
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure-based de novo drug design using 3D deep generative models†
Yibo
Li
 a,
Jianfeng
Pei
*b and
Luhua
Lai
*abc
a,
Jianfeng
Pei
*b and
Luhua
Lai
*abc
aCenter for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China. E-mail: ybli@pku.edu.cn
bCenter for Quantitative Biology, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China. E-mail: jfpei@pku.edu.cn
cBNLMS, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: lhlai@pku.edu.cn
First published on 9th September 2021
Abstract
Deep generative models are attracting much attention in the field of de novo molecule design. Compared to traditional methods, deep generative models can be trained in a fully data-driven way with little requirement for expert knowledge. Although many models have been developed to generate 1D and 2D molecular structures, 3D molecule generation is less explored, and the direct design of drug-like molecules inside target binding sites remains challenging. In this work, we introduce DeepLigBuilder, a novel deep learning-based method for de novo drug design that generates 3D molecular structures in the binding sites of target proteins. We first developed Ligand Neural Network (L-Net), a novel graph generative model for the end-to-end design of chemically and conformationally valid 3D molecules with high drug-likeness. Then, we combined L-Net with Monte Carlo tree search to perform structure-based de novo drug design tasks. In the case study of inhibitor design for the main protease of SARS-CoV-2, DeepLigBuilder suggested a list of drug-like compounds with novel chemical structures, high predicted affinity, and similar binding features to those of known inhibitors. The current version of L-Net was trained on drug-like compounds from ChEMBL, which could be easily extended to other molecular datasets with desired properties based on users' demands and applied in functional molecule generation. Merging deep generative models with atomic-level interaction evaluation, DeepLigBuilder provides a state-of-the-art model for structure-based de novo drug design and lead optimization.
1 Introduction
Searching for molecules with good bioactivity and druggability is the central task of de novo drug discovery, which is complicated by the enormous size and complexity of the chemical space.1–4 Virtual screening (VS) approaches are widely used to search large-scale compound libraries for potential hits based on either molecular docking with a known target structure or similarity to known active compounds. Although VS-based drug discovery projects are able to identify active compounds,5 they are limited by the chemical libraries used. In contrast, de novo drug design approaches can build molecular structures from scratch based on given objectives and have the advantage of exploring a much wider chemical space beyond existing compound libraries. Since the early 1990s,2 various structure-based de novo design programs have been developed, including LEGEND,6 LUDI,7 CONCEPTS,8 LigBuilder,9–11 BOMB,12etc. Some of them have been used successfully in practical drug discovery projects.3,4,13–16 In theory, these de novo drug design approaches could search the large chemical space, but in practice, the space explored is usually limited by computational constraints. In addition, the synthesis of designed compounds is usually a demanding task.More recently, various deep generative models have been introduced to the field of de novo design. Compared to traditional methods, which usually require expert crafted rules to guarantee that the output molecules are practical, deep generative models are mostly “rule-free” and can be trained in a fully data-driven manner with minimal requirement for expert knowledge. A wide range of deep learning architectures, including SMILES-based17 and graph-based18 language models, VAE,19 and GAN,20 have been used. Various methods have been developed to address different needs during the drug discovery process, such as property-based,19,21 target-based,17,21,22 pharmacophore-based,23 and scaffold-based molecular design approaches.18,24,25
However, unlike the majority of traditional de novo design methods, which construct 3D ligands directly inside the 3D structure of binding pockets, most deep learning-based methods only generate 1D (SMILES) or 2D (graph) representations of molecules without using the structural and interaction information of targets. As a result, most of those methods rely on ligand-based objectives,21,22,26 which may be subject to biases related to the ligands in the training set. This also makes the models difficult to be applied to cases with limited bioactivity data. Structure-based information provides direct guidance to optimize the interactions between the ligand and the target and reduces the dependency on existing bioactivity information. It is therefore highly desirable to include 3D conditions in deep molecule generative models.
Recently, Nigam et al. developed algorithms to explore the topological space of molecules with a docking-based scoring function for structure-based de novo drug design. They developed STONED27 which performs molecule optimization by manipulating SELFIES, a sequential representation of molecular structures similar to SMILES but is guaranteed to be 100% valid. An advantage of STONED is that it does not require deep learning or expert knowledge to explore the chemical space. This method can be used in structure-based drug design by combining it with the docking score. They further developed JANUS28 which combines the genetic algorithm with a docking program to optimize the topological structures of molecules. However, both methods still operate on topological structures of molecules and require an external docking program to optimize the 3D poses and obtain docking scores.
In this work, we developed DeepLigBuilder, a novel program for de novo molecular design that enjoys the benefits of both traditional and deep learning-based methods. Specifically, DeepLigBuilder uses a deep generative model to construct and optimize the 3D structures of ligands directly inside 3D binding pockets. We divided this goal into the following two tasks. First, we trained a deep generative model that can produce drug-like molecules with valid 3D structures. Second, we introduced target-based information into the model so that molecules with good predicted binding affinity could be obtained.
For the first task, although there are existing methods that use deep learning to generate 3D molecular structures,29–31 most of them focus on structurally simple molecules like those in the QM9 dataset. Those molecules are usually not sufficient for applications in drug discovery. We introduced Ligand Neural Network (L-Net), a graph generative model that is specifically designed to generate 3D drug-like molecules. L-Net contains two major features that are important for its performance. First, it is built from a new graph convolution architecture, which combines features like graph pooling and rotational covariance, increasing the size of the receptive field for the network while making it more efficient for training and sampling. Second, L-Net is trained using a novel scheme that makes it resistant to 3D errors during generation. As a result, the model can generate chemically correct, conformationally valid, and highly drug-like molecules. Compared to G-SchNet, a previous state-of-the-art model in 3D generation tasks, L-Net achieves significantly better chemical validity while maintaining an improved quality for the generated conformers.
To accomplish the second task, L-Net was combined with Monte Carlo tree search (MCTS), a widely applied technique in reinforcement learning, to optimize molecules directly inside the binding pocket. To our knowledge, this work presents the first attempt to combine deep generative models with MCTS for 3D ligand generation. There are other deep learning-based methods for the conditioned generation of 3D molecules based on binding pocket structures, most notably liGAN.32,33 However, liGAN generates molecules indirectly using atomic density maps, and subsequent rule-based inference of atom location and bond types is necessary. It also requires protein–ligand complexes as training data. On the other hand, DeepLigBuilder only needs a structure-based scoring function and can generate full 3D structures of ligands, including hydrogens, without additional processing, making it much easier to use and extend.
DeepLigBuilder is unique in that it directly operates on 3D molecular structures and optimizes both the topological and 3D structures of molecules at the same time directly inside the binding pocket using MCTS. Due to its ability to directly operate 3D structures, DeepLigBuilder is more flexible and can easily achieve more advanced capabilities, such as anchoring the generated molecules in a spatial location or performing generation based on a privileged 3D fragment.
As a case study, we used DeepLigBuilder to design molecules targeting the main protease of SARS-CoV-2, a key target for anti-COVID-19 drug design.34,35 DeepLigBuilder recaptures the structural and chemical features of known inhibitors and generates potential drug-like inhibitors with novel chemical structures.
2 Methods
2.1 A brief overview of L-Net
L-Net generates 3D molecules by iteratively refining existing structures, using a state encoder and a policy network (Fig. 1a). At each step, the state encoder is first used to analyze the existing structure and encode the information into a continuous representation, which is then used by a policy network to decide how the molecule should be edited. The policy network then determines how many atoms should be added to the molecule, the type of each new atom and bond, and the 3D positions of new atoms. Details of the generation process are given in Fig. S1.† As a visualized demonstration, the full trajectory for generating a pyridine molecule is given in Fig. S2.† | ||
| Fig. 1 The architecture of DeepLigBuilder, which contains two components: a 3D molecule generative network called L-Net (a) and an optimization module based on MCTS (b). (a) The architecture of L-Net. L-Net generates 3D molecular structures by iteratively editing the structure. A state encoder is used to analyze the existing structure, and a policy network is used for decision-making. (b) A schematic diagram of L-Net combined with MCTS to optimize drug-like molecules inside the protein binding pocket. | ||
The architectures of the state encoder and the policy network are shown in Fig. S3 and S7,† respectively. The state encoder is mainly composed of MPNN layers36 organized into DenseNet blocks,37 which use graph pooling and unpooling layers (see Section S1.5†) to reduce the memory cost during training. The policy network uses MADE (masked autoencoder for distribution estimation38) to efficiently model various decision variables, which explicitly considers the valence constraints and local geometries of molecules to improve the performance of L-Net. The policy network is also rotationally covariant (or equivariant) thanks to the introduction of a local coordinate system (see Section S1.3†). The architectures of the state encoder and the policy network are discussed in detail in Sections S1.2 and S1.6.†
2.2 Training L-Net
The goal of L-Net is to generate 3D molecular structures with drug-like properties. To achieve this goal, we compiled a drug-like dataset (QED39 > 0.5) containing 1 million molecules filtered from ChEMBL40 with conformations generated using RDKit.41 Compared to previous methods, like G-SchNet that only recognizes atom types in the QM9 dataset, L-Net uses a full set of commonly encountered atom types (i.e., {C, H, O, N, P, S, F, Cl, Br, I}, see Section S1.7†) covering most of the drug-like molecules in the ChEMBL dataset. During training, the model was required to “imitate” the molecules inside the dataset. Specifically, an “expert trajectory” was created for each molecule to produce its structure, such as the one shown in Fig. S2,† and L-Net was asked to imitate the generation path. Several techniques, including SoftMADE (Section S1.6†), ring-first traversal (Section S1.7†), and random input errors (Section S1.7†), are used to improve the robustness of the training process. The dataset was split into the training, validation, and test sets in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. Hyperparameters were tuned manually using the validation set, targeting the 3D maximum mean discrepancy value (3D MMD, discussed in Section 2.3). The optimized set of hyperparameters is referred to as the “standard configuration” of L-Net and is recommended for future use. Detailed information about data collection and model training is available in Section S1.7.† Information related to hyperparameters is available in Section S1.9.†
:1:1 ratio. Hyperparameters were tuned manually using the validation set, targeting the 3D maximum mean discrepancy value (3D MMD, discussed in Section 2.3). The optimized set of hyperparameters is referred to as the “standard configuration” of L-Net and is recommended for future use. Detailed information about data collection and model training is available in Section S1.7.† Information related to hyperparameters is available in Section S1.9.†
2.3 Evaluation metrics
There are several widely cited benchmarks for evaluating molecule generative models,42,43 but none is designed for 3D generation tasks. Thus, we assembled a set of evaluation metrics to comprehensively assess the performance of L-Net and place special emphasis on the quality of 3D structures.The following aspects of L-Net are examined during the evaluation:
(1) The ability to generate chemically valid compounds. This was measured using the percentage of valid outputs.
(2) The diversity of the generated samples. This was measured by the percentage of unique outputs, as well as internal diversity values of 2D and 3D structures, which were calculated using the Morgan fingerprint and USRCAT (Ultrafast Shape Recognition with CREDO Atom Types),44 respectively.
(3) The ability to correctly learn the distributions of important molecular properties. We report the distributions of several important 2D and 3D molecular properties of the generated molecules, including molecular weight (MW), logP, the number of hydrogen bond donors (HBD) and acceptors (HBA), the number of rotatable bonds (ROT), QED,39 the normalized principal moment of inertia ratios (NPR1 and NPR2)45 and solvent accessible surface areas (total and polar SASA46), and compare the results to those of the test set.
(4) Whether L-Net can correctly model the chemical space (2D and 3D) of the training set. This is measured using 2D and 3D maximum mean discrepancy (MMD) calculated using the Morgan fingerprint and USRCAT. We also introduced precision and recall47 for a more interpretable evaluation. Precision measures the quality of the samples, while recall measures the number of samples in the target distribution that can be covered by the generative model.
(5) The ability to generate high-quality conformers. The quality of local structures is checked by examining the distribution of bond lengths, bond angles, and torsion angles. MMD is used to quantitatively measure whether the distribution of torsion angles is correctly modeled. Global conformation quality is measured using RMSD of heavy atoms after optimization using MMFF94s.48
Detailed descriptions of each metric are given in Section S1.11.† We also trained L-Net using QM9 to enable comparison to G-SchNet on the percentage of valid output and the median RMSD after optimization (details are given in Section S1.11†).
2.4 Using L-Net for structure-based molecule design
Compared to previous 3D molecular generative models,32,33 a major advantage of L-Net is its ability to generate a 3D molecular structure end-to-end. This feature makes L-Net convenient to be used and extended. In fact, L-Net can be combined with many techniques in a “plug and play” manner for objective-directed molecule design. In this work, we showcased its application in structure-based drug design by combining it with MCTS and directly generating ligands with high predicted affinity inside a protein binding pocket. We call this new method DeepLigBuilder. To our knowledge, this is the first time that a 3D generative model is combined with MCTS to address problems related to structure-based drug discovery (SBDD).Briefly, MCTS finds promising solutions of a reward function by iteratively constructing a search tree (see Fig. 1b). Each node in the tree represents an intermediate state during molecule generation. At each iteration, the model first chooses a promising state from the search tree (the selection step), enumerates possible actions for that state (the expansion step), and performs a rollout to generate the rest of the molecular structure (the simulation step). The reward is collected for that structure, and the information is backpropagated through the tree to update the Q-values of each node. More technically, our implementation of MCTS adopts the framework of MENTS (Maximum Entropy for Tree Search49), with custom modifications detailed in Section S1.12.† The L-Net has two roles during this process. First, it is used as a rollout policy during the simulation step. Second, it is used to compute a “prior” Q-value for each node in the search tree. In other words, MCTS is responsible for searching for molecules with high binding affinity, while L-Net is used to encourage the structure to be valid, drug-like, easy to synthesize, and diverse.
We used the docking score provided by smina50 as the reward function. A major advantage of DeepLigBuilder is that the docking score can be evaluated much faster than previous 2D MCTS models (such as ChemTS51) because 3D structures of molecules are already generated inside the binding pocket. This attribute allows the use of the fast local optimization capability provided by smina to calculate the docking score (using the minimize option, with a time cost of less than 0.1 seconds per evaluation) without the need for global optimization (costing on average several minutes per evaluation), which affords an orders of magnitude increase in speed.
For the case study, DeepLigBuilder was used to design potential inhibitors for the main protease (Mpro, also referred to as 3CL protease) of SARS-CoV-2, the virus causing the COVID-19 pandemic.52 We first investigated the ability of DeepLigBuilder to perform lead optimization based on a fragment derived from existing peptidomimetic covalent inhibitors. We then tested its performance on de novo designs of noncovalent inhibitors, with the goal of generating highly potent inhibitors with novel scaffolds.
3 Results and discussion
3.1 Molecule generation using L-Net
Several randomly generated samples from L-Net are shown in Fig. S11.† Visual inspection shows that the molecules have correct local geometry. For example, sp3 and sp2 hybridized atoms correctly adopt tetrahedral and planar geometries, and aromatic systems correctly form planar structures. More quantitative evaluation of L-Net is shown in Fig. 2. Each metric was calculated using 50000 randomly generated samples from L-Net. A dataset containing 10 million 3D molecules generated using L-Net (L-Net-10M) is provided as supplementary data, and it can be used as a regular compound library for virtual screening or as a benchmark to compare other 3D molecule generation methods with L-Net in the future.
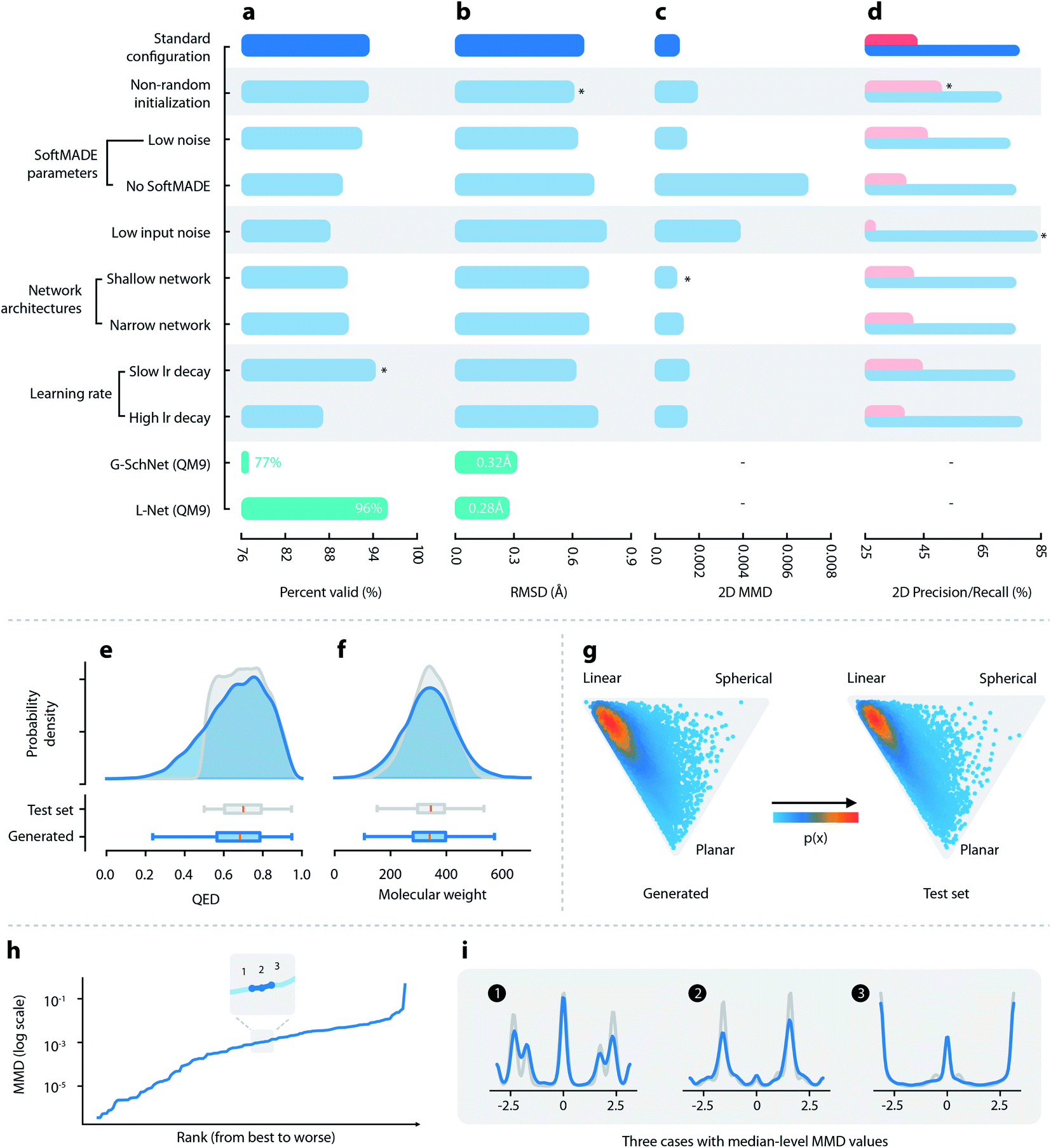 | ||
| Fig. 2 Quantitative evaluations of L-Net. (a–d) Performance of L-Net measured by (a) the percentage of valid outputs; (b) RMSD values after optimization; (c) 2D MMD; and (d) 2D precision (pink) and recall (blue). Rows indicate different hyperparameter selections. Comparison with G-SchNet is shown in green (temperature set to 1.0 during comparison). (e and f) Distribution of (e) QED and (f) molecular weight among generated (blue) and test set (grey) molecules. (g) Shape distribution of generated (left) and test set (right) molecules, visualized using NPR descriptors. (h) The MMD values of the torsion distribution for each torsion pattern, ranked from lowest to highest. (i) Torsion distributions with median-level MMD values, blue: generated molecules, grey: test set molecules. | ||
P, HBA, HBD, and ROT (see Fig. 2f and S12†). Although a small percentage of molecules show QED lower than 0.5, most of them (83.9%) have QED greater than 0.5 (Fig. 2e), which is in accordance with the dataset that used a cutoff value of 0.5. The 3D shape distributions are given in Fig. 2. For both the generated and the test set molecules, the shape distributions tend to gather around the “linear” corner of the triangle, while being slightly tilted towards the “planar” corner. The distributions of the total and polar SASA (shown in Fig. S13†) also show good matches. Overall, L-Net was able to correctly model the distribution of important 3D and 2D molecular properties. A quantitative report of the means and standard deviations of these properties is given in Table S3.†
Finally, we discuss the ability of L-Net to generate ring structures with valid conformations. We place our focus on aromatic rings. Most aromatic rings in test set molecules adopt a strict planar structure. We first investigate the issue related to the generation of non-cyclic atoms inside aromatic rings. Non-closed aromatic rings will be marked as invalid molecules by RDKit, and since the percentage of invalid output molecules is low (5.7% as shown in Section 3.1.1), we assume that this issue is relatively minor for L-Net. The occurrence of the noncyclic conformation of rings can also be indicated by incorrect bond lengths. The average and standard deviation for the length of aromatic bonds from test set molecules is 1.39 Å and 0.051 Å, while the values for the generated molecules are 1.39 Å and 0.067 Å. The average bond length matches well between the generated and the test set molecules. Although the standard deviation is a little bit larger among the generated samples, the difference of 0.016 Å is small and would not cause significant problems for applications in de novo drug design.
To evaluate the severity of wrong torsion angles, the standard deviation of torsion angles for aromatic rings is also calculated. The standard deviation is 1.72° for the generated molecules and 0.46° for the test set molecules. Again, the standard deviation is slightly larger for molecules generated by the model. However, the increased deviation of 1.27° is tolerable and could be minimized by the following relaxation, indicating that for most of the time the model correctly generates a planar structure for aromatic rings.
To summarize, although the ring structures generated by L-Net may contain some imperfections, the problem is relatively small and should have a limited impact on the application of the model. If we need to correct the structures, the fast local relaxation utility provided by RDKit can be used. This would improve the quality of ring conformation without causing significant changes to the global conformation of the molecule (with the average RMSD equal to 0.613 Å as shown in Section 3.1.2).
3.2 Structure-based molecule design using DeepLigBuilder
To apply L-Net to structure-based design, we developed DeepLigBuilder by combining L-Net with MCTS to directly generate 3D molecules inside the binding pocket of the target protein. Taking the SARS-CoV-2 main protease (Mpro) as a test case, we used DeepLigBuilder to perform the following tasks: (1) lead optimization of known covalent inhibitors targeting Mpro and (2) de novo design of noncovalent inhibitors.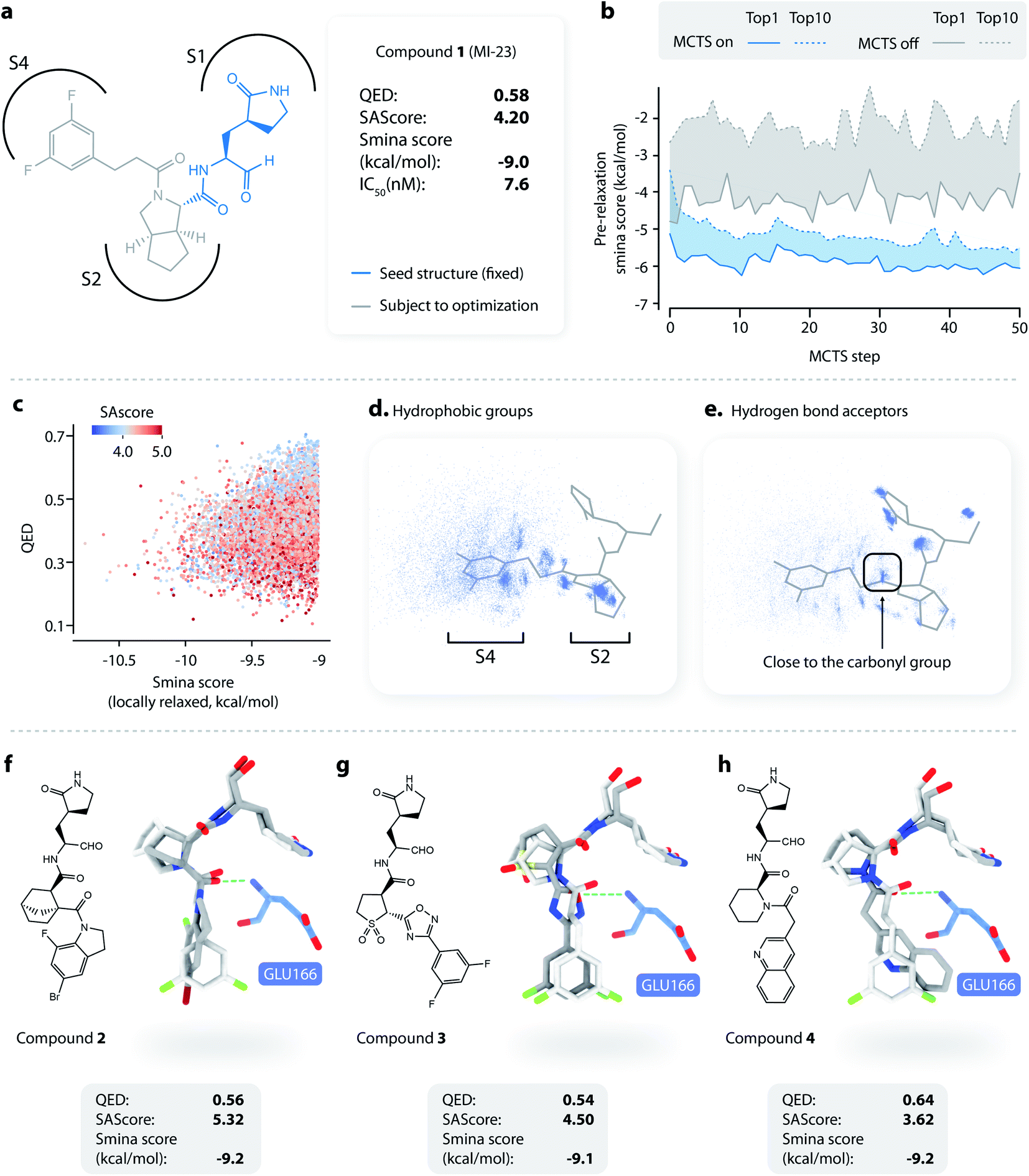 | ||
| Fig. 3 Lead optimization using DeepLigBuilder. (a) The topological structure of MI-23, a reported covalent inhibitor targeting SARS-CoV-2 Mpro.54 The blue part of the molecule is used as a seed structure by DeepLigBuilder for molecule generation. (b) The range of the scores for the 10 best molecules sampled at each step during the Monte Carlo tree search. Blue: MCTS turned on; grey: MCTS turned off. (c) The distribution of the smina score, QED, and SAscore of high-quality samples. (d) The distribution of hydrophobic groups in high-quality samples. (e) The distribution of hydrogen bond acceptors in high-quality samples. (f–h) Examples of molecules generated by DeepLigBuilder. The binding poses of the generated molecules (grey) are aligned with MI-23 (light grey) for comparison. The residue Glu166 is shown in blue. Molecular properties and predicted binding affinities are listed in the grey boxes. | ||
The structure of Mpro complexed with MI-23 was used for molecular generation (PDB ID: 7D3I). We only kept the P1 fragment (Fig. 3a, blue) as the starting point for generation. Note that MI-23 is not contained in the training set of L-Net, so the model does not know the structure in advance. A total of 64 MCTS runs were performed to ensure the diversity of the generated molecules. After the MCTS runs, we applied the following operations to filter for high-quality compounds. The general idea of the workflow is similar to that used in CovDock.56 (1) First, Cys145 was mutated to alanine, and the generated molecules were locally relaxed and scored using smina. (2) Second, molecules with a smina score worse than −9.0 kcal mol−1 were removed. This procedure retained molecules with high predicted binding affinity. (3) Third, molecules with warhead groups displaced by more than 1 Å after optimization were removed. (4) Finally, the molecules were clustered by their BM-scaffold.57
We first investigated whether MCTS had helped to find molecules with better scores compared with the random search. Fig. 3b shows the range of the smina scores (before structure relaxation) for the 10 best molecules sampled at each step, with MCTS turned on (blue area) and off (grey area). Molecules sampled using MCTS showed significantly lower smina scores than those sampled with the random search (for smina scores, the lower the better), indicating that MCTS can indeed help with the exploration of molecules with a higher predicted binding affinity compared with the random search.
Next, we investigated the distribution of the high-quality molecules generated with DeepLigBuilder. A total of 7,508 high-quality molecules were obtained, with an overall total of 4,016 BM-scaffolds.57 None of these molecules were present in our training and testing datasets, demonstrating the ability of DeepLigBuilder to find novel structures. The distributions of the smina score (after local relaxation) and QED are shown in Fig. 3c, and the color of each record corresponds to the synthetic accessibility score (SAscore,58 the lower the better). All molecules selected had an improved smina score compared with MI-23 (−9.0 kcal mol−1). The mean QED value of the generated molecule was 0.41, which was lower than that of MI-23 (0.57). However, the value was higher compared to those of several other wide-spectrum, peptide-like inhibitors of coronavirus Mpro, such as N1 (ref. 59) (QED = 0.131), N3 (ref. 34 and 59) (QED = 0.123), and N9 (ref. 59) (QED = 0.127). The average synthetic accessibility score was 4.26, which is similar to that of MI-23 (SAscore = 4.20).
The MI-23 molecule contains three features that are relevant to its high binding affinity: (1) a hydrophobic group filling the S2 site, (2) a hydrogen bond acceptor (the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the linker) interacting with the backbone NH group of Glu166, and (3) a hydrophobic group filling the S3 and S4 sites. To examine whether the generated molecules have captured these features, pharmacophore models were generated using the software Align-It.60 The spatial distributions of hydrophobic groups and hydrogen bond acceptors are shown in Fig. 3d and e. Visual inspection of Fig. 3d shows a dense cluster inside the S2 site, indicating that most generated molecules occupy the S2 site with a hydrophobic group, similar to that of MI-23. Although the distribution of hydrophobic groups extends to the S4 site, their distribution at this site is more spread out than that inside S2, possibly because the S4 site is more open than the S2 site. The distribution of hydrogen bond acceptors (Fig. 3e) showed a cluster (although smaller) around the carbonyl group, indicating that the interaction with Glu166 can be captured by some of the generated molecules.
O group in the linker) interacting with the backbone NH group of Glu166, and (3) a hydrophobic group filling the S3 and S4 sites. To examine whether the generated molecules have captured these features, pharmacophore models were generated using the software Align-It.60 The spatial distributions of hydrophobic groups and hydrogen bond acceptors are shown in Fig. 3d and e. Visual inspection of Fig. 3d shows a dense cluster inside the S2 site, indicating that most generated molecules occupy the S2 site with a hydrophobic group, similar to that of MI-23. Although the distribution of hydrophobic groups extends to the S4 site, their distribution at this site is more spread out than that inside S2, possibly because the S4 site is more open than the S2 site. The distribution of hydrogen bond acceptors (Fig. 3e) showed a cluster (although smaller) around the carbonyl group, indicating that the interaction with Glu166 can be captured by some of the generated molecules.
Several generated molecules (compounds 2–4) are shown in Fig. 3f–h. Searching the full PubChem61 and ChEMBL datasets did not reveal any existing molecules similar to compounds 2–4 (Tanimoto score > 95%), indicating that they are indeed novel structures. The interaction patterns of these molecules with Mpro resemble that of MI-23. Notably, compound 3 has recovered the fluorine substituted phenyl ring in M-23 with a similar position and orientation.
To summarize, we show that given a known privileged fragment, DeepLigBuilder was capable of generating molecules with good predicted binding affinity, reasonable drug-likeness, and binding features similar to known inhibitors. These attributes demonstrate the practical applicability of DeepLigBuilder to lead optimization problems.
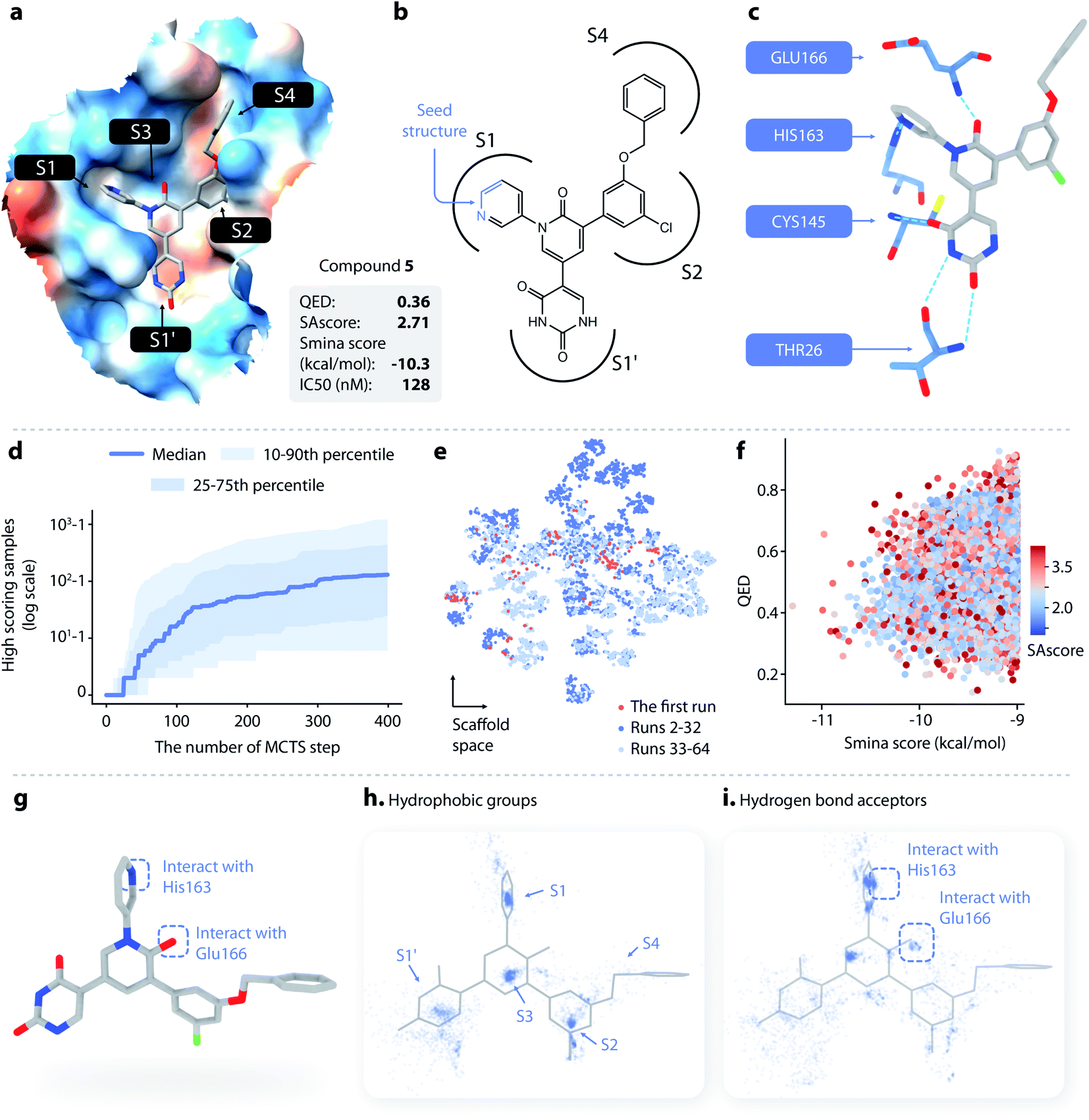 | ||
| Fig. 4 (a–c) The structure and binding pose of compound 5, a known noncovalent inhibitor of SARS-CoV-2 Mpro. (a) The binding pose of compound 5 inside Mpro. (b) The topological structure of compound 5; the part of the molecule colored in blue is used as a seed structure. (c) Key interaction between Mpro and compound 5. (d) The cumulative number of generated samples with smina score < −9 kcal mol−1 as a function of the number of MCTS steps performed: blue line, the median value across successful runs; light blue areas, the 10–90th and 25–75th percentiles. (e) The distributions of scaffolds generated from the first run (red), the 2nd to 32nd runs (dark blue), and the 33rd to 64th runs (light blue). (f) The distribution of the smina score, QED, and SAscore of the molecules generated with DeepLigBuilder. (g) Several important structural features of compound 5. (h) The distribution of hydrophobic groups of the generated molecules. (i) The distribution of hydrogen bond acceptors of the generated molecules. | ||
DeepLigBuilder succeeded in generating molecules with good binding affinities (smina score < −9 kcal mol−1) in 50 of the 64 runs. This result translates to a success rate of 78.1% (standard error 7%). When turning off the MCTS algorithm, the success rate drops to 0% (in 64 runs), demonstrating the ability of MCTS to improve the efficiency of finding molecules with high scores. Fig. 4d shows how the cumulative number of generated samples with good smina scores changed as MCTS proceeded for successful runs, with the blue line indicating the median and the light blue areas indicating the 10–90th and 25–75th percentiles. A typical MCTS run will produce about 129 unique molecules with good binding affinity within 400 runs, which may take 3–5 h to complete on an NVIDIA GeForce RTX 3070 graphics card.
Although the MCTS already includes an entropy-based penalty that encourages exploration, we found that multiple runs are still necessary to achieve better diversity. Fig. 4e shows the t-SNE visualization of the distributions of scaffolds generated from the first run (red), the 2nd to 32nd runs (dark blue), and the 33rd to 64th runs (light blue). Visual inspection shows that there was a large portion of chemical space unexplored after the first run, indicating that multiple runs are needed. We also found that a major portion of the chemical space of the generated molecules was visited within the first 32 runs, as indicated by the large overlap between the light blue and dark blue points. This analysis shows that 32 runs are generally sufficient for this system.
A total of 19014 molecules with smina scores better than −9.0 kcal mol−1 were obtained; none of these molecules were present in our training and testing datasets. The distributions of the smina score, QED, and SAscore are shown in Fig. 4f. The MCTS model manages to maintain an average QED of 0.53, which is higher than the QED value of compound 5. The average SAscore is 2.75, at a similar level to that of compound 5. This shows that MCTS can properly balance between the requirement for a high binding affinity and the requirement for high drug-likeness and synthesis accessibility, even without explicitly including QED and SAscore in the reward function (these properties have already been captured by L-Net during its training using the drug-like ChEMBL subset).
Compound 5 contains several major features contributing to its high inhibitory activity (as shown in Fig. 4g), namely, (1) five rings in the compound occupy the five sub-binding sites (S1′, S1, S2, S3, and S4) in the pocket, providing good steric complementarity between the ligand and the protein; (2) the carbonyl group in the pyridinone ring forms a hydrogen bond with the backbone NH of Glu166; and (3) the pyridine ring forms a hydrogen bond with His163. We investigated the spatial distribution of pharmacophores of the generated molecules to check whether they also contain these structural features. The distributions of hydrophobic groups and hydrogen bond acceptors are shown in Fig. 4h and i, respectively. For hydrophobic groups, it can be seen that there are dense clusters inside the S1′, S1, S2, and S3, with a similar location to the four corresponding rings inside compound 5, showing that those sub-pockets have been well occupied. From the distribution of hydrogen bond acceptors, we can see that the interactions with Glu166 and His163 are well covered by the generated molecule, and the location of the corresponding hydrogen bond donors aligns well with that of compound 5. To summarize, important pharmacophore features for protein–ligand interaction can be observed in the generated molecules.
We then analyzed the chemical scaffolds of the generated molecules. The 19014 generated molecules contained 2,984 BM-scaffolds, examples of which are shown in Fig. 5a. A full list of the top 50 scaffolds is given in the ESI (Fig. S17 and S18†). These scaffolds highly resemble compound 5, with the basic structure of an aromatic core connected with three other rings, filling sites S1′, S1, and S2. This result indicates that DeepLigBuilder can recover the privileged structural patterns. Encouragingly, we also found that the model recalls fragments of compound 5 that are important for binding. For example, scaffold 2 contains a pyridazinone core that mimics the pyridinone ring in compound 5 that interacts with Glu166. Scaffold 3 contains the uracil-like ring in compound 5 that interacts with Thr26 and Cys145.
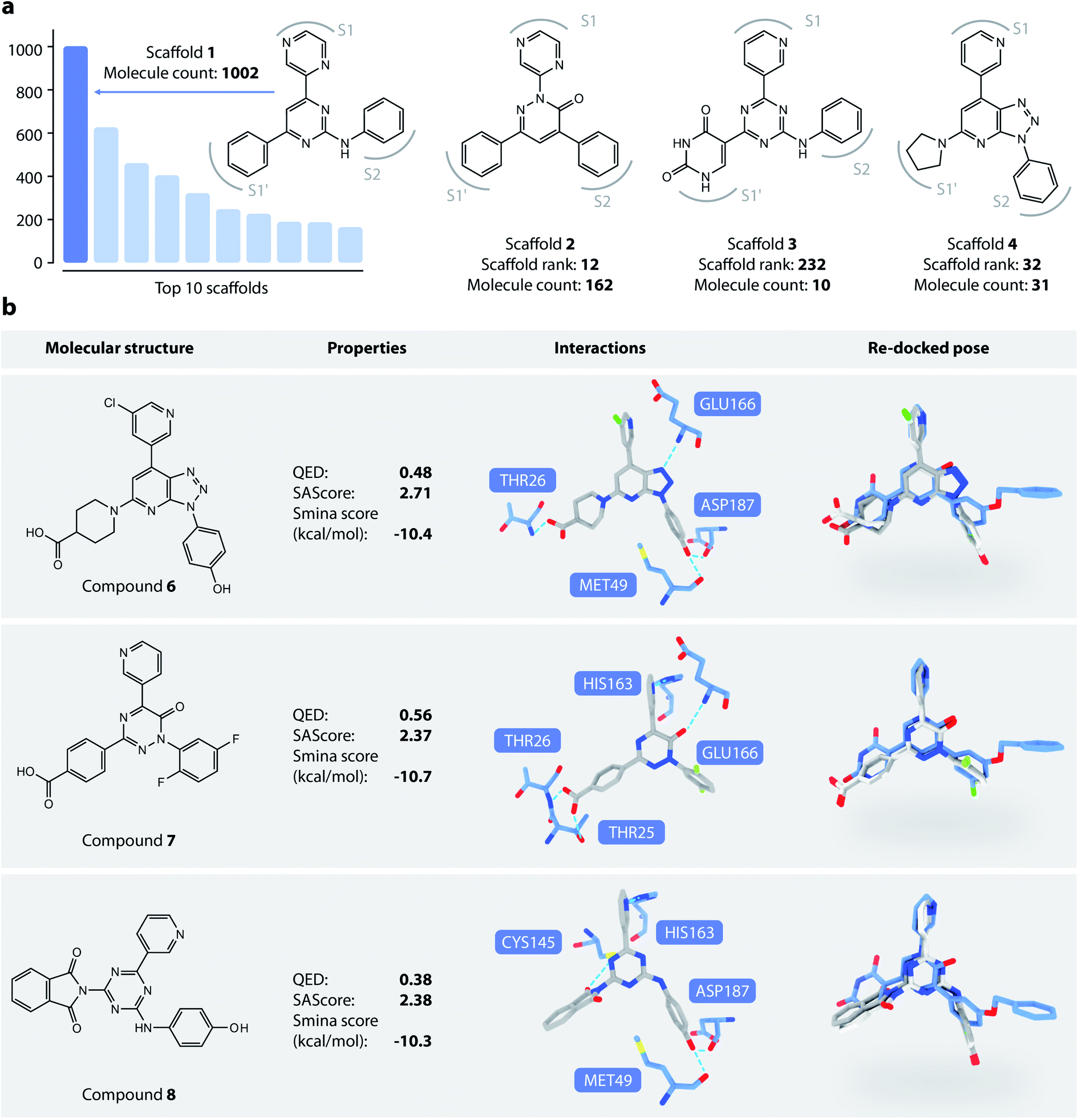 | ||
| Fig. 5 (a) Examples of DeepLigBuilder generated scaffolds. (b) Analysis of three promising compounds generated. The first column: topological structures of generated molecules. The second column: molecular properties (QED, SAscore) and predicted binding affinity. The third column: interactions between the generated molecules and the protein. The last column: comparison between the locally minimized (dark grey) and globally minimized poses (light grey) of the generated molecules, as well as the binding pose of compound 5 (blue). | ||
We selected three compounds with improved QED, SAscore, and smina score compared to compound 5 for further analysis (Fig. 5b). These are novel structures, and searches in the PubChem and ChEMBL databases do not result in molecules with Tanimoto scores larger than 95%. All three compounds show similar interaction patterns to that of compound 5. Interestingly, compounds 6 and 8 contain an additional hydroxyl group in the S2 site phenyl ring that forms hydrogen bonds with the backbone CO groups of Met49 and Asp187, which also makes the phenyl ring dive deeper into the S2 site to offer better steric complementarity. These findings highlight the ability of DeepLigBuilder to discover novel interactions with the target protein. Of course, determining whether these designed compounds potently inhibit Mpro needs further experimental study.
4 Conclusion
We have developed DeepLigBuilder, a novel deep learning-based method for de novo drug design that generates 3D molecular structures in the binding site of the target protein. DeepLigBuilder uses L-Net to generate valid 3D drug-like molecules and MCTS to search for strong binding molecules. Our L-Net is a novel deep generative model that directly outputs 3D and topological structures of drug-like molecules without the need for additional atom placement or bond order inference.L-Net provides a powerful tool for 3D molecule deep generation, which can be extended or fine-tuned with other datasets. For example, L-Net could be trained with organic material datasets to generate compounds with desired properties. L-Net could also be used to explore the drug-like chemical space. A previous study63 shows that SMILES-based generative models trained on a small subset of GDB-13 can cover a major portion of the GDB-13 chemical space. Similarly, we expect that L-Net can be used to enumerate molecules in drug-like and synthetically accessible chemical space with 3D structures, thereby enabling a better understanding of the scale of the chemical space and its underlying structure.
In the present study, we used docking scores that reflect the interactions between the target and the ligand as the reward function in the MCTS step, which can be changed to address a variety of design needs, such as similarity and pharmacophore-based rewards. Conditional generative models can also be built using L-Net to further reduce the search time of MCTS.
We used DeepLigBuilder to design potential inhibitors for the main protease of SARS-CoV-2 as a case study. DeepLigBuilder was able to produce promising drug-like compounds with novel chemical structures and high predicted binding affinity, which capture important pharmacophore features of known inhibitors for both lead optimization and de novo generation tasks. DeepLigBuilder is flexible enough to accept a user-defined seed structure, even starting from one atom. Automatic seed generation methods could be included in the future to further extend its capability. Graphical interfaces for user interactions during the generation process may also be developed. To summarize, L-Net and DeepLigBuilder provide promising new tools for structure-based de novo drug design, lead optimization, and design of other functional molecules.
Data availability
The L-Net-10M dataset is available from https://disk.pku.edu.cn:443/link/FA7AD4D5E57C4134EC7869225DB4063F.Author contributions
Y. L. designed and implemented the deep learning model and performed the model training. J. P. and L. L. supervised the project. Y. L., J. P. and L. L. discussed and analysed the data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (Grants 22033001, 21673010 and 21633001), the National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Grant 2018ZX09711002), and the Ministry of Science and Technology of China (Grant 2016YFA0502303).Notes and references
- R. S. Bohacek, C. McMartin and W. C. Guida, Med. Res. Rev., 1996, 16, 3–50 CrossRef CAS PubMed.
- G. Schneider and U. Fechner, Nat. Rev. Drug Discovery, 2005, 4, 649–663 CrossRef CAS PubMed.
- G. Schneider, J. Comput. Aided Mol. Des., 2012, 26, 115–120 CrossRef CAS PubMed.
- G. Schneider and D. E. Clark, Angew. Chem., Int. Ed., 2019, 58, 10792–10803 CrossRef CAS PubMed.
- J. J. Irwin and B. K. Shoichet, J. Med. Chem., 2016, 59, 4103–4120 CrossRef CAS PubMed.
- Y. Nishibata and A. Itai, Tetrahedron, 1991, 47, 8985–8990 CrossRef CAS.
- H.-J. Böhm, J. Comput. Aided Mol. Des., 1992, 6, 61–78 CrossRef PubMed.
- D. A. Pearlman and M. A. Murcko, J. Comput. Chem., 1993, 14, 1184–1193 CrossRef CAS.
- R. Wang, Y. Gao and L. Lai, Molecular Modeling Annual, 2000, 6, 498–516 CrossRef CAS.
- Y. Yuan, J. Pei and L. Lai, J. Chem. Inf. Model., 2011, 51, 1083–1091 CrossRef CAS PubMed.
- Y. Yuan, J. Pei and L. Lai, Front. Chem., 2020, 8, 142 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Ruiz-Caro, J. Tirado-Rives, A. Basavapathruni, K. S. Anderson and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2006, 16, 663–667 CrossRef CAS PubMed.
- S. Ni, Y. Yuan, J. Huang, X. Mao, M. Lv, J. Zhu, X. Shen, J. Pei, L. Lai, H. Jiang and J. Li, J. Med. Chem., 2009, 52, 5295–5298 CrossRef CAS PubMed.
- E. Shang, Y. Yuan, X. Chen, Y. Liu, J. Pei and L. Lai, J. Chem. Inf. Model., 2014, 54, 1235–1241 CrossRef CAS PubMed.
- H. Park, S. Hong, J. Kim and S. Hong, J. Am. Chem. Soc., 2013, 135, 8227–8237 CrossRef CAS PubMed.
- H. Park, Y. Shin, H. Choe and S. Hong, J. Am. Chem. Soc., 2015, 137, 337–348 CrossRef CAS PubMed.
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2017, 4, 120–131 CrossRef PubMed.
- Y. Li, J. Hu, Y. Wang, J. Zhou, L. Zhang and Z. Liu, J. Chem. Inf. Model., 2019, 60, 77–91 CrossRef PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, ChemRxiv, 2017 Search PubMed.
- Y. Li, L. Zhang and Z. Liu, J. Cheminformatics, 2018, 10, 33 CrossRef PubMed.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, J. Cheminformatics, 2017, 9, 48 CrossRef PubMed.
- P. Pogány, N. Arad, S. Genway and S. D. Pickett, J. Chem. Inf. Model., 2018, 59, 1136–1146 CrossRef PubMed.
- J. Lim, S.-Y. Hwang, S. Moon, S. Kim and W. Y. Kim, Chem. Sci., 2019, 11, 1153–1164 RSC.
- J. Arús-Pous, A. Patronov, E. J. Bjerrum, C. Tyrchan, J.-L. Reymond, H. Chen and O. Engkvist, J. Cheminformatics, 2020, 12, 38 CrossRef PubMed.
- W. Jin, R. Barzilay and T. Jaakkola, International Conference on Machine Learning, 2020 Search PubMed.
- A. Nigam, R. Pollice, M. Krenn, G. d. P. Gomes and A. Aspuru-Guzik, Chem. Sci., 2021, 12, 7079–7090 RSC.
- A. Nigam, R. Pollice and A. Aspuru-Guzik, arXiv:2106.04011, 2021.
- N. W. A. Gebauer, M. Gastegger and K. T. Schütt, Advances in Neural Information Processing Systems, 2019, vol. 32, pp. 7566–7578 Search PubMed.
- V. Nesterov, M. Wieser and V. Roth, arXiv:2010.06477, 2020.
- G. N. C. Simm, R. Pinsler, G. Csányi and J. M. Hernández-Lobato, International Conference on Learning Representations, 2021 Search PubMed.
- M. Ragoza, T. Masuda and D. R. Koes, NeurIPS 2020 Workshop on Machine Learning for Structural Biology, 2020 Search PubMed.
- T. Masuda, M. Ragoza and D. R. Koes, NeurIPS 2020 Workshop on Machine Learning for Structural Biology, 2020 Search PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- C. Li, Q. Sun, Y. Lu, Y. Liu and L. Lai, Sci. Sin.: Chim., 2020, 50, 1250–1279 Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, International Conference on Machine Learning, 2017 Search PubMed.
- G. Huang, Z. Liu, L. v. d. Maaten and K. Q. Weinberger, Conference on Computer Vision and Pattern Recognition, 2017 Search PubMed.
- M. Germain, K. Gregor, I. Murray and H. Larochelle, International Conference on Machine Learning, 2015 Search PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. Magariños, J. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. Radoux, A. Segura-Cabrera, A. Hersey and A. Leach, Nucleic Acids Res., 2018, 47, D930–D940 CrossRef PubMed.
- G. Landrum, 2013, http://www.rdkit.org.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, Front. Pharmacol., 2020, 11, 565644 CrossRef CAS PubMed.
- N. Brown, M. Fiscato, M. H. S. Segler and A. C. Vaucher, J. Chem. Inf. Model., 2019, 59, 1096–1108 CrossRef CAS PubMed.
- A. M. Schreyer and T. Blundell, J. Cheminformatics, 2012, 4, 27 CrossRef CAS PubMed.
- W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comp. Sci., 2003, 43, 987–1003 CrossRef CAS PubMed.
- S. Mitternacht, F1000Research, 2016, 5, 189 Search PubMed.
- T. Kynkäänniemi, T. Karras, S. Laine, J. Lehtinen and T. Aila, arXiv:1904.06991, 2019.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- C. Xiao, R. Huang, J. Mei, D. Schuurmans and M. Muller, Advances in Neural Information Processing Systems, 2019, 32, 9520–9528 Search PubMed.
- D. R. Koes, M. P. Baumgartner and C. J. Camacho, J. Chem. Inf. Model., 2013, 53, 1893–1904 CrossRef CAS PubMed.
- X. Yang, J. Zhang, K. Yoshizoe, K. Terayama and K. Tsuda, Sci. Technol. Adv. Mat., 2017, 18, 972–976 CrossRef CAS PubMed.
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes and Y.-Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS PubMed.
- S. Riniker and G. A. Landrum, J. Chem. Inf. Model., 2015, 55, 2562–2574 CrossRef CAS PubMed.
- J. Qiao, Y.-S. Li, R. Zeng, F.-L. Liu, R.-H. Luo, C. Huang, Y.-F. Wang, J. Zhang, B. Quan, C. Shen, X. Mao, X. Liu, W. Sun, W. Yang, X. Ni, K. Wang, L. Xu, Z.-L. Duan, Q.-C. Zou, H.-L. Zhang, W. Qu, Y.-H.-P. Long, M.-H. Li, R.-C. Yang, X. Liu, J. You, Y. Zhou, R. Yao, W.-P. Li, J.-M. Liu, P. Chen, Y. Liu, G.-F. Lin, X. Yang, J. Zou, L. Li, Y. Hu, G.-W. Lu, W.-M. Li, Y.-Q. Wei, Y.-T. Zheng, J. Lei and S. Yang, Science, 2021, 371, 1374–1378 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam, V. Namasivayam, Y. Hayashi and S.-H. Jung, J. Med. Chem., 2016, 59, 6595–6628 CrossRef CAS PubMed.
- D. T. Warshaviak, G. Golan, K. W. Borrelli, K. Zhu and O. Kalid, J. Chem. Inf. Model., 2014, 54, 1941–1950 CrossRef PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminformatics, 2009, 1, 8 CrossRef PubMed.
- H. Yang, W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei, J. Ziebuhr, R. Hilgenfeld, K. Y. Yuen, L. Wong, G. Gao, S. Chen, Z. Chen, D. Ma, M. Bartlam and Z. Rao, PLoS Biol., 2005, 3, e324 CrossRef PubMed.
- J. Taminau, G. Thijs and H. D. Winter, J. Mol. Graph. Model., 2008, 27, 161–169 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2020, 49, D1388–D1395 CrossRef PubMed.
- C.-H. Zhang, E. A. Stone, M. Deshmukh, J. A. Ippolito, M. M. Ghahremanpour, J. Tirado-Rives, K. A. Spasov, S. Zhang, Y. Takeo, S. N. Kudalkar, Z. Liang, F. Isaacs, B. Lindenbach, S. J. Miller, K. S. Anderson and W. L. Jorgensen, ACS Cent. Sci., 2021, 7, 467–475 CrossRef CAS PubMed.
- J. Arús-Pous, T. Blaschke, S. Ulander, J.-L. Reymond, H. Chen and O. Engkvist, J. Cheminformatics, 2019, 11, 20 CrossRef PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Supplementary methods, figures and tables. See DOI: 10.1039/d1sc04444c. |
| This journal is © The Royal Society of Chemistry 2021 |