
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of nahuoic acid A via a putative biogenetic intramolecular Diels–Alder (IMDA) reaction†
Lucía
Guillade
,
Paula
Mora
,
Pedro
Villar
 ,
Rosana
Alvarez
* and
Angel
R. de Lera
*
,
Rosana
Alvarez
* and
Angel
R. de Lera
*
Departamento de Química Orgánica, Facultade de Química, CINBIO, IIS Galicia Sur, Universidade de Vigo, 36310 Vigo, Spain. E-mail: qolera@uvigo.es; rar@uvigo.es
First published on 27th October 2021
Abstract
Inspired by the biogenetic proposal of an intramolecular Diels–Alder (IMDA) cycloaddition, the total synthesis of natural product nahuoic acid A, a cofactor-competitive inhibitor of the epigenetic enzyme lysine methyl transferase SETD8, has been carried out. A non-conjugated pentaenal precursor was synthesized with high levels of stereoselectivity at seven stereogenic centers and with the appropriate control of double bond geometries. Although the IMDA reaction of the non-conjugated pentaenal using Me2AlCl for catalysis at −40 °C selectively afforded the trans-fused diastereomer corresponding to the Re-endo mode of cycloaddition, under thermal reaction conditions it gave rise to a mixture of diastereomers, that preferentially formed through the exo mode, including the cis-fused angularly-methylated octahydronaphthalene diastereomer precursor of nahuoic acid A. The natural product could be obtained upon oxidation and overall deprotection of the hydroxyl groups present in the Si-exo IMDA diastereomer.
A Introduction
Nahuoic acid A (1a) was first isolated in 2013 from cultures of Streptomyces sp. (isolate RJA2928) in a marine sediment collected near the passage Padana Nahua in Papua New Guinea1a using a chemical genetics approach.1b The structure of 1a was shown to contain an octahydronaphthalene core with seven contiguous stereocentres and unsaturated substituents of E geometry at C4 and C13, namely a 2-methylpropenoic acid, and a 2,4,10-trimethylundec-9-ene-3,5,7-triol side chain with four additional stereogenic centers, respectively. The relative configuration of the stereogenic elements of this polyketide natural product was determined1a through comprehensive analysis of 1H-NMR spectroscopic data using a combination of trNOESY and gCOSY60 correlations, and the absolute configuration was assigned by NMR analysis of the acetonides2 and modified Mosher ester derivatives.3Additional family members, termed nahuoic acids B-E (1b–d; 2a–c; Fig. 1), were further isolated from the same genus.4 Structural differences among the series relative to parent nahuoic acid A (1a) rely on the level and positional oxidation of the octahydronaphthalene core (1b–c) and on the length and number of hydroxyl-containing stereocenters on the polyhydroxylated side chain at C13 (2a–c). A unified nomenclature of the natural nahuoic acid family members was later proposed by A. B. Smith III et al., as depicted in Fig. 1.5
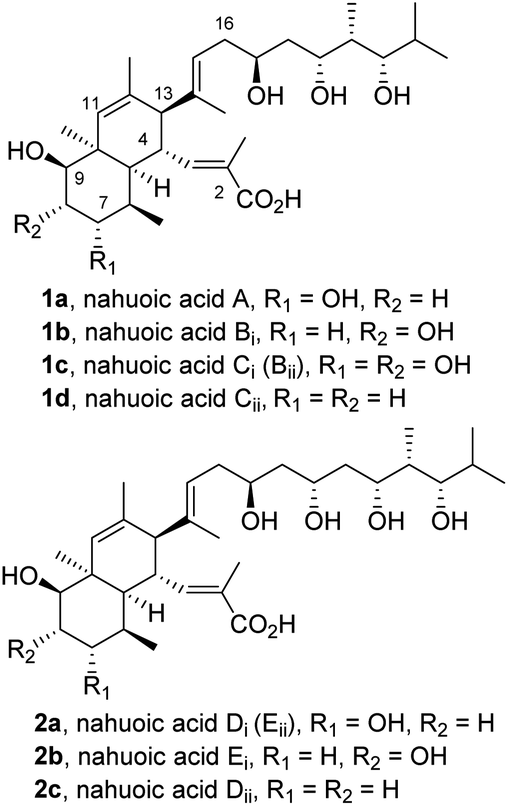 | ||
| Fig. 1 Structure and unified nomenclature of nahuoic acids.5 | ||
A significant decrease in cell proliferation of the osteosarcoma U2OS cells (IC50 = 65 ± 2 μM), and the SUM159 (IC50 = 45 μM), and MDA-MB-436 (IC50 = 85 μM) breast cancer cells was noticed after treatment with nahuoic acid A (1a).1a,4b In contrast to parent 1a, structurally similar analogs B-E (1b–d and 2a–c) were found to be inactive in the same assay.4a However, the polyacetylated derivative of 1a (not shown) was reported to inhibit the proliferation of certain cancer cell lines with moderate potency.4b
The inhibition of proliferation of several cancer cell lines by 1a1a was causally linked to the competitive inhibition (Ki = 2 μM) with the binding of biological cofactor S-adenosylmethionine (SAM or AdoMet 3, Scheme 1) to the epigenetic enzyme SETD8.4b,6 SETD8, also known as SET8/PR-Set7/KMT5A, is a member of the histone methyltransferase (HMT) family,7 which is implicated, along with other enzymes, in the chemical modifications of histones, a group of highly basic proteins that pack the DNA into the nucleosomes in eukaryotic cells.
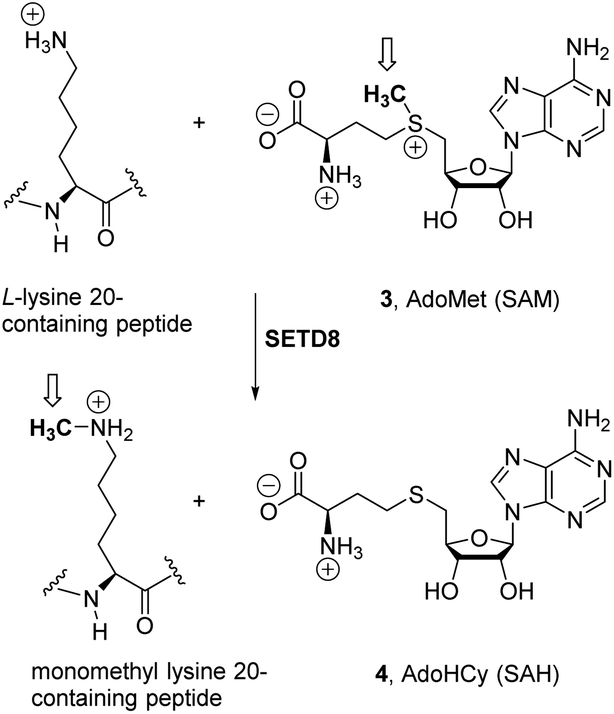 | ||
| Scheme 1 Histone lysine methylation catalyzed by lysine methyl transferases (KMTs). | ||
In particular, the methylation status of specific lysine residues in histone proteins is tightly regulated by the competitive actions of the lysine methyl transferase (KMT) and lysine demethylase (KDM) families of epigenetic enzymes.7c–f,8a,b Since the methylation status of histones plays important roles in the regulation of transcription and the maintenance of genomic integrity in eukaryotes, the lack of control of this epigenetic modification from its normal status appears to be related to inflammation and to several diseases, including leukaemia and breast and prostate cancer.9a,b
Mechanistically, ca. 100 KMT family members7b promote an SN2-type reaction (Scheme 1) through an early transition state10 for the transfer of the methyl group of cofactor SAM (3) to a partially deprotonated terminal ε-amine lysine group, releasing S-adenosylhomocysteine (SAH, AdoHCy, 4, Scheme 1).11 SETD8 (ref. 12) has been shown to monomethylate the ε-amino group of lysine 20 on histone 4 (H4K20me)13 and also specific lysines of proliferating cell nuclear antigen (PCNA)14 and p53/TP53.15 This KMT enzyme has been found to be overexpressed in several types of cancer,12a,15 and appears to play an important role in the progression from the S phase of the cell cycle.16
Since nahuoic acid A (1a) is the first reported cofactor-competitive inhibitor of SETD8, and its promising biological activities are putatively linked to SETD8 inhibition,6,7d–f,17 its structure can be considered as a challenging lead for further development. In fact, our interest in the synthesis and biological evaluation of epigenetic modulators18a–d inspired by the structure of natural products made nahuoic acid A (1a) an attractive target candidate for synthesis.
While our efforts to synthesize nahuoic acid A (1a) were in progress,19 the total synthesis of nahuoic acid Ci (1c, Scheme 2) was reported.5
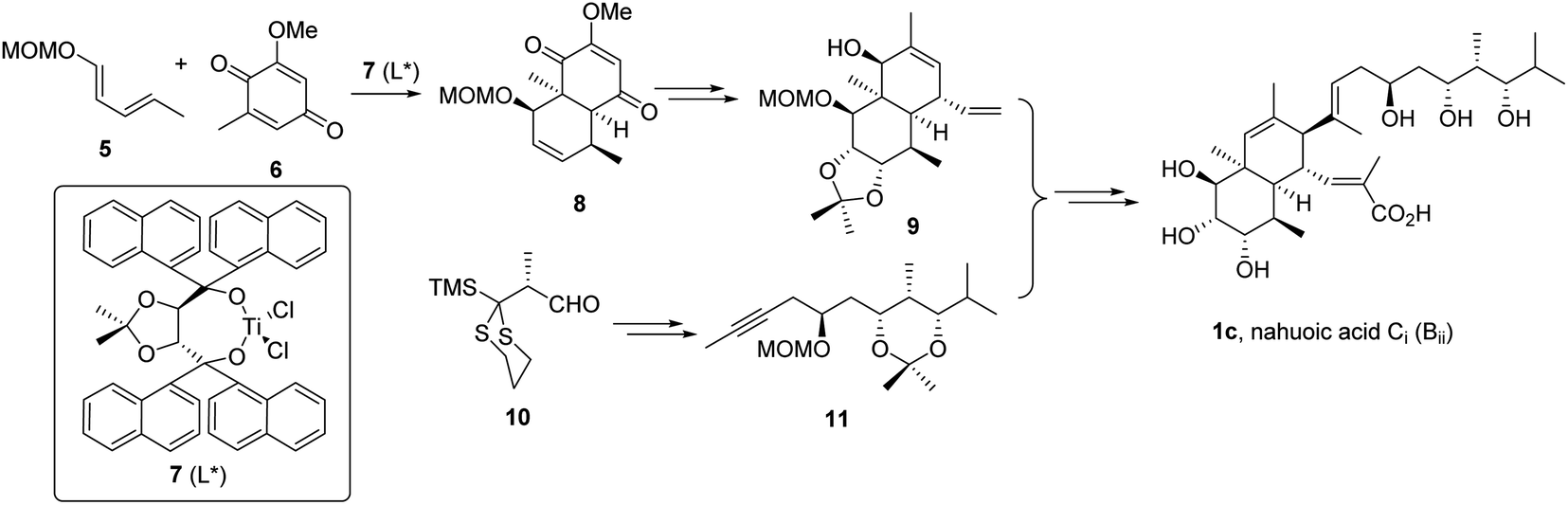 | ||
| Scheme 2 Smith's total synthesis of nahuoic acid Ci (Bii, 1c).5 | ||
The cis-octahydronaphthalene fragment 9 was obtained by the intermolecular Diels–Alder cycloaddition reaction of components 5 and 6 promoted by enantiopure TADDOL-derived titanium complex 7 to afford intermediate 8, followed by functional group interconversion and generation of additional stereocenters. The trihydroxylated side chain functionalized as internal alkyne 11 was constructed from intermediate 10 by the anion relay chemistry (ARC) approach20 (Scheme 2), and connected to 9 by application of the Micalizio protocol.21
Our retrosynthetic analysis of the nahuoic acid skeleton (Scheme 3) was instead inspired by the hypothetical biogenesis of the octahydronaphthalene core structure of 1a through an intramolecular Diels–Alder (IMDA) reaction22 of an appropriate pentaenoate precursor 12. In order to afford the desired diastereomer, the pentaenoate of E geometry of 12 should undergo intramolecular cycloaddition following an exo orientation towards the diene of the reacting Si face of the dienophile. The central conjugated triene fragment of 12 would instead be obtained by a Suzuki–Miyaura cross-coupling23 of two components of similar complexity, namely internal boronic acid 13 and terminal non-conjugated iodotetraenoate 14. Several approaches could be envisaged for the construction of these polypropionate-like polyols, and one of them is illustrated in Scheme 3. The alkenylboronic acid 13 was proposed to be generated by acyclic cross-metathesis of an alkenyl boronate and homoallylic alcohol 15 containing a formal protected 1,3,5-triol fragment. The latter would be prepared by diastereoselective allylation of aldehyde 16 containing three contiguous stereocenters, which could arise from protected aldol 17, the product expected from a diastereo- and enantioselective aldol reaction of isobutyraldehyde 18 and propanal surrogates. Iodotetraenoate 14 would instead be obtained by unsaturated chain extension of precursor iodotrienal 19 and the latter from protected aldol 20, which could be generated following similar protocols already described for 16. The synthesis of the aldol precursor 21 could be based on the enantioselective allylation of δ-iododienal 22 followed by ozonolysis. The combination of ligand-dependent enantioselective reactions and substrate-promoted diastereoselective bond-forming reactions should allow us to set up the relative and absolute configurations of the intermediates on route to the natural product.
 | ||
| Scheme 3 Biogenetically-inspired retrosynthetic analysis of nahuoic acid A (1a). | ||
An additional interest of the synthetic proposal was the exploration of the intramolecular Diels–Alder reaction (IMDA)22 of 12 for the construction of the octahydronaphthalene core of nahuoic acid A (1a). This biogenetic route appears to be feasible in nature and a series of putative Diels–Alderase enzymes have been structurally and functionally characterized,24 and grouped under the general family of “biosynthetic pericyclases”.25a,b Members of natural [4 + 2]-cyclases25c,d including enzymes involved in inverse electron-demand Diels–Alder reactions25e have been identified in nature, in some cases as part of enzymatic cascade reactions that generate further biosynthetic complexity.25f–i
We have already reported19 the synthesis of functionalized tetraenal 25 and tetraenoates 26–27 (Scheme 4), which are shorter unsaturated analogues of pentaenoate 12 (Scheme 3), as model systems to test the feasibility of the IMDA tactic22 to achieve 1a. The IMDA reaction of tetraenal 25 led, under both purely thermal and Lewis acid-promoted IMDA reaction conditions, to the octahydronaphthalenes as mixtures of two cis-octahydronaphthalenes and a trans-diastereoisomer, thus confirming the formation of products through both exo approaches and through one of the putative endo alternatives, the latter being of lower energy.19
 | ||
Scheme 4 Synthesis of non-conjugated tetraenals, tetraenoates and pentaenals for the preparation of the octahydronaphthalene core of nahuoic acid model systems. Reagents and reaction conditions: (a) 28 or 29, Pd(PPh3)4 (cat), 10% aq. TlOH, THF, 25 °C, 3 h (97% for 26; 99% for 27; 83% for 25; 92% for 34a; 99% for 34b). (b) DIBAL-H, THF, −78 °C, 6 h (99% for 24, 32a and 32b). (c) MnO2, Na2CO3, CH2Cl2, 25 °C, 16–24 h (92% for 19; 92% for 33a; 95% for 33b). (d) (EtO)2P(O)CH2CO2Et 30a or (EtO)2P(O)CH(CH3)CO2Et 30b, n-BuLi, DMPU, THF, −78 to 25 °C, 16 h (77% for 31a, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 E/Z ratio; 89% for 31b, 1:0.1 E/Z ratio). :0.2 E/Z ratio; 89% for 31b, 1:0.1 E/Z ratio). | ||
We wish to report herein the full account of our synthesis of nahuoic acid A (1a) based on the putative biogenetic proposal using the corresponding pentaenals related to 12 (Scheme 3), which required the construction of the entire unsaturated acyclic skeleton in a highly diastereo- and enantioselective fashion.
B Results and discussion
Non-conjugated tetraenal and pentaenal model systems and IMDA reaction
For the preparation of functionalized tetraenes 25, 26 and 27 (Scheme 4) containing the acyclic skeletons required for the IMDA reaction,19 a Suzuki–Miyaura cross-coupling23 was chosen as the last step of the synthesis. Under the conditions developed by Kishi for alkene–alkene cross-coupling (catalysis by Pd(PPh3)4 in THF and 10% aqueous TlOH)26 tetraenes 25, 26 and 27 were synthesized19 starting from iodotrienoate 23 or iodotrienal 19 and either commercial alkenylpinacol boronate 28 or analogue 29, itself obtained by regio- and stereoselective borylation of protected but-2-yn-1-ol.27In addition, model pentaenals 34a and 34b were constructed by unsaturated chain extension of iodotrienal 19.19 The Horner–Wadsworth–Emmons (HWE) reaction28 of 19 (ref. 19) with the corresponding anions of phosphonates 30a and 30b, generated with n-BuLi and DMPU in THF, afforded conjugated dienoates 31a and 31b, respectively (Scheme 4), whereas the reaction with phosphonate 30a afforded a 5:1 E/Z mixture of isomers of 31a in 77% yield, and using the methyl-substituted analogue 30b both the isomer ratio (10:1) of pentaenoate 31b and the yield of the HWE reaction (89%) were higher. Reduction of 31a and 31b27 with DIBAL-H afforded allylic alcohols 32a and 32b, respectively, in quantitative yield, from which conjugated dienals 33a and 33b, respectively, were obtained uneventfully (92 and 95% yield, respectively) upon allylic oxidation with MnO2 and Na2CO3. The Suzuki–Miyaura cross-coupling reaction23 of 33a or 33b with 28 under the conditions indicated above gave rise to non-conjugated pentaenals 34a and 34b in 92 and 99% yield, respectively (Scheme 4).
No conversion was noticed when solutions of ethyl tetraenoates 26 or 27 in toluene-d8 where heated up to 120 °C in the presence of BHT.29 Further temperature increase up to 165 °C led to extensive decomposition. Strikingly, although tetraenal 25 proved to be unreactive when treated with Me2AlCl in CH2Cl2 at −78 °C for 24 h,30 a smooth and quantitative conversion to a cyclic derivative was observed upon increasing the temperature to −40 °C and stirring for 40 h. Exhaustive NMR studies suggested structure 37 (Scheme 5) for the octahydronaphthalene diastereoisomer obtained in the IMDA reaction, which was further confirmed by X-ray diffraction analysis.19 This compound must likely originate from the Re-endo orientation of the dienophile fragment relative to the reacting diene in non-conjugated tetraenal 25.
 | ||
| Scheme 5 IMDA reactions of non-conjugated tetraenal 25 (A) and pentaenals 34a and b (B) under different reaction conditions. | ||
Heating instead a solution of tetraenal 25 in o-xylene at 200 °C for 40 h afforded a mixture of three major compounds, which were identified after chromatographic separation as the octahydronaphthalenes Si-exo35, Re-exo36, and Re-endo37 diastereomers, in a 40:40:20 isomer ratio, respectively, and 81% overall yield (Scheme 5). The structure of 35 was obtained by X-ray single crystal diffraction analysis,19 which allowed us to confirm the absolute configuration of the octahydronaphthalene core present in natural product 1a. Although 1H-NMR analysis revealed the presence of minor components (<10% yield) in the reaction mixture, signals putatively assigned to the Si-endo diastereomer were not clearly identified. The use of stronger and bulkier Lewis acid B(C6F5)3 in DA reactions of acyclic dienes with unsaturated aldehydes has been found to alter the innate diastereoselectivity of these processes, leading predominantly to the formation of the exo adducts.31
The effect has been proposed to be merely steric in origin, as the bulky B(C6F5)3 complexed to the aldehyde carbonyl group would disfavor the endo transition state.32 In our case, it was found that upon stirring a solution of tetraenal 25 in CH2Cl2 with B(C6F5)3 (0.5 mol equiv.) at −10 °C for 48 h, a mixture of the Si-exo35, Re-exo36 and Re-endo37 octahydronaphthalenes, in a 38:34:28 isomer ratio, respectively, was isolated in 74% overall yield (Scheme 5).
Unfortunately, all attempts to perform the unsaturated chain extension of aldehyde Si-exo35 either using the HWE reaction28 with phosphonate 30b (Scheme 4, by heating up to 40 °C), the Wittig reaction28a,33,34 with the phosphonium salt analogue or the less-sterically demanding Peterson olefination35 with the trialkylsilyl derivative as the reagent, proved to be fruitless, and the alkenyloctahydronaphthalene carboxaldehyde Si-exo35 was recovered.
Vinylogous pentaenals 34a and 34b were alternatively treated under thermal or Lewis-acid catalyzed reaction conditions (Scheme 5). Upon activation with B(C6F5)3 in CH2Cl2 containing 4 Å MS at −10 °C (ref. 31) pentaenal 34a afforded an octahydronaphthalene skeleton, which was identified through NOESY-1d experiments as the Re-endo cycloadduct 38a (21% yield) and unreacted substrate 34a (32% yield), accompanied by an additional compound (15% yield), which could not be fully characterized. The methyl-substituted pentaenal analogue 34b proved to be unreactive under the same reaction conditions, suggesting a limitation of the B(C6F5)3-based activation procedure for more sterically hindered aldehydes. As an alternative, heating 34b to 200 °C in o-xylene containing BHT as the radical inhibitor afforded a mixture of products, which were further characterized by NMR spectroscopy following HPLC purification (Scheme 5). Octahydronaphthalenes resulting from Re-exo39b (31%) and Si-exo40b (17%) orientations of the reacting fragments were identified, accompanied by a polycyclic compound that could not be fully characterized and might be derived, when compared with the reactivity of 25, from further rearrangements of the putative Re-endo diastereomer.
Computational studies on the IMDA reaction of model systems
DFT calculations at the ωB97XD/Def2SVP level36 using the Gaussian 09 suite of programs37 of the non-conjugated tetraenal model system I (Scheme 6) lacking the trihydroxylated side chain of nahuoic acid 1a in toluene or MeOH as solvent were carried out in order to analyze and thus justify the diastereoselectivity of the IMDA reaction. Conformational analysis of model reactant I indicates s-cis,s-cis to be the most stable conformer as it reduces the destabilizing interactions between the methyl substituents of the formal 1,3,5,6-tetramethylhexatriene fragment, whereas conformers showing a single bond rotation of these Csp2–Csp2 connections are destabilized by only 0.5–0.7 kcal mol−1, and the s-trans,s-trans extended conformation is destabilized by 2.3 kcal mol−1 (Scheme 6A). Further destabilization was expected for the model reactant to adapt to the folded conformation required for the IMDA reaction. The energy values computed for the reacting conformations (Scheme 6B) vary between 2.7 and 5.8 kcal mol−1 from the most stable extended s-cis,s-cis conformer of I. | ||
| Scheme 6 (A) Lowest energy conformations of non-conjugated tetraenal model system I. (B) Lowest energy approaches computed for the thermal IMDA reaction to afford the cis-octahydronaphthalene II. (C) Representation of the Si-exo approach. All computations were carried out at the ωB97XD/DefTZVP (SMD, toluene) level of theory. Relative energies are given in kcal mol−1. | ||
All IMDA reactions were characterized as concerted but asynchronous cycloadditions, with the formed C3–C8 shorter than the C2–C11 bond length (ESI†). Although the lowest energy of activation (ΔG# = 30.5 kcal mol−1, Scheme 6B) was computed for the transition state leading to the Si-exo product II from the s-cis,s-trans conformer of I in the folded conformation (itself disfavored by 5.4 kcal mol−1), those for the Re-endo and Re-exo transition states showed similar energy values (ΔG# = 30.7 and 31.0 kcal mol−1, respectively, Scheme 6B) and therefore the octahydronaphthalene diastereomers should also be present in the reaction mixture as IMDA reaction products. The IMDA Re-endo approach exhibited the highest energy of activation of the series (ΔG# = 33.0 kcal mol−1, Scheme 6B) despite being the less disfavored (by 2.7 kcal mol−1) starting conformation. Therefore, the computational data confirms the lack of selectivity of the IMDA reaction under thermal reaction conditions for this system and provides a theoretically estimated diastereomeric ratio at 200 °C (39:25:33 dr Si-exo35/Re-exo36/Re-endo37) that is consistent with the experimental results.
To summarize, an efficient biogenetic approach to the acyclic pentaene fragment of nahuoic acid A was carried out, and the limitations were noted for the construction of the bicyclic octahydronaphthalene core using the IMDA reaction. However upon using Me2AlCl to activate tetraenal model system 25 in CH2Cl2 at −78 °C for 24 h, a single diastereomer (Re-endo37) was obtained under thermal cyclization reaction conditions (BHT, o-xylene, 200 °C), and a mixture of three diastereomers (Si-exo35/Re-exo36/Re-endo37 40:40:20 dr) was generated, being the major components formed through the exo mode of cycloaddition of diene and dienophile (exo/endo ratio of ca. 3.76:1) with opposite facial selectivity. The IMDA reaction catalyzed by the bulky Lewis acid B(C6F5)3 provided these octahydronaphthalenes with lower diastereoselectivity (2.5:1 exo/endo ratio), and moreover this protocol could not be extended to the pentaenal model system 34b, which underwent the IMDA reaction under thermal reaction conditions to afford a mixture of the Si-exo40b and Re-exo39b octahydronaphthalenes in a ca. 1:1 ratio.
Total synthesis of nahuoic acid A (1a)
Since iodotetraenal 33b (Scheme 4) contains the appropriate fragment to generate the octahydronaphthalene core of nahouic acid A (1a) through the IMDA reaction, the stereoselective synthesis of the trihydroxylated side chain with four stereocenters present in natural products 1a–1d was addressed, as summarized in Scheme 3.The synthesis started from the aldol reaction of the enolates derived from N-acyloxazolidine-2-thione 41 (ref. 38) containing Nagao's auxiliary39 and isobutyraldehyde 18. The combination of TiCl4 and DIPEA at −78 °C (ref. 40) afforded the syn “non-Evans” aldol product 42 in 80% yield with very high diastereoselectivity.41 Transformation of 42 into the corresponding aldehyde 17 involved the formation of the Weinreb amide [Me(OMe)HN·HCl, imidazole],42 protection of the alcohol as silyl ether (TBDMSOTf, 2,6-lutidine) and reduction with DIBAL-H (THF, −78 °C), which resulted in an overall 71% yield (Scheme 7).
 | ||
| Scheme 7 Reagents and reaction conditions: (a) TiCl4, i-Pr2NEt, CH2Cl2, 0 to −78 °C, 3 h (80%). (b(i)) Me(OMe)NH·HCl, imidazole, CH2Cl2, 25 °C, 24 h. (ii) TBDMSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 0.5 h. (iii) DIBAL-H, THF, −78 °C, 6 h (71%, three steps); (c) (S,S)-43, Sc(OTf)3, CH2Cl2, 0 to 25 °C, 22 h (99%). (d) TBDMSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 0.5 h (94%). (e(i)) O3, CH2Cl2, −78 °C, 1 min. (ii) PPh3, 25 °C, 1 h (16, 92%). (f) (−)-Ipc2Ball 46, Et2O, −78 to −30 °C, 5 h, 90% (70:30 dr). (g) MOMCl, DIPEA, CH2Cl2, 25 °C, 22 h (85%). (h(i)) O3, CH2Cl2, −78 °C, 3 min. (ii) PPh3, 25 °C, 1 h (94% for 49; 81% for 53). (i) Allyl magnesium bromide 50, THF, 0 to 25 °C, 6 h (70% for 51, 76% for 54, 50:50 dr). (j) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C, 1 h (86%). (k) AcOH/H2O (4:1 v/v), THF, 25 °C, 12 h (99%). (l) 2,2-DMP, PPTS, CH2Cl2, 25 °C, 10 h (86%). (m) 58, Hoveyda–Grubbs II cat. 59, CH2Cl2, reflux, 17.5 h (5:1 E/Z ratio, 60%). | ||
Allylation of 17 using the optimized protocol of Leighton et al.43 with (S,S)-43 and Sc(OTf)3 at 0 °C for 24 h afforded the homoallylic alcohol 44 in quantitative yield as a single diastereomer.44 Despite this promising result, further allylation of aldehyde 16, which was obtained by protection of 44 as silyl ether and ozonolysis (upon treatment first with O3 in CH2Cl2 at −78 °C for 1 min and then with PPh3) with the enantiomeric reagent (R,R)-43 (not shown) was unsuccessful and mixtures of compounds were obtained, from which the deprotected and dehydrated derivative of the homoallylic alcohol reactant (compound 45) could be identified (Scheme 7). No further improvement was observed upon modification of the work-up procedure using a saturated aqueous solution of NaHCO3 instead of TBAF, or freshly-opened bottles of the reagent. Conformational effects likely play a role in the reactivity of this intermediate,45 given the formal destabilizing 1,3-syn-pentane-type interactions46 between the methyl groups and also between the two silyloxy substituents (see the ESI†)
When using instead the allyl bis-isopinocampheylborane [(−)Ipc2BCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 or (−)-Ipc2Ball 46],47 itself prepared by treatment of (−)-Ipc2BCl [derived from hydroboration of (+)-α-pinene with chloroborane etherate (H2BCl·OEt2)] with allylmagnesium bromide,48 the resulting homoallylic alcohol 47 (Scheme 7) was obtained from 16 in high yield (90%) but, unfortunately, as a mixture of diastereomers (70:30 dr).
CH2 or (−)-Ipc2Ball 46],47 itself prepared by treatment of (−)-Ipc2BCl [derived from hydroboration of (+)-α-pinene with chloroborane etherate (H2BCl·OEt2)] with allylmagnesium bromide,48 the resulting homoallylic alcohol 47 (Scheme 7) was obtained from 16 in high yield (90%) but, unfortunately, as a mixture of diastereomers (70:30 dr).
Considering that the size of the protecting group might interfere with the diastereoselective allylation, alternative protecting groups were assayed. The p-methoxybenzyl ether, which could eventually be further modified by oxidation with DDQ in order to achieve the double protection of the 1,3-diol,49 was first selected. However, using a variety of reaction conditions,50 from the more classical NaH, p-methoxybenzyl chloride and n-Bu4NI to modified protocols changing the base to KH and adding DMF,51 led to complex mixtures of silyl ethers resulting from trans-silylation reactions of reactant 44. Treatment with the trichloroacetimidate derivative PMBOC(NH)CCl3 and Cu(OTf)2 instead led to the recovery of the substrate. Attempts to alternatively protect the Weinreb amide intermediate as a p-methoxybenzyl ether were also unsuccessful.
Although benzylation of the homoallylic alcohol was feasible (but in low yields), the subsequent ozonolysis led to product degradation. Lastly, protection as the mixed acetal upon treatment with MOMCl (DIPEA and CH2Cl2)52 followed by ozonolysis of 48 as described above for 16 afforded 49 in a combined 80% yield (Scheme 7). Using Leighton's reagent (R,R)-43 (Scheme 7)43 the outcome was unfortunately the same as obtained with the silyl ether. The allylation with allyltributylstannane mediated by Ti(OiPr)4 and (S)-BINOL following the protocol described by Keck et al.,53 did not take place. The same result was observed using (−)-Ipc2Ball 46 (Scheme 7).54 However, the allylation of 49 with an excess of allylmagnesium bromide 50 in THF55 did afford the homoallylic alcohol 51 in 76% yield but unfortunately as a 50:50 mixture of diastereomers (Scheme 7). The relative and absolute configurations of 51 were determined by the analysis of the 13C-NMR chemical shifts of the corresponding acetal derivatives following Rychnovsky's protocol.2 The same approach was adopted for the triethylsilyl ether 53, which was prepared from monoprotected 1,3-diol 44 following a similar sequence.56 For characterization purposes, deprotection of an equimolar mixture of allylic alcohol 54 with AcOH and H2O (4:1 v/v) in THF for 12 h (ref. 57) afforded diastereomeric diols 55 and 56, which were separated and treated with 2,2-dimethoxypropane and PPTS (Scheme 8).58 The structure of the anti diastereomer 55 could be easily confirmed based on the similar 13C-NMR chemical shift values measured for the methyl groups of the acetal.2
 | ||
| Scheme 8 Reagents and reaction conditions: (a) TBAF, THF, 0 to 25 °C, 4 h, 99%. (b) p-Anisaldehyde dimethylacetal 62, CSA, CH2Cl2, 25 °C, 4 h, 86%. (c) K2OsO2(OH)4, NaIO4, 2,6-lutidine, 1,4-dioxane, H2O, 25 °C, 3 h, 98%. (d) [(−)-Ipc2]Ball 46, Et2O, −78 °C, 4 h, 86%. (e) NaBH4, MeOH, −10 °C, 30 min, 99%. (f) 67, (S)-BINAP 68, [Ir(cod)Cl]2, 4-Cl,3-NO2-C6H3CO2H 69, Cs2CO3, THF, 100 °C, 40 h, 82%. (g) TBDMSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 5 h, 99%; (h) 65 or 70, Hoveyda–Grubbs II 59 (cat.), degassed CH2Cl2, 40 °C, 24 h (70% for 71; 57% for 72). | ||
Cross-metathesis reaction of anti-55 and propenylpinacolboronate 57 using Hoveyda–Grubbs 2nd generation catalyst 59,59a,b in CH2Cl2 under reflux conditions59c led to a 5:1 E/Z mixture of trisubstituted alkenylpinacolboronate isomer 60 in 60% yield (Scheme 7). Given the lack of stereocontrol in the last two C–C bond-formation steps, the sequence was reconsidered using alternative protecting groups.
Straightforward deprotection of the silyl ether of 44 with TBAF (99% yield) was followed by protection of the syn-1,3-diol 61 as a cyclic acetal.60 Since the anisylidene protecting group is ca. 10 times more labile to acid than isopropylidene or benzylidene counterparts, dioxolane 63 was chosen to protect the syn-1,3-diol 61, and the process was carried out using p-anisaldehyde dimethylacetal 62 and camphorsulfonic acid.61 Oxidative cleavage through a modified procedure,62 involving treatment with catalytic OsO4, 2,6-lutidine and NaIO4, generated aldehyde 64 in 98% yield (Scheme 9). Using Antilla's method for allylboration of aldehydes using (R)-TRIP-PA and the allyltetramethyldioxaborolane reagents,63 a ca. 1:1 mixture of diastereomers was obtained. The result was comparable to that obtained using allyltributylstannane and MgBr2·Et2O. Alternative methods for the allylation of aldehyde 64 which proved to be inefficient or capricious, were Leighton's protocol,43b Keck's Ti(OiPr)4-(S)-BINOL system,53 and Brown's (−)-Ipc2Ball generated in situ from (−)-Ipc2BCl and allyl magnesium bromide.62 However, when recently purchased [(−)-(Ipc2)]Ball reagent 46 was used, the desired homoallylic alcohol 65 was obtained in 86% yield accompanied by very minor amounts of the diastereomer (1:0.06 dr). Despite the success, the instability of the reagent and its use in stoichiometric amounts made necessary to search for a more robust and efficient procedure.
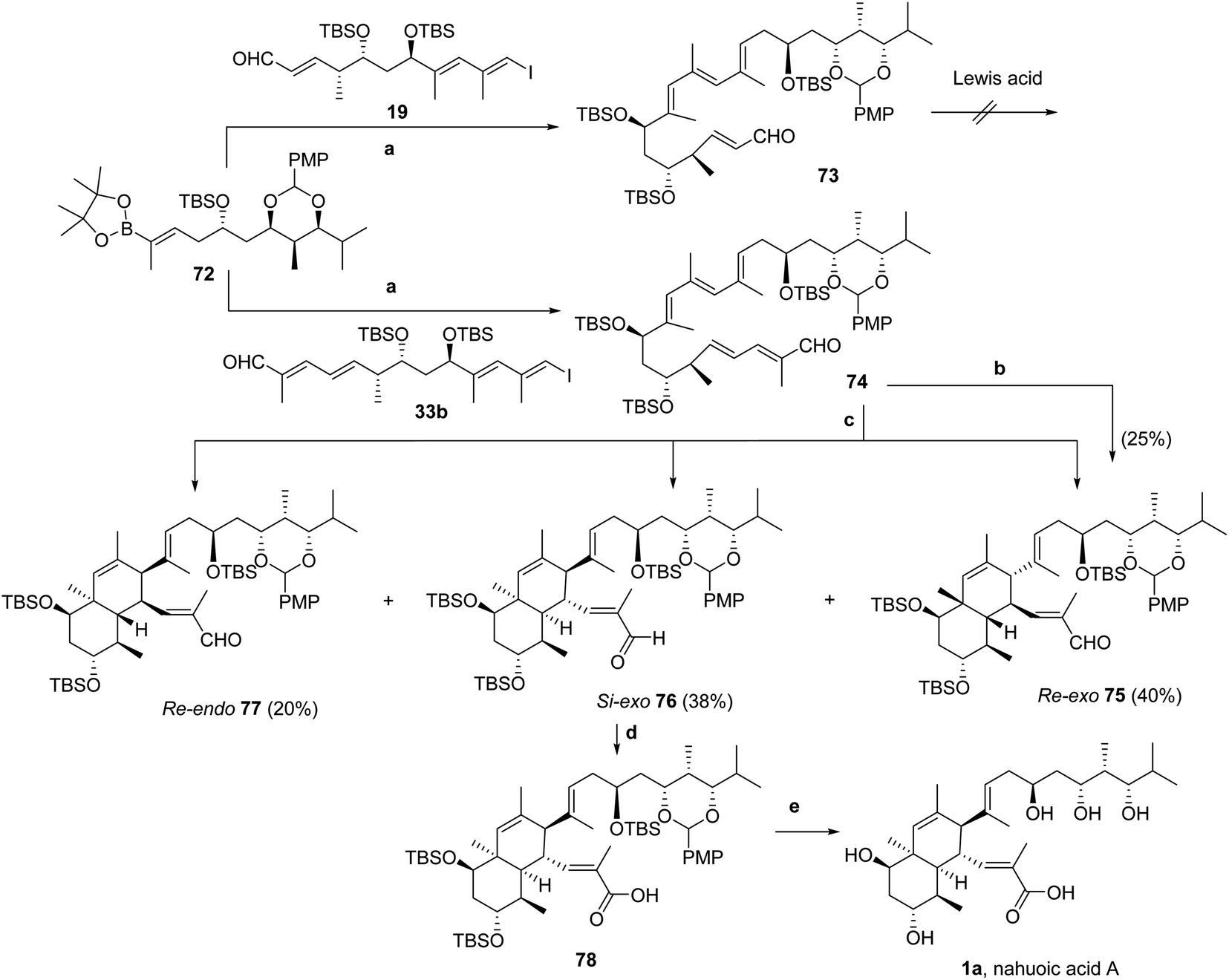 | ||
| Scheme 9 Reagents and reaction conditions: (a) Pd(PPh3)4, 10% aq. TlOH, THF, 25 °C, 3 h (93% for 73; 86% for 74). (b) BHT, o-xylene, 200 °C, 18 h (25% for Re-exo75). (c) Eu(fod)3, BHT, toluene, 160 °C, 16 h or CD3OD, BHT, 170 °C, 14 h (40% for Re-exo75; 38% for Si-exo76; 20% for Re-endo77). (d) NaClO2, NaH2PO4, 2-methyl-2-butene, THF, tBuOH/H2O, 25 °C, 12 h, 84%. (e) 4 M HCl, THF, 25 °C, 64 h, 22%. | ||
To this end, we focused on Krische's enantioselective carbonyl allylation via transfer hydrogenation coupling with allylic acetates catalyzed by chiral non-racemic iridium complexes generated from [Ir(cod)Cl]2 and an enantiopure biphosphine ligand.64 Following this procedure, alcohol 66, which was efficiently generated upon reduction of aldehyde 64 with NaBH4 in MeOH at −10 °C for 30 min,65 was treated with allyl acetate 67, (S)-BINAP 68, [Ir(cod)Cl]2, 4-chloro-3-nitrobenzoic acid 69 and Cs2CO3 at 100 °C for 40 h (ref. 66) to provide homoallylic alcohol 65 as a single diastereomer in 82% yield (Scheme 8).
Cross-metathesis as described above using Hoveyda–Grubbs 2nd generation catalyst 59 (5 mol%) and excess (5 equiv.) of isopropenyldioxaborolane 58 in refluxing benzene for 18 h67 led to the desired unsaturated alkenylpinacolboronate 71 although in a disappointing 23% yield, together with the dimer and other unidentified by-products. However, adopting the variant68 based on the addition of 10 equivalents of boronate 58 to a solution of 65 in dichloromethane and portionwise (3×) addition of the catalyst (5% + 2.5% + 2.5%) with removal of the also generated ethylene from the frozen flask under high vacuum conditions, the yield of 71 could be increased to 70% (Scheme 8).
The generated δ-hydroxyboronic acid 71 was used in the Suzuki–Miyaura cross-coupling reaction23 with dienyl iodide 15 (Scheme 2), promoted by Pd(PPh3)4 as the catalyst and 10% aqueous TlOH in THF at room temperature, but the yields of the resulting triene were lower than 30%. In turn, protection of the free alcohol of 65 as a silyl ether in quantitative yield using TBDMSOTf and 2,6-lutidine in CH2Cl2 allowed us to carry out the same cross-metathesis connection of 58 with 70 catalyzed by 59 to afford internal alkenyl pinacolboronate 72 in 57% yield (Scheme 8).
Suzuki–Miyaura cross-coupling23 of components 72 and 19 (Scheme 2) was in this case highly effective, and non-conjugated tetraenal 73 was obtained in 93% yield (Scheme 9). Non-conjugated pentaenal 74 was likewise prepared by Suzuki–Miyaura cross-coupling23 of 72 and 33b in 86% yield (Scheme 9).
The IMDA reaction22 was first attempted with non-conjugated tetraenal 73 under Lewis acid-catalyzed conditions. However, all trials led to the recovery of 73 when using Me2AlCl in toluene at low temperatures (−78 to −40 °C)30 or B(C6F5)3 in CH2Cl2 (with powdered 4 Å MS) at −10 °C.31 Product degradation and formation of unidentified mixtures of by-products were noticed upon increasing the reaction temperature following these protocols to either −10 °C or 23 °C, respectively.
Using instead the non-conjugated pentaenal 74, IMDA cycloaddition (Scheme 9) was promoted by heating the solution in o-xylene at 200 °C for 18 h (longer reaction periods were detrimental to the stability of the products and extensive decomposition was also noted). Purification by HPLC (Synergi MAX-RP column, C18-silica gel, 4 μm, 250 × 4.6 mm; MeOH, flow rate = 2 mL min−1) allowed us to characterize the major product (tR = 19 min), obtained in 25% yield, as the cycloadduct Re-exo75, by analysis of 1H- and 13C-NMR spectra in CD2Cl2 solution, DEPT data, NOE effects, and two-dimensional NMR experiments (HSQC, HMBC, COSY, DQF-COSY, and TOCSY1D) as well as MS data. Additional reaction products (10–15% yield) could not be identified, but the NMR data resembled those of Re-exo75.
Moreover, computational studies (see the ESI†) predicted a reduction in activation energies for the IMDA reaction of the s-trans conformer (up to ΔΔG‡ ≈ 4 kcal mol−1), when using MeOH as solvent relative to toluene, and a greater selectivity for formation of octahydronaphthalenes through the Si-exo relative to the Re-endo (ΔΔG‡ ≈ 1.8 kcal mol−1) approach. Monitoring the IMDA reaction process by 1H-NMR spectroscopy using solutions of 74 in CD3OD with BHT as a radical scavenger allowed us to confirm the prediction, since the reaction was completed after heating at 170 °C for 14 h. Separation of the products by HPLC as described above allowed us to identify, in order of elution, the Re-exo75 (40% yield), Si-exo76 (38% yield) and Re-endo77 (20% yield) diastereomers in excellent overall yield. A similar result was obtained upon alternatively heating solutions of 74 in toluene-d8 with BHT using a weak Lewis acid [Eu(fod)3, Resolve-Al®] as the catalyst at 160 °C for 16 h (Scheme 9).
Therefore, the use of the entire pentaenal 74 under the IMDA thermal cyclization reaction conditions allowed us to improve the exo/endo ratio to 4:1 (cf. 3.76:1 in the case of 25, Scheme 5) when compared to the results obtained (2.5:1) with the bulky Lewis acid B(C6F5)3 for model system 34b (Scheme 5). In addition, the results agree with the computational predictions on the tetraenal model system under thermal conditions.
The Si-exo diastereomer 76 was oxidized under the Pinnick–Lindgren reaction conditions69 modified by adding THF in addition to tBuOH/H2O as the solvent mixture,70 which afforded carboxylic acid 78 in 80% yield after 12 h stirring (Scheme 9). Being acid-sensitive protecting groups, further treatment of solutions of 78 in THF with 4 N HCl at ambient temperature for 72 h led to overall deprotection.71 Work-up using NaHCO3 followed by the addition of 1 M TFA until pH ≈ 1 and freeze-drying provided a solid that was dissolved in ethyl acetate, filtered with Celite®, and evaporated to dryness. Finally, HPLC purification of the residue (Synergi MAX-RP column, C12-silica gel, 4 μm, 250 × 4.6 mm; linear gradient elution from 30% MeCN in water to 100% MeCN over 30 min, with a flow rate of 2.5 mL min−1) provided nahuoic acid A (1a) in an overall 22% yield (Scheme 9). The 1H-NMR data matched those previously reported for the natural product.1a
C Conclusions
To summarize, the bioinspired synthesis of nahuoic acid A (1a) was achieved using the IMDA reaction of non-conjugated pentaenal 74 to construct the octahydronaphthalene core structure of the natural product. The synthesis of pentaenal 74 made use of Suzuki cross-coupling of a non-conjugated iodotetraenal 33b and trisubstituted alkenylboronate 72, themselves prepared through enantio- and diastereoselective reactions including the Evans aldol condensation reaction, and a sequence of the Leighton and Krische allylation reactions, as well as the cross-metathesis for the generation of the alkenylboronate. IMDA reaction upon heating solutions of 74 in CD3OD with BHT as the radical scavenger at 170 °C for 14 h afforded a mixture of three diastereomers (Re-exo, Si-exo and Re-endo) in a 2:2:1 ratio and high yield, favoring the exo mode of cycloaddition with one stereoisomer (Si-exo) showing the relative and absolute configurations present in nahuoic acid A (1a). The preference for the exo-mode of cycloaddition in an uncatalyzed thermal process has been computationally predicted. By contrast, catalysis by Me2AlCl at −40 °C of model tetraenal 25 was highly selective for the trans-fused angularly-methylated octahydronaphthalene model fragment 37 corresponding to the Re-endo mode of cycloaddition.
Given the reactivity of the synthetic non-conjugated pentaenal 74, it is tempting to suggest that the putative DNAse enzyme of Streptomyces sp. (isolate RJA2928) might be providing an active site environment that allows us to select the conformation of the unprotected and natural component leading to the cis decalin stereoisomer of nahuoic acid A (1a).24e,f,72 In line with this assumption, recent studies by Houk and co-workers on a model system of an electrocyclic reaction catalyzed by the MycB protein revealed that although the activation free energy for the spontaneous reaction (catalyzed by p-cresol) was lower for the cis-decalin exo adduct, the model MycB-catalyzed reaction, likewise a synchronous and concerted process, led to the trans-decalin through the endo transition state approach.24f,73
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: A. R. de L.; funding acquisition: A. R. de L. and R. A.; methodology: A. R. de L. and R. A.; investigation: L. G., P. M., and P. V.; methodology: A. R. de L. and R. A.; project administration: A. R. de L. and R. A.; supervision: A. R. de L. and R. A.; computations: P. V. and R. A.; visualization: P. V. and R. A.; writing-original draft: A. R. de L.; writing, review and editing: L. G.; P. M., P. V.; R. A., and A. R. de L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funds from the Spanish MINECO (SAF2016-77620-R-FEDER; PID2019-107855RB-I00-FEDER), and Xunta de Galicia (Consolidación GRC ED431C 2017/61 from DXPCTSUG; ED-431G/02 INBIOMED-FEDER “Unha maneira de facer Europa”). We are indebted to Dr Susana Alvarez and Dr Adán B. González-Pérez for preliminary results in this project. We thank Centro de Apoio Científico-Tecnolóxico á Investigación (CACTI) for invaluable help in structural elucidation.Notes and references
- (a) D. E. Williams, D. S. Dalisay, F. Li, J. Amphlett, W. Maneerat, M. A. G. Chavez, Y. A. Wang, T. Matainaho, W. Yu, P. J. Brown, C. H. Arrowsmith, M. Vedadi and R. J. Andersen, Org. Lett., 2013, 15, 414–417 CrossRef CAS PubMed; (b) D. E. Williams and R. J. Andersen, Nat. Prod. Rep., 2020, 37, 617–633 RSC.
- S. D. Rychnovsky, B. Rogers and G. Yang, J. Org. Chem., 1993, 58, 3511–3515 CrossRef CAS.
- (a) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS; (b) T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- (a) X.-H. Nong, X.-Y. Zhang, X.-Y. Xu, J. Wang and S.-H. Qi, J. Nat. Prod., 2016, 79, 141–148 CrossRef CAS PubMed. None of these natural products (Fig. 1) showed inhibitory activity against acetylcholinesterase, cytotoxicity towards the H1975, K562, BGC 823, MCF-7, HL-60, and Huh-7 cancer cell lines, antibacterial activity against Staphylococcus aureus and Shewanella oneidensis MR-1 or antibiofilm activity against S. aurueus. (b) D. E. Williams, F. Izard, S. Arnould, D. S. Dalisay, C. Tantapakul, W. Maneerat, T. Matainaho, E. Julien and R. J. Andersen, J. Org. Chem., 2016, 81, 1324–1332 CrossRef CAS PubMed.
- Q. Liu, Y. Deng and A. B. Smith III, J. Am. Chem. Soc., 2017, 139, 13668–13671 CrossRef CAS PubMed.
- A. Ma, W. Yu, Y. Xiong, K. V. Butler, P. J. Brown and J. Jin, MedChemComm, 2014, 5, 1892–1898 RSC.
- (a) H. ú. Kaniskan, K. D. Konze and J. Jin, J. Med. Chem., 2015, 58, 1596–1629 CrossRef CAS PubMed; (b) M. Schapira, Cell Chem. Biol., 2016, 23, 1067–1076 CrossRef CAS PubMed; (c) Z. A. Wang and W. R. Liu, Chem.–Eur. J., 2017, 23, 11732–11737 CrossRef CAS PubMed; (d) V. Veschi, Z. Liu, T. C. Voss, L. Ozbun, B. Gryder, C. Yan, Y. Hu, A. Ma, J. Jin, S. J. Mazur, N. Lam, B. K. Souza, G. Giannini, G. L. Hager, C. H. Arrowsmith, J. Khan, E. Appella and C. J. Thiele, Cancer Cell, 2017, 31, 50–63 CrossRef CAS PubMed; (e) M. Luo, Chem. Rev., 2018, 118, 6656–6705 CrossRef CAS PubMed; (f) S. Chen, K. Kapilashrami, C. Senevirathne, Z. Wang, J. Wang, J. A. Linscott and M. Luo, J. Am. Chem. Soc., 2019, 141, 8064–8067 CrossRef CAS PubMed.
- (a) S. M. Kooistra and K. Helin, Nat. Rev. Mol. Cell Biol., 2012, 13, 297–311 CrossRef CAS PubMed; (b) J. W. Hojfeldt, K. Agger and K. Helin, Nat. Rev. Drug Discovery, 2013, 12, 917–930 CrossRef CAS PubMed.
- (a) J. C. Black, C. Van Rechem and J. R. Whetstine, Mol. Cell, 2012, 48, 491–507 CrossRef CAS PubMed; (b) L. Morera, M. Lübbert and M. Jung, Clin. Epigenet., 2016, 8, 57 CrossRef PubMed.
- J. A. Linscott, K. Kapilashrami, Z. Wang, C. Senevirathne, I. R. Bothwell, G. Blum and M. Luo, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E8369–E8378 CrossRef CAS PubMed.
- (a) X. Cheng, R. E. Collins and X. Zhang, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 267–294 CrossRef CAS PubMed; (b) M. B. Poulin, J. L. Schneck, R. E. Matico, P. J. McDevitt, M. J. Huddleston, W. Hou, N. W. Johnson, S. H. Thrall, T. D. Meek and V. L. Schramm, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1197–1201 CrossRef CAS PubMed.
- (a) M. Takawa, H.-S. Cho, S. Hayami, G. Toyokawa, M. Kogure, Y. Yamane, Y. Iwai, K. Maejima, K. Ueda, A. Masuda, N. Dohmae, H. I. Field, T. Tsunoda, T. Kobayashi, T. Akasu, M. Sugiyama, S.-i. Ohnuma, Y. Atomi, B. A. J. Ponder, Y. Nakamura and R. Hamamoto, Cancer Res., 2012, 72, 3217–3227 CrossRef CAS PubMed; (b) C. Milite, A. Feoli, M. Viviano, D. Rescigno, A. Mai, S. Castellano and G. Sbardella, ChemMedChem, 2016, 11, 1680–1685 CrossRef CAS PubMed.
- J. Fang, Q. Feng, C. S. Ketel, H. Wang, R. Cao, L. Xia, H. Erdjument-Bromage, P. Tempst, J. A. Simon and Y. Zhang, Curr. Biol., 2002, 12, 1086–1099 CrossRef CAS PubMed.
- X. Shi, I. Kachirskaia, H. Yamaguchi, L. E. West, H. Wen, E. W. Wang, S. Dutta, E. Appella and O. Gozani, Mol. Cell, 2007, 27, 636–646 CrossRef CAS PubMed.
- F. Yang, L. Sun, Q. Li, X. Han, L. Lei, H. Zhang and Y. Shang, EMBO J., 2012, 31, 110–123 CrossRef CAS PubMed.
- S. Jørgensen, I. Elvers, M. B. Trelle, T. Menzel, M. Eskildsen, O. N. Jensen, T. Helleday, K. Helin and C. S. Sørensen, J. Cell Biol., 2007, 179, 1337–1345 CrossRef PubMed.
- K. W. Kuntz, J. E. Campbell, H. Keilhack, R. M. Pollock, S. K. Knutson, M. Porter-Scott, V. M. Richon, C. J. Sneeringer, T. J. Wigle, C. J. Allain, C. R. Majer, M. P. Moyer, R. A. Copeland and R. Chesworth, J. Med. Chem., 2016, 59, 1556–1564 CrossRef CAS PubMed.
- (a) C. Pérez-Balado, P. Rodríguez-Graña, A. Nebiosso, A. Minichiello, M. Miceli, L. Altucci and A. R. de Lera, J. Med. Chem., 2007, 50, 2497–2505 CrossRef PubMed; (b) J. A. Souto, E. Vaz, I. Lepore, A.-C. Pöppler, G. Franci, R. Álvarez, L. Altucci and Á. R. de Lera, J. Med. Chem., 2010, 53, 4654–4667 CrossRef CAS PubMed; (c) J. García, G. Franci, R. Pereira, R. Benedetti, A. Nebbioso, F. Rodríguez-Barrios, H. Gronemeyer, L. Altucci and A. R. de Lera, Bioorg. Med. Chem., 2011, 19, 3637–3649 CrossRef PubMed; (d) P. García-Domínguez, I. Lepore, C. Erb, H. Gronemeyer, L. Altucci, R. Álvarez and A. R. de Lera, Org. Biomol. Chem., 2011, 6979–6987 RSC.
- L. Guillade, A. B. Gonzalez-Perez and A. R. de Lera, Org. Biomol. Chem., 2017, 15, 7430–7438 RSC.
- Y. Deng and A. B. Smith, Acc. Chem. Res., 2020, 53, 988–1000 CrossRef CAS PubMed.
- (a) H. A. Reichard and G. C. Micalizio, Angew. Chem., Int. Ed., 2007, 46, 1440–1443 CrossRef CAS PubMed; (b) G. C. Micalizio and S. B. Hale, Acc. Chem. Res., 2015, 48, 663–673 CrossRef CAS PubMed; (c) N. F. O'Rourke, M. J. Kier and G. C. Micalizio, Tetrahedron, 2016, 72, 7093–7123 CrossRef PubMed.
- (a) W. R. Roush, in Comprehensive Organic Synthesis, ed. I. Fleming, B. M. Trost and L. A. Paquette, Pergamon Press, Oxford, 1991, ch. 4.4, vol. 5, pp. 513–550 Search PubMed; (b) K.-i. Takao, R. Munakata and K.-i. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed; (c) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983–2992 RSC; (d) M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- (a) E. M. Stocking and R. M. Williams, Angew. Chem., Int. Ed., 2003, 42, 3078–3115 CrossRef CAS PubMed; (b) K. Klas, S. Tsukamoto, D. H. Sherman and R. M. Williams, J. Org. Chem., 2015, 80, 11672–11685 CrossRef CAS PubMed; (c) A. Minami and H. Oikawa, J. Antibiot., 2016, 69, 500–506 CrossRef CAS PubMed; (d) T. Hashimoto and T. Kuzuyama, Curr. Opin. Chem. Biol., 2016, 35, 117–123 CrossRef CAS PubMed; (e) M. J. Byrne, N. R. Lees, L.-C. Han, M. W. van der Kamp, A. J. Mulholland, J. E. M. Stach, C. L. Willis and P. R. Race, J. Am. Chem. Soc., 2016, 138, 6095–6098 CrossRef CAS PubMed; (f) L. Li, P. Yu, M.-C. Tang, Y. Zou, S.-S. Gao, Y.-S. Hung, M. Zhao, K. Watanabe, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 15837–15840 CrossRef CAS PubMed.
- (a) D. Tan, C. S. Jamieson, M. Ohashi, M.-C. Tang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 769–773 CrossRef CAS PubMed; (b) C. S. Jamieson, M. Ohashi, F. Liu, Y. Tang and K. N. Houk, Nat. Prod. Rep., 2019, 36, 698–713 RSC; (c) B.-s. Jeon, S.-A. Wang, M. W. Ruszczycky and H.-w. Liu, Chem. Rev., 2017, 117, 5367–5388 CrossRef CAS PubMed; (d) B. R. Lichman, S. E. O'Connor and H. Kries, Chem.–Eur. J., 2019, 25, 6864–6877 CrossRef CAS PubMed; (e) Z. Zhang, C. S. Jamieson, Y.-L. Zhao, D. Li, M. Ohashi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 5659–5663 CrossRef CAS PubMed; (f) C. T. Walsh and Y. Tang, Natural Product Biosynthesis. Chemical Logic and Enzymatic Machinery, Croydon, UK, 2017 Search PubMed; (g) C. T. Walsh and Y. Tang, Biochemistry, 2018, 57, 3087–3104 CrossRef CAS PubMed; (h) C. T. Walsh and B. S. Moore, Angew. Chem., Int. Ed., 2019, 58, 6846–6879 CrossRef CAS PubMed; (i) B. Zhang, K. B. Wang, W. Wang, X. Wang, F. Liu, J. Zhu, J. Shi, L. Y. Li, H. Han, K. Xu, H. Y. Qiao, X. Zhang, R. H. Jiao, K. N. Houk, Y. Liang, R. X. Tan and H. M. Ge, Nature, 2019, 568, 122–126 CrossRef CAS PubMed.
- J. Uenishi, J.-M. Beau, R. W. Armstrong and Y. Kishi, J. Am. Chem. Soc., 1987, 109, 4756–4758 CrossRef CAS.
- A. L. Moure, R. Gómez Arrayás, D. J. Cárdenas, I. Alonso and J. C. Carretero, J. Am. Chem. Soc., 2012, 134, 7219–7222 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, M. W. Härter, J. L. Gunzner and A. Nadin, Liebigs Ann., 1997, 1997, 1283–1301 CrossRef; (b) K. Kobayashi, K. Tanaka and H. Kogen, Tetrahedron Lett., 2018, 59, 568–582 CrossRef CAS.
- M. Ramanathan, C.-J. Tan, W.-J. Chang, H.-H. G. Tsai and D.-R. Hou, Org. Biomol. Chem., 2013, 11, 3846–3854 RSC.
- J. P. Burke, M. Sabat, D. A. Iovan, W. H. Myers and J. J. Chruma, Org. Lett., 2010, 12, 3192–3195 CrossRef CAS PubMed.
- J.-H. Zhou, B. Jiang, F.-F. Meng, Y.-H. Xu and T.-P. Loh, Org. Lett., 2015, 17, 4432–4435 CrossRef CAS PubMed.
- Ab initio computations appear to be consistent with the lower energy cost associated with deformation of reagents to reach the transition state, where they get stabilized by a combination of stronger electrostatic interactions, dispersion forces and orbital interactions. In fact, non-covalent CH–Fortho interactions in the activated model substrate were found to contribute to the stabilization of the exo-like transition state; see: D. Yepes, P. Pérez, P. Jaque and I. Fernández, Org. Chem. Front., 2017, 4, 1390–1399 RSC.
- R. W. Hoffmann, Angew. Chem., Int. Ed., 2001, 40, 1411–1416 CrossRef CAS PubMed.
- P. Farfán, S. Gómez and A. Restrepo, J. Org. Chem., 2019, 84, 14644–14658 CrossRef PubMed.
- (a) D. J. Ager, J. Chem. Soc., Perkin Trans. 1, 1986, 183–194 RSC; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (c) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, in Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) M. Nerz-Stormes and E. R. Thornton, J. Org. Chem., 1991, 56, 2489–2498 CrossRef CAS; (b) Y. Zhang and T. Sammakia, Org. Lett., 2004, 6, 3139–3141 CrossRef CAS PubMed; (c) Y. Zhang, A. J. Phillips and T. Sammakia, Org. Lett., 2004, 6, 23–25 CrossRef CAS PubMed.
- Y. Nagao, Y. Hagiwara, T. Kumagai, M. Ochiai, T. Inoue, K. Hashimoto and E. Fujita, J. Org. Chem., 1986, 51, 2391–2393 CrossRef CAS.
- Á. González, J. Aiguadé, F. Urpí and J. Vilarrasa, Tetrahedron Lett., 1996, 37, 8949–8952 CrossRef.
- (a) D. A. Evans, F. Urpi, T. C. Somers, J. S. Clark and M. T. Bilodeau, J. Am. Chem. Soc., 1990, 112, 8215–8216 CrossRef CAS; (b) M. T. Crimmins, B. W. King and E. A. Tabet, J. Am. Chem. Soc., 1997, 119, 7883–7884 CrossRef CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- (a) J. W. A. Kinnaird, P. Y. Ng, K. Kubota, X. Wang and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 7920–7921 CrossRef CAS PubMed; (b) H. Kim, S. Ho and J. L. Leighton, J. Am. Chem. Soc., 2011, 133, 6517–6520 CrossRef CAS PubMed.
- X. Zhang, K. N. Houk and J. L. Leighton, Angew. Chem., Int. Ed., 2005, 44, 938–941 CrossRef CAS PubMed.
- R. Chen, Y. Shen, S. Yang and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14198–14210 CrossRef CAS PubMed.
- G. Stork and S. D. Rychnovsky, J. Am. Chem. Soc., 1987, 109, 1565–1567 CrossRef CAS.
- H. C. Brown and P. K. Jadhav, J. Am. Chem. Soc., 1983, 105, 2092–2093 CrossRef CAS.
- S. Das, D. Paul and R. K. Goswami, Org. Lett., 2016, 18, 1908–1911 CrossRef CAS PubMed.
- B. M. Trost, B. M. O'Boyle and D. Hund, J. Am. Chem. Soc., 2009, 131, 15061–15074 CrossRef CAS PubMed.
- L. Brewitz, J. Llaveria, A. Yada and A. Fürstner, Chem.–Eur. J., 2013, 19, 4532–4537 CrossRef CAS PubMed.
- M. W. Andersen, B. Hildebrandt, G. Dahmann and R. W. Hoffmann, Chem. Ber., 1991, 124, 2127–2139 CrossRef CAS.
- S. BouzBouz and J. Cossy, Tetrahedron Lett., 2006, 47, 901–904 CrossRef CAS.
- G. E. Keck, K. H. Tarbet and L. S. Geraci, J. Am. Chem. Soc., 1993, 115, 8467–8468 CrossRef CAS.
- P. Álvarez-Bercedo, J. Murga, M. Carda and J. A. Marco, J. Org. Chem., 2006, 71, 5766–5769 CrossRef PubMed.
- M. C. Warner, G. A. Shevchenko, S. Jouda, K. Bogár and J.-E. Bäckvall, Chem.–Eur. J., 2013, 19, 13859–13864 CrossRef CAS PubMed.
- P. Wang, Y.-J. Kim, M. Navarro-Villalobos, B. D. Rohde and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 3256–3257 CrossRef CAS PubMed.
- K. Kobayashi, Y. Fujii, I. Hayakawa and H. Kigoshi, Org. Lett., 2011, 13, 900–903 CrossRef CAS PubMed.
- R. Perla, A. Ramisetti and R. Atla, Tetrahedron Lett., 2016, 57, 2100–2102 CrossRef CAS.
- (a) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS; (b) A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed; (c) C. Morrill, T. W. Funk and R. H. Grubbs, Tetrahedron Lett., 2004, 45, 7733–7736 CrossRef CAS.
- S. S. Dachavaram, K. B. Kalyankar and S. Das, Tetrahedron Lett., 2014, 55, 5629–5631 CrossRef CAS.
- P. Radha Krishna and M. Narsingam, Synthesis, 2007, 3627–3634 CrossRef.
- R. G. Reddy, R. Venkateshwarlu, K. V. S. Ramakrishna, J. S. Yadav and D. K. Mohapatra, J. Org. Chem., 2017, 82, 1053–1063 CrossRef CAS PubMed.
- P. Jain and J. C. Antilla, J. Am. Chem. Soc., 2010, 132, 11884–11886 CrossRef CAS PubMed.
- I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891–14899 CrossRef CAS PubMed.
- H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567–607 CrossRef CAS.
- J. Cui, M. Morita, O. Ohno, T. Kimura, T. Teruya, T. Watanabe, K. Suenaga and M. Shibasaki, Chem.–Eur. J., 2017, 23, 8500–8509 CrossRef CAS PubMed.
- M. Kretschmer, M. Dieckmann, P. Li, S. Rudolph, D. Herkommer, J. Troendlin and D. Menche, Chem.–Eur. J., 2013, 19, 15993–16018 CrossRef CAS PubMed.
- R. J. Armstrong, C. García-Ruiz, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 786–790 CrossRef CAS PubMed.
- B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888–890 CrossRef CAS.
- A. B. Smith III, K. Basu and T. Bosanac, J. Am. Chem. Soc., 2007, 129, 14872–14874 CrossRef PubMed.
- (a) A. Arefolov and J. S. Panek, J. Am. Chem. Soc., 2005, 127, 5596–5603 CrossRef CAS PubMed; (b) Y. Zhang, C. C. Arpin, A. J. Cullen, M. J. Mitton-Fry and T. Sammakia, J. Org. Chem., 2011, 76, 7641–7653 CrossRef CAS PubMed.
- K. Klas, S. Tsukamoto, D. H. Sherman and R. M. Williams, J. Org. Chem., 2015, 80, 11672–11685 CrossRef CAS PubMed.
- K. Black, P. Liu, L. Xu, C. Doubleday and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12860–12865 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04524e |
| This journal is © The Royal Society of Chemistry 2021 |