
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWeaving host matrices with intermolecular hydrogen bonds for high-efficiency white thermally activated delayed fluorescence†
Yuee
Tian‡
,
Huiqin
Wang‡
,
Yi
Man
,
Nan
Zhang
,
Jing
Zhang
*,
Ying
Li
,
Chunmiao
Han
and
Hui
Xu
 *
*
Key Laboratory of Functional Inorganic Material Chemistry, Ministry of Education & School of Chemistry and Material Science, Heilongjiang University, 74 Xuefu Road, Harbin 150080, P. R. China. E-mail: hxu@hlju.edu.cn; zhangjing@hlju.edu.cn
First published on 12th October 2021
Abstract
A thermally activated delayed fluorescence (TADF) white organic light-emitting diode (WOLED) holds great promise for low-cost, large-scale lighting applications. Nevertheless, manipulating exciton allocation in a white TADF single layer is still a challenge. Herein, we demonstrate that the exciton kinetic process of dually doped white TADF films is strongly dependent on the grid regularity of the host matrix. Intermolecular hydrogen bonds (IHBs) are used to weave the matrices of two host molecules DPEQPO and DPSQPO featuring four phosphine oxide (PO) groups and different IHB orientations. The DPSQPO matrix forms regular grids to uniformly disperse and separate dopants, while DPEQPO exhibits chaotic IHBs, in turn inducing a heterogeneous dopant distribution. As a consequence, in both photoluminescence and electroluminescence processes, in contrast to DPEQPO hosted systems with comparable singlet Förster resonance energy transfer and triplet Dexter energy transfer, DPSQPO provides a FRET-predominant exciton allocation between blue and yellow dopants, which markedly suppresses triplet quenching and improves the white color purity, resulting in a state-of-the-art external quantum efficiency up to 24.2% of its single-emissive-layer pure-white TADF diode, in contrast to 16.0% for DPEQPO based analogs. These results indicate the significance of host engineering for exciton kinetics and suggest the feasibility of host grid design for developing high-performance TADF lighting.
1. Introduction
Thermally activated delayed fluorescence (TADF) holds immense potential for application in white organic light-emitting diodes (WOLEDs) for diverse daily lighting applications,1–3 owing to its merits of 100% theoretical internal quantum efficiency (IQE, ηIQE), low cost and environmental friendliness.4–7 One of the intrinsic advantage of TADF materials is that they are pure-organic donor–acceptor (D–A) systems featuring strong intramolecular charge transfer (CT), giving rise to negligible singlet–triplet splitting (ΔEST)8,9 and feasible reverse intersystem crossing (RISC) for up-conversion from nonradiative triplets to radiative singlets.10 Therefore, despite showing potentially 100% exciton harvesting and desired sustainability, TADF molecules have high polarity and strong intermolecular interactions, causing serious collision and dipole-interaction induced exciton quenching and marked efficiency roll-off. Consequently, doped emissive layers (EMLs) featuring TADF emitters dispersed in an inertia host matrix were widely used in high-efficiency monochromic diodes, partially leading to the rapid improvement of external quantum efficiencies (ηEQE) beyond 25%.11–18 These advances lead to developing high-performance pure-TADF WOLEDs,19–25 especially based on single-EML structures26–33 to maximize the advantages of organic materials and devices in low cost and large-scale production.However, with CT-featuring first singlet (S1) states (1CT), the emission colors of TADF emitters are directly proportional to molecular polarities. Therefore, dipole–dipole interactions34,35 between blue and other high-polarity emitters worsen the quenching of multicomponent white TADF systems. Furthermore, compared to fluorescence (FL) and phosphorescence (PH) counterparts, the exciton allocation between different TADF emitters is markedly more complicated through not only singlet Förster resonance energy transfer (FRET), but also charge-exchange based triplet Dexter energy transfer (DET) and intermolecular CT.36,37 These processes should be accurately balanced to rationalize exciton allocation for achieving both high efficiencies and satisfactory white color purity.38,39 Moreover, the short-range feature of DET makes it often accompanied by intermolecular interaction induced triplet quenching,40e.g. singlet–triplet (STA) and triplet–triplet annihilation (TTA).41 Therefore, combining the state-of-the-art monochromatic efficiencies into a single-EML TADF WOLED is still a formidable challenge, because of mutual quenching between different emitters and their competition in the exciton allocation process.42–45
Considering the different distance dependences of energy transfer,46,47 CT and quenching effect,48 the rational dispersion of TADF molecules in host matrices is crucial to overcome this challenge, since intermolecular interactions and contributions of FRET, DET and intermolecular CT to exciton allocation can be accurately modulated and optimized by adjusting dispersive uniformity in the host matrix. Compared to FL and PH counterparts, most blue/white TADF diodes adopted high doping concentrations of blue emitters (≥10%) to take use of intermolecular CT channels for RISC enhancement.49–52 In EMLs of these devices, hosts with too weak intermolecular interactions cannot effectively disperse blue dopants. Conversely, too strong intermolecular interactions of host matrices cause the concurrent heterogeneous distributions of both host and dopant molecules. It means the “ideal” host materials for TADF WOLEDs should have appropriate intermolecular interactions to form regular gridding matrices uniformly filled with dopant molecules. In our previous studies, compared to too strong π–π stacking, intermolecular hydrogen bond (IHB) networks in host matrices were more effective to separate dopants and avoid worsening host–host and host–dopant quenching.53–56 It is also noted that IHB modes directly influenced the electroluminescence (EL) performance of the devices, which actually provides a desired platform to get a deep insight into the accurate relationship between the regularity of host grids and exciton-involved PL and EL processes in single-layer white TADF systems.
In this contribution, we construct two host molecules named DPEQPO and DPSQPO respectively featuring diphenylether (DPE) and diphenylsulfide (DPS) skeletons substituted with four diphenylphosphine oxide (DPPO) groups (Scheme S1†). The bond angle reduction from ether to sulfide induces the orientations of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups to change from four different directions for DPEQPO to more concentrated but sufficient two directions for DPSQPO. Consequently, in contrast to the chaotic and complicated IHB network of DPEQPO, DPSQPO exhibits high regularity with ordered IHB grids, giving rise to the uniform dispersion of both blue and yellow TADF dopants in its dually doped single-layer film. The rationally weaved DPSQPO matrix endowed the single-EML pure-white and warm-white diodes with a state-of-the-art ηEQE up to 24.2% and 22.4%, respectively, which are improved by 70% and 40%, compared to DPEQPO based analogs. For both PL and EL processes, in addition to dramatically improved quantum efficiencies, the delayed fluorescence (DF) lifetimes (τDF) of DPSQPO based white TADF systems are far shorter than those of DPEQPO based analogs. These results suggest that uniform dopant dispersion and separation in the DPSQPO matrix make FRET predominant in exciton allocation, and the effective restraint of DET actually accelerates exciton radiation, and simultaneously suppresses triplet quenching. Thus, host intermolecular interaction optimization and host-dopant synergism are crucial for developing high-performance single-layer white TADF systems.
O groups to change from four different directions for DPEQPO to more concentrated but sufficient two directions for DPSQPO. Consequently, in contrast to the chaotic and complicated IHB network of DPEQPO, DPSQPO exhibits high regularity with ordered IHB grids, giving rise to the uniform dispersion of both blue and yellow TADF dopants in its dually doped single-layer film. The rationally weaved DPSQPO matrix endowed the single-EML pure-white and warm-white diodes with a state-of-the-art ηEQE up to 24.2% and 22.4%, respectively, which are improved by 70% and 40%, compared to DPEQPO based analogs. For both PL and EL processes, in addition to dramatically improved quantum efficiencies, the delayed fluorescence (DF) lifetimes (τDF) of DPSQPO based white TADF systems are far shorter than those of DPEQPO based analogs. These results suggest that uniform dopant dispersion and separation in the DPSQPO matrix make FRET predominant in exciton allocation, and the effective restraint of DET actually accelerates exciton radiation, and simultaneously suppresses triplet quenching. Thus, host intermolecular interaction optimization and host-dopant synergism are crucial for developing high-performance single-layer white TADF systems.
2. Results and discussion
2.1 Design, structures and morphological properties
To form continuous IHB networks, host molecules should contain more than one hydrogen-bond donor group. In the cases of DPEQPO and DPSQPO, collectively named DPXQPO, their four PO groups make two-dimensionally structural weaving feasible. The plain weave models show that the grid structure is directly dependent on the knot arrangement, which provides two conventional four-knot models (Fig. 1a): (i) Model I: wefts with four random and scattered knots linked with warps to constitute a complicated but dense cross structure; (ii) Model II: pairwise centralizing knots on wefts improves weft and warp regularity, and relatively enlarges grid gaps. In DPXQPO matrices, PO groups serve as knots on DPE and DPS skeletons (wefts) to form PO⋯H IHBs (warps) between adjacent molecules (Fig. 1b). Single-crystal structures show that the angles of the C–O–C bond in DPEQPO and the C–S–C bond in DPSQPO are respectively 118.7° and 101.8°, respectively, close to the typical values of 120° and 103° (Fig. 1c). The larger C–O–C bond angle corresponds to a dihedral angle of 79.9° for the DPE skeleton, in contrast to 68.7° of the DPS skeleton. Therefore, the larger mutual steric hindrance between two DPPOs at the ortho positions of more linear DPE gives rise to the different orientations of one inward and the other outward PO bonds. Due to a similar steric effect, the PO orientations of two para-DPPOs in DPEQPO are also different. As a result, all PO groups of DPEQPO point to different directions. In contrast, a more folded DPS skeleton provides more space to the two ortho-DPPO groups of DPSQPO, leading to two inward PO bonds with the same orientation. The identical meta-position steric hindrances of these ortho-DPPOs further induce the same PO orientations of two para-DPPOs.
 | ||
| Fig. 1 Design strategy of host matrices with different weaving modes. (a) Weaving modes of a quadruple system featuring a skeleton substituted with four hydrogen-bond donors. Two plain weave models are borrowed to illustrate the correlation between hydrogen-bond donor arrangements and intermolecular hydrogen-bond (IHB) network formation: (I) four hydrogen-bond donors (red dot, corresponding to knots) randomly disperse on the skeleton, corresponding to the weft, leading to diverse and multidirectional IHB networks; (II) two sets of two hydrogen-bond donors respectively locate on each side of the skeleton, constituting IHB grids but reducing bond directions. (b) Chemical structures of DPXQPO (X = S for DPSQPO and O for DPEQPO), in which skeletons diphenyl sulfide (DPS) and diphenyl ether (DPE) serve as the weft, and PO acceptors are like knots to form IHBs, corresponding to warps. (c) Single-crystal structures of DPXQPO, in which the dihedral angle and C–X–C angle respectively decrease from 79.9° and 118.7° of DPEQPO to 68.7° and 101.8° of DPSQPO. (d) Packing diagrams of DPXQPO single crystals. Adjacent four molecules are included to show intermolecular interactions. Distances between adjacent molecules are marked with arrows. IHBs are highlighted as blue dashed lines. (e) Atom force microscopy (AFM) images of neat and doped films based on DPXQPO matrices. DPXQPO:30% DMAC-DPS and DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu are blue and white-emitting films, in which DMAC-DPS and 4CzTPNBu are blue and yellow TADF emitters, respectively. | ||
Consequently, in accord with Model I, DPEQPO has four differently orientated PO groups alternately on both sides of the DPE skeleton. The PO arrangement in DPSQPO is similar to that in Model II, namely two ortho-substituted PO groups centralized on one side of the DPS skeleton, accompanied by two PO groups with the same orientation at the para-positions on the other side. As predicated by plain weave models, the single-crystal packing diagram of DPEQPO reveals that strong IHBs are formed between adjacent molecules with bond lengths in a range of 2.5–2.9 Å and especially diverse bond directions, which further constitute a complicated, disordered and interlaced IHB network (Fig. 1d). In contrast, DPSQPO displays grid-like IHB networks with simply two main bond directions and elongated bond lengths of 2.6–3.6 Å. The strong and accumulated IHB interactions markedly shorten the intermolecular distances of the DPEQPO matrix. More notably, DPEQPO exhibits four different molecular orientations. Each molecule of the DPEQPO tetramer contributes one phenyl to form a quasi-J-aggregate along the b axis with an average distance of 3.5 Å in the inner space (Fig. S1†). Not enough gaps between adjacent molecules actually reflect the remarkable self-aggregation tendency of DPEQPO. In comparison, owing to the regular intermolecular interactions, only two molecular orientations are observed in the DPSQPO packing diagram (Fig. S2†). Despite the slightly shorter intermolecular distance of DPSQPO dimer, the closest centroid–centroid distance between phenyls reaches 4.4 Å, beyond the critical value for π–π stacking. Meanwhile, the dimer–dimer distance in the tetramer is elongated by 0.5 Å. Thus, the DPSQPO tetramer constitutes a grid gap unit, which is further connected by a regular IHB network. It is obvious that compared to DPEQPO with a dense and intricate packing mode, DPSQPO is undoubtedly superior in alleviating local aggregation and improving doping uniformity.
The decomposition temperatures of DPXQPO are equally beyond 420 °C, indicating their similar device fabricability by vacuum evaporation (Fig. S3 and Table S1†). But, the melting point (Tm) of DPEQPO is 46 °C lower than that of DPSQPO, since an ordered and uniform molecular arrangement is more effective to improve packing stability than locally enhanced intermolecular interactions. The atom force microscopy (AFM) images of the vacuum evaporated films show that the surface roughness of the neat DPEQPO film (0.59 nm) is twice that of the neat DPSQPO film (0.25 nm) (Fig. 1e and S4†). The granular patterns further indicate local aggregations of the neat DPEQPO film. Singly doped blue-emitting and dually doped white-emitting films were prepared based on a blue TADF emitter bis[4-(9,9-dimethyl-9,10-dihydroacridine)phenyl]sulfone (DMAC-DPS) and a yellow TADF emitter 2,3,5,6-tetrakis(3,6-di-(tert-butyl)carbazol-9-yl)-1,4-dicyanobenzene (4CzTPNBu). DPEQPO:30% DMAC-DPS and DPEQPO:30% DMAC-DPS:0.5% 4CzTPNBu films reveal nearly identical roughness and granular patterns, indicating unchanged aggregation. Similarly, DMAC-DPS and 4CzTPNBu doped DPSQPO films preserve the good surface morphology, verifying the uniform dopant dispersion in DPSQPO grids.
2.2 WOLED performance
To figure out the influence of host matrix regularity on EL performance, we fabricated single-EML pure-TADF WOLEDs with a conventional four-layer configuration of ITO|MoO3 (6 nm)|NPB (50 nm)|mCP (5 nm)|DPXQPO:x% DMAC-DPS:y% 4CzTPNBu (20 nm)|DPXQPO (5 nm)|BPhen (45 nm)|LiF (1 nm)|Al, in which N,N′-bis-(1-naphthalenyl)-N,N′-bis-phenyl-(1,1′-biphenyl)-4,4′-diamine (NPB) and 4,7-diphenyl-1,10-phenanthroline (BPhen) were hole and electron transporting layers, and 5-nm thin layers of 1,3-bis(carbazol-9-yl)benzene (mCP) and DPXQPO were used to block exciton migration, respectively (Fig. 2a). x and y were respectively tuned in ranges of 20–40 and 0.2–1.5 (Fig. S5–S7†). The singly doped blue OLEDs achieved the best performance at x = 30, in which DPSQPO based devices realized the maximum ηEQE reaching 18.8%, 1.4 fold that of DPEQPO based analogs (Fig. S5 and Table S2†). Simultaneously, at x = 30, DPSQPO effectively reduced EQE roll-off to 20% at 1000 cd m−2, compared to 25% of DPEQPO based devices. It is noted that the efficiencies of DPSQPO:x% DMAC-DPS based devices were markedly reduced by increasing x from 30 to 40, in contrast to the nearly unchanged efficiencies of DPEQPO based analogs. Furthermore, the EL spectra of DPSQPO based blue devices were nearly independent on x, but the EL emissions of DPEQPO based analogs gradually shifted red when increasing x. It means that the intermolecular interaction-induced concentration quenching of DMAC-DPS can be hardly alleviated in DPEQPO based devices, but can be effectively suppressed in the EMLs of DPSQPO:x% DMAC-DPS.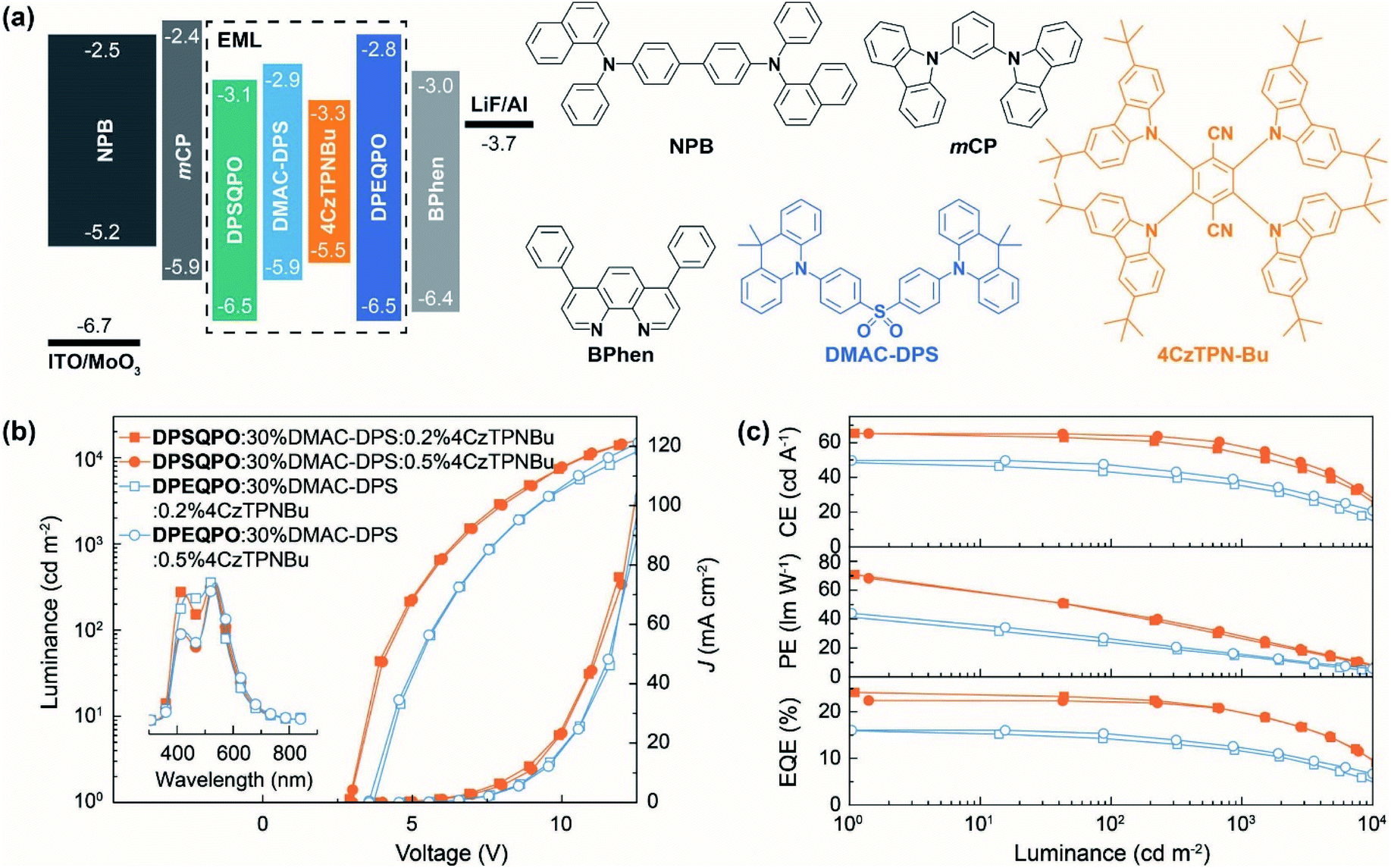 | ||
| Fig. 2 EL performance of white TADF devices. (a) Structures of single-emissive layer WOLEDs, ITO|MoOx (6 nm)|NPB (50 nm)|mCP (5 nm)|DPXQPO:30 wt% DMAC-DPS:y% 4CzTPNBu (20 nm)|Bphen (35 nm)|LiF (1 nm)|Al (y = 0.2 for pure-white and 0.5 for warm-white), and chemical structures of the employed materials. (b) Current density (J)-voltage–luminance characteristics and EL spectra of the devices. (c) Relationship between the current efficiency (CE), power efficiency (PE) and external quantum efficiency (EQE) and luminance of the WOLEDs. | ||
The EL spectra of all the devices based on dually doped EMLs containing DMAC-DPS and 4CzTPNBu showed blue and yellow components respectively at ∼470 and ∼550 nm (Fig. 2b, S6a and S7a†). EL chromaticity was readily tuned by gradually increasing the 4CzTPNBu concentration (y%), causing emission color change from pure white to warm white, corresponding to Commission Internationale de l'Eclairage (CIE) coordinates and correlated color temperature (CCT) close to standard Illuminants D65 and A (Fig. S6b and S7b†). In the practical brightness range of 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2, the EL emissions of these WOLEDs were early unchanged, corresponding to the negligible variations of CIE coordinates (x, y) and CCT values were within 0.02 for x + y and 300 K, respectively. The excellent chromatic stabilities make these devices competent for diverse lighting applications. It is noteworthy that the relative blue intensities of DPSQPO based WOLEDs were higher than those of DPEQPO based analogs, reflecting more excitons allocated to 4CzTPNBu in DPEQPO hosted EMLs due to enhanced blue-to-yellow energy transfer.
000 cd m−2, the EL emissions of these WOLEDs were early unchanged, corresponding to the negligible variations of CIE coordinates (x, y) and CCT values were within 0.02 for x + y and 300 K, respectively. The excellent chromatic stabilities make these devices competent for diverse lighting applications. It is noteworthy that the relative blue intensities of DPSQPO based WOLEDs were higher than those of DPEQPO based analogs, reflecting more excitons allocated to 4CzTPNBu in DPEQPO hosted EMLs due to enhanced blue-to-yellow energy transfer.
Compared to the corresponding blue diodes, 4CzTPNBu incorporation in DPEQPO based EMLs largely increased the brightness of the WOLEDs (Fig. 2b and S6c and Table S2†). The maximum luminance of DPEQPO based WOLEDs was directly proportional to y when y ≤1.0 and then kept stable at y = 1.5. Furthermore, codoping 4CzTPNBu hardly influences driving voltages and current densities (J). It means that in the EMLs of DPEQPO:30% DMAC-DPS:y% 4CzTPNBu, energy transfer from DMAC-DPS to 4CzTPNBu actually alleviated the self-quenching of the former, thereby facilitating exciton radiation. The driving voltages and J of DPSQPO based WOLEDs were also comparable at y ≤0.5 (Fig. 2b and S7c†). At the critical concentration (y = 1.0), direct carrier injection, capture and recombination by 4CzTPNBu made a significant contribution to exciton formation and utilization, leading to markedly reduced driving voltages and increased J and luminance. So, it implies that 4CzTPNBu molecules in DPEQPO hosted films were mostly embedded by DMAC-DPS aggregates; meanwhile, for DPSQPO:30% DMAC-DPS:y% 4CzTPNBu, both the dopants are dispersed uniformly and separated from each other in the DPSQPO grids.
DPSQPO:30% DMAC-DPS:0.2% 4CzTPNBu endowed its diode with the state-of-the-art efficiencies, e.g. a maximum current efficiency (CE, ηCE), power efficiency (PE, ηPE) and ηEQE of 65.3 cd A−1, 70.8 lm W−1 and 24.2%, which are among the best results from single-EML pure-TADF WOLEDs (Fig. 2c and S7d and Table S2†). At y = 0.5, DPSQPO still supported the highest efficiencies with the maxima of 65.2 cd A−1, 68.2 lm W−1 and 22.4% for its WOLED. At 1000 cd m−2, both these two WOLEDs still preserved the favorable efficiencies beyond 55 cd A−1, 30 lm W−1 and 20%. In contrast, DPEQPO based WOLEDs achieved the highest efficiencies at y = 1.0 with the maxima of 49.7 cd A−1, 44.0 lm W−1 and 16.1%, which rapidly decreased to 39.0 cd A−1, 16.2 lm W−1 and 12.5% at 1000 cd m−2 (Fig. 2c and S6d†). It is noted that compared to the corresponding blue diodes, codoping 0.2% 4CzTPNBu increased the maximum ηEQE by 29% for the DPSQPO hosted WOLED, but had negligible influence on the ηEQE of the DPEQPO based device. So, the DPSQPO matrix is superior to DPEQPO, in terms of improving the synergism between blue and yellow dopants in exciton utilization.
Furthermore, in accord with the situations of the volt–ampere characteristics for these WOLEDs, the efficiencies of DPEQPO hosted WOLEDs were nearly independent on the 4CzTPNBu concentration (Fig. 2c and S6d†). The variation percentage of the maximum ηEQE was within 11% at y = 0.2–1.5. It is convincing that codoping 4CzTPNBu in the DPEQPO matrix had a positive effect on alleviating triplet concentration quenching on DMAC-DPS by triplet energy transfer, but was roughly counteracted by dopant–dopant quenching. In comparison, the efficiencies of DPSQPO hosted WOLEDs at y = 0.2 and 0.5 were also nearly identical. However, further increasing y to 1.0 led to a sharp efficiency decrease by more than 20%. At y = 1.5, the ηEQE values of both DPXQPO hosted WOLEDs were already comparable. Obviously, in the DPSQPO matrix, at y <1.0, 4CzTPNBu is markedly more effective in quenching alleviation and exciton utilization improvement. But, at y ≥1.0, dramatic efficiency reduction revealed that dopant–dopant quenching and competition in exciton allocation became dominant.
The different dependencies of efficiencies on dopant concentrations for DPXQPO hosted diodes indicates that DPSQPO is much more efficient to prevent the concentration quenching of DMAC-DPS and mutual quenching between DMAC-DPS and 4CzTPNBu; meanwhile, even at a low doping concentration, dopant concentration quenching was already serious in the DPEQPO matrix. These results suggest that the device performances of DPXQPO are directly correlated with their dispersive abilities. Within a rational range of doping concentration, the DPSQPO matrix can uniformly disperse and effectively separate dopants to alleviate mutual interaction induced quenching. In contrast, the DPEQPO matrix seemed incapable of preventing intermolecular interactions between dopants. It is consistent with its self-aggregation tendency as shown in the single-crystal packing diagram, which in turn leads to the heterogeneous distributions of dopants and consequently worsened concentration quenching. Furthermore, owing to effective quenching suppression, DPSQPO simultaneously endowed its WOLEDs with a doubled device duration time, compared to its DPE based analogs (Fig. S8†).
2.3 Theoretical simulation
Because intermolecular interactions markedly influence excited-state properties, it is indispensable to figure out the relationship between the transition characteristics of DPXQPO matrices and their different intermolecular interactions. Models of monomers and tetramers for density functional theory (DFT) and time-dependent DFT (TDDFT) simulations are established according to single-crystal data (Fig. 3 and S8†). | ||
| Fig. 3 Variation of ground and excited-state characteristics after host weaving estimated by Gaussian simulations. (a) Contours and energy levels of the Frontier molecular orbitals for ground states (S0) and “holes” and “particles” for the first singlet (S1) and triplet (T1) excited states of DPXQPO single molecules and tetramers. The geometries and packings are generated according to single crystal data. ES, ET and fS refer to the excited energy levels of the S1 and T1 states and the singlet oscillator strength. (b) Variation of the centroid distance (dH–L), overlap integral (〈ΨH|ΨL〉) of the Frontier orbitals and electronic cloud overlap probabilities (〈Ψ2H|Ψ2L〉) of the Frontier orbitals for the S0, S1 and T1 states of single molecules and tetramers. | ||
The DFT simulation of ground states (S0) shows that in addition to the high electronegativity of ether oxygen atoms, the combined electron-withdrawing effects of the four PO groups of DPEQPO on the DPE skeleton result in the major contribution of DPE to unoccupied molecular orbitals, and the occupied molecular orbitals are mainly localized on the phenyls of electron-deficient DPPO groups (Fig. 3a and S9†). In contrast, the highest occupied (HOMO) and the lowest unoccupied (LUMO) molecular orbitals of DPSQPO are mainly contributed by the DPS skeleton, but slightly dispersed to PO groups, due to the ambipolar characteristics of the sulfur-containing moiety.57 Cyclic voltammetry (CV) analysis shows that the lowest occupied molecular orbital (LUMO) energy level of DPSQPO (−3.11 eV) is 0.28 eV deeper than that of DPEQPO, manifesting the stronger electron injecting ability of the former (Fig. S10a†). However, the ultraviolet photoelectron spectra (UPS) of DPXQPO films show that the ionization potential and electron affinity of DPSQPO are 7.48 and 4.46 eV, which are 0.83 and 0.57 eV lower than those of DPEQPO (Fig. S11†). Therefore, in solid states, the DPEQPO matrix improves electron injection, based on its stronger intermolecular interactions, but its hole injection ability is weakened, due to the embedment of the hole-transporting skeleton. In contrast, the DPSQPO matrix provides balanced hole and electron injecting ability (Table S3†).
Both DPXQPO exhibit locally excited singlet and triplet states. For S0 → S1 excitation, the “holes” and “electrons” of DPXQPO are localized on their DPE/DPS skeletons. Nonetheless, the calculated first singlet (S1) energy level of DPEQPO reaches 4.58 eV, markedly higher than that of DPSQPO (3.74 eV). In contrast, the singlet oscillator strength (fS) of DPSQPO (0.4471) is one order of magnitude larger than that of DPEQPO. Therefore, compared to DPEQPO, DPSQPO is superior in forming singlet excitons and transferring to the dopant by FRET. In contrast, the first triplet (T1) energy levels of DPXQPO (∼−3.1 eV) are nearly identical. But, the “hole” and “electron” of DPEQPO for S0 → T1 excitation are centralized on one phenyl of its peripheral DPPO group, in contrast to the T1 location of DPSQPO still on the embedded DPS skeleton. The exposed T1 state of DPEQPO is more beneficial to charge-exchange-based triplet DET. So, FRET would be predominant for DPSQPO, in contrast to comparable FRET and DET for DPEQPO.
The S1 states of DPXQPO tetramers are mainly and equally contributed by two molecules, decreasing fS by 1–2 orders of magnitude. Different to the S1 state of the DPSQPO tetramer like the simply duplicated S1 state of the monomer, the singlet “hole” and “particle” of the DPEQPO tetramer are separately distributed on two phenyls and two DPE skeletons, which is similar to the FMO distributions. Thus, the S1 energy levels of the DPSQPO monomer and tetramer are identical, but the DPEQPO tetramer reveals an S1 energy level 0.18 eV lower than that of the monomer. Furthermore, despite similar fS values, the high singlet-state similarity of the DPSQPO monomer and tetramer makes the mutual transformation between the S1 states feasible, thereby combining the advantages of high transition probability for exciton formation and uniform dispersion for FRET. Meanwhile, identical to monomers, the T1 states of DPXQPO tetramers are centralized on a single molecule, accompanied by unchanged T1 energy levels. As a consequence, the respective predominance of DPXQPO molecules in FRET and DET is further enhanced in their matrices.
The quantitative analysis of Frontier orbital parameters shows that at the S0 states, despite the reduced HOMO–LUMO centroid distance (dH–L), the FMO wave-function overlap integral (〈ΨH|ΨL〉) and electron-cloud-density overlap integral (〈Ψ2H|Ψ2L〉) of the DPXQPO tetramer are reduced, in accord with their enhanced FMO separation. However, in contrast to the equal singlet dH–L of the DPEQPO monomer and tetramer, the dH–L of the DPSQPO tetramer is shorter than that of its monomer. Therefore, although the singlet “holes” and “electrons” of DPXQPO tetramers are more dispersive than monomers, the S1 state of the DPSQPO tetramer basically preserves the 〈ΨH|ΨL〉 and 〈Ψ2H|Ψ2L〉 of its monomer, compared to markedly decreased 〈ΨH|ΨL〉 and 〈Ψ2H|Ψ2L〉 of the DPEQPO tetramer. Therefore, the DPSQPO tetramer can form more stable singlet excitons for facilitating FRET to dopants. In contrast, the triplet-state properties of the monomers and tetramers are almost identical. In this case, the triplet dH–L of the DPEQPO tetramer is shorter than that of the DPSQPO tetramer, and the corresponding 〈ΨH|ΨL〉 and 〈Ψ2H|Ψ2L〉 of the former are larger than those of the latter. Therefore, DET is dominant in the DPEQPO matrix with a more stable T1 state.
2.4 Photophysical investigation
The electronic absorption spectra of DPXQPO in dilute CH2Cl2 solution (10−6 mol L−1) consist of three main bands peaked around 228, 262 and 310 nm, attributed to the π → π* transitions of phenyls and n → π* transitions of PO and ether/sulfide moieties (Fig. 4a). The absorption tail of DPSQPO extends to 410 nm, corresponding to the S1 energy level of 3.01 eV, which is lower than 3.28 eV of DPEQPO (Table S1†). Furthermore, the intensities of n → π* transitions for DPSQPO are markedly higher than those of DPEQPO. As estimated with Mulliken's relationship,58 the single-molecular fS of DPSQPO is 0.2441, 1.5 fold that of DPEQPO (0.1594), which is in accord with TDDFT results. Compared to DPEQPO, the steady-state photoluminescence (PL) spectrum of the solution of DPSQPO peaking at 365 nm is also red shifted by 54 nm. Moreover, the full width at half maximum (FWHM) of FL emission from DPEQPO is as small as 33 nm, which is less than half that of DPSQPO (68 nm), due to the high molecular rigidity of DPEQPO enhanced by intramolecular π–π interaction between its ortho-DPPO and DPE skeleton, as shown in the single crystal structure (Fig. 1c). The PH spectra of DPXQPO in dilute solutions were recorded with time-resolved technology at 77 K (inset of Fig. 4a). As estimated from the first PH peak wavelengths, the T1 energy levels of DPXQPO are beyond 2.9 eV, and are high enough to support positive DET to blue TADF emitters. The T1 difference between DPXQPO is only 0.07 eV, about one fourth of their S1 difference. Thus, DPSQPO simultaneously achieves a comparably high T1 energy level and improved electrical properties.
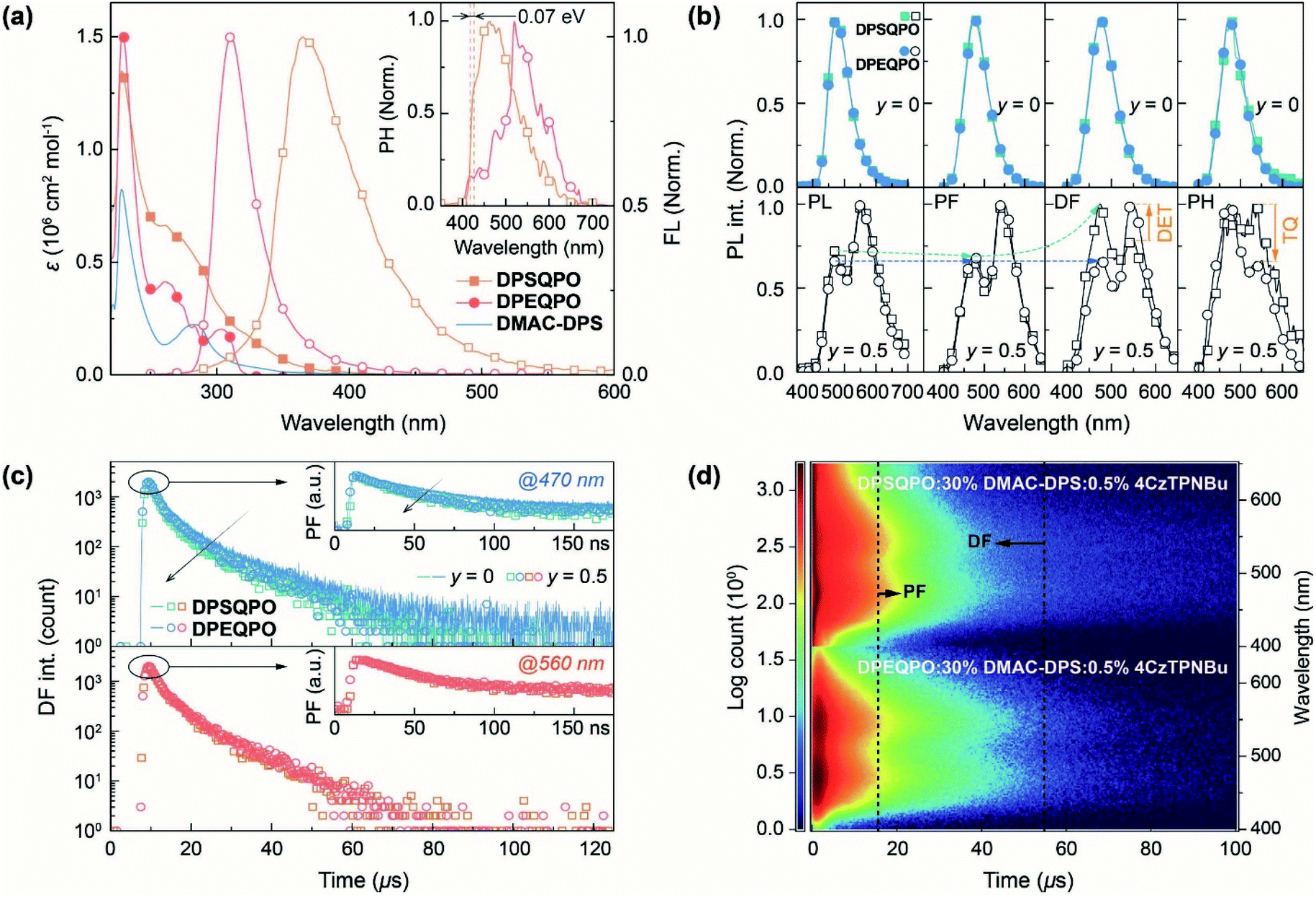 | ||
| Fig. 4 Photophysical properties of DPXQPO. (a) Electronic absorption and photoluminescence (PL) spectra of DPXQPO and absorption spectrum of DMAC-DPS in dilute CH2Cl2 solutions (10−6 mol L−1). Fluorescence (FL) spectra were recorded at room temperature. Phosphorescence (PH) spectra were recorded at 77 K with time resolution technology after a delay of 100 μs to exclude delay fluorescence. (b) PL, prompt (PF) (≤1 μs) and delayed fluorescence (DF) (1–100 μs) and PH (>100 μs) spectra of blue and white-emitting films of DPXQPO:30% DMAC-DPS:y% 4CzTPNBu (y = 0 for blue; 0.5 for white). Film thickness is 100 nm. DET and TQ refer to Dexter energy transfer and triplet quenching. (c) Time decay curves of blue and yellow PF and DF components from DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu films, respectively, recorded at 470 and 560 nm. PF and DF decays of DPXQPO:30% DMAC-DPS are included for comparison. (d) Transient emission contours of DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu films. PF and DF differences are marked with arrows. | ||
It is noteworthy that in addition to the single-molecular main peak at 313 nm, the vacuum-evaporated DPEQPO neat film reveals an aggregation-originated shoulder peaked at 370 nm and another long emission tail centered 470 nm, which manifests the heterogeneous distribution of intermolecular interactions and local self-aggregation in the DPEQPO matrix (Fig. S12†). In contrast, the DPSQPO neat film exhibits a single-peak emission with a smaller redshift of 34 nm, reflecting the homogeneous and weaker intermolecular interactions in the DPSQPO matrix. This result suggests the preservation of packing regularity and grid-like uniformity in DPSQPO based thin films. Despite aggregation, the T1 energy levels of DPXQPO films are equal to 3.03 eV, and are high enough to support positive energy transfer to DMAC-DPS (Fig. S13†). In addition, despite the small singlet–triplet splitting energy, the time decay curve of the DPSQPO film reveals a monoexponential fitting with a lifetime value of ∼2 ns (Fig. S14†). Therefore, emission from the DPSQPO film corresponds to fluorescence rather than TADF.
DMAC-DPS was further doped in the DPXQPO matrix by vacuum evaporation. The PL spectra of the resultant DPXQPO:x% DMAC-DPS films (x = 10–40, with an interval of 10) correspond to typical blue emissions from the dopants (Fig. S15 and S16†). However, it is noted that increasing x induces the marked emission-peak redshift of DPEQPO hosted films by 16 nm from 463 to 479 nm, which is obviously induced by gradually worsened aggregation (Fig. S14a†). Due to aggregation, intermolecular CT and concentration quenching is simultaneously in direct proportion to x, giving rise to elongated prompt fluorescence (PF) and shortened delayed fluorescence (DF) time decays of DPEQPO:x% DMAC-DPS films (Fig. S15b†). In comparison, the emission redshift of DPSQPO:x% DMAC-DPS films is limited to 7 nm at x = 40, accompanied by x-independent PF and DF time decays (Fig. S16†). Therefore, the DPSQPO matrix can indeed effectively alleviate dopant aggregation and concentration quenching. Furthermore, the PF and DF decays of a conventional blue host bis{2-[di(phenyl)phosphino]-phenyl}ether oxide (DPEPO) (T1 > 3.2 eV) were also measured for comparison (Fig. S17†), and are almost identical to those of DPEQPO. This in turn demonstrates the positive energy transfer from DPXQPO to DMAC-DPS.
At x = 30, the PL, PF, DF and PH spectra of DPXQPO:30% DMAC-DPS films are almost identical; however, the corresponding dually doped DPXQPO:30% DMAC-DPS:y% 4CzTPNBu films (y = 0.2–1.5) reveal totally different emission spectra (Fig. 4b and S18†). At the same y, the intensity ratios of blue and yellow peaks in the PL spectra of DPSQPO:30% DMAC-DPS:y% 4CzTPNBu films are markedly larger than those for DPEQPO hosted analogs. More importantly, for DPSQPO hosted films, yellow PF components are relatively stronger than yellow DF components; meanwhile, the blue/yellow ratios of PF and DF components for DPEQPO hosted films are nearly the same. A more detailed comparison of the emission spectra of DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu films indicates that despite their PL and PF spectra with the same blue/yellow ratios, the DF spectra of the DPSQPO hosted film is remarkably blue-peak dominant, in contrast to the yellow-peak dominant DF emission of the DPEQPO hosted analog (Fig. 4b). In contrast, the relative intensity of the yellow PH component for the DPSQPO hosted film is higher than that for the DPEQPO hosted analog. Compared to DPEQPO:30% DMAC-DPS, codoping 0.5% 4CzTPNBu markedly reduces both the PF and DF lifetimes of DMAC-DPS, due to blue-to-yellow FRET and DET (Fig. 4c). DPSQPO:30% DMAC-DPS:0.5% 4CzTPNBu reveals a similar blue PF lifetime reduction, but its blue DF decay is basically unchanged. Therefore, compared to DPEQPO, the DPSQPO matrix limits triplet DET to 4CzTPNBu, which should shorten the yellow DF lifetime. However, both dually doped white-emitting films display similar yellow PF and DF decay curves. It means that the DPSQPO matrix simultaneously alleviates triplet quenching induced lifetime reduction.
The time-resolved emission spectra (TRES) of DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu films further indicate that in the DPSQPO matrix, PF emissions are predominant for both blue and yellow components, whose intensities are far beyond DF emissions; meanwhile, the DPEQPO matrix slightly reduces the PF proportion, but significantly increases the DF contribution. Considering the short-range feature of the DET mechanism and the collision-inducible character of triplet quenching, the DPSQPO matrix effectively separates dopant molecules through grid-like uniform dispersion, thereby simultaneously suppressing intermolecular-interaction based triplet energy transfer and concentration quenching. In contrast, the heterogeneous distribution of DMAC-DPS in the DPEQPO matrix increases the probability of doping 4CzTPNBu directly into or close to DMAC-DPS aggregates, thus enhancing DET and worsening triplet quenching.
2.5 Exciton kinetics
The steady-state PL and EL spectra of DPEQPO:30% DMAC-DPS:0.5% 4CzTPNBu are nearly overlapped, manifesting the absolute predominance of energy transfer in the exciton allocation of DPEQPO hosted devices (Fig. 5a). However, the blue EL component of DPSQPO:30% DMAC-DPS:0.5% 4CzTPNBu is weaker than its blue PL component, indicating the considerable incorporation of direct charge capture and recombination by 4CzTPNBu into exciton allocation. It is rational that in the DPEQPO matrix, directly interacting DMAC-DPS and 4CzTPNBu both with ambipolar characteristics mutually compete in terms of charge capture. Therefore, the effective separation and uniform dispersion of DMAC-DPS and 4CzTPNBu in the unipolar DPSQPO matrix amplify the advantage of 4CzTPNBu with the deepest LUMO and the shallowest HOMO in direct charge capture. | ||
| Fig. 5 Exciton allocation in DPXQPO based WOLEDs. (a) Comparison of the PL and EL spectra of DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu systems. (b) EL transient emission contours of DPXQPO:30% DMAC-DPS:0.5% 4CzTPNBu based devices (left) and the comparison of exciton formation and transfer in PL and EL processes. (c and d) Exciton allocation mechanisms of binary complementary white-emitting systems in a uniformly weaved matrix like DPSQPO (c) and a locally aggregated matrix like DPEQPO (d) regarding energy transfer. FRET and DET refer to Förster resonance energy transfer and Dexter energy transfer. ISC and RISC are intersystem crossing and reverse ISC. | ||
Transient EL emission spectra further show that compared to the DPEQPO:30% DMAC-DPS:0.5% 4CzTPNBu based device, the DPSQPO matrix simultaneously and dramatically shortened the blue and yellow emission decays of its WOLED by about 100 and 700 μs, respectively (Fig. 5b left). Obviously, the DPSQPO matrix effectively accelerated exciton formation and radiation, which is beneficial to quenching suppression. Furthermore, compared to the blue PL decay (Fig. 4d), the EL decay of the blue component for DPSQPO:30% DMAC-DPS:0.5% 4CzTPNBu was slightly elongated by 10–20 μs, due to delays for carrier injection, transportation and recombination. In contrast, the time decay of its yellow EL DF was largely increased by 200 μs, further reflecting the predominance of charge transfer and capture in triplet allocation to 4CzTPNBu rather than DET. In contrast, compared to PL decays, the blue and yellow EL components of DPEQPO:30% DMAC-DPS:0.5% 4CzTPNBu were markedly elongated by 200 and 900 μs, respectively. Obviously, DMAC-DPS aggregation in the DPEQPO matrix enhanced intermolecular charge transfer, which significantly extended carrier recombination and blue EL time decay. Furthermore, effective DET from DMAC-DPS markedly enhanced triplet allocation to 4CzTPNBu, further increasing the yellow DF EL proportion and elongating the yellow EL decay.
Different to simultaneous photoexcitation processes in DPXQPO hosted films, the EL kinetic process shows that carrier recombination in the DPEQPO based device occurred earlier than that in the DPSQPO based analog (Fig. 5b right). It is because TADF molecules with low concentrations are uniformly dispersed, carriers mainly transport in inertia DPSQPO matrix, which postponed carrier capture and recombination by TADF emitters; meanwhile, in the DPEQPO matrix, dopant aggregation actually increased the probability of exciton formation. For both DPXQPO hosted EMLs, exciton formation is completely determined by the statistical advantage for photoexcitation at the first, but then carrier capture ability for electro-excitation. Therefore, excitons were firstly generated on DMAC-DPS with a high concentration in the PL process, and then on 4CzTPNBu due to its advantageous FMOs in the EL process. Although exciton formation on DMAC-DPS in the DPEQPO based device was also earlier than that in the DPSQPO based analog, in accord with the PL process, the blue emission intensity of the former was always markedly lower than that of the latter, manifesting the synchronism of exciton formation and blue-to-yellow energy transfer in the DPEQPO based device. Compared to long-distance FRET, short-range DET is more dependent on high exciton concentration; therefore, markedly higher blue exciton concentration in the DPSQPO based WOLED suggests limited incorporation of DET in exciton allocation, compared to the DPEQPO based device. Therefore, carrier injection in EMLs would still be mainly supported by DPXQPO, rather than DMAC-DPS or 4CzTPNBu. Exciton formation through either energy transfer or carrier capture occurs during the carrier transportation process. In this sense, the driving voltages of the devices should be in accord with the FMO energy levels of DPXQPO, leading to the lower driving voltages of DPSQPO based devices.
So, as illustrated in Fig. 5c and d, the DPSQPO matrix provides high grid regularity to uniformly disperse and separate blue and yellow dopants, inducing FRET-predominant exciton allocation. The suppression of dopant–dopant interactions not only alleviates exciton quenching, but also avoids excessive exciton allocation to yellow dopants by DET. In contrast, the local aggregations of the DPEQPO matrix and TADF dopants enhance intermolecular charge transfer and blue–yellow interactions, accelerating DET and worsening mutual quenching. Therefore, rationalizing the intermolecular interaction network to weave host grids is effective for optimizing the exciton kinetic process in high-efficiency single-layer white TADF systems.
3. Conclusions
We demonstrate the significance of host grid regularity with two similar host molecules DPEQPO and DPSQPO, whose IHB networks are differentiated by their DPE and DPS skeleton induced different orientations of PO hydrogen-bond donors. Compared to DPEQPO with a complicated IHB network-induced heterogeneous dispersion, DPSQPO featuring highly regular IHB grids supported a state-of-the-art ηEQE up to 24.2% for its single-EML pure-white TADF diode, which is 1.5 fold the maximum ηEQE for DPEQPO based analogs. Both steady-state and time-resolved PL and EL spectra indicate different exciton allocation and kinetic processes in DPXQPO hosted films and WOLEDs. Compared to the DPEQPO matrix, DPSQPO uniformly disperses and effectively separates TADF dopants, and therefore preserves more blue DF components for its white TADF systems by limiting blue-to-yellow DET. FRET-predominant exciton allocation in the DPSQPO matrix not only shortens emission decays and restrains dopant–dopant interactions for quenching suppression, but also improves white emission purity. These results suggest that host grid regularization is a feasible strategy for optimizing exciton kinetics, establishing a fundamental basis for developing high-performance TADF WOLEDs for daily lighting applications.
Data availability
All the data are available from the authors.Author contributions
H. X. and J. Z. conceived the project. Y. T., H. W., Y. M., N. Z., and Y. L. performed the experiments. H. X., Y. T., H. W. and J. Z. analyzed the data and wrote the paper. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
YT and HW contributed equally to this work. This study was supported by the National Natural Science Foundation of China (92061205, U1801258, 62175060, 51873056, 61905070 and 22005088), National Postdoctoral Program for Innovative Talents (BX20180092), Young Innovative Team Supporting Projects of Heilongjiang Province, and National Science Fund for Excellent Young Scholars of Heilongjiang Province (YQ2020B006).Notes and references
- J.-i. Nishide, H. Nakanotani, Y. Hiraga and C. Adachi, Appl. Phys. Lett., 2014, 104, 233304 CrossRef
.
- B. S. Kim, K. S. Yook and J. Y. Lee, Sci. Rep., 2014, 4, 6019 CrossRef CAS PubMed
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 160544 Search PubMed
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS PubMed
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed
- W. Zeng, H.-Y. Lai, W.-K. Lee, M. Jiao, Y.-J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K.-T. Wong, C.-C. Wu and C. Yang, Adv. Mater., 2018, 30, 1704961 CrossRef PubMed
- Y. Xiang, P. Li, S. Gong, Y.-H. Huang, C.-Y. Wang, C. Zhong, W. Zeng, Z. Chen, W.-K. Lee, X. Yin, C.-C. Wu and C. Yang, Sci. Adv., 2020, 6, eaba7855 CrossRef CAS PubMed
- X. Tang, L.-S. Cui, H.-C. Li, A. J. Gillett, F. Auras, Y.-K. Qu, C. Zhong, S. T. E. Jones, Z.-Q. Jiang, R. H. Friend and L.-S. Liao, Nat. Mater., 2020, 19, 1332–1338 CrossRef CAS PubMed
- Y.-L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hänisch, S. Reineke, J. Fan and L.-S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef CAS PubMed
- B. Zhao, H. Wang, C. Han, P. Ma, Z. Li, P. Chang and H. Xu, Angew. Chem., Int. Ed., 2020, 59, 19042–19047 CrossRef CAS PubMed
- Y. Li, X.-L. Li, D. Chen, X. Cai, G. Xie, Z. He, Y.-C. Wu, A. Lien, Y. Cao and S.-J. Su, Adv. Funct. Mater., 2016, 26, 6904–6912 CrossRef CAS
- J. Li, D. Ding, Y. Tao, Y. Wei, R. Chen, L. Xie, W. Huang and H. Xu, Adv. Mater., 2016, 28, 3122–3130 CrossRef CAS PubMed
- X. Lv, W. Zhang, D. Ding, C. Han, Z. Huang, S. Xiang, Q. Zhang, H. Xu and L. Wang, Adv. Opt. Mater., 2018, 6, 1800165 CrossRef
- Z. Wu, Y. Liu, L. Yu, C. Zhao, D. Yang, X. Qiao, J. Chen, C. Yang, H. Kleemann, K. Leo and D. Ma, Nat. Commun., 2019, 10, 2380 CrossRef PubMed
- J. Rao, X. Liu, X. Li, L. Yang, L. Zhao, S. Wang, J. Ding and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 1320–1326 CrossRef CAS PubMed
- J. Zeng, J. Guo, H. Liu, Z. Zhao and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 2000019 CrossRef CAS
- C. Zhang, Y. Lu, Z. Liu, Y. Zhang, X. Wang, D. Zhang and L. Duan, Adv. Mater., 2020, 32, 2004040 CrossRef CAS PubMed
- W. Li, B. Li, X. Cai, L. Gan, Z. Xu, W. Li, K. Liu, D. Chen and S.-J. Su, Angew. Chem., Int. Ed., 2019, 58, 11301–11305 CrossRef CAS PubMed
- R. Du, C. Duan, Y. Li, J. Zhang, C. Han and H. Xu, Chem. Eng. J., 2019, 374, 471–478 CrossRef CAS
- F. Gao, R. Du, Y. Wei and H. Xu, Opt. Lett., 2019, 44, 5727–5730 CrossRef CAS PubMed
- R. Du, C. Duan, Y. Li, J. Zhang, C. Han and H. Xu, Chem. Eng. J., 2020, 392, 124870 CrossRef CAS
- C. Han, W. Yang and H. Xu, Chem. Eng. J., 2020, 401, 126049 CrossRef CAS
- J. Sun, J. Zhang, Q. Liang, Y. Wei, C. Duan, C. Han and H. Xu, Adv. Funct. Mater., 2020, 30, 1908568 CrossRef CAS
- J. Zhang, C. Han, F. Du, C. Duan, Y. Wei and H. Xu, Adv. Funct. Mater., 2020, 30, 2005165 CrossRef CAS
- Y. Li, Z. Li, J. Zhang, C. Han, C. Duan and H. Xu, Adv. Funct. Mater., 2021, 2011169 CrossRef CAS
- C. Han, Z. Zhang, D. Ding and H. Xu, Chem, 2018, 4, 2154–2167 CAS
- Z. Zhang, D. Ding, Y. Wei, J. Zhang, C. Han and H. Xu, Chem. Eng. J., 2020, 382, 122485 CrossRef CAS
- Q. Wang and D. Ma, Chem. Soc. Rev., 2010, 39, 2387–2398 RSC
- L. Gan, Z. Xu, Z. Wang, B. Li, W. Li, X. Cai, K. Liu, Q. Liang and S.-J. Su, Adv. Funct. Mater., 2019, 29, 1808088 CrossRef
- H. Nakanotani, T. Furukawa, K. Morimoto and C. Adachi, Sci. Adv., 2016, 2, e1501470 CrossRef PubMed
- Y. R. Sun, N. C. Giebink, H. Kanno, B. W. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef CAS PubMed
- S. Reineke, K. Walzer and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125328 CrossRef
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS
- X. Tang, Y. Li, Y.-K. Qu, C.-C. Peng, A. Khan, Z.-Q. Jiang and L.-S. Liao, Adv. Funct. Mater., 2020, 30, 1910633 CrossRef CAS
- D. Ding, Z. Wang, C. Li, J. Zhang, C. Duan, Y. Wei and H. Xu, Adv. Mater., 2020, 32, 1906950 CrossRef CAS PubMed
- P. Wei, D. Zhang and L. Duan, Adv. Funct. Mater., 2019, 29, 1907083 Search PubMed
- C. Han, R. Du, H. Xu, S. Han, P. Ma, J. Bian, C. Duan, Y. Wei, M. Sun, X. Liu and W. Huang, Nat. Commun., 2021, 12, 3640 CrossRef CAS PubMed
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS
- J. Kalinowski, W. Stampor, J. Mecedilzdotyk, M. Cocchi, D. Virgili, V. Fattori and P. Di Marco, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 235321 CrossRef
- G. X. Yifan Li, S. Gong, K. Wu and C. Yang, Chem. Sci., 2016, 7, 5441–5447 RSC
- X.-L. Chen, J.-H. Jia, R. Yu, J.-Z. Liao, M.-X. Yang and C.-Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 15006–15009 CrossRef CAS PubMed
- G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong and C. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef PubMed
- S. Shao, J. Hu, X. Wang, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2017, 139, 17739–17742 CrossRef CAS PubMed
- H. Yang, Q. Liang, C. Han, J. Zhang and H. Xu, Adv. Mater., 2017, 29, 1700553 CrossRef PubMed
- F. Gao, R. Du, F. Jiao, G. Lu, J. Zhang, C. Han and H. Xu, Adv. Opt. Mater., 2020, 8, 2000052 CrossRef CAS
- Y. Shi, K. Wang, Y. Tsuchiya, W. Liu, T. Komino, X. Fan, D. Sun, G. Dai, J. Chen, M. Zhang, C. Zheng, S. Xiong, X. Ou, J. Yu, J. Jie, C.-S. Lee, C. Adachi and X. Zhang, Mater. Horiz., 2020, 7, 2734–2740 RSC
- Y.-Z. Shi, K. Wang, X.-C. Fan, J.-X. Chen, X.-M. Ou, J. Yu, J.-S. Jie, C.-S. Lee and X.-H. Zhang, Adv. Opt. Mater., 2021, 9, 2100461 CrossRef CAS
- S.-C. Dong, C.-H. Gao, X.-D. Yuan, L.-S. Cui, Z.-Q. Jiang, S.-T. Lee and L.-S. Liao, Org. Electron., 2013, 14, 902–908 CrossRef CAS
- R. S. Mulliken, J. Chem. Phys., 1939, 7, 14–20 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental detail, single-crystal packing diagrams, morphological properties, theoretical simulation results, optical properties, device performance, and electrochemical analysis. CCDC 2100434 and 2100433. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04188f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |