
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperfluorescent polymers enabled by through-space charge transfer polystyrene sensitizers for high-efficiency and full-color electroluminescence†
Jun
Hu
ab,
Yinuo
Wang
ab,
Qiang
Li
ab,
Shiyang
Shao
 *a,
Lixiang
Wang
*ab,
Xiabin
Jing
a and
Fosong
Wang
a
*a,
Lixiang
Wang
*ab,
Xiabin
Jing
a and
Fosong
Wang
a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, China. E-mail: ssyang@ciac.ac.cn; lixiang@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 3rd September 2021
Abstract
Fluorescent polymers are suffering from low electroluminescence efficiency because triplet excitons formed by electrical excitation are wasted through nonradiative pathways. Here we demonstrate the design of hyperfluorescent polymers by employing through-space charge transfer (TSCT) polystyrenes as sensitizers for triplet exciton utilization and classic fluorescent chromophores as emitters for light emission. The TSCT polystyrene sensitizers not only have high reverse intersystem crossing rates for rapid conversion of triplet excitons into singlet ones, but also possess tunable emission bands to overlap the absorption spectra of fluorescent emitters with different bandgaps, allowing efficient energy transfer from the sensitizers to emitters. The resultant hyperfluorescent polymers exhibit full-color electroluminescence with peaks expanding from 466 to 640 nm, and maximum external quantum efficiencies of 10.3–19.2%, much higher than those of control fluorescent polymers (2.0–3.6%). These findings shed light on the potential of hyperfluorescent polymers in developing high-efficiency solution-processed organic light-emitting diodes and provide new insights to overcome the electroluminescence efficiency limitation for fluorescent polymers.
1. Introduction
Fluorescent polymers have attracted much attention of researchers in developing low-cost, solution-processed organic light-emitting diodes (OLEDs) owing to their promising photoluminescence efficiencies and rich emission colors expanding from the deep blue to near-infrared region.1,2 Given that the recombination of hole and electron carriers forms singlet and triplet excitons with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 according to the spin statistic rule, traditional fluorescent polymers often suffer from low device efficiencies since only singlet excitons can be utilized for light emission, which limits the internal quantum efficiency (IQE) to 25%.3 To overcome this limitation, two main approaches involving the use of phosphorescent polymers and thermally activated delayed fluorescence (TADF) polymers have been developed to utilize triplet excitons.4 Phosphorescent polymers, which can harvest both singlet and triplet excitons to achieve unit IQEs, however, rely on the use of expensive heavy-metal complexes.5 Recently, TADF polymers2,6,7 based on twisted donor–acceptor architectures have emerged as attractive luminescent materials because they can convert triplet excitons into singlet ones through reverse intersystem crossing (RISC)8,9 owing to small singlet-triplet energy splitting (ΔEST) by separating frontier molecular orbitals (FMOs). However, the separation of FMOs is often accompanied by a decrease in radiative decay rate, leading to contradiction between exciton utilization and luminescent efficiency.6 To address these issues, new design strategies to simultaneously enhance exciton utilization and radiative decay for polymers are highly demanded.
:3 according to the spin statistic rule, traditional fluorescent polymers often suffer from low device efficiencies since only singlet excitons can be utilized for light emission, which limits the internal quantum efficiency (IQE) to 25%.3 To overcome this limitation, two main approaches involving the use of phosphorescent polymers and thermally activated delayed fluorescence (TADF) polymers have been developed to utilize triplet excitons.4 Phosphorescent polymers, which can harvest both singlet and triplet excitons to achieve unit IQEs, however, rely on the use of expensive heavy-metal complexes.5 Recently, TADF polymers2,6,7 based on twisted donor–acceptor architectures have emerged as attractive luminescent materials because they can convert triplet excitons into singlet ones through reverse intersystem crossing (RISC)8,9 owing to small singlet-triplet energy splitting (ΔEST) by separating frontier molecular orbitals (FMOs). However, the separation of FMOs is often accompanied by a decrease in radiative decay rate, leading to contradiction between exciton utilization and luminescent efficiency.6 To address these issues, new design strategies to simultaneously enhance exciton utilization and radiative decay for polymers are highly demanded.
Hyperfluorescence, also termed “TADF-sensitized fluorescence”, has been developed as an attractive strategy for high-efficiency small-molecule OLEDs by vacuum deposition in the last couple of years.10–27 In this motif, TADF materials and fluorescent chromophores are co-doped in host matrices to act as sensitizers and emitters respectively. Under electrical excitation, triplet excitons formed on TADF sensitizers can be up-converted into singlet ones by the RISC process, which are then transferred to the singlet (S1) states of fluorescent chromophores via Förster resonance energy transfer (FRET) for light emission. Therefore, hyperfluorescence has the advantage of combining high exciton utilization of TADF materials and the high radiative decay rate of fluorescent chromophores in one emissive system.18 By developing novel TADF sensitizers with high RISC rate constants22,23,25 and fluorescent emitters with bulk substituents,15,17,19 high-efficiency and stable hyperfluorescent OLEDs have been reported. However, hyperfluorescent OLEDs have focused on small molecules so far and are dominated by vacuum-deposition manufacturing technology.18 Hyperfluorescent polymers for solution-processed OLEDs that are compatible with low-cost and large-area wet-processing technologies are rarely reported.
To develop hyperfluorescent polymers, the key is to exploit suitable TADF polymer sensitizers for harvesting triplet excitons.28 For this purpose, at least two requirements should be considered. First, polymer sensitizers should have high RISC rate constants (kRISC) for rapidly converting triplet excitons into singlet ones.25 Second, their emission bands should have good overlap with the absorption spectra of fluorescent emitters for efficient FRET from the sensitizer to emitter.10 However, the majority of TADF polymers so far are based on conjugated donor–acceptor architectures with through-bond charge transfer (TBCT) emissions,29–41 exhibiting kRISC less than 106 s−1 (see Fig. S1 and Table S1†). Meanwhile, strong electron coupling between donors and acceptors in TBCT-based TADF polymers could lower the energy levels of excited states and induce redshifting of emission, undesired for energy transfer from polymer sensitizers to emitters, especially for blue emitters with wide bandgaps.
Here we demonstrate the design of high-efficiency hyperfluorescent polymers with full-color electroluminescence by using through-space charge transfer (TSCT) polymers as sensitizers and classic fluorescent chromophores as emitters for light emission (Fig. 1a). TSCT polymers42–47 with separated frontier molecular orbitals and small ΔESTs are able to exhibit TADF effect with large kRISC values higher than 106 s−1 (Table S2†), as has been demonstrated in a previous report.43 By incorporating fluorescent chromophores as emitters, this study utilizes singlet excitons generated from these TSCT TADF polymers through the FRET pathway for light emission. Meanwhile, the emission colors of TSCT polymers can be easily and continuously tuned from the deep blue to red region through the substituent effect of the donor or acceptor,43 making it facile to realize good overlap of emission bands with absorption of fluorescent emitters with different bandgaps. By choosing suitable combination of TSCT polymer sensitizers and fluorescent emitters, complete energy transfer from polymers to emitters can be achieved via the FRET process. Solution-processed OLEDs based on hyperfluorescent polymers exhibit blue, green and red emissions with peak wavelengths expanding from 466 to 640 nm, as well as maximum external quantum efficiencies of 10.3–19.2% that are much higher than those of control fluorescent polymers (2.0–3.6%), which sheds light on the potential of hyperfluorescent polymers in developing high-efficiency solution-processed OLEDs.
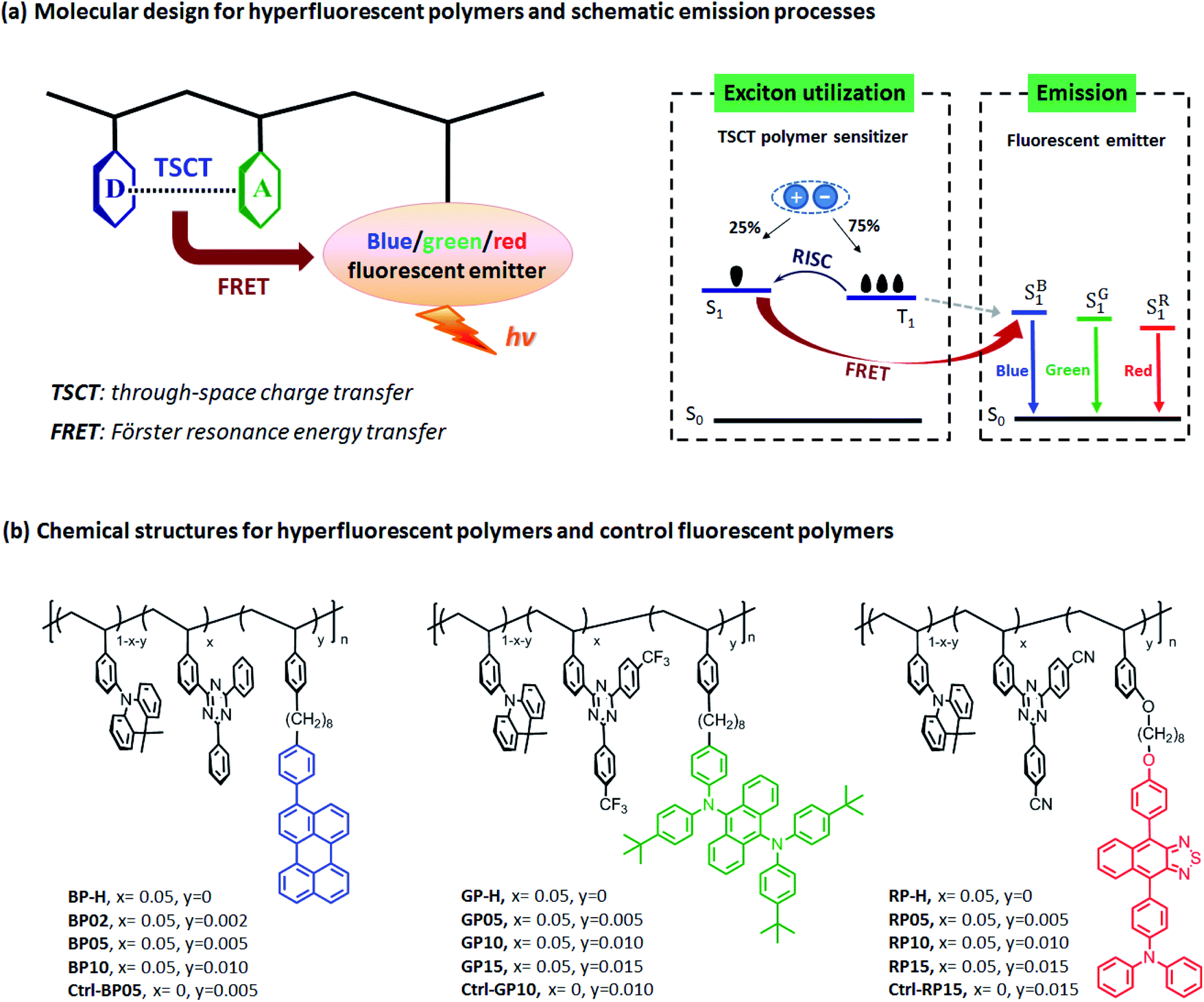 | ||
| Fig. 1 Molecular design and chemical structures of hyperfluorescent polymers employing through-space charge transfer polystyrene sensitizers. (D: donor, A: acceptor, RISC: reverse intersystem crossing, S0: ground state, S1: lowest singlet state, T1: lowest triplet state, S1B, S1G and S1R: lowest singlet states for blue, green and red emitters respectively.) | ||
2. Results and discussion
2.1 Molecular design and synthesis
To design hyperfluorescent polymers, non-conjugated polystyrene-based TSCT polymers (BP-H, GP-H and RP-H, Fig. 1b) containing an acridan donor and triazine acceptors are employed as sensitizers because of their high kRISC rates (3.6–8.5 × 106 s−1, Table S2†) and easily tuned emission wavelengths by regulating charge transfer (CT) strength between the donor and acceptor.43 For full-color emission, three fluorescent chromophores 3-phenylperylene (PhPe),48 tetrakis(4-tert-butylphenyl)anthracene-9,10-diamine (TBPAD)17 and 4-(9-(4-methoxyphenyl)naphthothiadiazol-4-yl)-N,N-diphenylaniline (NTTPA)49 with blue, green and red emission respectively, are selected as emitters because of their high photoluminescence quantum yields (94%, 90% and 75% for PhPe, TBPAD and NTTPA respectively, Table S3†). To guarantee efficient energy transfer from sensitizers to emitters, combinations of TSCT polymers and fluorescent chromophores are screened to ensure overlap between sensitizer emission and emitter absorption. For blue hyperfluorescent polymers (BP02 ∼ BP10) using PhPe as the emitter, BP-H containing the 1,3,5-triphenyltriazine acceptor is used as the sensitizer because it has the bluest emission that can overlap with PhPe absorption. However for green and red hyperfluorescent polymers (GP05 ∼ GP15 and RP05 ∼ RP15) containing TBPAD and NTTPA as emitters respectively, GP-H and RP-H consisting of trifluoromethyl- and cyano-substituted 1,3,5-triphenyltriazine acceptors are selected as sensitizers because their longer emission wavelengths can better overlap with the absorption of TBPAD and NTTPA respectively. All the emitters are incorporated with corresponding TSCT polymers by saturated octyl chains to keep their electronic structures independent to each other. Meanwhile, the calculated centre-to-centre distances from the donor/acceptor pair of the TSCT polymer to the emitter are 20.7, 19.9 and 24.4 Å for blue, green and red hyperfluorescent polymers respectively (Fig. S4†), which are in the range for the FRET process (10–100 Å).50 Moreover, to explore the role of TSCT polymer sensitizers, three control fluorescent polymers (Ctrl-BP05, Ctrl-GP10 and Ctrl-RP15) having only an acridan unit and fluorescent emitter, and thus without a TSCT polymer sensitizer are also designed for comparison.Synthetic procedures for hyperfluorescent polymers and control fluorescent polymers are presented in Schemes S1 and S2.† The hyperfluorescent polymers were synthesized by copolymerization of vinyl functionalized acridan donors, triazine acceptors and corresponding emitter-containing monomers using 2-azoisobutyronitrile (AIBN) as an initiator and tetrahydrofuran (THF) as solvent through free radical polymerization in yields of 63–74%. The mole content of the triazine acceptor in copolymerization was fixed at 5 mol% because this content led to the best device efficiency for these TSCT polymers.42,43 The contents of fluorescent emitters were controlled at relatively low levels (0.2–1.0 mol% for BP02 ∼ BP10, 0.5–1.5 mol% for GP05 ∼ GP15 and RP05 ∼ RP15), aiming to suppress Dexter energy transfer from the sensitizer to emitter to avoid exciton loss.18 The control fluorescent polymers were synthesized by similar copolymerization procedures except that triazine acceptor monomers were not included. The number-average molecular weights (Mn) of the polymers were in the range of 21.4–28.1 kDa with a polydispersity index (PDI) of 1.60–1.99. The polymers showed decomposition temperatures (Td) of 371–375 °C, together with glass transition temperatures (Tg) in the range of 192–195 °C (Fig. S5†), indicative of their good thermal stability. All the polymers can be easily dissolved in common organic solvents such as toluene, dichloromethane, THF and chlorobenzene, favorable for fabrication of OLEDs by a solution process.
2.2 Energy transfer from the sensitizer to emitter
Energy transfer from the sensitizer to emitter plays the critical role in determining the electroluminescence performance of hyperfluorescent OLEDs.10 To explore the energy transfer process for hyperfluorescent polymers, steady-state PL spectra were first measured. As shown in Fig. 2a–c, TSCT polystyrene sensitizers BP-H, GP-H and RP-H exhibit emission bands at 486, 542 and 577 nm respectively, which can overlap with the absorption bands of PhPe, TBPAD and NTTPA respectively. As a result, the corresponding hyperfluorescent polymers exhibit emissions mainly from their fluorescent emitters. For example, the PL spectra of BP05, GP10 and RP15 exhibit emission bands at 466, 533 and 646 nm respectively, with emission profiles similar to those of the corresponding emitters (Fig. 2d–f). As the emitter content increases, the relative emission intensity between the emitter and TSCT polymer increases, indicating that Förster energy transfer from the sensitizer to emitter is more efficient at a higher emitter content.11,51 Meanwhile, it is found that emissions of TSCT polymers are completely quenched at an emitter content of 1.0 mol% for blue and green polymers, but there is still emission residue from the TSCT polymer for red polymers at the same emitter content (Fig. 2g–i). In fact, the FRET integral (JDA) between the absorption of the emitter and the emission of the sensitizer can be calculated using the following equation: | (1) |
 | ||
| Fig. 2 Absorption (Abs) spectra of fluorescent emitters and PL spectra of TSCT polymer sensitizers in toluene solution (1 × 10−5 M) (a–c), the PL spectra of hyperfluorescent polymers, TSCT polymer sensitizers and emitters (d–f), and the PL spectra of hyperfluorescent polymers with different emitter contents in a solid state (g–i). | ||
The energy transfer from the TSCT polymer to fluorescent emitter is also confirmed from transient PL decay spectra. As shown in Fig. 3, the parent TSCT polymers BP-H, GP-H and RP-H exhibit both nanosecond-scale prompt fluorescence and microsecond-scale delayed fluorescence with lifetimes in the range of 36.0–47.0 ns and 1.14–1.28 μs, respectively (Table S7†). By introducing emitters into TSCT polymers, the lifetimes of prompt fluorescence (τp) are reduced to 10.1–12.1, 14.5–17.4 and 18.0–24.4 ns for BP02 ∼ BP10, GP05 ∼ GP15 and RP05 ∼ RP15, respectively. Moreover, as the content of fluorescent emitter increases, the prompt fluorescence lifetimes are shortened. For instance, upon the emitter content increase from 0.2% (BP02) to 1.0% (BP10), τp is decreased from 12.1 ns to 10.1 ns. These observations suggest that, in addition to the ISC process from the S1 to T1 state for TSCT polymers, the FRET process from the S1 state of the TSCT polymer to that of the emitter provides an additional singlet state decay channel. On the other hand, despite all the emitters exhibiting only prompt fluorescence (4.0–14.4 ns), the hyperfluorescent polymers exhibit both prompt and delayed fluorescence upon monitoring the emission of emitters (Fig. S8†), indicating that emissions of fluorescent emitters in hyperfluorescent polymers are derived from the long-lived triplet states of TSCT polymers which are first up-converted to single states and then transferred to the S1 state of emitters. For comparison, the control fluorescent polymers (Ctrl-BP05, Ctrl-GP10 and Ctrl-RP15) exhibit no delayed fluorescence although their PL spectra are similar to those of hyperfluorescent polymers (Fig. S9†), indicating that no triplet states participate in the emission process in this case.
 | ||
| Fig. 3 Transient PL decay characteristics of TSCT polymer sensitizers and hyperfluorescent polymers in toluene solution. (a) BP-H and BP02 ∼ BP10, monitored at 430 nm; (b) GP-H and GP05 ∼ GP15, monitored at 480 nm; (c) RP-H and RP05 ∼ RP15, monitored at 550 nm. | ||
To gain insight into the energy transfer process, FRET rate constants (kFRET) are calculated for the hyperfluorescent polymers. In general, the lifetime of prompt fluorescence for sensitizers (τSensp) can be defined as
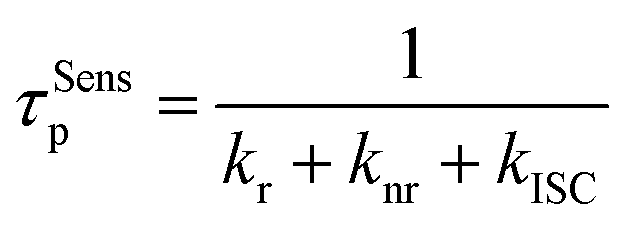 | (2) |
 | (3) |
Therefore kFRET can be calculated as54
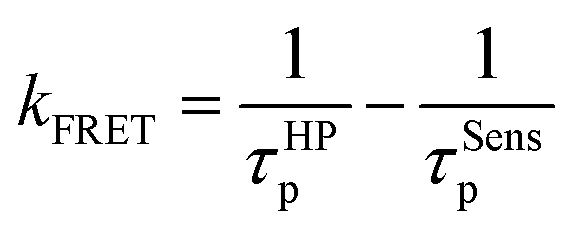 | (4) |
According to these equations, kFRET can be calculated from the lifetime of prompt fluorescence for the TSCT polymer and hyperfluorescent polymer. As listed in Table S7,†kFRET for BP02 is 5.5 × 107 s−1, which increased to 6.3 × 107 s−1 and 7.2 × 107 s−1 for BP05 and BP10 respectively. Similarly, kFRET for the green polymer increased from 3.4 × 107 s−1 for GP05 to 3.8 × 107 s−1 for GP10 and 4.6 × 107 s−1 for GP15, while that for the red polymer increased from 2.0 × 107 s−1 for RP05 to 2.6 × 107 s−1 for RP10 and 3.4 × 107 s−1 for RP15. These kFRET values are comparable with those of small-molecule TADF-sensitized fluorescence emitters through vacuum deposition,11 supporting the efficient FRET process in hyperfluorescent polymers.
2.3 Electroluminescent properties
To investigate the electroluminescence (EL) performance of hyperfluorescent polymers, solution-processed OLEDs were fabricated with device configuration of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS, 40 nm)/polymer (40 nm)/TSPO1 (8 nm)/TmPyPB (42 nm)/LiF (1 nm)/Al (100 nm) (Fig. 4a). Herein TSPO1 stands for diphenyl(4-(triphenylsilyl)phenyl)-phosphine oxide which is an exciton blocking layer,55 while TmPyPB is 1,3,5-tri(m-pyrid-3-yl-phenyl) benzene which acts as an electron-transporting layer.56 As shown in Fig. 4b, the EL spectra of the hyperfluorescent polymers are similar to the PL spectra of the corresponding emitters, indicating that the electroluminescence of the polymers is coming from the fluorescent emitter. The polymers containing PhPe as an emitter exhibit blue emissions with peaks located at 466, 466 and 467 nm, giving CIE coordinates of (0.16, 0.23), (0.16, 0.24) and (0.16, 0.25) for BP02, BP05 and BP10 respectively. However the TBPAD-based polymers GP05 ∼ GP15 exhibit green emissions at 529–530 nm with CIE coordinates of (0.34, 0.58) for GP05, (0.34, 0.59) for GP10 and (0.34, 0.59) for GP15. For NTTPA-containing polymers (RP05 ∼ RP15), the emissions are at 590–640 nm, with the emission residue from TSCT polymer RP-H observed for RP05 and RP10. The polymer RP15 with a higher NTTPA content exhibits red emission mainly from the emitter, accompanied by the CIE coordinates of (0.60, 0.38). Notably, the hyperfluorescent polymers reveal stable EL spectra at different driving voltages. As shown in Fig. 5, upon increasing the voltage from 4 V to 8 V, the EL spectra of BP05, GP10 and RP15 almost remained unchanged, with CIE variations less than (±0.01, ±0.01). | ||
| Fig. 4 Device configuration and energy level alignment diagram (a), EL spectra (b), J–V–L (c–e) and EQE-L characteristics (f–h) for solution-processed OLEDs based on hyperfluorescent polymers. | ||
 | ||
| Fig. 5 EL spectra for solution-processed OLEDs based on BP05, GP10 and RP15 at different driving voltages. | ||
The current density (J) – voltage (V) – luminance (L) characteristics for the solution-processed OLEDs are shown in Fig. 4c–e and S10–S12.† The devices show low turn-on voltages (Von) of 2.8–3.2 V at a luminance of 1 cd m−2, indicating smooth carrier injection from adjacent layers to hyperfluorescent polymers. Moreover, Vons are independent of emitter contents of polymers, suggesting that energy transfer from the TSCT polymer to emitter, rather than direct charge trapping by the emitter, plays a dominant role in exciton formation under electrical excitation.51 This observation is consistent with energy level alignment for TSCT polymers and emitters. As shown in Fig. 4a, the HOMO levels of PhPe and NTTPA are lower than those of the corresponding TSCT polymers (BP-H and RP-H), while the LUMO level of TBPAD is higher than that of GP-H. That is, energy levels of emitters are not located in the gap between the HOMO and LUMO of TSCT polymers, which can avoid charge trapping by emitters.57 The absence of direct charge trapping by the emitter is crucial for hyperfluorescence because the direct formation of excitons (including 75% triplet excitons) on fluorescent emitters, which is an exciton loss pathway, can be avoided.18
The dependence of device efficiency on luminance for the polymers is shown in Fig. 4f–h, and summarized in Table 1. The blue polymer BP02 containing 0.2 mol% PhPe emitter reveals the maximum LE of 24.4 cd A−1 and maximum EQE of 13.5%, which are enhanced to 26.6 cd A−1 and 14.6% respectively as the emitter content increased to 0.5 mol% for BP05. Further increasing the emitter content to 1.0 mol%, however, leads to lower device efficiency with a maximum LE/EQE of 23.3 cd A−1/12.2%. For green and red hyperfluorescent polymers, the optimized device efficiencies are obtained at emitter contents of 1.0 mol% (GP10) and 1.5 mol% (RP15) respectively. In particular, GP10 exhibits a remarkable maximum LE of 66.5 cd A−1 and EQE of 19.2%. Meanwhile, the efficiency roll-off for GP10 at high luminance is small, with LE/EQE remained at 66.5 cd A−1/19.2% at 100 cd m−2, and 60.3 cd A−1/17.4% at 1000 cd m−2. RP15 reveals a maximum LE/EQE of 10.6 cd A−1/10.3%, which decreased to 9.4 cd A−1/9.2% and 5.0 cd A−1/4.8% at luminance values of 100 and 1000 cd m−2 respectively. It is worth noting that the efficiencies of hyperfluorescent polymers are much superior to those of control fluorescent polymers based on the conventional fluorescence emission mechanism. For instance, the maximum EQE of GP10 (19.2%) is 5.3 fold higher than that of Ctrl-GP10 (3.6%). Similarly, the EQEs of BP05 and RP15 are enhanced by 4.1–4.9 folds relative to their corresponding control fluorescent polymers (Ctrl-BP05 and Ctrl-RP15). The much higher EQEs of hyperfluorescent polymers are in accordance with their capability to utilize triplet exciton through a rapid RISC process. In general, the EQE for an OLED is expressed as
| EQE = IQE × ηout = γ × ηST × ηPL × ηout | (5) |
| Polymers | V on (V) | LEb (cd A−1) | EQEc (%) | L max (cd m−2) | CIEe (x,y) | |
|---|---|---|---|---|---|---|
| Maximum value/@100 cd m−2/@1000 cd m−2 | ||||||
| a Voltage at 1 cd m−2. b Luminous efficiency. c External quantum efficiency. d Maximum luminance. e CIE coordinates at 6 V. | ||||||
| Blue | BP02 | 3.2 | 24.4/23.0/17.0 | 13.5/13.0/9.4 | 5860 | 0.16, 0.23 |
| BP05 | 3.2 | 26.6/25.2/19.0 | 14.6/13.9/10.5 | 6367 | 0.16, 0.24 | |
| BP10 | 3.2 | 23.3/22.8/16.0 | 12.2/11.9/8.3 | 6379 | 0.16, 0.25 | |
| Ctrl-BP05 | 3.6 | 5.7/5.4/2.7 | 3.0/2.9/1.4 | 1879 | 0.18, 0.25 | |
| Green | GP05 | 2.8 | 61.2/61.2/55.0 | 18.2/18.2/16.3 | 14280 |
0.34, 0.58 |
| GP10 | 2.8 | 66.5/66.5/60.3 | 19.2/19.2/17.4 | 16061 |
0.34, 0.59 | |
| GP15 | 2.8 | 62.3/62.0/55.1 | 17.6/17.5/15.6 | 16408 |
0.34, 0.59 | |
| Ctrl-GP10 | 3.2 | 13.1/11.8/5.2 | 3.6/3.3/1.4 | 1337 | 0.30, 0.59 | |
| Red | RP05 | 3.0 | 20.7/20.0/13.0 | 8.8/8.4/5.4 | 8858 | 0.50, 0.48 |
| RP10 | 3.0 | 18.0/15.1/8.9 | 9.6/8.1/4.8 | 6373 | 0.54, 0.44 | |
| RP15 | 3.0 | 10.6/9.4/5.0 | 10.3/9.2/4.8 | 4603 | 0.60, 0.38 | |
| Ctrl-RP15 | 3.8 | 2.3/1.1/— | 2.0/1.0/— | 330 | 0.56, 0.34 | |
3. Conclusion
Hyperfluorescent polymers have been developed by incorporating through-space charge transfer polystyrenes as sensitizers and fluorescent chromophores as emitters in one macromolecule for achieving efficient full-color electroluminescence by a solution process. The TSCT polystyrene sensitizers play a role in harvesting triplet excitons by converting them into singlet excitons via a rapid RISC process and then transferring energy to fluorescent emitters. By tuning the emission wavelength of TSCT polymer sensitizers to overlap the absorption of fluorescent emitters with different bandgaps, efficient FRET from the sensitizer to emitter can be achieved. Solution-processed OLEDs based on hyperfluorescent polymers exhibit full-color emissions covering blue, green and red regions, as well as high maximum EQEs of 10.3–19.2% that are much superior to those of control fluorescent polymers having no TSCT polymer sensitizers (EQE: 2.0–3.6%). This work sheds light on the potential of hyperfluorescent polymers for overcoming the internal quantum efficiency limitation of classical fluorescent polymers under electrical excitation, and provides an approach to develop high-efficiency solution-processed OLEDs based on pure organic materials.4. Experimental section
4.1 Synthesis of the polymers
The polymers were synthesized by free radical polymerization using azodiisobutyronitrile (AIBN) as the radical initiator and THF as the solvent. Specifically, the vinyl-functionalized acridan, triazine and emitter-containing monomers and AIBN (2 mol% of the total amount of monomers) were dissolved in THF with a total concentration of ∼0.3 g mL−1, which were then stirred at 50 °C for 48 hours under an argon atmosphere. After cooling to room temperature, the mixture was precipitated into cold acetone and methanol subsequently. The mixture was filtered and dried under vacuum to give the desired polymers in yields of 63–74% (see the ESI†).4.2 Fabrication and characterization of solution-processed white organic light-emitting diodes
Glass substrates coated with indium tin oxide (ITO) (15 Ω per square) were cleaned by ultrasound in acetone, isopropanol and water, which were then treated by ultraviolet-ozone for 25 minutes. Subsequently PEDOT:PSS (Clevious P Al4083) was spin-coated on the substrate to form the hole-injection layer with a thickness of 40 nm, which was then dried at 120 °C for 45 min. After the substrate was transferred to a N2-filled glove box, solution of the hyperfluorescent polymer dissolved in chlorobenzene (10 mg L−1) was spin-coated with a thickness of 40 nm. After annealing the polymer film at 100 °C for 30 min, the substrate was transferred to a vacuum chamber with pressure less than 4 × 10−4 Pa. TSPO1 and TmPyPB were then successively evaporated on top of the polymer film with thicknesses of 8 nm and 42 nm respectively. Finally, LiF (1 nm) and Al (100 nm) were deposited for use as a cathode through a shadow mask with an emissive area of 3.5 × 4.0 mm2. The J–V–L characteristics of the device were measured under an ambient atmosphere at room temperature using a Keithley 2400/2000 source meter equipped with a calibrated silicon photodiode. The electroluminescence spectra and CIE coordinates were tested with a PR650 spectra colorimeter.Data availability
Data associated with this article, including experimental procedures and compound characterization, are available in the ESI.†Author contributions
J. Hu, Y. Wang and Q. Li synthesized the polymers. J. Hu carried out the photophysical property measurements as well as fabrication and characterization of solution-processed OLEDs. S. Shao and L. Wang conceived the idea, designed the experiments, and wrote the manuscript. L. Wang supervised and directed this study. X. Jing and F. Wang initiated the research project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 51833009 and 52073282), the Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm2021017) and the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2015180).References
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed.
- F. Huang, Z. Bo, Y. Geng, X. Wang, L. Wang, Y. Ma, J. Hou, W. Hu, J. Pei, H. Dong, S. Wang, Z. Li, Z. Shuai, Y. Li and Y. Cao, Acta Polym. Sin., 2019, 50, 988 CAS.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- S. Jhulki, M. W. Cooper, S. Barlow and S. R. Marder, Mater. Chem. Front., 2019, 3, 1699 RSC.
- F. Xu, H. U. Kim, J.-H. Kim, B. J. Jung, A. C. Grimsdale and D.-H. Hwang, Prog. Polym. Sci., 2015, 47, 92 CrossRef CAS.
- Q. Wei, Z. Y. Ge and B. Voit, Macromol. Rapid Commun., 2019, 40, 1800570 CrossRef.
- Y. Zou, S. L. Gong, G. H. Xie and C. L. Yang, Adv. Opt. Mater., 2018, 6, 1800568 CrossRef.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS.
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050 CrossRef CAS.
- T. Higuchi, H. Nakanotani and C. Adachi, Adv. Mater., 2015, 27, 2019 CrossRef CAS.
- D. Chen, X. Cai, X. L. Li, Z. He, C. Cai, D. Chen and S. J. Su, J. Mater. Chem. C, 2017, 5, 5223 RSC.
- D. H. Ahn, J. H. Jeong, J. Song, J. Y. Lee and J. H. Kwon, ACS Appl. Mater. Interfaces, 2018, 10, 10246 CrossRef CAS.
- S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 1504 RSC.
- S. K. Jeon, H. J. Park and J. Y. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 5700 CrossRef CAS.
- D. Zhang, X. Song, M. Cai and L. Duan, Adv. Mater., 2018, 30, 1705250 CrossRef PubMed.
- S. Y. Byeon, D. R. Lee, K. S. Yook and J. Y. Lee, Adv. Mater., 2019, 31, 1803714 CrossRef PubMed.
- D. Liu, K. Sun, G. Zhao, J. Wei, J. Duan, M. Xia, W. Jiang and Y. Sun, J. Mater. Chem. C, 2019, 7, 11005 RSC.
- X. Song, D. Zhang, H. Li, M. Cai, T. Huang and L. Duan, ACS Appl. Mater. Interfaces, 2019, 11, 22595 CrossRef CAS PubMed.
- H. Abroshan, Y. Zhang, X. Zhang, C. Fuentes-Hernandez, S. Barlow, V. Coropceanu, S. R. Marder, B. Kippelen and J. L. Bredas, Adv. Funct. Mater., 2020, 30, 2005898 CrossRef CAS.
- L. S. Cui, A. J. Gillett, S. F. Zhang, H. Ye, Y. Liu, X. K. Chen, Z. S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J. L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636 CrossRef CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643 CrossRef CAS.
- C. Zhang, Y. Lu, Z. Liu, Y. Zhang, X. Wang, D. Zhang and L. Duan, Adv. Mater., 2020, 32, 2004040 CrossRef CAS.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Adv. Mater., 2020, 32, 1908355 CrossRef CAS.
- C. Y. Chan, M. Tanaka, Y. T. Lee, Y. W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203 CrossRef CAS.
- S. O. Jeon, K. H. Lee, J. S. Kim, S. G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208 CrossRef CAS.
- A. M. Polgar, C. M. Tonge, C. J. Christopherson, N. R. Paisley, A. C. Reyes and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 38602 CrossRef CAS.
- A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad and M. Roberts, Adv. Mater., 2015, 27, 7236 CrossRef CAS PubMed.
- S. Y. Lee, T. Yasuda, H. Komiyama, J. Lee and C. Adachi, Adv. Mater., 2016, 28, 4019 CrossRef CAS PubMed.
- J. J. Luo, G. H. Xie, S. L. Gong, T. H. Chen and C. L. Yang, Chem. Commun., 2016, 52, 2292 RSC.
- R. S. Nobuyasu, Z. J. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. K. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597 CrossRef CAS.
- Z. J. Ren, R. S. Nobuyasu, F. B. Dias, A. P. Monkman, S. Yan and M. R. Bryce, Macromolecules, 2016, 49, 5452 CrossRef CAS.
- Q. Wei, P. Kleine, Y. Karpov, X. P. Qiu, H. Komber, K. Sahre, A. Kiriy, R. Lygaitis, S. Lenk, S. Reineke and B. Voit, Adv. Funct. Mater., 2017, 27, 1605051 CrossRef.
- G. H. Xie, J. J. Luo, M. L. Huang, T. H. Chen, K. L. Wu, S. L. Gong and C. L. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef PubMed.
- Y. Y. Hu, W. Q. Cai, L. Ying, D. J. Chen, X. Y. Yang, X. F. Jiang, S. J. Su, F. Huang and Y. Cao, J. Mater. Chem. C, 2018, 6, 2690 RSC.
- H. J. Kim, C. Lee, M. Godumala, S. Choi, S. Y. Park, M. J. Cho, S. Park and D. H. Choi, Polym. Chem., 2018, 9, 1318 RSC.
- C. S. Li, Z. J. Ren, X. L. Sun, H. H. Li and S. K. Yan, Macromolecules, 2019, 52, 2296 CrossRef CAS.
- X. Zhou, M. L. Huang, X. Zeng, T. H. Chen, G. H. Xie, X. J. Yin and C. L. Yang, Polym. Chem., 2019, 10, 4201 RSC.
- Y. Liu, L. Hua, S. Yan and Z. Ren, Nano Energy, 2020, 73, 104800 CrossRef CAS.
- J. C. Rao, X. R. Liu, X. F. Li, L. Q. Yang, L. Zhao, S. M. Wang, J. Q. Ding and L. X. Wang, Angew. Chem., Int. Ed., 2020, 59, 1320 CrossRef CAS.
- S. Y. Shao, J. Hu, X. D. Wang, L. X. Wang, X. B. Jing and F. S. Wang, J. Am. Chem. Soc., 2017, 139, 17739 CrossRef CAS PubMed.
- J. Hu, Q. Li, X. D. Wang, S. Y. Shao, L. X. Wang, X. B. Jing and F. S. Wang, Angew. Chem., Int. Ed., 2019, 58, 8405 CrossRef CAS PubMed.
- C. M. Tonge and Z. M. Hudson, J. Am. Chem. Soc., 2019, 141, 13970 CrossRef CAS PubMed.
- Q. Li, J. Hu, J. Lv, X. Wang, S. Shao, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 20174 CrossRef CAS PubMed.
- S. Shao and L. Wang, Aggregate, 2020, 1, 45 CrossRef.
- J. Poisson, C. M. Tonge, N. R. Paisley, E. R. Sauvé, H. McMillan, S. V. Halldorson and Z. M. Hudson, Macromolecules, 2021, 54, 2466 CrossRef CAS.
- X. Cui, A. Charaf-Eddin, J. Wang, B. Le Guennic, J. Zhao and D. Jacquemin, J. Org. Chem., 2014, 79, 2038 CrossRef CAS PubMed.
- L. Chen, H. Tong, Z. Xie, L. Wang, X. Jing and F. Wang, J. Mater. Chem., 2011, 21, 15773 RSC.
- L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Chem. Soc. Rev., 2020, 49, 5110 RSC.
- X. Song, D. Zhang, Y. Zhang, Y. Lu and L. Duan, Adv. Opt. Mater., 2020, 8, 2000483 CrossRef CAS.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- D. Zhang, C. Zhao, Y. Zhang, X. Song, P. Wei, M. Cai and L. Duan, ACS Appl. Mater. Interfaces, 2017, 9, 4769 CrossRef CAS PubMed.
- M. Mamada, S. Ergun, C. Perez-Bolivar and P. Anzenbacher Jr, Appl. Phys. Lett., 2011, 98, 073305 CrossRef.
- S. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125 CrossRef CAS.
- H. Li, C. Li, L. Duan and Y. Qiu, Isr. J. Chem., 2014, 54, 918 CrossRef CAS.
- G. Gomard, J. B. Preinfalk, A. Egel and U. Lemmer, J. Photonics Energy, 2016, 6, 030901 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Measurements and characterization details, synthetic procedures and supplementary physical properties of the polymers. See DOI: 10.1039/d1sc04389g |
| This journal is © The Royal Society of Chemistry 2021 |