
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalysed low-temperature water–gas shift reaction for selective deuteration of aryl halides†
Wu
Li
 ,
Ruiyang
Qu
,
Weiping
Liu
,
Florian
Bourriquen
,
Stephan
Bartling
,
Nils
Rockstroh
,
Kathrin
Junge
* and
Matthias
Beller
*
,
Ruiyang
Qu
,
Weiping
Liu
,
Florian
Bourriquen
,
Stephan
Bartling
,
Nils
Rockstroh
,
Kathrin
Junge
* and
Matthias
Beller
*
Leibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Kathrin.Junge@catalysis.de; matthias.beller@catalysis.de
First published on 17th September 2021
Abstract
The introduction of deuterium atoms into organic compounds is of importance for basic chemistry, material sciences, and the development of drugs in the pharmaceutical industry, specifically for identification and quantification of metabolites. Hence, methodologies for the synthesis of selectively labelled compounds continue to be a major area of interest for many scientists. Herein, we present a practical and stable heterogeneous copper catalyst, which permits for dehalogenative deuteration via water–gas shift reaction at comparably low temperature. This novel approach allows deuteration of diverse (hetero)aryl halides with good functional group tolerance, and no reduction of the aromatic rings or other easily reducible formyl and cyano groups. Multi-gram experiments show the potential of this method in organic synthesis and medicinal chemistry.
Introduction
Isotope labeling processes play an essential role in the investigation of basic reaction mechanism, e.g. determination of kinetic isotope effects (KIE).1 In addition, they are used in most discovery processes in life sciences for metabolism studies. Specifically for drug development, they are crucial as it is well known that metabolites show significantly altered biological activity, clearance rates, and toxicity.2 Hence, isotopes of active drugs are commonly prepared as internal standards for bioanalytical characterizations in the pharmaceutical and agrochemical industries. Among the various isotopes, deuterium atoms are the most common ones and deuterated compounds are the primary source for the preparation of internal standards for liquid chromatography-mass spectrometry (LC-MS) analysis of environmental, animal, and human samples.3 More recently, several deuterated Active Pharmaceutical Ingredient (APIs) such as Austedo have entered the clinical trials4 due to the potentially improved pharmacokinetic and pharmacological properties. In this context, specific and practical deuterium labelling methodologies are of increasing importance.5 However, practical procedures for deuteration with high quality control to produce isotopically pure compounds remain challenging.6 Although C–H/C–D exchange (H/D exchange) reactions, are considered as the most convenient methods to produce D-labelled compounds,7 alternative transformations such as dehalogenative deuterations are also essential components of the isotope labelling toolkit. Advantageously, dehalogenative deuteration can be easily controlled at specific positions and generally gives >99% D-incorporation. Thus, dehalogenative deuterium incorporation of widely available C–X (e.g. X is Cl, Br) bonds is a convenient way to access versatile deuterated products with a high deuterium content. Traditional methodologies for this transformation are based on strong bases and/or reductants and need special homogenous transition metal complexes (Fig. 1a).8 In those cases, CD3CN, CH3OD, CD3OD, THF-d8etc. are mainly used as deuterium source. Recently, also photoinduced deuteration using sacrificial agents and electrochemical reductive deuteration of aryl halides with D2O have been established with high D-incorporation (Fig. 1b).9 However, those methods are so far applicable only on mg-scale. | ||
| Fig. 1 Illustration of various selectivities available with distinct dehalogenative deuteration reactions. (a) Traditional approach using homogeneous transition metal complexes. (b) Photo- and electrocatalytic approach. (c) This work: heterogeneous copper-catalysed water–gas shift reaction. | ||
While most methods described above make use of homogeneous catalyst systems, more practical heterogeneous materials have been scarcely studied in dehalogenative labeling reactions.2a The clear advantage of heterogeneous catalysts is the ease of product separation by simple filtration, as well as their convenient catalyst recovery and recycling. In addition, a suitable heterogeneous material might be more stable in aqueous conditions. Therefore, developing a general heterogeneous catalyst for deuterium labelling with broad substrate scope, selectivity along with excellent D-incorporation is highly desirable.
While most currently known protocols for deuterium labelling make use of deuterium gas, the use of D2O would be advantageous with respect to price and handling; however, it is much less investigated. In this respect, the well-known water–gas shift (WGS) reaction is of interest, as it allows to produce high purity hydrogen gas from carbon monoxide (CO) and water. So far, this concept has been used both in homogeneous and heterogeneous catalysis, yet mainly in hydroformylation and hydrogenation reactions including nitro reduction, reductive amination, hydrogenation of carbonyls and alkenes.10 However, due to the high-temperature requirement (>180 °C), applications in organic synthesis remain largely underdeveloped.11
Herein, we present a heterogeneous copper catalyst, which enables water–gas shift reaction at milder conditions. Thereby, selective deuterodehalogenation of more than 20 diverse aryl bromides is achieved with high deuterium incorporation using of D2O/CO (Fig. 1c).
Results and discussion
Reaction development
Initially, we used commercially available 2-bromo-6-methoxynaphthalene as the benchmark substrate in D2O under CO pressure for deuteration at 140 °C (Tables 1 and S1 in ESI†).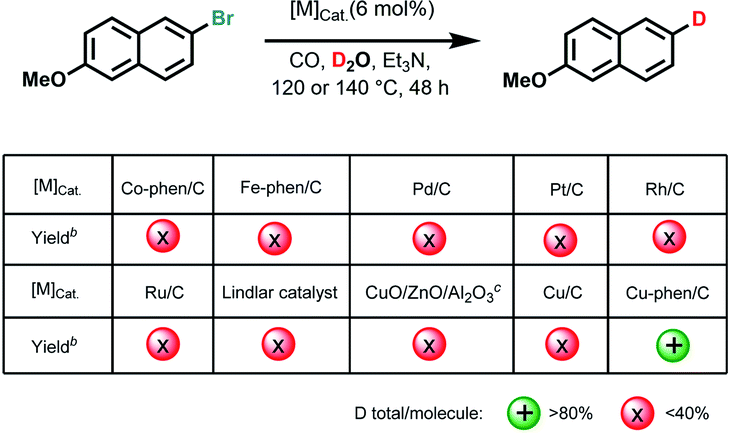
Based on our previous work on water–gas shift reactions for hydrogenation12 selected noble and non-noble metal precursors with phosphorous and nitrogen ligands were first examined. However, no deuterated products were observed with or without ligands (Table S1,† entries 1–6), except for Cu(OAc)2, Cu(OTf)2 as well as CuO providing trace amounts of the desired products (Table S1,† entries 7–12). Recently, we introduced a variety of heterogeneous 3d-metal catalysts for advanced organic synthesis.13 Typically, these nano-structured materials are prepared by pyrolysis of N-ligated complexes in the presence of carbon (Vulcan XC72R) or common inorganic supports. Unfortunately, none of them including Cu-phen/Al2O3, Co-phen/C-800 (800 refers to the pyrolysis temperature), Co-mel/TiO2 (mel = melamine), Fe-phen/C-800, Mn-phen/C-800, Ni-phen/SiO2-800, Ni-phen/SiO2-1000 (phen = 1,10-phenanthroline) showed labelling activity for our benchmark system (Table S1,† entries 14–20). Much to our surprise, pyrolysis of copper(II) acetate and 1,10-phenanthroline on Vulcan XC72R resulted in a highly active material, whereas Cu/C-800 and Cu/Al2O3-800 only furnished much smaller amounts of D-product (Table S1,† entries 13, 21–22). Similarly, materials pyrolyzed at 600, 900, and 1000 °C as well as the ones obtained with a different pyrolysis temperature ramp (5 °C min−1 instead of 25 °C min−1) provided lower yields for dehalogenative deuteration (Table S1,† entries 23–26). Performing the benchmark reaction in D2O in the presence of CO at 120 °C gave 92% deuterium-labelled product after 48 h (Table S1,† entries 27 and 28). In contrast to our novel material, commercially available noble metal catalysts such as Pd/C, Pt/C, Rh/C, Ru/C, and Lindlar catalyst did not show significant activities (Table S1,† entries 29–33). For example, the classic Lindlar catalyst provided 1b in 13% deuteration yield (Tables 1 and S1,† entries 29–33). Comparing the catalytic activity with Cu/ZnO/Al2O3, the methanol synthesis catalyst, the new material outperforms this benchmark catalyst, too (Table S1,† entry 34). As expected, a control experiment under identical conditions revealed no labelled product in the absence of the catalyst (Table S1,† entry 35).
Catalyst characterisation
The optimal catalyst Cu-phen/C-800 was prepared by impregnation of commercially available Vulcan XC72R with in situ generated complexes of copper(II) acetate and 1,10-phenanthroline and subsequent pyrolysis at 800 °C under inert conditions (see ESI†). For this material, a Cu content of 2.4 wt% was determined by elemental analysis.To further understand the structural features of Cu-phen/C-800, X-ray photoelectron spectroscopy (XPS), and scanning transmission electron microscopy (STEM) measurements were performed. XPS measurements were carried out to obtain information about the valence states of copper on the surface of the material. As a result, a mixture of Cu2+ and Cu1+/Cu0 was detected by XPS analysis at the surface of the fresh catalyst with Cu2+ as dominating valence state.
In addition, the ratio between both species changed from the fresh to the used catalyst so that Cu1+/Cu0 becomes dominating after the fifth run (see Section S.7 Fig. S1 in the ESI† for detailed discussion), and the total concentration of Cu was reduced slightly in the used catalyst.
STEM measurements of the fresh catalyst show a few very large Cu-containing particles (Fig. 2a), while in most parts of the material no Cu particles are visible at all (Fig. 2b). Notably, after the first and the fifth run (Fig. S2 and S4†), smaller Cu particles can be detected, which may either arise from sintering effects of finely dispersed copper in the fresh catalyst or are created by reorganization of the larger Cu particles during the reaction. An indication for the latter case is the decreased amount of Cu after the reaction, which points to a partial dissolution of Cu. Moreover, it is known that copper oxides can undergo reversible dissolution–precipitation steps.14 Analysis of the EDX spectra further reveal a transition from mainly oxidic copper species after the first run to mainly metallic copper after five runs (Fig. S3 and S5†). This is in accordance with the results found by XPS. Summarizing the obtained characterization data, we conclude that the freshly pyrolyzed catalyst consists of larger copper particles, which are reformatted and converted to fine dispersed nanoparticles with a lower oxidation state during the reaction.
 | ||
| Fig. 2 Selected HAADF-STEM images of fresh Cu-phen/C-800. The fresh catalyst contains a few very large Cu containing particles (a), while in most parts of the material no Cu particles are visible at all (b). | ||
Nitrogen dopants are known to tailor the properties of supported materials and render their potential use for catalysis.15 Thus, the role of nitrogen was studied in more details. Firstly, we compared the structural features of Cu-phen/C-800, Cu/C-800 and Cu-melamine/C-800. STEM measurements on this Cu-melamine/C-800 material as well as on the material without any nitrogen source revealed that there is virtually no difference on the appearance of the materials. We observed the typical Vulcan XC72R support and, some nitrogen in the melamine derived material, similar to the 1,10-phenanthroline derived material. Copper was, as in the other materials, not visible or just as huge separate entities. To sum up, nitrogen does not seem to influence the morphology and appearance of the material. It is more likely providing sites that are involved in the mechanism. This is also reflected by catalytic performance. As shown in Table S2,† the nitrogen source is crucial for the reaction to proceed. We propose from the differences in catalytic activity of Cu-phen/C-800, Cu-melamine/C-800 and Cu/C-800 that nitrogen might provide basic sites for either D-abstraction or intermediate D “storage” in this catalytic process.
KIE investigations
To prove the proposed WGS reaction, the gas composition during the catalytic process was analyzed by GC (for details see ESI, Section 6†), confirming formation of CO2. Moreover, kinetic studies utilizing 3-bromoanisole were performed to investigate this deuterodehalogenation reaction (Fig. 3 and ESI, Section 8†). As shown in Fig. S6,† no induction period is observed and 15% of the deuterated product was formed after the first hour. Next, to get a better mechanistic understanding two parallel kinetic isotope effect experiments were carried out using D2O and H2O (Fig. S7†). These KIE experiments showed basically no difference (kH/kD: 1.01; Fig. 3a), which rules out the cleavage of the D–OD bond as the rate-limiting step. Interestingly, by comparing the deuterodehalogenation of 4-bromoaniline and 4-bromo-2,6-d2-aniline, secondary KIE is observed (Fig. 3b). This kinetic isotope effect between the deuterated and non-deuterated substrates may be derived from the weaker coordination of the aromatic rings on the catalyst surface.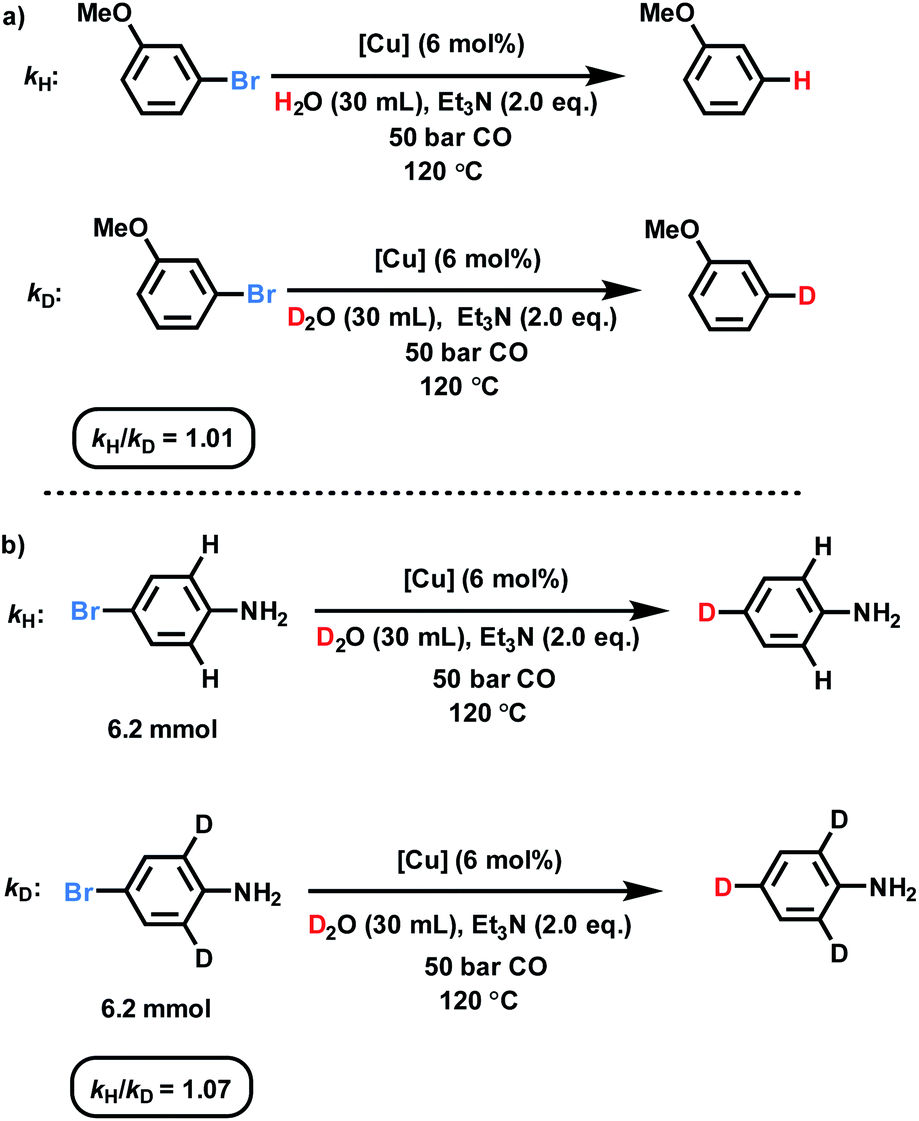 | ||
| Fig. 3 KIE investigations over the dehalogenative deuteration of (a) 3-bromoanisole and (b) 4-bromoaniline. | ||
Synthetic applications
With the optimized conditions for deuteration of 2-bromo-6-methoxynaphthalene in hand, substrate scope and tolerance towards functional groups were evaluated. Without further optimization Cu-phen/C-800 permitted smooth deuteration of 16 different compounds to furnish the corresponding products with high chemo- and regioselectivity (Scheme 1, 1b–16b). As many important biological compounds including drugs contain amide moieties, deuteration of benzamides and acetanilides is interesting and the labelled products are isolated in high yields by simple filtration from corresponding bromides (2a–6a). However, small amount of D-incorporation on the acetyl groups are observed, too. As an example, fluoro-containing acetanilide afforded the deuterated product 6b. Besides, no hydrolysis and hydrogenation of cyano groups were observed in 7b and 8b. Similarly, pyridine-containing bromides provided the dehalogenative deuteration products in good yields (9b and 10b). | ||
| Scheme 1 Synthetic applications of Cu-phen/C-800 in deuteration of (hetero)aryl bromides. Standard conditions: substrate (0.25 mmol, 1 equiv.), copper catalyst (6 mol%), D2O (1.5 mL), Et3N (2.0 equiv.), 40 bar CO, 120 °C, 48 h. Isolated yields. The black circles and numbers denote the positions of the C–H bonds that are labelled and the percent incorporation of the hydrogen isotope, respectively. | ||
Next, deuteration of different oxygen, and/or nitrogen-containing heterocyclic aryl bromides were investigated, and the corresponding D-labelled O-heterocycles, 3,4-dihydro-2(1H)-chinolinone, pyridine, carbazole, indole, and quinoline were isolated (11b–16b). These heterocycles were used as they represent common structural components of pharmaceuticals. Indeed, nitrogen-based heterocyclic motives are included in 59% of FDA-approved drugs.16 As shown in Scheme 1, catalytic labeling with D2O/CO provided the corresponding products following our standard protocol in 84–96% yields. However, aryl chlorides gave no significant dehalogenative deuteration under standard conditions.
Having a convenient approach for deuterodehalogenation of a variety of substrates in hand, some practical aspects of the D2O/CO system were evaluated. It is possible to run such labelling reactions smoothly on >20 g-scale as shown in Scheme 2 albeit with a longer reaction time and slightly higher temperatures. Notably, the catalyst system could be re-used for different substrates. For example, aryl bromides 17a–19a (>20 g scale) are converted to deuterated-products using the same, recycled copper catalyst (see more details in the ESI†). Moderate isolated yields resulted due to the relatively low conversion (48 h at 140 °C, 17b–19b). This method also exhibited high chemoselectivity with the easily reducible formyl group even on large scale (19b). 4-Bromoanisole, aniline, and phenol gave the corresponding deuterated products in more than 90% yields (>30 g scale). In case of 4-bromo-2,6-dideuterophenol 21a and 2-acetamido-4-fluoro-1-bromobenzene 23a H/D exchange products were obtained at elevated temperature (130 °C) after extended reaction time (6 d) along with hydrogen deuterium exchange on the methyl group in 23b.
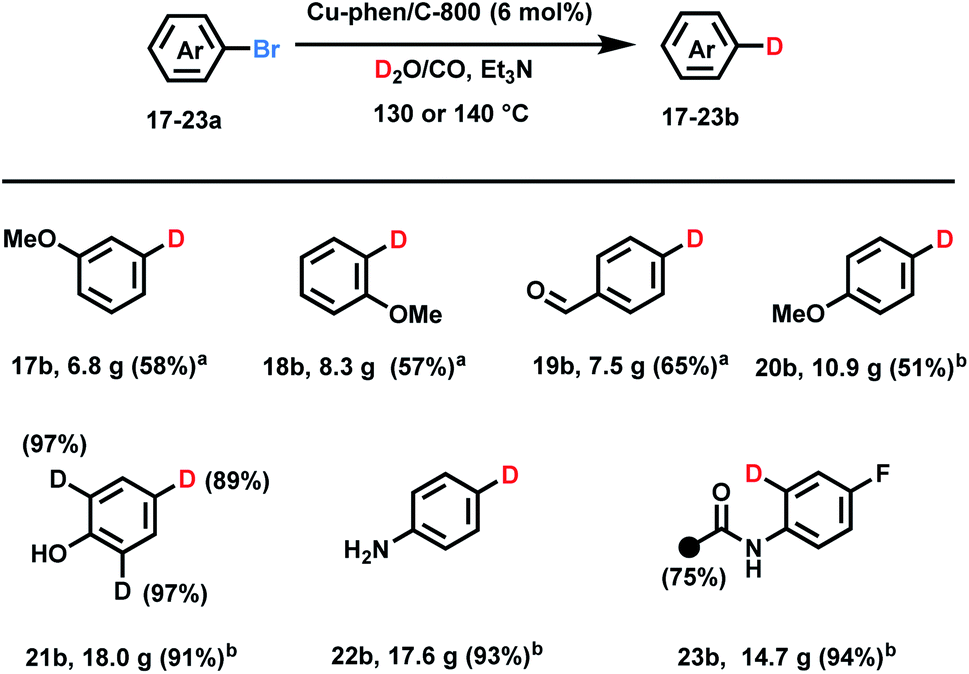 | ||
| Scheme 2 Synthetic applications of Cu-phen/C-800 in deuteration of aryl bromides with various functionalities. Standard conditions: substrate (100 or 200 mmol, 1 equiv.), copper catalyst (6 mol%), D2O (100 mL), Et3N (2.0 equiv.), 50 bar CO. (a) 140 °C, 48 h. (b) 130 °C, 6 days. Isolated yields. The black circles and numbers denote the positions of the C–H bonds that are labelled and the percent incorporation of the hydrogen isotope, respectively. | ||
Finally, the heterogeneous copper catalyst was recycled up to five times for the benchmark reaction. As depicted in Fig. S10 and S11,† no significant loss of activity was observed at the first three runs. The low yields of the fourth and fifth runs are resulted from the low conversion of the substrate. ICP-OES measurements proved that small amounts of copper leached after first run (17 ppm) and after the second run (42 ppm, Table S3 in the ESI†). Concerning the activity loss, we suppose that the copper leaching and the reduction of the metal could be the main reason. Based on the STEM analysis, we observed that copper from 5th run is more reduced after the 1st run. Additionally, we observed smaller copper species in the recycled catalysts. These originate most likely from reorganization, i.e. dissolution/re-precipitation, of the bigger copper particles in the fresh sample. This dissolution can also lead to a certain degree of leaching.
Conclusions
In conclusion, we present a novel labelling approach taking advantage of a catalytic domino water gas shift-deuterodehalogenation sequence utilizing inexpensive D2O under CO pressure. In the presence of a heterogeneous nitrogen-doped copper catalyst several aryl bromides could be effectively labelled in high yields and with good functional group tolerance. This procedure represents an easily scalable deuterodehalogenation methodology and the presented catalyst is air- and water-stable.Data availability
All the experimental data have been included in the ESI.†Author contributions
M. B. and W. Li conceived and designed the project; W. Li, R. Q., W. Liu and F. B. performed the experiments and analysed the data; S. B. was responsible for XPS experiments; N. R. was responsible for STEM measurements; K. J. supervised the research activities, supported the project with funding acquisition, and coedited the manuscript; W. Li. and M. B. co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the analytical staff of the Leibniz-Institute for Catalysis, Rostock, for their excellent service, specifically Dr Henrik Junge and Anja Kammer for GC analysis, David K. Leonard for assistance in the preparation of the manuscript. Funding: We gratefully acknowledge the support from the Federal Ministry of Education and Research (BMBF), the State of Mecklenburg-Vorpommern, EU (FLIX project), the European Research Council (ERC; project NoNaCat 670986), and the Alexander von Humboldt Foundation.References
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed; (b) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed; (c) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- H. Yang and D. Hesk, J. Labelled Compd. Radiopharm., 2020, 63, 296–307 CrossRef CAS PubMed.
- (a) C. Schmidt, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS PubMed; (b) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed; (c) A. Katsnelson, Nat. Med., 2013, 19, 656 CrossRef CAS PubMed; (d) C. Elison, H. Rapoport, R. Laursen and H. W. Elliott, Science, 1961, 134, 1078–1079 CrossRef CAS PubMed.
- (a) M. Zhang, X. A. Yuan, C. Zhu and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 312–316 CrossRef CAS PubMed; (b) M. Wang, Y. Zhao, Y. Zhao and Z. Shi, Sci. Adv., 2020, 6, eaba0946 CrossRef CAS PubMed; (c) H. Geng, X. Chen, J. Gui, Y. Zhang, Z. Shen, P. Qian, J. Chen, S. Zhang and W. Wang, Nat. Catal., 2019, 2, 1071–1077 CrossRef CAS PubMed; (d) L. Neubert, D. Michalik, S. Bahn, S. Imm, H. Neumann, J. Atzrodt, V. Derdau, W. Holla and M. Beller, J. Am. Chem. Soc., 2012, 134, 12239–12244 CrossRef CAS PubMed; (e) Z. Zhang, C. Qiu, Y. Xu, Q. Han, J. Tang, K. P. Loh and C. Su, Nat. Commun., 2020, 11, 4722 CrossRef CAS PubMed; (f) Y. Sawama, A. Nakano, T. Matsuda, T. Kawajiri, T. Yamada and H. Sajiki, Org. Process Res. Dev., 2019, 23, 648–653 CrossRef CAS; (g) G. Pieters, C. Taglang, E. Bonnefille, T. Gutmann, C. Puente, J. C. Berthet, C. Dugave, B. Chaudret and B. Rousseau, Angew. Chem., Int. Ed., 2014, 53, 230–234 CrossRef CAS PubMed; (h) C. Taglang, L. M. Martinez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. Sam Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., Int. Ed., 2015, 54, 10474–10477 CrossRef CAS PubMed; (i) M. Valero, D. Bouzouita, A. Palazzolo, J. Atzrodt, C. Dugave, S. Tricard, S. Feuillastre, G. Pieters, B. Chaudret and V. Derdau, Angew. Chem., Int. Ed., 2020, 59, 3517–3522 CrossRef CAS PubMed; (j) A. Kurimoto, R. S. Sherbo, Y. Cao, N. W. X. Loo and C. P. Berlinguette, Nat. Catal., 2020, 3, 719–726 CrossRef CAS; (k) R. P. Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef PubMed; (l) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed; (m) J. A. Smith, K. B. Wilson, R. E. Sonstrom, P. J. Kelleher, K. D. Welch, E. K. Pert, K. S. Westendorff, D. A. Dickie, X. Wang, B. H. Pate and W. D. Harman, Nature, 2020, 581, 288–293 CrossRef CAS PubMed; (n) F. Legros, P. Fernandez-Rodriguez, A. Mishra, R. Weck, A. Bauer, M. Sandvoss, S. Ruf, M. Mendez, H. Mora-Rado, N. Rackelmann, C. Poverlein and V. Derdau, Chem.–Eur. J., 2020, 26, 12738–12742 CrossRef CAS PubMed; (o) V. Müller, R. Weck, V. Derdau and L. Ackermann, ChemCatChem, 2019, 12, 100–104 CrossRef; (p) L. V. A. Hale and N. K. Szymczak, J. Am. Chem. Soc., 2016, 138, 13489–13492 CrossRef CAS PubMed; (q) J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990–1993 CrossRef CAS PubMed; (r) A. Palazzolo, S. Feuillastre, V. Pfeifer, S. Garcia-Argote, D. Bouzouita, S. Tricard, C. Chollet, E. Marcon, D. A. Buisson, S. Cholet, F. Fenaille, G. Lippens, B. Chaudret and G. Pieters, Angew. Chem., Int. Ed., 2019, 58, 4891–4895 CrossRef CAS PubMed.
- B. Czeskis, C. S. Elmore, A. Haight, D. Hesk, B. D. Maxwell, S. A. Miller, T. Raglione, K. Schildknegt, J. F. Traverse and P. Wang, J. Labelled Compd. Radiopharm., 2019, 62, 690–694 CrossRef CAS PubMed.
- (a) D. Hesk, J. Labelled Compd. Radiopharm., 2020, 63, 247–265 CrossRef CAS PubMed; (b) W. J. Kerr, G. J. Knox and L. C. Paterson, J. Labelled Compd. Radiopharm., 2020, 63, 281–295 CrossRef CAS PubMed.
- (a) X. Wang, M. H. Zhu, D. P. Schuman, D. Zhong, W. Y. Wang, L. Y. Wu, W. Liu, B. M. Stoltz and W. B. Liu, J. Am. Chem. Soc., 2018, 140, 10970–10974 CrossRef CAS PubMed; (b) D. Y. Ong, C. Tejo, K. Xu, H. Hirao and S. Chiba, Angew. Chem., Int. Ed., 2017, 56, 1840–1844 CrossRef CAS PubMed; (c) Y. Miura, H. Oka, E. Yamano and M. Morita, J. Org. Chem., 1997, 62, 1188–1190 CrossRef CAS; (d) M. Kuriyama, N. Hamaguchi, G. Yano, K. Tsukuda, K. Sato and O. Onomura, J. Org. Chem., 2016, 81, 8934–8946 CrossRef CAS PubMed.
- (a) C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G. H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q. H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef PubMed; (b) C. Liu, S. Han, M. Li, X. Chong and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18527–18531 CrossRef CAS PubMed; (c) L. Lu, H. Li, Y. Zheng, F. Bu and A. Lei, CCS Chem., 2020, 2, 2669–2675 CrossRef; (d) K. Mitsudo, T. Okada, S. Shimohara, H. Mandai and S. Suga, Electrochem, 2013, 81, 362–364 CrossRef CAS; (e) Y. Li, Z. Ye, Y.-M. Lin, Y. Liu, Y. Zhang and L. Gong, Nat. Commun., 2021, 12, 2894 CrossRef CAS PubMed.
- (a) A. Ambrosi and S. E. Denmark, Angew. Chem., Int. Ed., 2016, 55, 12164–12189 CrossRef CAS PubMed; (b) M. Y. S. Ibrahim and S. E. Denmark, Angew. Chem., Int. Ed., 2018, 57, 10362–10367 CrossRef CAS PubMed.
- C. M. Crudden, Y. Maekawa, J. J. Clarke, T. Ida, Y. Fukumoto, N. Chatani and S. Murai, Org. Lett., 2020, 22, 8747–8751 CrossRef CAS PubMed.
- (a) A. Tlili, J. Schranck, H. Neumann and M. Beller, Chem.–Eur. J., 2012, 18, 15935–15939 CrossRef CAS PubMed; (b) J. Liu, C. Kubis, R. Franke, R. Jackstell and M. Beller, ACS Catal., 2016, 6, 907–912 CrossRef CAS; (c) S. Gulak, L. Wu, Q. Liu, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 7320–7323 CrossRef PubMed.
- (a) R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed; (b) X. Cui, Y. Li, S. Bachmann, M. Scalone, A. E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137, 10652–10658 CrossRef CAS PubMed; (c) R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brückner, ACS Catal., 2014, 4, 1845–1849 CrossRef CAS.
- (a) H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS; (b) L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04259a |
| This journal is © The Royal Society of Chemistry 2021 |