 18, 2244–2247 CrossRef CAS PubMed; (e) Y. Zhou, J. Chem. Res., 2017, 41, 591–593 CrossRef CAS; (f) M. Reitti, R. Gurubrahamam, M. Walther, E. Lindstedt and B. Olofsson, Org. Lett., 2018, 20, 1785–1788 CrossRef CAS PubMed.
- (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed; (b) W. Liu, J. Li, C. Huang and C. Li, Angew. Chem., Int. Ed., 2020, 59, 1786–1796 CrossRef PubMed.
- (a) J. F. Bunnett and J. K. Kim, J. Am. Chem. Soc., 1970, 92, 7463–7464 CrossRef CAS; (b) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71–168 CrossRef CAS PubMed; (c) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- (a) C. Amatore, J. Badoz-Lambling, C. Bonnel-Huyghes, J. Pinson, J. M. Saveant and A. Thiebault, J. Am. Chem. Soc., 1982, 104, 1979–1986 CrossRef CAS; (b) M. T. Baumgartner, A. B. Pierini and R. A. Rossi, Tetrahedron Lett., 1992, 33, 2323–2326 CrossRef CAS; (c) J. M. Saveant, J. Phys. Chem., 1994, 98, 3716–3724 CrossRef CAS.
- For an example of an SRN1 reaction with oxime anions and alkyl radicals initiated by the photochemical activation of a CTC, see: Z. Chen, H. Liang, R. Chen, L. Chen, X. Tang, M. Yan and X. Zhang, Adv. Synth. Catal., 2019, 361, 3324–3330 CrossRef CAS.
- (a) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS; (b) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed; (c) M. A. Fox, J. Younathan and G. E. Fryxell, J. Org. Chem., 1983, 48, 3109–3112 CrossRef CAS; (d) S. V. Rosokha and J. K. Kochi, New J. Chem., 2002, 26, 851–860 RSC.
- (a) P. S. Fier and K. M. Maloney, Angew. Chem., Int. Ed., 2017, 56, 4478–4482 CrossRef CAS PubMed; (b) P. S. Fier and K. M. Maloney, Org. Lett., 2017, 19, 3033–3036 CrossRef CAS PubMed.
- (a) A. L. J. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983–4992 CrossRef CAS; (b) M. R. Heinrich, A. Wetzel and M. Kirschstein, Org. Lett., 2007, 9, 3833–3835 CrossRef CAS PubMed.
- All calculations were performed using the ORCA 4.2.0 software package: (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed ; Unless stated otherwise, calculations were carried out using the ωB97X-D3(BJ) functional with the ma-def2-TZVP basis set and the CPCM continuum solvation model (DMSO); (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (e) A. Najibi and L. Goerigk, J. Chem. Theory Comput., 2018, 14, 5725 CrossRef CAS PubMed; (f) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (g) J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed; (h) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- Dielectric continuum solvation models are known to obscure weak radical–anion interactions. For example, see: A. Cardinale, A. A. Isse, A. Gennaro, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2002, 124, 13533–13539 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- Although no other regioisomers were observed, the possibility of benzyne intermediates in some reactions cannot be excluded. For relevant work, see: (a) A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 CrossRef CAS PubMed; (b) Y. Dong, M. I. Lipschutz and T. D. Tilley, Org. Lett., 2016, 18, 1530–1533 CrossRef CAS PubMed; (c) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044–9056 RSC.
- (a) A. B. Pierini and D. M. A. Vera, J. Org. Chem., 2003, 68, 9191–9199 CrossRef CAS PubMed; (b) C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2004, 126, 16051–16057 CrossRef CAS PubMed; (c) N. Takeda, P. V. Poliakov, A. R. Cook and J. R. Miller, J. Am. Chem. Soc., 2004, 126, 4301–4309 CrossRef CAS PubMed.
- For examples of regio- and chemoselective radical anion C–F bond fragmentation, see: (a) D. E. Mashkantsev, I. V. Beregovaya and L. N. Shchegoleva, J. Fluorine Chem., 2016, 188, 171–176 CrossRef CAS; (b) R. V. Andreev, I. V. Beregovaya and L. N. Shchegoleva, J. Fluorine Chem., 2020, 234, 109513 CrossRef CAS.
- For an example of an alternative polar pathway enabled by donor–acceptor interactions, see: S. Senaweera and J. D. Weaver, Chem. Commun., 2017, 53, 7545–7548 RSC.
- C. Costentin, P. Hapiot, M. Médebielle and J. M. Savéant, J. Am. Chem. Soc., 1999, 121, 4451–4460 CrossRef CAS.
- (a) J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402–7410 CrossRef CAS PubMed; (b) D. A. Caminos, M. Puiatti, J. I. Bardagí and A. B. Peñéñory, RSC Adv., 2017, 7, 31148–31157 RSC; (c) G. Nocera and J. A. Murphy, Synthesis, 2020, 52, 327–336 CrossRef CAS.
- For pioneering examples of 2c, 3e σ bonds in synthesis, see: (a) C. J. Evoniuk, G. D. P. Gomes, S. P. Hill, S. Fujita, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2017, 139, 16210–16221 CrossRef CAS PubMed; (b) Q. Elliott, G. dos Passos Gomes, C. J. Evoniuk and I. V. Alabugin, Chem. Sci., 2020, 11, 6539–6555 RSC.
- Due to the lower electron donating ability of phenolates relative to oxime anion 1d we anticipate that radical–anion coupling with oxime anion 1d will be substantially faster than with the phenolate products. This selectivity will be further enhanced by the relatively high concentration of the oxime anion.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, computational details, and copies of 1H, 13C and 19F NMR spectra for all compounds featured in this manuscript. CCDC 2102632 and 2102633. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04748e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |