DOI:
10.1039/D1SC04296C
(Edge Article)
Chem. Sci., 2021,
12, 13037-13044
Substituted aromatic pentaphosphole ligands – a journey across the p-block†
Received
5th August 2021
, Accepted 3rd September 2021
First published on 14th September 2021
Abstract
The functionalization of pentaphosphaferrocene [Cp*Fe(η5-P5)] (1) with cationic group 13–17 electrophiles is shown to be a general synthetic strategy towards P–E bond formation of unprecedented diversity. The products of these reactions are dinuclear [{Cp*Fe}2{μ,η5:5-(P5)2EX2}][TEF] (EX2 = BBr2 (2), GaI2 (3), [TEF]− = [Al{OC(CF3)3}4]−) or mononuclear [Cp*Fe(η5-P5E)][X] (E = CH2Ph (4), CHPh2 (5), SiHPh2 (6), AsCy2 (7), SePh (9), TeMes (10), Cl (11), Br (12), I (13)) complexes of hetero-bis-pentaphosphole ((cyclo-P5)2R) or hetero-pentaphosphole ligands (cyclo-P5R), the aromatic all-phosphorus analogs of prototypical cyclopentadienes. Further, modifying the steric and electronic properties of the electrophile has a drastic impact on its reactivity and leads to the formation of [Cp*Fe(μ,η5:2-P5)SbICp′′′][TEF] (8) which possesses a triple-decker-like structure. X-ray crystallographic characterization reveals the slightly twisted conformation of the cyclo-P5R ligands in these compounds and multinuclear NMR spectroscopy confirms their integrity in solution. DFT calculations shed light on the bonding situation of these compounds and confirm the aromatic character of the pentaphosphole ligands on a journey across the p-block.
Introduction
Electrophilic aromatic substitution is one of the most basic and widely applied reactions for the functionalization of aromatic organic compounds. While Friedel–Crafts alkylation and acylation are in fact textbook examples for the reactivity of benzene derivatives,1 they can also be applied to smaller ring systems.2 In contrast, derivatives of the carbocyclic aromatic cyclopentadienide anion (Cp−, C5H5−) form non-aromatic cyclopentadiene derivatives (CpR, C5H5R) upon salt metathesis with element halogenides. Compared to their alkali metal salts, transition metal (TM) bound Cp− ligands (e.g. in Cp2Fe)3 exhibit a different reactivity towards electrophiles mimicking that of benzene derivatives (e.g. (C6H6)Cr(CO)3).4 The isolobal relationship between the CH fragment and the P atom5 and the diagonal relationship between carbon and phosphorus suggest a comparable reactivity for the pentaphospholide anion cyclo-P5−.6 However, investigations on the reactivity of the salts of cyclo-P5− towards alkylhalogenides showed further aggregation to polyphosphides,7 leaving the chemistry of pentaphospholes cyclo-P5R to theoretical studies for decades.8 Similar to Cp−, cyclo-P5− can be stabilized within the coordination sphere of different TMs9 and is thus closely associated with the TM-mediated conversion of P4.10 One of the most prominent examples of such complexes is pentaphosphaferrocene [Cp*Fe(η5-P5)] (1).9a While 1 readily reacts with various monovalent metal salts to form coordination compounds,11 we could also demonstrate both its redox reactivity12 and its behavior towards anionic13 and neutral nucleophiles.14 These reactions yielded complexes with bent cyclo-P5R ligands (I, Scheme 1),13,14 which are also accessible by the reaction of a niobium phosphorus triple bond complex with P4.15 Disubstituted cyclo-P5R2 moieties (II and III, Scheme 1) could be obtained via the coordination of the respective [P5R2]+ cations16 to low-valent transition metal fragments.17 The formation of the structural motif II was also observed upon condensation of an anionic cyclo-P4 complex with chlorophosphines.18 The introduction of germylene substituents has recently been achieved by the reaction of 1 with a digermylene.19 While their structural motifs are remarkable, the respective cyclo-P5Rn ligands (n = 1, 2) within types I–III do not show any aromatic character, since they do not represent pentaphospholes (IV).8 Only recently, we succeeded in the synthesis of the first transition metal complexes [Cp*Fe(η5-P5R)][B(C6F5)4] (IV, R = H, Me, SiEt3) featuring such pentaphosphole ligands.20 While our previous approach is well-suited to prepare the parent compound with a cyclo-P5H ligand, it is limited to group 14 electrophiles (such as CH3/SiEt3) as introducible substituents.21
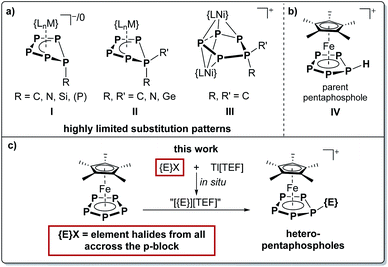 |
| | Scheme 1 (a) Known substituted cyclo-P5 ligand architectures I ({LnM} = {(MesneoPentN)2Nb} or {Cp*Fe}), II ({LnM} = {Cp*Fe}, {CpPEtCo}+, {(BIAN)Co}), III (L = CpPEt); (b) the parent pentaphosphole complex IV; (c) targeted functionalization of an iron bound pentaphospholide ion with in situ generated cationic electrophiles from across the p-block to yield unprecedented coordinatively stabilized hetero-pentaphospholes. | |
Thus, we sought a more general strategy to electrophilically functionalize 1 which may also be applicable beyond the synthesis of the targeted hetero-pentaphosphole complexes to other heteroaromatic ligands. Such a strategy would represent a valuable contribution to electrophilic substitution reactions in general. We hypothesized that the in situ generation of cationic electrophiles from p-block element halogenides and a suitable halide-abstracting reagent, as has for example been utilized for the functionalization of coordinated chlorophosphines,22 and subsequent reaction with 1 could provide this reactivity. Such an approach would allow P–E bond formation with a nearly unlimited choice of electrophiles and would thus overcome the limitations of known synthetic strategies towards the P–E bond formation in the vicinity of transition metals.14,15,23 Indeed, the major drawback of the so far used strategies is their immensely limited applicability to a very narrow range of electrophile/nucleophile combinations. This is usually reflected in the use of rather exotic low-valent main group species such as tetrylenes,14b,18 {Cp*Al}4,24 or the utilization of highly reactive, e.g. anionic, polyphosphorus ligand complexes.23e,f While our goal is the functionalization of 1 to obtain unprecedented hetero-pentaphosphole ligands, namely the aromatic all-P congeners of cyclopentadienes, we expect our approach to also be applicable to polyphosphorus ligand complexes of various sizes. Furthermore, this idea merges the functionalization of TM polyphosphorus complexes with the concept of electrophilic substitution,2 a classic organic or organometallic (for e.g. Cp2Fe (ref. 3) or (C6H6)Cr(CO)3 (ref. 4)) reaction. Thus, it may be transferred back to these fields and allow the electrophilic functionalization of still challenging substrates such as multifunctionalized CpR derivatives.25 To achieve the in situ generation of the desired electrophiles from the respective halogenides, Tl[TEF] ([TEF]− = [Al{OC(CF3)3}4]−)26 was chosen to facilitate efficient halide abstraction and simultaneously introduce the weakly coordinating [TEF]− anion. However, for some cases, we also provide an easily accessible alternative avoiding Tl+ salts by the reaction of simple element hydrides with [Ph3C][B(C6F5)4] for in situ electrophile generation. This method has been employed e.g. for the generation of the highly reactive silylium ion precursor [(Et3Si)2(μ-H)][B(C6F5)4].21 However, the question arises as to how broad, if successful, the applicability of this approach would actually be, and which electrophiles could be introduced by employing it. We herein report on a synthetic strategy allowing the functionalization and P–E bond formation of 1 with twelve distinct cationic electrophiles all across the p-block elements and compare their structural and electronic properties.
Results and discussion
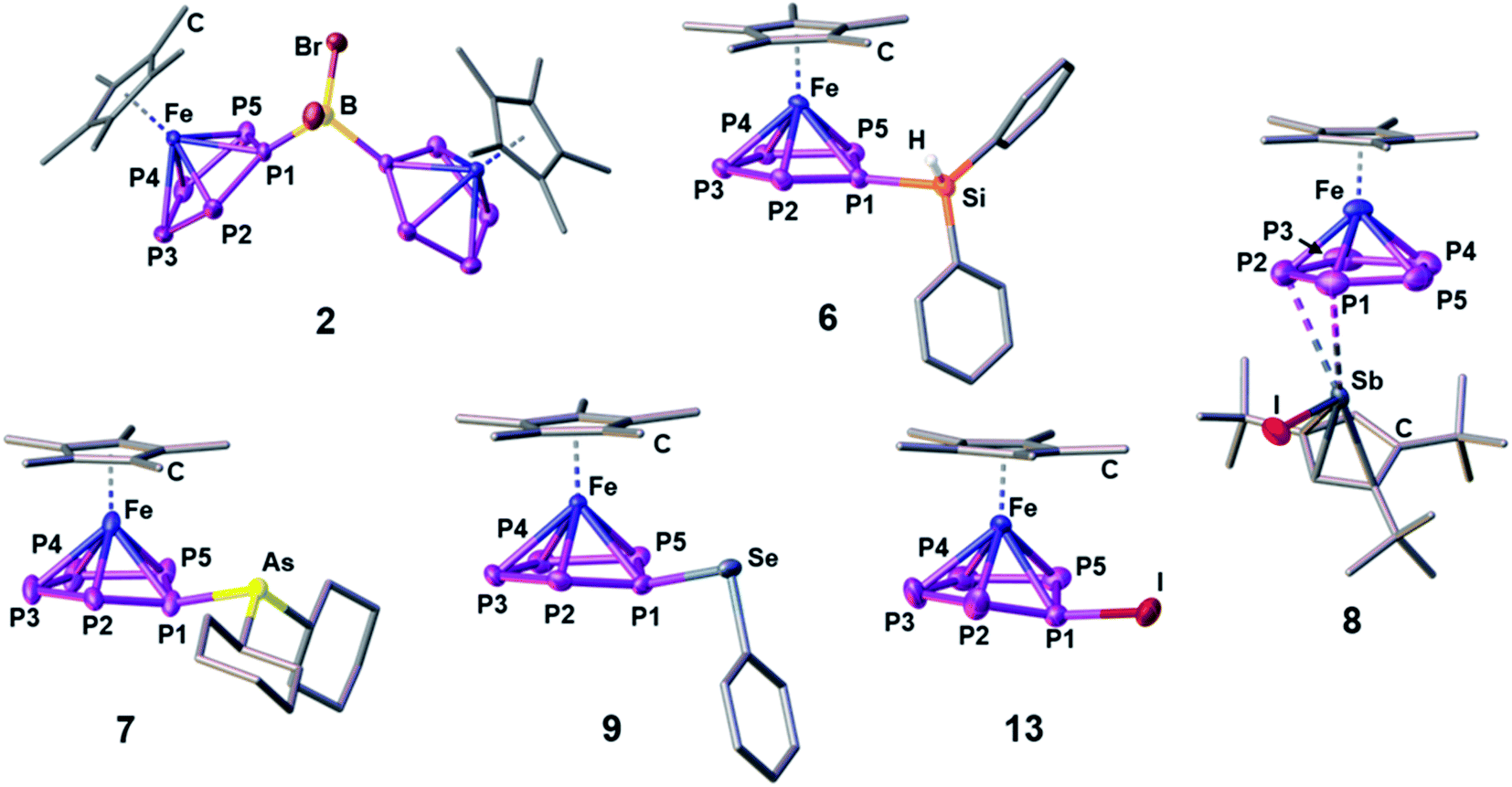
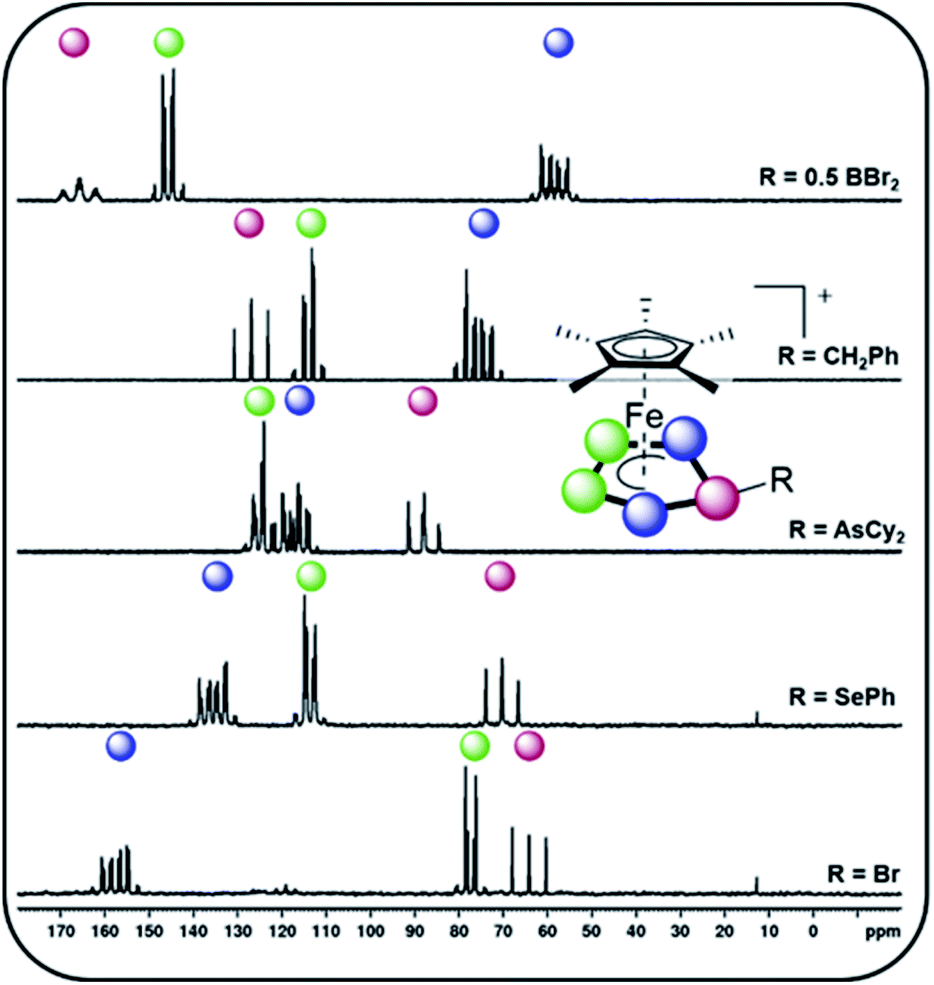
Starting with group 13 electrophiles, we investigated the reactivity of 1 towards BBr3 in the presence of Tl[TEF]. However, performing the reaction in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometry leads to a reaction of the in situ prepared borinium cation [BBr2]+ with the solvent, resulting in a mixture of products (see ESI†). Repeating the reaction in the presence of a second equivalent of 1 afforded the bimetallic compound [{Cp*Fe}2{μ,η5:5-(P5)2BBr2}][TEF] (2), featuring an unprecedented{(cyclo-P5)2BBr2} ligand (Scheme 2). After workup, 2 could be isolated in 70% yield. To our pleasure, exchanging BBr3 for the heavier analog GaI3 affords the isostructural compound [{Cp*Fe}2{μ,η5:5-(P5)2GaI2}][TEF] (3) in 32% yield. While we had already been able to demonstrate the silylation and methylation of 1,20 we now wanted to broaden the scope of group 14 functionalized pentaphosphole derivatives, by using better accessible and easier-to-handle electrophile precursors. In both cases, the reaction of 1 with PhCH2Br and Ph2CHCl in the presence of Tl[TEF] affords the pentaphosphole complexes [Cp*Fe(η5-P5CH2Ph)][TEF] (4) and [Cp*Fe(η5-P5CHPh2)][TEF] (5) in a yield of 45% and 80%, respectively. To demonstrate an easily accessible alternative pathway towards the electrophile generation avoiding the use of Tl+ salts, we chose to react 1 with Ph2SiH2 in the presence of one equivalent of [Ph3C][B(C6F5)4]. A rapid reaction is indicated by the color change of the solution from green to greenish red and, after simple workup, the product [Cp*Fe(η5-P5SiHPh2)][B(C6F5)4] (6) could be isolated in an astonishing 85% yield. In principle, it should be possible to prepare phosphino-pentaphosphole complexes similarly well as they benefit from the additional stabilization caused by P–P bond formation. However, attempts to prepare such phosphino-functionalized species are seriously affected by the inherently high tendency of halogenophosphines to form phosphinophosphonium ions under Lewis acidic conditions.27 We were able to observe the desired cation [Cp*Fe(η5-P5PBr2)]+ spectroscopically only after reacting 1 with PBr3 and Tl[TEF] at −80 °C (see ESI†). This cation, however, undergoes rapid fragmentation and rearrangement processes above −60 °C, affording a complex mixture of several polyphosphorus compounds and thus prohibiting its isolation even at low temperatures. In contrast, the reaction of 1, Cy2AsBr and Tl[TEF] proceeds smoothly, and from the filtered solution, the arsino-pentaphosphole complex [Cp*Fe(η5-P5AsCy2)][TEF] (7) could be isolated in 68% yield. When both the electronic and steric properties of the pnictogenium cation were changed and 1 was reacted with Cp′′′SbI2 (Cp′′′ = 1,2,4-tBu3C5H2) and Tl[TEF], the unique triple-decker-like complex [Cp*Fe(μ,η5:2-P5)SbICp′′′][TEF] (8) with a P5 middle deck could be obtained in 24% yield. The molecular structure of 8 may provide insight into the mechanism of the electrophilic functionalization of 1 which initially seems to occur through the π-system and not by the lone pairs of one of the P atoms of the cyclo-P5 ligand. Functionalization of 1 with group 16 electrophiles could be achieved by the reaction with Tl[TEF] and PhSeBr or MesTeBr, respectively. The products [Cp*Fe(η5-P5ER)][TEF] (9: E = Se, R = Ph; 10: E = Te, R = Mes) could be isolated in 65% and 57% yield, respectively, and reveal novel seleno- or telluro-pentaphosphole ligands. To date, there is no synthetic pathway for a rational and selective monohalogenation of unsubstituted polyphosphorus frameworks, although the products would provide both insight into the mechanism of Pn halogenation reactions28 and a high potential for further functionalization. After recently investigating the complex iodination chemistry of 1 (ref. 29) and of the diphosphorus complex [{CpMo(CO)2}2(μ,η2:2-P2)],30 we were wondering if electrophilic halogen transfer could lead to the desired reactivity. Indeed, when a solution of [PCl4][TEF] is added to 1 in o-DFB, the NMR spectra of the crude solution indicate the clean conversion to [Cp*Fe(η5-P5Cl)][TEF] (11) and PCl3. After workup, 11 can be isolated in 65% yield. Similarly, the addition of X2 to mixtures of 1 and Tl[TEF] leads to the formation of [Cp*Fe(η5-P5X)][TEF] (12: X = Br; 13: X = I), which could be isolated in 65% and 62% yield, respectively. Notably, these halogenation reactions can be scaled up to at least 2 mmol, allowing the gram scale preparation of 11–13, which opens broad perspectives for their further functionalization. While the starting material 1 is comparably robust, the pentaphosphole complexes 2–13 are highly sensitive towards both moisture and air, while retaining decent thermal stability. All compounds 2–13 can be crystallized from mixtures of o-DFB or CH2Cl2 and n-hexane either at room temperature or at −30 °C. However, incommensurate modulation (11) or extreme disorder of anion and cation (12) within 11 and 12 prohibit a satisfactory refinement of their crystal structure. Compound 8 reveals a triple-decker-like arrangement (Fig. 1) of the cation [Cp*Fe(μ,η5:2-P5)SbICp′′′]+ with elongated P–Sb interactions (3.236(2)/3.400(2) Å) and a planar P5 ligand. The cations in 4–7 and 9–13 show the anticipated pentaphosphole complex structure with a slightly bent cyclo-P5R ligand (Fig. 1). In contrast, the cations in 2 and 3 are dinuclear complexes in which two {Cp*Fe} units are bridged by the respective {(cyclo-P5)2EX2} (2: E = B, X = Br, 3: E = Ga, X = I) ligand. The P–P bond lengths within 2–13 are similar and in between the sum of the covalent radii of P–P single and double bonds.31 As observed for the parent compound [Cp*Fe(η5-P5H)]+,20 the substituents in the hetero-pentaphosphole complexes 2–7 and 9–13 are oriented in exo-fashion regarding the envelope of the P5 ring (Fig. 1). The respective P–E bond lengths (2: 1.985(7) Å, 4: 1.853(4) Å, 5: 1.866(4) Å, 7: 2.348(1) Å, 9: 2.2234(7) Å, 10: 2.438(2) Å, 13: 2.385(1) Å) are in the range of single bonds, whereas those in 3 (2.410(1)/2.387(1) Å) and 6 (2.3053(8) Å) are elongated compared to the sum of the respective covalent radii (P–Ga: 2.35 Å, P–Si: 2.27 Å).31 The slight folding of the P5 ring in these pentaphosphole complexes is far less pronounced than in anionic or neutral substituted cyclo-P5R ligands possessing an envelope structure (type I, Scheme 1).13,14 Interestingly, this slight folding, represented by the pyramidalization at the P1 atom (359.94°(2) – 350.41°(13)), gradually increases when going from group 13 to group 17 substituents at the pentaphosphole ligand. To elaborate the molecular structure of the obtained pentaphosphole complexes 2–13 in CD2Cl2 solution, they were investigated by multinuclear NMR spectroscopy. In general, all of them reveal the expected signals for the Cp* ligand and the respective substituents in the 1H NMR spectra. Furthermore, the signals for the benzylic hydrogen atoms in 4 (δ = 4.42 ppm) and 5 (δ = 5.93 ppm) show a coupling to the P5 moiety (4: 2JH–P = 11.2 Hz, 3JH–P = 3.2 Hz, 5: 2JH–P = 17.3 Hz). Interestingly, the isostructural 6 does not show a similar coupling for the hydrogen atom bound to silicon, which we attribute to the dynamic behavior of this compound in solution. Accordingly, the 13C{1H} NMR spectra of 4 and 5 both reveal signals for the benzylic carbon atoms, which show 1JC–P coupling of 23 Hz for both compounds. The 31P{1H} NMR spectra of most of the obtained products reveal complex AMM′XX′ (4, 5), AA′BB′X (7), AA′MXX′ (11), AA′MM′X (6, 9, 12) or even AA′M2M′2X2X′2 (2) spin systems (see ESI†). However, 3, 8, 10 and 13 show a highly dynamic behavior in CD2Cl2 solution which cannot be resolved, not even at −80 °C. Similar dynamic broadening of signals has been observed for the parent compound20 and might be caused by a tumbling process of the respective substituent around the P5 ring. When comparing the 31P{1H} NMR spectra (Fig. 2), the signal assigned to P1 (according to Fig. 1) gradually shifts to higher fields going from group 13 to group 17 substituents at the pentaphosphole moiety. Similarly, the signal assigned to P2/5 is downfield shifted following the same order, whereas the effect on the P3/4 signal is rather small (for assignment see Fig. 1). While this trend should depend on various electronic and steric factors, it correlates well with the folding of the P5 moiety (vide supra). The nJP–P (n = 1–2) coupling constants for all compounds are within the expected range (see ESI†). Additional 1JP–B coupling of 64 Hz is clearly visible in the 31P NMR spectrum of 2 and is confirmed in its 11B{1H} NMR spectrum. While the dynamic behavior of the Si-substituted compound 6 in CD2Cl2 solution, even at −80 °C, does not allow for determination of P–P coupling constants, its 29Si(DEPT 135) NMR spectrum shows a clear doublet of multiplets at δ = −20 ppm with a 1JSi–P = 239 Hz coupling constant. Compared to the SiEt3-substituted derivative (1JP–Si = 61 Hz), this rather large 1JP–Si coupling constant may be the result of the difference in the substitution at Si and the resulting change in the orbital contribution to the P–Si bonding interaction. Furthermore, the 31P NMR spectrum of 9 reveals 77Se satellites for the PX signal and a clear doublet (1JP–Se = 418 Hz) at δ = 287.3 ppm in the 77Se{1H} NMR spectrum.
:1:1 stoichiometry leads to a reaction of the in situ prepared borinium cation [BBr2]+ with the solvent, resulting in a mixture of products (see ESI†). Repeating the reaction in the presence of a second equivalent of 1 afforded the bimetallic compound [{Cp*Fe}2{μ,η5:5-(P5)2BBr2}][TEF] (2), featuring an unprecedented{(cyclo-P5)2BBr2} ligand (Scheme 2). After workup, 2 could be isolated in 70% yield. To our pleasure, exchanging BBr3 for the heavier analog GaI3 affords the isostructural compound [{Cp*Fe}2{μ,η5:5-(P5)2GaI2}][TEF] (3) in 32% yield. While we had already been able to demonstrate the silylation and methylation of 1,20 we now wanted to broaden the scope of group 14 functionalized pentaphosphole derivatives, by using better accessible and easier-to-handle electrophile precursors. In both cases, the reaction of 1 with PhCH2Br and Ph2CHCl in the presence of Tl[TEF] affords the pentaphosphole complexes [Cp*Fe(η5-P5CH2Ph)][TEF] (4) and [Cp*Fe(η5-P5CHPh2)][TEF] (5) in a yield of 45% and 80%, respectively. To demonstrate an easily accessible alternative pathway towards the electrophile generation avoiding the use of Tl+ salts, we chose to react 1 with Ph2SiH2 in the presence of one equivalent of [Ph3C][B(C6F5)4]. A rapid reaction is indicated by the color change of the solution from green to greenish red and, after simple workup, the product [Cp*Fe(η5-P5SiHPh2)][B(C6F5)4] (6) could be isolated in an astonishing 85% yield. In principle, it should be possible to prepare phosphino-pentaphosphole complexes similarly well as they benefit from the additional stabilization caused by P–P bond formation. However, attempts to prepare such phosphino-functionalized species are seriously affected by the inherently high tendency of halogenophosphines to form phosphinophosphonium ions under Lewis acidic conditions.27 We were able to observe the desired cation [Cp*Fe(η5-P5PBr2)]+ spectroscopically only after reacting 1 with PBr3 and Tl[TEF] at −80 °C (see ESI†). This cation, however, undergoes rapid fragmentation and rearrangement processes above −60 °C, affording a complex mixture of several polyphosphorus compounds and thus prohibiting its isolation even at low temperatures. In contrast, the reaction of 1, Cy2AsBr and Tl[TEF] proceeds smoothly, and from the filtered solution, the arsino-pentaphosphole complex [Cp*Fe(η5-P5AsCy2)][TEF] (7) could be isolated in 68% yield. When both the electronic and steric properties of the pnictogenium cation were changed and 1 was reacted with Cp′′′SbI2 (Cp′′′ = 1,2,4-tBu3C5H2) and Tl[TEF], the unique triple-decker-like complex [Cp*Fe(μ,η5:2-P5)SbICp′′′][TEF] (8) with a P5 middle deck could be obtained in 24% yield. The molecular structure of 8 may provide insight into the mechanism of the electrophilic functionalization of 1 which initially seems to occur through the π-system and not by the lone pairs of one of the P atoms of the cyclo-P5 ligand. Functionalization of 1 with group 16 electrophiles could be achieved by the reaction with Tl[TEF] and PhSeBr or MesTeBr, respectively. The products [Cp*Fe(η5-P5ER)][TEF] (9: E = Se, R = Ph; 10: E = Te, R = Mes) could be isolated in 65% and 57% yield, respectively, and reveal novel seleno- or telluro-pentaphosphole ligands. To date, there is no synthetic pathway for a rational and selective monohalogenation of unsubstituted polyphosphorus frameworks, although the products would provide both insight into the mechanism of Pn halogenation reactions28 and a high potential for further functionalization. After recently investigating the complex iodination chemistry of 1 (ref. 29) and of the diphosphorus complex [{CpMo(CO)2}2(μ,η2:2-P2)],30 we were wondering if electrophilic halogen transfer could lead to the desired reactivity. Indeed, when a solution of [PCl4][TEF] is added to 1 in o-DFB, the NMR spectra of the crude solution indicate the clean conversion to [Cp*Fe(η5-P5Cl)][TEF] (11) and PCl3. After workup, 11 can be isolated in 65% yield. Similarly, the addition of X2 to mixtures of 1 and Tl[TEF] leads to the formation of [Cp*Fe(η5-P5X)][TEF] (12: X = Br; 13: X = I), which could be isolated in 65% and 62% yield, respectively. Notably, these halogenation reactions can be scaled up to at least 2 mmol, allowing the gram scale preparation of 11–13, which opens broad perspectives for their further functionalization. While the starting material 1 is comparably robust, the pentaphosphole complexes 2–13 are highly sensitive towards both moisture and air, while retaining decent thermal stability. All compounds 2–13 can be crystallized from mixtures of o-DFB or CH2Cl2 and n-hexane either at room temperature or at −30 °C. However, incommensurate modulation (11) or extreme disorder of anion and cation (12) within 11 and 12 prohibit a satisfactory refinement of their crystal structure. Compound 8 reveals a triple-decker-like arrangement (Fig. 1) of the cation [Cp*Fe(μ,η5:2-P5)SbICp′′′]+ with elongated P–Sb interactions (3.236(2)/3.400(2) Å) and a planar P5 ligand. The cations in 4–7 and 9–13 show the anticipated pentaphosphole complex structure with a slightly bent cyclo-P5R ligand (Fig. 1). In contrast, the cations in 2 and 3 are dinuclear complexes in which two {Cp*Fe} units are bridged by the respective {(cyclo-P5)2EX2} (2: E = B, X = Br, 3: E = Ga, X = I) ligand. The P–P bond lengths within 2–13 are similar and in between the sum of the covalent radii of P–P single and double bonds.31 As observed for the parent compound [Cp*Fe(η5-P5H)]+,20 the substituents in the hetero-pentaphosphole complexes 2–7 and 9–13 are oriented in exo-fashion regarding the envelope of the P5 ring (Fig. 1). The respective P–E bond lengths (2: 1.985(7) Å, 4: 1.853(4) Å, 5: 1.866(4) Å, 7: 2.348(1) Å, 9: 2.2234(7) Å, 10: 2.438(2) Å, 13: 2.385(1) Å) are in the range of single bonds, whereas those in 3 (2.410(1)/2.387(1) Å) and 6 (2.3053(8) Å) are elongated compared to the sum of the respective covalent radii (P–Ga: 2.35 Å, P–Si: 2.27 Å).31 The slight folding of the P5 ring in these pentaphosphole complexes is far less pronounced than in anionic or neutral substituted cyclo-P5R ligands possessing an envelope structure (type I, Scheme 1).13,14 Interestingly, this slight folding, represented by the pyramidalization at the P1 atom (359.94°(2) – 350.41°(13)), gradually increases when going from group 13 to group 17 substituents at the pentaphosphole ligand. To elaborate the molecular structure of the obtained pentaphosphole complexes 2–13 in CD2Cl2 solution, they were investigated by multinuclear NMR spectroscopy. In general, all of them reveal the expected signals for the Cp* ligand and the respective substituents in the 1H NMR spectra. Furthermore, the signals for the benzylic hydrogen atoms in 4 (δ = 4.42 ppm) and 5 (δ = 5.93 ppm) show a coupling to the P5 moiety (4: 2JH–P = 11.2 Hz, 3JH–P = 3.2 Hz, 5: 2JH–P = 17.3 Hz). Interestingly, the isostructural 6 does not show a similar coupling for the hydrogen atom bound to silicon, which we attribute to the dynamic behavior of this compound in solution. Accordingly, the 13C{1H} NMR spectra of 4 and 5 both reveal signals for the benzylic carbon atoms, which show 1JC–P coupling of 23 Hz for both compounds. The 31P{1H} NMR spectra of most of the obtained products reveal complex AMM′XX′ (4, 5), AA′BB′X (7), AA′MXX′ (11), AA′MM′X (6, 9, 12) or even AA′M2M′2X2X′2 (2) spin systems (see ESI†). However, 3, 8, 10 and 13 show a highly dynamic behavior in CD2Cl2 solution which cannot be resolved, not even at −80 °C. Similar dynamic broadening of signals has been observed for the parent compound20 and might be caused by a tumbling process of the respective substituent around the P5 ring. When comparing the 31P{1H} NMR spectra (Fig. 2), the signal assigned to P1 (according to Fig. 1) gradually shifts to higher fields going from group 13 to group 17 substituents at the pentaphosphole moiety. Similarly, the signal assigned to P2/5 is downfield shifted following the same order, whereas the effect on the P3/4 signal is rather small (for assignment see Fig. 1). While this trend should depend on various electronic and steric factors, it correlates well with the folding of the P5 moiety (vide supra). The nJP–P (n = 1–2) coupling constants for all compounds are within the expected range (see ESI†). Additional 1JP–B coupling of 64 Hz is clearly visible in the 31P NMR spectrum of 2 and is confirmed in its 11B{1H} NMR spectrum. While the dynamic behavior of the Si-substituted compound 6 in CD2Cl2 solution, even at −80 °C, does not allow for determination of P–P coupling constants, its 29Si(DEPT 135) NMR spectrum shows a clear doublet of multiplets at δ = −20 ppm with a 1JSi–P = 239 Hz coupling constant. Compared to the SiEt3-substituted derivative (1JP–Si = 61 Hz), this rather large 1JP–Si coupling constant may be the result of the difference in the substitution at Si and the resulting change in the orbital contribution to the P–Si bonding interaction. Furthermore, the 31P NMR spectrum of 9 reveals 77Se satellites for the PX signal and a clear doublet (1JP–Se = 418 Hz) at δ = 287.3 ppm in the 77Se{1H} NMR spectrum.
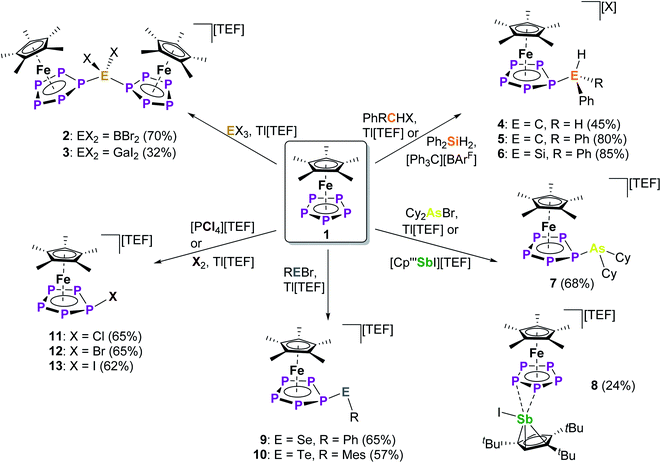 |
| | Scheme 2 Reactivity of pentaphosphaferrocene 1 with different cationic p-block electrophiles; reactions were carried out in o-DFB and stirred at room temperature for several hours (for details see ESI†); while all other compounds are obtained as their [TEF]− salt, 6 has a [B(C6F5)4]− counterion; [TEF]− = [Al{OC(CF3)3}4]−; while all displayed reactions are quantitative (by NMR), the yields provided are those for the crystalline compounds. | |
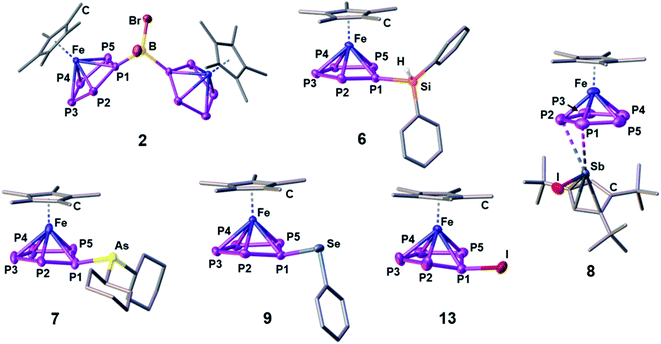 |
| | Fig. 1 Molecular structures of the cations in 2, 6, 7, 8, 9 and 13 in the solid state; Structural models for the cations in 3, 4, 5, 10 and 12 as well as a list of selected structural parameters (bond lengths and angles) for all compounds can be found in the ESI.† | |
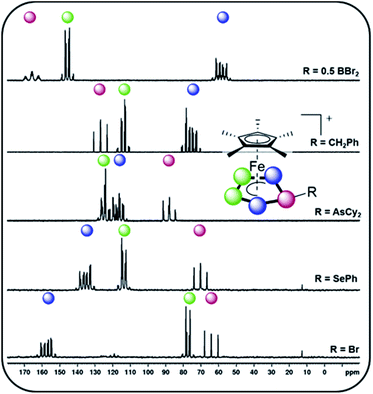 |
| | Fig. 2
31P{1H} NMR spectra of 2, 4, 7, 9 and 12 (from top to bottom, representing substitution with electrophiles from each group within the p-block) recorded in CD2Cl2 at room temperature and signal assignment according to the color code; respective NMR spectra of all other compounds are given in the ESI.† | |
Computational studies
To attain a better understanding of the electronic structure of the obtained complexes 2–13, computational analyses at the B3LYP32/def2-TZVP33 level of theory were performed.
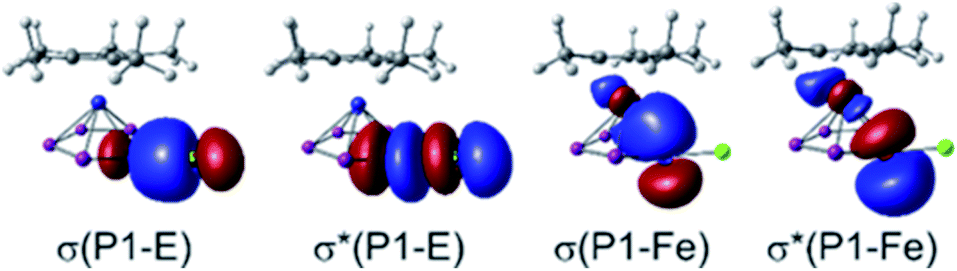
Employing solvent correction,34 the molecular structures, determined in the solid state, are well reproduced by these computations. NBO analysis35 indicates the interaction between the [Cp′′′SbI]+ fragment and 1 in 8 to be of a mostly electrostatic and dispersive nature as shown by the absence of bonding MOs and the low charge transfer from the electrophile (see ESI†). Furthermore, the Wiberg Bond Indices (WBIs) for the P1–E bonds in 3 (E = Ga, WBI = 0.58/0.57) and 6 (E = Si, WBI = 0.72) suggest these bonds to be of polar single bond character. This is substantiated by the respective orbital contributions to the P1–E bonding (3: 79% P/21% Ga, 6: 72% P/28% Si) and unoccupied antibonding (3: 21% P/79% Ga, 6: 28% P/72% Si) molecular orbitals and agrees with the respective bond lengths determined for 3 (2.410(1)/2.387(1) Å) and 6 (2.3053(8) Å). In contrast, the WBIs of all other pentaphosphole complexes corroborate the P1–E single bonds (WBI = 0.82–0.99) determined from their respective solid-state structures (Table 1, column 3). Accordingly, the orbital contributions to the respective P1–E bonding and antibonding molecular orbitals (Fig. 3, left) are more balanced (Table 1, column 2), suggesting more covalent bonding in these cases. Analyzing the summed up natural charges for the fragments {Cp*Fe(η5-P5)} ({1}) and {RnE} ({E}, n = 1–3) (see ESI†) of 2–13 reveals correlation between the positive charge accumulation at {1} and the electronegativity of the central atom of the electrophiles. As 3 and 6 incorporate the least electronegative central atoms in the respective electrophile, they show the lowest charge accumulation at {1} in the series of the herein reported pentaphosphole complexes (see Table S13†). The charge transfer from {1} to {E} (Table 1, column 5) obtained by an extended charge decomposition analysis (ECDA)36 on the optimized structures of 2–13 shows a similar trend but hints towards other factors to be of relevance as well. Thus, effective charge transfer seems to be at least one of the governing factors for the formation of covalent P1–E bonds and consequently less labile pentaphosphole ligands, which is in line with the highly dynamic behavior of 3 and 6 in CD2Cl2 solution (vide supra). Similar to the parent compound [Cp*Fe(η5-P5H)]+,20 the pentaphosphole complexes 2–7 and 9–13 exhibit a pronounced P1–Fe bonding interaction (Fig. 3, right), which is represented by WBIs of 0.34–0.36. This interaction manifests their η5-binding mode and hints towards the aromatic character of the pentaphosphole ligands. To clarify the latter point, we computed the NICS(0/1/–1)zz37 values for the ligand geometries from the optimized structures of the cations 2–7 and 9–13 on the PBE0 (ref. 38)/aug-pcSseg-1 (ref. 39)/def2-TZVPPD33,40 level of theory. Consequently, these NICS(±1)zz values (Table 1) corroborate the aromaticity of the cyclo-P5R ligands but are smaller than the ones for the cyclo-P5− ligand in 1 (NICS(±1)zz = −40.5). However, they compare well with that determined for the parent pentaphosphole complex [Cp*Fe(η5-P5H)]+ (NICS(±–1)zz = −32.1/−31.2).20 A rough correlation between the pyramidalization at P1 and the computed NICS values is observed, although this effect is not much accentuated and has minor exceptions (see ESI†).
Table 1 Selected computational and experimental parameters for the pentaphosphole complexes 2–7 and 9–13
| Compound |
WBI (P1–E) |
Orbital contribution P1–E |
d(P1–E)/Å |
Charge transfer {1} → {E}b |
NICS(±1)zzc |
| Exp. |
Theo. |
|
As 2 and 3 are dinuclear complexes the average of the actual values is provided for clarity, NICS values have only been obtained on one the P5 rings.
The charge transfer from fragment {1} to the respective fragments {E} is obtained by ECDA.
NICS(±1)zz values are computed on the ligand geometries from the optimized structures of the complexes 2–7 and 9–13 after removal of the {Cp*Fe}+ fragments.
The charge transfer is given per fragment {1}.
|
|
2
|
0.89 |
57/43 |
1.985(7) |
2.01 |
0.57d |
−31.2/−30.5 |
|
3
a
|
0.58 |
79/21 |
2.399(1) |
2.46 |
0.56d |
−34.7/−34.3 |
|
4
|
0.92 |
45/55 |
1.853(4) |
1.86 |
0.85 |
−33.7/−32.8 |
|
5
|
0.88 |
45/55 |
1.866(4) |
1.89 |
0.84 |
−31.2/−32.1 |
|
6
|
0.72 |
72/28 |
2.3053(8) |
2.35 |
0.68 |
−34.3/−34.7 |
|
7
|
0.82 |
67/33 |
2.348(1) |
2.38 |
0.63 |
−34.0/−33.4 |
|
9
|
0.98 |
53/47 |
2.2234(7) |
2.25 |
0.83 |
−33.5/−32.6 |
|
10
|
0.92 |
62/38 |
2.438(2) |
2.48 |
0.74 |
−34.9/−34.0 |
|
11
|
0.96 |
36/64 |
— |
2.03 |
1.09 |
−31.0/−30.9 |
|
12
|
0.98 |
41/59 |
— |
2.21 |
1.05 |
−31.8/−31.7 |
|
13
|
0.99 |
51/49 |
2.385(1) |
2.42 |
0.94 |
−34.3/−34.2 |
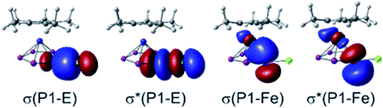 |
| | Fig. 3 Selected representative NBOs for the bonding and antibonding molecular orbitals for the P1–E and P1–Fe interactions found in the pentaphosphole complexes 2–7 and 9–13 exemplified at 11; the σ* orbitals are unoccupied. | |
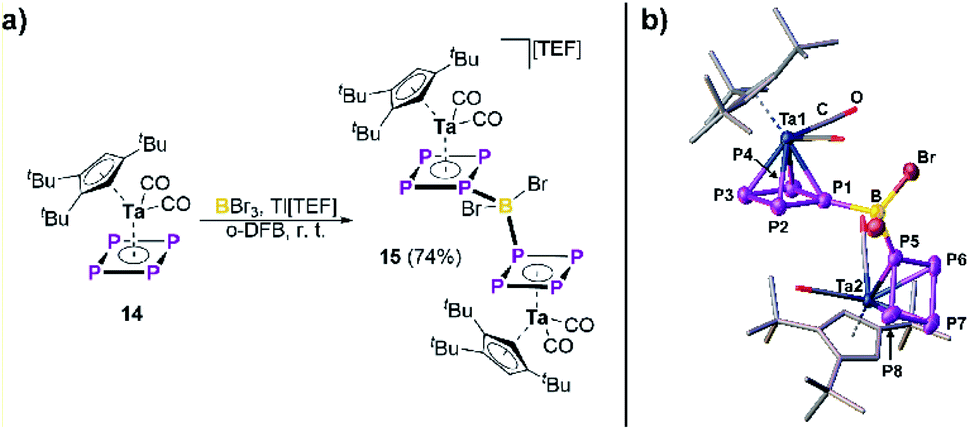
Thus, we demonstrated the in situ electrophilic functionalization of 1 to be a highly versatile approach towards a variety of unprecedented hetero-pentaphosphole complexes. Moreover, to provide a first insight into the generality of this synthetic strategy, we extended our investigations to the electrophilic functionalization of [Cp′′′Ta(CO)2(η4-P4)] (14)41 bearing an aromatic tetra-phospha-cyclo-butadiendiide (cyclo-P42−) ligand. First reactivity studies reveal that two equivalents of 14 react with BBr3 in the presence of Tl[TEF] to afford [{Cp′′′Ta(CO)2}2{μ,η4:4-((P4)2BBr2})][TEF] (15) in 74% yield (Fig. 4). Compound 15 crystallizes as orange blocks and shows a similar dinuclear structure as found in 2, which was confirmed by X-ray structural analysis (Fig. 4). Interestingly, the metal fragments in 15 are rotated by nearly 180° compared to those in 2 and the P1/P5 atoms are bent out of the former P4 plane by only 1.2(2)/0.7(2)°, respectively. The P–P bond lengths (2.124(4) – 2.171(4) Å) in the {(cyclo-P4)2BBr2} ligand are slightly shortened compared to the starting material 14 (2.1555(15) – 2.1800(15) Å),41 but still in-between covalent P–P single (2.22 Å) and double bonds (2.04 Å).31 The P1/P5–B bond lengths (1.997(10)/1.996(9) Å) are within the expected range for P–B single bonds (1.96 Å) and the P1–B–P5 angle of 112.3(5)° leads to a slightly distorted tetrahedral geometry around the B atom. The 31P NMR spectrum of 15 in CD2Cl2 reveals a complicated A2A′2MM′XX′Z (Z = B) spin system with resonances located at −15.4, −37.1 and −80.8 ppm, agreeing with its structure being retained in solution. 1JP–B coupling is clearly visible for the PM/M′ resonance and consequently the respective 11B{1H} NMR spectrum shows a triplet at −10.2 ppm with a 1JP–B coupling constant of 60 Hz.
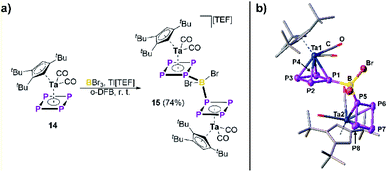 |
| | Fig. 4 (a) Reactivity of 14 towards the in situ formed dibromoborinium ion “[BBr2][TEF]”; (b) molecular structure of 15 in the solid state (hydrogen atoms and the counter ion are omitted for clarity and thermal ellipsoids are drawn at the 50% probability level). | |
The solid-state structure of 15 is well reproduced by DFT calculations (B3LYP32/def2-TZVP,33 PCM solvent correction for CH2Cl2) which give further insight into its electronic structure. The WBIs for the P1/P5–B bonds (0.88/0.86) in 15 are in agreement with covalent P–B single bonds. Furthermore, the orbital contributions to these P–B bonds (58% P/42% B%) are in line with this formulation. While the positive charge within 15 is distributed across both {14} fragments, the B atom shows negative charge accumulation (natural charge of −0.44), labelling the {(cyclo-P4)2BBr2} ligand in 15 a borate-bridged bis-tetra-phospha-butadiendiide. At last, the respective NICS(±1)zz values indicate the aromaticity of the cyclo-P4 ligand of 14 (−3.3/−3.5) to be preserved within the ligand geometry in 15 (−9.5/−9.7, −10.8/−9.6), although it is less pronounced than in the pentaphosphole complexes 2–7 and 9–13.
Conclusions
In conclusion, we demonstrated a general synthetic strategy to obtain a broad range of hetero-pentaphosphole coordination complexes bearing substituents from across the whole p-block of the periodic table. While the group 14–17 substituted species [Cp*Fe(η5-P5E)]+ (E = CH2Ph (4), CHPh2 (5), SiHPh2 (6), AsCy2 (7), SePh (9), TeMes (10), Cl (11), Br (12), I (13)) reveal the desired mononuclear aromatic pentaphosphole complexes, the group 13 functionalized species result in unexpected dinuclear complexes [{Cp*Fe}2{μ,η5:5-(P5)2EX2}][TEF] (EX2 = BBr2 (2), GaI2 (3)) with bridging bis-pentaphosphole {(cyclo-P5)2EX2} ligands. In contrast, the alternation of the electronic and steric properties of the employed electrophile, as in [Cp′′′SbI]+, leads to the formation of the triple-decker-like arrangement observed in [Cp*Fe(μ,η5:2-P5)SbICp′′′][TEF] (8). This paves the way towards the investigation of the chemical, physical and electronic properties of these all-P congeners of prototypical cyclopentadienes, which have raised considerable theoretical interest over the past decades.8 Furthermore, this report highlights the versatility of the electrophilic functionalization of transition metal-stabilized polyphosphorus frameworks. While some P–E bond formation processes at such compounds have been observed, these are highly specific towards certain electrophile/nucleophile combinations (vide supra). In contrast, our approach shows an unprecedented diversity and thus allows P–E bond formation for elements all across the p-block. The generality of this approach is proven by the additional electrophilic aromatic functionalization of the aromatic cyclo-P4 ligand of 14, which furnishes [{Cp′′′Ta(CO)2}2(μ,η4:4-(P4)2BBr2)][TEF] (15), bearing a unique borate-bridged bis-tetra-phospha-butadiendiide ligand. Finally, the obtained cationic pentaphosphole complexes 2–13 represent highly interesting starting materials (e.g. the halogen derivatives 11–13) towards even further functionalized polyphosphorus frameworks, which is underlined by the gram scale synthesis of selected representatives (5, 11–13).
Data availability
Crystallographic data for 2–10, 13 and 15 has been deposited at the CCDC/ICSD under 2083554 (2), 2083555 (3), 2083556 (4), 2083557 (5), 2083558 (6), 2083559 (7), 2083560 (8), 2083561 (9), 2083562 (10), 2083563 (13) and 2101248 (15). All other data are provided in this article or in the ESI.†
Author contributions
C. R. performed the experimental work and wrote the original draft. G. B. assisted in the performance of DFT calculations. M. Seidl assisted in the interpretation of X-ray structural data and M. Scheer supervised and acquired funding for the project. All authors contributed in preparing the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, Sche 384/36-1). C. R. is grateful to the Studienstiftung des Deutschen Volkes for his PhD fellowship. This paper is dedicated to Professor Werner Thiel on the occasion of his 60th birthday.
References
-
(a) C. Friedel and J. M. Crafts, Compt. Rend., 1877, 84, 1392 Search PubMed;
(b) C. Friedel and J. M. Crafts, Compt. Rend., 1877, 84, 1450 Search PubMed.
-
(a)
R. Brückner, Reaktionsmechanismen, Springer, Berlin, Heidelberg, 2004, pp. 203–235 Search PubMed;
(b)
P. Y. Bruice, Organische Chemie, Pearson Studium, Hallbergmoos, 2013, pp. 627–642 Search PubMed.
-
(a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS;
(b) R. B. Woodward, M. Rosenblum and M. C. Whiting, J. Am. Chem. Soc., 1952, 74, 3458–3459 CrossRef CAS.
- E. O. Fischer and K. Öfele, Chem. Ber., 1957, 90, 2532–2535 CrossRef CAS.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
-
(a) M. Baudler and T. Etzbach, Chem. Ber., 1991, 124, 1159–1160 CrossRef CAS;
(b) V. A. Milyukov, A. V. Kataev, O. G. Sinyashin and E. Hey-Hawkins, Russ. Chem. Bull., 2006, 55, 1297–1299 CrossRef CAS;
(c) M. Jo, A. Dragulescu-Andrasi, L. Z. Miller, C. Pak and M. Shatruk, Inorg. Chem., 2020, 59, 5483–5489 CrossRef CAS.
- M. Baudler, S. Akpapoglou, D. Ouzounis, F. Wasgestian, B. Meinigke, H. Budzikiewicz and H. Münster, Angew. Chem., Int. Ed. Engl., 1988, 27, 280–281 CrossRef.
-
(a) L. Nyulászi, Inorg. Chem., 1996, 35, 4690–4693 CrossRef;
(b) M. N. Glukhovtsev, A. Dransfeld and P. v. R. Schleyer, J. Phys. Chem., 1996, 100, 13447–13454 CrossRef CAS;
(c) A. Dransfeld, L. Nyulászi and P. v. R. Schleyer, Inorg. Chem., 1998, 37, 4413–4420 CrossRef CAS PubMed;
(d) M. K. Cyrański, P. v. R. Schleyer, T. M. Krygowski, H. Jiao and G. Hohlneicher, Tetrahedron, 2003, 59, 1657–1665 CrossRef;
(e) W. P. Ozimiński and J. C. Dobrowolski, Chem. Phys., 2005, 313, 123–132 CrossRef;
(f) L. Wang, H. J. Wang, W. B. Dong, Q. Y. Ge and L. Lin, Struct. Chem., 2007, 18, 25–31 CrossRef CAS;
(g) W.-Q. Li, L.-L. Liu, J.-K. Feng, Z.-Z. Liu, A.-M. Ren, G. Zhang and C.-C. Sun, J. Theor. Comput. Chem., 2008, 07, 1203–1214 CrossRef CAS;
(h) D. Josa, A. Peña-Gallego, J. Rodríguez-Otero and E. M. Cabaleiro-Lago, J. Mol. Model., 2011, 17, 1267–1272 CrossRef CAS PubMed.
-
(a) O. J. Scherer and T. Brück, Angew. Chem., Int. Ed. Engl., 1987, 26, 59 Search PubMed;
(b) M. Baudler and T. Etzbach, Angew. Chem., Int. Ed. Engl., 1991, 30, 580–582 CrossRef;
(c) A. R. Kudinov, D. A. Loginov, Z. A. Starikova, P. V. Petrovskii, M. Corsini and P. Zanello, Eur. J. Inorg. Chem., 2002, 2002, 3018–3027 CrossRef;
(d) E. Urnius, W. W. Brennessel, C. J. Cramer, J. E. Ellis and P. v. R. Schleyer, Science, 2002, 295, 832–834 CrossRef PubMed;
(e) C. M. Knapp, B. H. Westcott, M. A. C. Raybould, J. E. McGrady and J. M. Goicoechea, Angew. Chem., Int. Ed., 2012, 51, 9097–9100 CrossRef CAS PubMed;
(f) S. Heinl, G. Balázs, M. Bodensteiner and M. Scheer, Dalton Trans., 2016, 45, 1962–1966 RSC.
-
(a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed;
(b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed;
(c) C. M. Hoidn, D. J. Scott and R. Wolf, Chem.– Eur. J., 2021, 27, 1886–1902 CrossRef CAS PubMed.
-
(a) J. Bai, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2002, 41, 1737–1740 CrossRef CAS;
(b) J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781–783 CrossRef CAS PubMed;
(c) M. Scheer, J. Bai, B. P. Johnson, R. Merkle, A. V. Virovets and C. E. Anson, Eur. J. Inorg. Chem., 2005, 2005, 4023–4026 CrossRef;
(d) M. Scheer, L. J. Gregoriades, A. V. Virovets, W. Kunz, R. Neueder and I. Krossing, Angew. Chem., Int. Ed., 2006, 45, 5689–5693 CrossRef CAS PubMed;
(e) M. Scheer, A. Schindler, R. Merkle, B. P. Johnson, M. Linseis, R. Winter, C. E. Anson and A. V. Virovets, J. Am. Chem. Soc., 2007, 129, 13386–13387 CrossRef CAS PubMed;
(f) S. Welsch, L. J. Gregoriades, M. Sierka, M. Zabel, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2007, 46, 9323–9326 CrossRef CAS PubMed;
(g) M. Scheer, L. J. Gregoriades, R. Merkle, B. P. Johnson and F. Dielmann, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 504–508 CrossRef CAS;
(h) M. Scheer, A. Schindler, C. Gröger, A. V. Virovets and E. V. Peresypkina, Angew. Chem., Int. Ed., 2009, 48, 5046–5049 CrossRef CAS PubMed;
(i) M. Scheer, A. Schindler, J. Bai, B. P. Johnson, R. Merkle, R. Winter, A. V. Virovets, E. V. Peresypkina, V. A. Blatov, M. Sierka and H. Eckert, Chem.– Eur. J., 2010, 16, 2092–2107 CrossRef CAS PubMed;
(j) F. Dielmann, A. Schindler, S. Scheuermayer, J. Bai, R. Merkle, M. Zabel, A. V. Virovets, E. V. Peresypkina, G. Brunklaus, H. Eckert and M. Scheer, Chem.– Eur. J., 2012, 18, 1168–1179 CrossRef CAS PubMed;
(k) E. Peresypkina, C. Heindl, A. Virovets, H. Brake, E. Mädl and M. Scheer, Chem.– Eur. J., 2018, 24, 2503–2508 CrossRef CAS PubMed.
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972–2976 CrossRef CAS PubMed.
- E. Mädl, M. V. Butovskii, G. Balázs, E. V. Peresypkina, A. V. Virovets, M. Seidl and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7643–7646 CrossRef PubMed.
-
(a) F. Riedlberger, S. Todisco, P. Mastrorilli, A. Y. Timoshkin, M. Seidl and M. Scheer, Chem.– Eur. J., 2020, 26, 16251–16255 CrossRef CAS PubMed;
(b) R. Yadav, T. Simler, S. Reichl, B. Goswami, C. Schoo, R. Köppe, M. Scheer and P. W. Roesky, J. Am. Chem. Soc., 2020, 142, 1190–1195 CrossRef CAS.
- D. Tofan, B. M. Cossairt and C. C. Cummins, Inorg. Chem., 2011, 50, 12349–12358 CrossRef CAS PubMed.
-
(a) J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295–298 CrossRef CAS PubMed;
(b) M. H. Holthausen and J. J. Weigand, Chem. Soc. Rev., 2014, 43, 6639–6657 RSC.
- A. K. Adhikari, C. G. P. Ziegler, K. Schwedtmann, C. Taube, J. J. Weigand and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 18584–18590 CrossRef CAS PubMed.
- C. G. P. Ziegler, T. M. Maier, S. Pelties, C. Taube, F. Hennersdorf, A. W. Ehlers, J. J. Weigand and R. Wolf, Chem. Sci., 2019, 10, 1302–1308 RSC.
- R. Yadav, B. Goswami, T. Simler, C. Schoo, S. Reichl, M. Scheer and P. W. Roesky, Chem. Commun., 2020, 56, 10207–10210 RSC.
- C. Riesinger, G. Balázs, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2020, 59, 23879–23884 CrossRef CAS PubMed.
- S. J. Connelly, W. Kaminsky and D. M. Heinekey, Organometallics, 2013, 32, 7478–7481 CrossRef CAS.
-
(a) A. Jayaraman, T. V. Jacob, J. Bisskey and B. T. Sterenberg, Dalton Trans., 2015, 44, 8788–8791 RSC;
(b) A. Jayaraman and B. T. Sterenberg, Organometallics, 2016, 35(14), 2367–2377 CrossRef CAS;
(c) A. Jayaraman, S. Nilewar, T. V. Jacob and B. T. Sterenberg, ACS Omega, 2017, 2, 7849–7861 CrossRef CAS PubMed.
-
(a) G. Capozzi, L. Chiti, M. Di Vaira, M. Peruzzini and P. Stoppioni, Chem. Commun., 1986, 24, 1799–1800 RSC;
(b) A. Barth, G. Huttner, M. Fritz and L. Zsolnai, Angew. Chem., Int. Ed. Engl., 1990, 29, 929–931 CrossRef;
(c) P. Barbaro, A. Ienco, C. Mealli, M. Peruzzini, O. J. Scherer, G. Schmitt, F. Vizza and G. Wolmershäuser, Chem.– Eur. J., 2003, 9, 5196–5210 CrossRef PubMed;
(d) M. Peruzzini, R. R. Abdreimova, Y. Budnikova, A. Romerosa, O. J. Scherer and H. Sitzmann, J. Organomet. Chem., 2004, 689, 4319–4331 CrossRef CAS;
(e) B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS PubMed;
(f) P. Barbaro, C. Bazzicalupi, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, Angew. Chem., Int. Ed., 2012, 51, 8628–8631 CrossRef CAS PubMed;
(g) C. Schwarzmaier, S. Heinl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13116–13121 CrossRef CAS;
(h) E. Mädl, G. Balázs, E. V. Peresypkina and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 7702–7707 CrossRef;
(i) A. Cavaillé, N. Saffon-Merceron, N. Nebra, M. Fustier-Boutignon and N. Mézailles, Angew. Chem., Int. Ed., 2018, 57, 1874–1878 CrossRef PubMed;
(j) C. M. Hoidn, T. M. Maier, K. Trabitsch, J. J. Weigand and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 18931–18936 CrossRef CAS PubMed;
(k) U. Chakraborty, J. Leitl, B. Mühldorf, M. Bodensteiner, S. Pelties and R. Wolf, Dalton Trans., 2018, 47, 3693–3697 RSC.
-
(a) C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1991, 30, 564–565 CrossRef;
(b) S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke and A. Kuhn, Angew. Chem., Int. Ed. Engl., 1993, 32, 1729–1731 CrossRef;
(c) R. Yadav, T. Simler, B. Goswami, C. Schoo, R. Köppe, S. Dey and P. W. Roesky, Angew. Chem., Int. Ed., 2020, 59, 9443–9447 CrossRef CAS PubMed.
- A. Frei, Chem.– Eur. J., 2019, 25, 7074–7090 CrossRef CAS PubMed.
-
(a) I. Krossing, Chem.– Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed;
(b) M. Gonsior, I. Krossing and N. Mitzel, Z. Anorg. Allg. Chem., 2002, 628, 1821 CrossRef CAS.
-
(a) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS PubMed;
(b) M. Gonsior, I. Krossing, L. Müller, I. Raabe, M. Jansen and L. van Wüllen, Chem.– Eur. J., 2002, 8, 4475–4492 CrossRef CAS;
(c) J. Possart, A. Martens, M. Schleep, A. Ripp, H. Scherer, D. Kratzert and I. Krossing, Chem.– Eur. J., 2017, 23, 12305–12313 CrossRef CAS PubMed.
- G. Manca and A. Ienco, Inorg. Chim. Acta, 2021, 517, 120205 CrossRef CAS.
- H. Brake, E. Peresypkina, A. V. Virovets, M. Piesch, W. Kremer, L. Zimmermann, C. Klimas and M. Scheer, Angew. Chem., Int. Ed., 2020, 59, 16241–16246 CrossRef CAS PubMed.
- A. Garbagnati, M. Seidl, G. Balázs and M. Scheer, Inorg. Chem., 2021, 60, 5163–5171 CrossRef CAS PubMed.
-
(a) P. Pyykkö and M. Atsumi, Chem.– Eur. J., 2009, 15, 186–197 CrossRef PubMed;
(b) P. Pyykkö and M. Atsumi, Chem.– Eur. J., 2009, 15, 12770–12779 CrossRef PubMed;
(c) P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
-
(a) P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1929, 123, 714–733 CrossRef CAS;
(b) J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS;
(c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS;
(d) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed;
(e) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed;
(f) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
-
(a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC;
(b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
-
(a) E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed;
(b) E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 2134 CrossRef CAS.
-
(a) S. Dapprich and G. Frenking, J. Phys. Chem., 1995, 99, 9352–9362 CrossRef CAS;
(b) M. Xiao and T. Lu, J. Adv. Phys. Chem., 2015, 04, 111–124 CrossRef CAS.
-
(a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed;
(b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed;
(c) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
-
(a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed;
(b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS;
(c) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2015, 11, 132–138 CrossRef CAS PubMed.
-
(a) B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef CAS;
(b) K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS;
(c) D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Dielmann, E. V. Peresypkina, B. Krämer, F. Hastreiter, B. P. Johnson, M. Zabel, C. Heindl and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 14833–14837 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*