
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHCOOH disproportionation to MeOH promoted by molybdenum PNP complexes†
Elisabetta
Alberico
 *ab,
Thomas
Leischner
a,
Henrik
Junge
*a,
Anja
Kammer
a,
Rui
Sang
a,
Jenny
Seifert
a,
Wolfgang
Baumann
a,
Anke
Spannenberg
a,
Kathrin
Junge
a and
Matthias
Beller
*a
*ab,
Thomas
Leischner
a,
Henrik
Junge
*a,
Anja
Kammer
a,
Rui
Sang
a,
Jenny
Seifert
a,
Wolfgang
Baumann
a,
Anke
Spannenberg
a,
Kathrin
Junge
a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e. V., Albert-Einstein Straße 29a, 18059 Rostock, Germany. E-mail: henrik.junge@catalysis.de; matthias.beller@catalysis.de
bIstituto di Chimica Biomolecolare, Consiglio Nazionale delle Ricerche, tr. La Crucca 3, 07100 Sassari, Italy. E-mail: elisabetta.alberico@cnr.it
First published on 31st August 2021
Abstract
Molybdenum(0) complexes with aliphatic aminophosphine pincer ligands have been prepared which are competent for the disproportionation of formic acid, thus representing the first example so far reported of non-noble metal species to catalytically promote such transformation. In general, formic acid disproportionation allows for an alternative access to methyl formate and methanol from renewable resources. MeOH selectivity up to 30% with a TON of 57 could be achieved while operating at atmospheric pressure. Selectivity (37%) and catalyst performance (TON = 69) could be further enhanced when the reaction was performed under hydrogen pressure (60 bars). A plausible mechanism based on experimental evidence is proposed.
Introduction
Formic acid1 is primarily used as an additive in the food industry, in grass silage, leather tanning, rubber production and anti-icing and as an intermediate in the chemical and pharmaceutical industries. Recently, it has also earned further interest as a hydrogen storage material.2 In this respect, HCOOH thermal decomposition releases the stored hydrogen together with carbon dioxide according to eqn (1).3 Depending on the reaction conditions, HCOOH dehydration to carbon monoxide may also occur (eqn (2)).From a thermodynamic point of view, formic acid with a slightly lower reduction potential than H2 (E = 0.17 V at pH = 0 vs. NHE) is well suited for the reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds.4,5 In addition to numerous examples of transfer hydrogenation reactions in organic synthesis, HCOOH can also act as a self-reductant and undergo disproportionation to MeOH according to eqn (3) in which one molecule of HCOOH is reduced by two other equivalents of the acid.
O and CN bonds.4,5 In addition to numerous examples of transfer hydrogenation reactions in organic synthesis, HCOOH can also act as a self-reductant and undergo disproportionation to MeOH according to eqn (3) in which one molecule of HCOOH is reduced by two other equivalents of the acid.
From a kinetic standpoint, the polarized C–H bond in HCOOH (BDE = 96 kcal mol−1) is 8 kcal mol−1 weaker than the H–H bond and therefore easier to activate at a metal centre. Notably, the MeOH yield, which reflects the relative selectivity of the processes (dehydrogenation of FA vs. disproportionation), is influenced by the hydricity (the ability to transfer a hydride ligand) of the metal centre.
In general, formic acid disproportionation suggests a pathway alternative to current ones to access MeOH, which does not rely on fossil fuel-based processes and has a reduced environmental impact, provided HCOOH is produced from CO2 reduction with renewable energy6 or oxidation of biomass.

MeOH is one of the most important chemicals from which formaldehyde, tert-butyl methyl ether (MTBE, fuel additive which serves as anti-knocking agent), tert-amyl methyl ether (MATE), acetic acid and higher hydrocarbons, among others, are produced on bulk scale.7 Methanol can be used as a fuel for internal combustion engines and direct methanol fuel cells (DMFC) to generate electricity. Like formic acid also methanol can be regarded as a liquid hydrogen carrier molecule and storage medium from which hydrogen can be released first through reforming8 and then applied either in proton exchange membrane (PEM) or reformed methanol fuel cells (RMFC) to advantageously combine the easy transportability of a liquid with the energy efficiency of PEM fuel cells.9
Currently, almost all methanol is produced from syngas in the presence of heterogeneous catalysts (copper/zinc/aluminum oxides) at elevated pressures (250–200 bars) and temperatures (350 °C). Syngas is in turn obtained from fossil carbon feedstocks through gasification processes. Alternative, preferably renewable, routes to MeOH are therefore highly sought after. One possibility lies in the direct reduction of CO2, captured from emission points as well as from dilute sources like ambient air, with hydrogen obtained from water electrolysis using renewable energy.10a The “George Olah CO2 to Renewable Methanol Plant” in Iceland is indeed a demonstration of the feasibility of such an approach made possible by the availability in loco of abundant geothermal energy for water electrolysis. A multicomponent heterogeneous catalyst (Cu/ZnO/Al2O3) is used to promote the reaction which however, due to the high thermodynamic stability and kinetic inertness of CO2, requires high temperatures (210–250 °C) and pressures (50–78 bars).
Compared to heterogeneous catalysts,10 homogeneous ones, while far from providing activities for practical application, can be more easily modified in search of improved efficiency under milder operational reaction conditions.11 The homogeneous systems so far reported which allow for a “direct” hydrogenation of CO2 to MeOH are based on the combination of either Ru12 or Co13 with a tridentate phosphorus ligand, 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos) or related ligands, and a Bronsted or Lewis acid cocatalyst. More recently an Fe catalyst supported by a C-scorpionate ligand hydrotris(1-pyrazolyl)methane has been reported: the catalyst does not require any additive to promote turnover but its efficiency is improved in the presence of an amine cocatalyst.14 While these homogeneous catalysts can operate at a temperature as low as 80 °C, overall high pressures between 60 and 120 bars are still required for significant conversions, highlighting the challenge posed by such a transformation. This challenge might be overcome through hydrogenation of more reactive CO2-derived intermediates such as methyl formate,15a dimethylcarbonate,15a methylcarbamate,15a ureas,15b formamides,15c,d or ethylene carbonate.15e These “indirect” approaches were performed in the presence of Ru and Fe catalysts supported by PNN and PNP pincer ligands. Moreover, if properly chosen, the base, either an amine or an alkali hydroxide, can perform a dual role, allowing CO2 capture and indirect CO2 hydrogenation through the resulting carbamate and/or carbonate, under the same conditions.16 While a single catalyst may catalyse all hydrogenation steps, compatible catalysts, one for each step of the cascade process, can be combined to improve the overall efficiency.17 Recently an alternative approach has been proposed based on a dinuclear iridium catalyst which operates in the absence of solvent: hydrogenation of CO2 takes place through contact of the gases with the solid catalyst which can activate hydrogen under mild conditions. The two iridium centres in close proximity allow for multiple intramolecular hydride transfer to CO2 and the following reduced intermediates, without HCOOH acid being released from the metal coordination sphere, thus preventing its reversible dehydrogenation. Because MeOH is produced in the gas phase, the catalyst can be easily recycled: after five cycles, under 40 bars H2/CO2 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 60 °C, a TON of 113 (0.507 mmol) in MeOH is achieved.18
:1) at 60 °C, a TON of 113 (0.507 mmol) in MeOH is achieved.18
Today, renewable formic acid is available either by oxidation of biomass19 or by direct hydrogenation of CO2 to formic acid or formate. Huge efforts have been undertaken in these fields and remarkable activities obtained. For the latter reaction,20a,b state of the art ruthenium- and iridium-based catalysts afford activities (TOF) up to 73000 and 1100000 and productivities (TON) up to 200000 and 3500000, respectively.20c,d Thus, the way to the development of processes for formic acid synthesis based on CO2 hydrogenation20e as well as biomass oxidation (OxFA-process) were paved.19d Therefore, the catalytic disproportionation of HCOOH represents an alternative indirect route to renewable MeOH which however does not require the use of either an alcohol or amine to proceed. Scheme 1 presents the homogenous catalytic systems so far developed for HCOOH disproportionation. It should be noted that the different reaction conditions and setups do not allow a direct comparison, yet a perusal of the related literature provides guidelines and benchmarks for the assessment of the herein presented Mo–PNP catalytic system (SI4, Scheme SI-1†).
 | ||
| Scheme 1 Homogenous catalytic systems proved to be effective in HCOOH disproportionation. | ||
The very first indirect evidence of methanol formation in the form of dimethoxymethane from thermolysis of formic acid over ThO2 was provided by Sabatier and Mailhe and dates back to 1911.21 More than one hundred years after, in 2013 Karen Goldberg's group reported the first example of a homogeneously catalysed disproportionation of HCOOH promoted by a cationic cyclopentadienyl iridium complex (A, Scheme 1) supported by a 2,2′-bipyridine ligand.3 The reaction was carried out in water, at 60 °C for 24 hours, in a closed vessel and therefore the pressure increased due to formic acid dehydrogenation and disproportionation. Under optimized conditions, a maximum MeOH selectivity of 12% was obtained including methyl formate formed as the follow up product of acid catalysed esterification. This corresponds to a TON for MeOH production of almost 58 and an average TOF of 2.4 h−1 (SI4, Scheme SI-1†). Here, HCOOH dehydrogenation was the predominant process. However, selectivity in MeOH was positively affected by lower temperature (ΔH0dehyd. = +31.6 kJ mol−1vs. ΔH0disprop. = −35.6 kJ mol−1),3 higher initial HCOOH concentration, low pH, achieved with HBF4, and H2 overpressure to disfavour HCOOH dehydrogenation. Later on, Himeda and Laurency introduced a much more efficient system based on a Cp*Ir complex with the 4,4′-dihydroxo-2,2′-bipyridine ligand (B, Scheme 1), thus obtaining 74% MeOH selectivity at almost full HCOOH conversion, 98%.22d The reaction was carried out in D2O in a closed vessel at 50 °C for 600 hours, with 3.75 molal H2SO4. In a single run the TON achieved 236 but the average TOF was 0.4 h−1 (SI4, Scheme SI-1†). Under comparable experimental conditions, the admission of increasing pressures of hydrogen allowed to improve selectivity towards MeOH and its final concentration in solution. In this case the MeOH yield exceeds the theoretical one based on sole HCOOH disproportionation, indicating that part of the MeOH arises from HCOOH reduction with H2 gas.22b Remarkably, the same Ir catalyst is able to promote the direct hydrogenation of CO2 to HCOOH in water without any additive, thus allowing the one-pot transformation of CO2 into MeOH. However the productivity for MeOH is inferior in this case because best performances in the two processes occur under different reaction conditions.22b Systematic investigations of the effect of various substituents at the 2,2′-bipyridine ligand other than –OH did not allow to identify a general trend correlating the substituent electronic properties and substitution pattern at the ligand with the selectivity towards MeOH.22a,c Evidence that disproportionation of HCOOH may proceed through intermediate formation of H2CO has been provided by Li and co-workers: hydrolysis and oxidation of wheat straw in the presence of vanadate salts affords an aqueous solution of HCOOH which can be used as such for the reduction and N-methylation of quinolines to the corresponding tetrahydroquinolines promoted by catalyst B (Scheme 1).22f
Heterogenized versions of the above mentioned Cp*Ir catalysts were also prepared by reacting suitable Ir precursors with bipyridine-silica nanotubes.23 Although activities were inferior to those provided by the homogeneous counterparts, a positive effect on MeOH selectivity even for reactions carried out at atmospheric pressure was attributed to the local confinement of the evolved gases in the pores of the support.23b,c
Beside Ir, Ru was shown to be competent for HCOOH disproportionation when bound to the ligand triphos.24a Cantat and co-workers developed a catalytic system (C, Scheme 1) which afforded a selectivity in MeOH higher than 50% at full HCOOH conversion, which was achieved within one hour (TON = 27.9). The reaction was carried out in THF at 150 °C in a closed vessel. Here methanesulfonic acid (1.5 mol% to HCOOH, 2.5 equivalents to the catalyst) was used as additive to promote the formation of Ru hydride species and thus enhance catalytic activity (SI4, Scheme SI-1†).
To by-pass competitive HCOOH dehydrogenation and allow only disproportionation, Cantat has devised the use of silyl formates as substrates in place of formic acid.24b Disproportionation was promoted by ruthenium complexes containing the HN(CH2CH2PPh2)2 ligand (catalyst C′, (SI4, Scheme SI-1†) and afforded methoxysilanes, which were subsequently hydrolysed to methanol. The by-product of hydrolysis, disilyl ethers, were recycled back to silyl formates by reaction with sulfuric acid and sodium formate.
Very recently a cooperative heterobimetallic catalyst (0.08 mol%), generated in situ from the reaction of the diruthenium complex [(Cp*Ru)2(μ-NPh)(μ-CH2)] and tin(II) oxide (D, Scheme 1 and SI4, Scheme SI-1†) was reported to be competent for HCOOH disproportionation in up to 28% selectivity at 150 °C.24c No methanol was formed when either of the two metal species was absent. By halving the catalyst amount to 0.04 mol% and raising the temperature to 185 °C, a maximum TON of 191 was achieved.
So far, the only example of HCOOH disproportionation promoted by a base metal is reported by Parkin and co-workers who investigated the performance of cyclopentadienyl molybdenum hydride compounds of general formula [CpRMo(PMe3)3−x(CO)xH] (CpR = Cp, Cp*; x = 0, 1,2 or 3).25 Compared to the above mentioned noble metal catalysts, these systems were less active and only stoichiometric amounts of MeOH could be achieved with [CpMo(CO)3H] (E, Scheme 1 and SI4, Scheme SI-1†) (HCOOH 0.34 M, E 7.3 mol%, C6D6, 100 °C).
In contrast, we report herein that molecularly-defined molybdenum complexes with aliphatic PNP pincer ligands are able to catalyse the disproportionation of formic acid to methanol, methyl formate, water, and CO2 with activity at least comparable to that of known precious metal systems.
Results and discussion
We recently introduced molybdenum PNP pincer complexes for highly selective hydrogenation reactions of various unsaturated compounds e.g. reduction of amides to alcohols and amines under relatively mild conditions.26 Based on this work and as part of our continuing interest in the development of catalysts for formic acid transformation,27 we set out to assess whether such Mo complexes may catalyse HCOOH dehydrogenation. In this respect, the relevant role played by molybdenum in formate dehydrogenases, which catalyse the reversible interconversion of formate and CO2, is interesting.28Scheme 2 shows the Mo–PNP pincer complexes which were initially tested for HCOOH dehydrogenation. Entries 1–8 in Table 1 report a preliminary catalyst screening in triglyme. This dipolar aprotic solvent was chosen due to its stability and complete miscibility with water and several organic solvents.29 The high boiling point of 218 °C allows for a broad range of operational temperatures. Reactions were carried out at 90 °C for a standard reaction time of 6 hours, using 10 mmol of HCOOH and 0.1 mol% catalyst (10 μmol) in 20 mL of solvent. We first explored the performance of Mo(II)-1 as well as Mo(I)-2 and Mo(I)-3 chloro derivatives having different substituents at the phosphorus atoms of the PNP ligand, either Ph 1, iPr 2 or Cy 3, (Table 1, entries 1 to 3). It is evident that with Mo(II) and less electron donating phenyl groups 1 a much lower catalytic activity, in terms of volume of evolved gas (entry 1, 12 mL), is obtained compared to Mo(I) and isopropyl substituents (2, entry 2, 154 mL). The improvement brought about by switching from Mo(II) to Mo(I) and to a more electron rich ligand is however far less significant with Cy groups (3, entry 3, 36 mL). Gas evolution rate in the presence of catalyst 2 becomes slightly faster after pretreatment with NaHBEt3 (154 mL, entry 2 vs. 181 mL, entry 4) which hints towards an active Mo(0) complex. Indeed, activity increased when the preformed catalyst 4 (Entry 5, 416 mL) was directly used. The corresponding high catalytic activity is retained and slightly improved when the preformed formate complex 5, obtained by stoichiometric reaction of 4 with HCOOH, (vide infra) is used instead of its acetonitrile precursor 4 (Entry 6, 431 mL).
 | ||
| Scheme 2 Mo–PNP complexes tested in the dehydrogenation of HCOOH. | ||
| Entry | Catalyst | Solvent | Vol2 h [mL] | Vol6 h [mL] | H2/CO2b |
|---|---|---|---|---|---|
| a General conditions: HCOOH 10 mmol, catalyst 10 μmol, solvent 20 mL, 90 °C, 6 h. The experiments were performed at least twice (except entries 1, 3 and 10), standard deviations are 1 to 12% of the average for experiments with volumes >75 mL and up to 44% for lower volumes. The volumes of evolved gas are as measured, values are not corrected for blank volumes (Fig. SI-12). b Calculated ratio based on a gas sample taken in between reflux condenser(s) and gas burette, unequal distribution of H2 and CO2 cannot be excluded (SI3). c Catalyst 2 was pretreated with one equivalent of NaHBEt3. | |||||
| 1 | 1 | Triglyme | 5.6 | 12 | 1.3 |
| 2 | 2 | Triglyme | 58 | 154 | 0.98 |
| 3 | 3 | Triglyme | 15 | 36 | 1.6 |
| 4c | 2 | Triglyme | 71 | 181 | 0.94 |
| 5 | 4 | Triglyme | 176 | 416 | 0.86 |
| 6 | 5 | Triglyme | 179 | 431 | 0.83 |
| 7 | 6 | Triglyme | 48 | 230 | 0.91 |
| 8 | 7 | Triglyme | 26 | 55 | 1.3 |
| 9 | 4 | NMP | 12 | 24 | 1.6 |
| 10 | 4 | DMSO | 1.1 | 2.0 | 2.7 |
| 11 | 5 | PC | 7.2 | 27 | 1.9 |
| 12 | 4 | Toluene | 226 | 300 | 0.70 |
| 13 | 5 | Toluene | 236 | 278 | 0.67 |
The amount of evolved gas is decreased by a factor of two when the Mo(0) catalyst with cyclohexyl substituents 6 instead of isopropyl ones 4 is used (entry 7, 230 mL). It is worth mentioning that the phenyl substituted analogue of 4 with a labile CH3CN ligand is not accessible through the same synthetic procedure from [Mo(CO)2(PPh3)2(CH3CN)2] and HN(CH2CH2PPh2)2 affording 7 instead, which retains one PPh3 ligand in the metal coordination sphere.26a This catalyst showed quite modest activity (entry 8, 55 mL). Based on these results, catalysts 4 and 5 were chosen for further screening. Because of the selected reaction temperature and the very poor solubility of catalyst 4 at room temperature, increasingly polar and high boiling solvents were tested such as N-methyl-2-pyrrolidone NMP (entry 9, 24 mL), dimethylsulfoxide DMSO (entry 10, 2.0 mL) and propylene carbonate PC (entry 11, 27 mL). However, almost no activity was recorded in these solvents. On the other hand, good activity, albeit lower than that in triglyme, was observed in toluene, where 4 (entry 12, 300 mL) and 5 (entry 13, 278 mL) were equally efficient. To our surprise, the ratios of the evolved gases H2 and CO2 in toluene differed from those obtained in triglyme and were well below 1 (0.70 with 4, entry 12 and 0.67 with 5, entry 13) suggesting that HCOOH dehydrogenation might not be the only process taking place in toluene solution. This prompted us to check the solution content for possible by-products after the reaction was stopped. Interestingly, methanol and methyl formate were found in solution to a significant extent. This can be explained by HCOOH disproportionation taking place besides dehydrogenation. From this point on, we focused on toluene as solvent and the reaction set up was reconsidered to minimize losses of the low boiling products with the stream of evolving gases. Therefore, the reactions were conducted in reactors fitted with two condensers, one on top of the other, both cooled to −5 °C with a circulating antifreezing liquid supplied by a cryostat. To assess the efficiency of this setup, for selected experiments a cooling trap was added to collect and quantify if any traces of products were lost with the stream of gases (Fig. SI-4†).
The results, including the volume of generated gas (H2 + CO2) and the amounts of produced MeOH and methyl formate, are reported in Table 2. The MeOH selectivity's obtained with 4 and 5 are 6.3 and 9.5%, respectively (Table 2, entries 1 and 2), suggesting a superior performance of the formate complex 5 as to HCOOH disproportionation. The MeOH selectivity provided by 5 is almost comparable to that reported by Goldberg with her iridium-based catalyst, 12%,3 but inferior to that reported by Parkin with stoichiometric amounts of molybdenum, 21%.25 However, in both cases, HCOOH decomposition was carried out in a closed vessel. Due to the developing hydrogen partial pressure, HCOOH dehydrogenation should be hampered. The turnover number (TON) for MeOH generation applying catalyst 5 was calculated to be 29, the turnover frequency (TOF) 4.8 h−1. Thus, already at this non-optimized state, catalyst 5 was outperforming the only reported non-noble metal complex used so far (Scheme 1, E). Moreover, it was also faster than the reported iridium catalysts (Scheme 1, A and B and Scheme SI-1†). Therefore, we deemed our initial results encouraging and we set out to find experimental conditions to improve selectivity and yield in MeOH/methyl formate. Remarkably, even catalyst 8, which can be formally derived from 4 by replacing acetonitrile with CO, a product of HCOOH dehydration and potential poison for the catalyst, showed activity (Table 2, entry 3) comparable to that of 4. The other available Mo–PNP complexes 1, 2, 3 and 6 were also tested in toluene (Table 2, entries 4 to 7): the same trend in triglyme with respect to the influence of the metal oxidation state, availability of an easily displaced ligand and substituents at phosphorus on catalyst performance was observed in toluene, confirming 4 and 5 as catalysts of choice for further investigations. Additionally, we considered whether the ability to promote HCOOH disproportionation beside dehydrogenation is characteristic for molybdenum or might be instead achieved with other metals supported by the aliphatic PNP ligand (Scheme 3). Thus, pincer complexes of Ru, 10,30 Ir, 11,31 Mn, 1232 as well as Fe, 1333a,b and 14,33 were selected which would require no additive to generate an active catalytic species (Scheme 3). However, under the applied reaction conditions, only the ruthenium catalyst 10 promoted HCOOH dehydrogenation (Table 2, entry 8, total evolved gas volume 347 mL) but in no case methanol or methyl formate were detected. Because of the positive results obtained by Cantat with the Ru-triphos,24a we also prepared a Mo(0)–triphos complex 15 analogous of 4, which however proved to be totally inactive (Table 2, entry 13).
| Entry | Catalyst | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | Conversione (%) |
|---|---|---|---|---|---|---|---|
|
a General conditions: HCOOH 10 mmol, catalyst 10 μmol, toluene 20 mL, 90 °C, 6 h.
b Calculated after correcting volumes of evolved gases by blank volume (5.4 mL).
c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/10 mmol HCOOH} × 100.
d See SI2 for calculation of selectivity.
e Based on recovered HCOOH. Each experiment was performed at least twice, standard deviations of yields and selectivity are 1 to 25% of the average (except for entry 5:37 and 38% for MeOH yield and selectivity respectively). Besides the listed products traces of CO were detected (Table SI-3 and Fig. SI-13).
|
|||||||
| 1 | 4 | 295 | <5 | 194 | 5.8 | 6.3 | 94 |
| 2 | 5 | 272 | 23 | 267 | 8.6 | 9.5 | 94 |
| 3 | 8 | 291 | 31 | 184 | 6.4 | 7.2 | 91 |
| 4 | 1 | 0 | n.d. | n.d. | — | — | 0 |
| 5 | 2 | 265 | <5 | 126 | 3.9 | <5.3 | 74 |
| 6 | 3 | 30 | n.d. | 5.0 | 0.15 | 1.5 | 9 |
| 7 | 6 | 299 | n.d. | 203 | 6.0 | 7.2 | 86 |
| 8 | 10 | 347 | n.d. | n.d. | — | — | 89 |
| 9 | 11 | 14 | n.d. | n.d. | — | — | 5.1 |
| 10 | 12 | 67 | n.d. | n.d. | — | — | 21 |
| 11 | 13 | 14 | n.d. | n.d. | — | — | 8.4 |
| 12 | 14 | 0 | n.d. | n.d. | — | — | 0 |
| 13 | 15 | 10 | n.d. | <3.4 | <0.10 | <1.5 | 7.4 |
 | ||
| Scheme 3 M-PNP complexes, M = Ru, Ir, Mn, Fe, Mo tested in the disproportionation of HCOOH. | ||
Based on these results we defined the standard reaction conditions (HCOOH 10 mmol, catalyst 4 or 5 10 μmol, toluene 20 mL, 90 °C, 6 h) which were then systematically varied to evaluate the response to reaction parameters.
We first addressed the effect of temperature, as this might have a significantly different influence on the two competing processes and HCOOH disproportionation should be favoured at lower temperature (eqn (1) vs. eqn (3)). As shown in Table 3, using 10 mmol of HCOOH in 20 mL toluene and 10 μmol of catalyst, the temperature was varied from 70 °C to 110 °C. In the latter case the reaction was carried out in p-xylene, to minimise solvent evaporation and product loss with the gas stream.
| Entry | T (°C) | Catalyst | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | Conversione (%) |
|---|---|---|---|---|---|---|---|---|
|
a General conditions: catalyst 10 μmol, HCOOH 10 mmol, toluene 20 mL, 90 °C, 6 h.
b Volumes of evolved gases are corrected by blank volume (5.4 mL).
c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/10 mmol HCOOH} × 100.
d See SI2 for calculation of selectivity.
e Based on recovered HCOOH.
f
p-Xylene used as solvent. Each experiment was performed at least twice, standard deviations of yields and selectivity are 1 to 31% of the average (except for entry 2:36% for yield MeOH) Besides the listed products, traces of CO were detected (Table SI-4 and Fig. SI-14).
|
||||||||
| 1 | 70 | 4 | 69 | n.d. | 79 | 2.3 | 5.6 | 46 |
| 2 | 80 | 4 | 173 | 11 | 136 | 4.4 | 7.5 | 59 |
| 3 | 90 | 4 | 295 | <5 | 194 | 5.8 | 6.3 | 94 |
| 4 | 90 | 5 | 272 | 23 | 267 | 8.6 | 9.5 | 94 |
| 5 | 100 | 4 | 297 | n.d. | 147 | 7.4 | 8.4 | 90 |
| 6 | 110 | 5 | 318 | n.d. | 407 | 12 | 13 | 95 |
As expected, formic acid dehydrogenation increased with increasing temperature going from 69 mL at 70 °C (Table 3, entry 1) to 318 mL at 110 °C (Table 3, entry 6). The same general trend was observed for MeOH yield, but, contrary to expectations, MeOH selectivity was highest at the highest temperature tested, 110 °C, 13% (Table 3, entry 6) corresponding to a 12% yield and a TON of 41. The trend in MeOH selectivity though is not clear cut. The reaction temperature also affected the formation of CO, as the detected ratio H2 to CO varied within the range of 133 (at 70 °C, Table SI-4,† entry 1) and 3539 (at 110 °C, Table SI-4,† entry 6).
We then studied the influence of the catalyst concentration on the reaction carried out at 90 °C (Table 4).
| Entry | Catalyst [mM] | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | TON | Conversione (%) |
|---|---|---|---|---|---|---|---|---|
|
a General conditions: catalyst 5, [HCOOH] = 0.5 M, HCOOH 10 mmol, toluene 20 mL, 90 °C, 6 h.
b Volumes of evolved gases are corrected by blank volume (5.4 mL).
c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/10 mmol HCOOH} × 100.
d See SI2 for calculation of selectivity.
e Based on recovered HCOOH. Each experiment was performed at least twice, standard deviations of yield and selectivity are 0.3 to 34% of the average (except for entry 1:42% for yield MeOH). Beside the listed products, traces of CO were detected (Table SI-5 and Fig. SI-15).
|
||||||||
| 1 | 0.25 | 259 | 13 | 136 | 4.5 | 6.0 | 30 | 74 |
| 2 | 0.5 | 272 | 23 | 267 | 8.6 | 9.5 | 29 | 94 |
| 3 | 1 | 325 | 28 | 338 | 11 | 12 | 18 | 92 |
| 4 | 1.5 | 303 | 17 | 410 | 13 | 14 | 14 | 94 |
| 5 | 2.5 | 298 | 24 | 473 | 15 | 16 | 10 | 96 |
The concentration of catalyst 5 was varied between 0.25 mM and 2.5 mM, corresponding to a HCOOH/catalyst ratio ranging from 2000 to 200 respectively. MeOH yield and MeOH selectivity greatly improved when the catalyst concentration was increased from 0.25 mM (Table 4, entry 1, 4.5% and 6.0% respectively) to 0.5 mM (Table 4, entry 2, 8.6% and 9.5% respectively). Both improved further, although less dramatically, up to 15% and 16% respectively, at 2.5 mM catalyst concentration (Table 4, entry 5). The variation in the total evolved gas (H2 + CO2) was instead modest, within the range 259 mL (Table 4, entry 1, 5 0.25 mM) – 298 mL (Table 4, entry 5, 5 2.5 M). An opposite trend as for MeOH selectivity and yield was observed for the turnover numbers which were highest at the lowest catalyst concentrations (Table 4, entries 1 and 2) being 30 and 29, respectively.
HCOOH concentration was varied between 0.125 M and 1.0 M at 90 °C while keeping catalyst 4 concentration constant at 0.5 mM (Table 5). Within this range MeOH yield and selectivity decreased, going from 13% and 15% respectively at [HCOOH] 0.125 M (Table 5, entry 1) to 2.1% and 6.1% at [HCOOH] 1 M (Table 5, entry 4) as expected because of the decreasing catalyst to substrate ratio (from 4 × 10−3 to 5 × 10−4).
| Entry | HCOOH [M] | Catalyst | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | Conversione (%) |
|---|---|---|---|---|---|---|---|---|
| a General conditions: catalyst 10 μmol, toluene 20 mL, 90 °C, 6 h. b Volumes of evolved gases are corrected by blank volume (5.4 mL). c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100. d See SI2 for calculation of selectivity. e Based on recovered HCOOH. Each experiment was performed at least twice, standard deviations of yields and selectivity are 1 to 25% of the average. Besides the listed products, traces of CO were detected (Table SI-6 and Fig. SI-16). | ||||||||
| 1 | 0.125 | 4 | 68 | 8.5 | 97 | 13 | 15 | 91 |
| 2 | 0.25 | 4 | 161 | <5 | 207 | 13 | 15 | 90 |
| 3 | 0.5 | 4 | 295 | <5 | 194 | 5.8 | 6.3 | 94 |
| 4 | 1.0 | 4 | 217 | n.d. | 138 | 2.1 | 6.1 | 34 |
| 5 | 0.25 | 5 | 138 | n.d. | 181 | 11 | 12 | 90 |
| 6 | 0.5 | 5 | 272 | 23 | 267 | 8.6 | 9.5 | 94 |
The same trend, although less pronounced, was observed applying catalyst 5: increasing the initial formic acid concentration from 0.25 M to 0.5 M the MeOH yield and selectivity both decreased from 11% and 12% respectively (Table 5, entry 5) to 8.6% and 9.5% (Table 5, entry 6). Such observation is opposite to previous results with catalyst A (Scheme 1) as reported by Goldberg.3 The highest TON (29) was achieved in the experiment applying 10 mmol formic acid (Table 5, entry 6).
To improve MeOH productivity it is necessary to favour HCOOH reduction by hydride transfer to a protonated and thus activated molecule of the acid. As documented in literature precedents, the addition of either HBF4,3 methanesulfonic acid24a or sulfuric acid22b,c indeed proved successful. In the present system, however, the addition of a Brønsted acid such as triflimide, HN[S(O)2CF3]234 shut down activity (Table SI-7,† entry 3).
The use of Al(OTf)3 was likewise detrimental.12a In both cases we deemed that the counter anion could, despite its low nucleophilicity, interact with molybdenum and therefore prevent catalytic turnover (Table SI-7,† entry 4). While some activity could be regained with B(C6F5)3 or LiBArF,33c,35 it was still poor compared to the standard conditions without additive (Table SI-7,† entries 5 and 6).
Regardless of the experimental conditions applied, the gas evolution curves show a sigmoidal trend, with an induction period with very little observable activity. This catalyst pre-activation becomes shorter at higher temperatures and at 110 °C it is almost completely erased (Fig. SI-14†). This raised the question whether the incubation period is required to generate a homogeneous catalytic active species from the catalyst precursor or to transform it into nanoparticles; thus, whether the process is homogeneous or heterogeneous.36 Visual inspection of the reaction did not reveal any noticeable precipitate formation during this time although onset of activity was marked by a colour change from the initial very pale yellow to orange (Fig. SI-22†). The solution turned yellow again when no further gas evolution could be measured. No activity was observed when either Mo(0) powder (Table 6, entry 2) or a combination of Mo(0) powder and one equivalent of the ligand HN(CH2CH2PiPr2)2 (Table 6, entry 3) were used. Addition of Hg did not halt gas evolution (Table SI-8,† entry 14).37 Nor was it stopped by the addition of 0.2 equivalent of PPh3 with respect to the catalyst to the reaction solution (Table 6, entry 4). All these observations suggest that nanoparticles are not formed during the induction period and no heterogenous catalyst is responsible for the observed activity.
| Entry | Catalyst | Additive (eqq. to catalyst) | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | Conversione (%) |
|---|---|---|---|---|---|---|---|---|
|
a General conditions: HCOOH 10 mmol, catalyst 5 10 μmol, toluene 20 mL, 90 °C, 6 h.
b Volumes of evolved gases are corrected by blank volume (5.4 mL).
c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100.
d See SI2 for calculation of selectivity.
e Based on recovered HCOOH.
f Mo(0) powder applied as catalyst (particle size < 100 nm).
g 50 μmol Mo(0) powder and ligand HN(CH2CH2PiPr2)2 in 1:1 molar ratio applied as catalyst (particle size <100 nm).
h Reaction time 4 h 30 min.
i Reaction performed either in glass reactor or closed autoclave; in both cases an additional cooling trap was applied to avoid loss of methyl formate during heating of the glass reactor and gas release from the autoclave.
j Entry 10 is included for the sake of comparison. Each experiment was performed at least twice, standard deviations of yields and selectivity are 1 to 34% of the average (except entries 6, 7 and 9: up to 68% and 6%, respectively for MeOH yield and selectivity). Besides the listed products, traces of CO were detected (Table SI-8 and Fig. SI-17).
|
||||||||
| 1 | 5 | — | 272 | 23 | 267 | 8.6 | 9.5 | 94 |
| 2f | Mo(0) | — | 0 | n.d. | n.d. | 0 | 0 | 2 |
| 3g | Mo(0) | HN(CH 2 CH 2 PiPr 2 ) 2 (1) | 4.6 | n.d. | n.d. | 0 | 0 | 0 |
| 4 | 5 | PPh 3 (0.2) | 269 | 21 | 347 | 11 | 12 | 93 |
| 5 | 5 | PPh 3 (2.0) | 245 | 24 | 226 | 7.5 | 8.2 | 93 |
| 6 | 5 | PPh 3 (20) | 234 | 16 | 213 | 6.8 | 7.7 | 90 |
| 7 | 5 | PPh 3 (100) | 202 | 8.5 | 144 | 4.6 | 5.5 | 82 |
| 8h | 5 | H 2 O (50) | 288 | <5 | 313 | 9.3 | 11 | 88 |
| 9i | 5 | HCOOMe (48.7) | 317 | 19 | 549 | 2.4 | 2.8 | 84 |
| 10j | 5 | — | 283 | 6.2 | 249 | 7.6 | 9.2 | 85 |
Interestingly, the addition of 0.2 equivalents of PPh3 improved MeOH yield (Table 6, entry 4, 11%), selectivity (12%) and TON (37 vs. 29) as to the use of 5 alone (Table 6, entry 1). On the other hand, the addition of excess PPh3, from 2 to 20, up to 100 equivalents (Table 6, entries 5, 6 and 7, respectively), negatively affected the overall activity, which, however, is not suppressed. In addition, selectivity in MeOH dropped as well, from 12% with 0.2 eq. PPh3 to 5.5% with 100 eq. (Table 6, entry 7). NMR evidence of the formation of a minor amount of a transient Mo species, very likely coordinated both to the pincer ligand and PPh3 (31P NMR (toluene-d8): δ = 57.33 (d, JPP = 16.7 Hz, 2P); δ = 41,76, (t, JPP = 16.9 Hz, 1P)) was obtained when the catalyst precursor, the Mo–formate complex 5, and PPh3 were heated at 90 °C in toluene-d8 (Fig. SI-23,† spectra b and c). Yet, even in the presence of 20 equivalents of PPh3 this species represents only 37% of the total phosphorus content (excluding excess PPh3), shared with [MoPNP(CO)3] 8 and a few molybdenum hydrides. This made its isolation or characterization impossible. Its synthesis starting from the reaction of the coordinatively labile Mo–ACN species 4 with PPh3 also failed in our hands. At this point we are not able to explain the origin of the positive influence of added phosphine, which, in the present case, preferentially affects disproportionation over dehydrogenation. However, similar positive effects have been reported for alcohol dehydrogenation and CO2 hydrogenation to dimethoxymethane promoted by Ru-MACHO38a and Ru-triphos catalysts respectively.38b In both cases the added phosphine was proposed to stabilize the catalyst in its active form, hampering the formation of less active carbonylated species.
The experimental results reported above highlight how MeOH selectivity is positively affected by high temperature, high catalyst concentration and low formic acid concentration, a dependence from reaction conditions opposite to that observed in literature precedents, although in those cases formic acid decomposition was carried out in closed vessels, under the influence of the increasing pressure developed by formic acid dehydrogenation and disproportionation. The unexpected behaviour was corroborated by the results obtained in an experiment carried out in order to monitor HCOOH dehydrogenation/disproportionation and product distribution as a function of time (Fig. 1 and Table 7). The reactions were carried out under the standard conditions, at 90 °C, in toluene, using 10 mmol of HCOOH and 10 μmol of catalyst 5, and were stopped at 90, 180, 270 and 360 minutes. It is evident that, under the applied conditions, the maximum variation in MeOH concentration, including that of methyl formate, is observed within the first 180 minutes (Table 7, entry 2). Within the first 90 minutes the TOF reached the highest value of 8.7 h−1. The yield in H2 was 54% after 180 minutes (Table 7, entry 2) after which it stayed substantially constant, while MeOH amount and selectivity further increased, very likely a consequence of the reduced concentration of formic acid. A similar trend was observed when the reaction was carried out with 50 μmoles of catalyst 5 (Table SI-10 and Fig. SI-18†). Here, because of the higher catalyst concentration, the highest variations in H2 and MeOH yield are observed within 45 minutes, with a maximum MeOH selectivity and TOF of 19% and 13 h−1, respectively (Table SI-10,† entry 1). At this time the proportion of free MeOH is quite high, afterwards it is converted to methyl formate while the overall selectivity did not change appreciably. Interestingly, the H2 yield does not increase above 56% applying catalyst 5.
 | ||
| Fig. 1 5-Promoted dehydrogenation and disproportionation of HCOOH: product distribution as a function of time using 10 μmol of catalyst. | ||
| Entry | Time (min) | H2b (mmol) | H2 yieldb (%) | MeOH [μmol] | HCOOMe [μmol] | MeOH Yieldc (%) | MeOH selectivityd (%) | TON | Conversione (%) |
|---|---|---|---|---|---|---|---|---|---|
|
a General conditions: HCOOH 10 mmol, catalyst 5 10 μmol, toluene 20 mL, 90 °C.
b For calculation see SI2.
c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100.
d See SI2 for calculation of selectivity.
e Calculated based on recovered HCOOH. Each experiment was performed at least twice, standard deviations of yields and selectivity are 0.1 to 19% of the average (except for entry 1:39% and 29%, respectively, for MeOH yield and selectivity). Besides the listed products traces of CO were detected (Table SI-9).
|
|||||||||
| 1 | 90 | 3.9 | 39 | 3.2 | 100 | 3.9 | 6.3 | 13 | 63 |
| 2 | 180 | 5.4 | 54 | 8.4 | 196 | 6.1 | 7.9 | 21 | 79 |
| 3 | 270 | 5.5 | 55 | 6.2 | 249 | 7.6 | 9.2 | 25 | 85 |
| 4 | 360 | 5.3 | 53 | 23 | 267 | 8.6 | 9.5 | 29 | 94 |
As both reactions, dehydrogenation as well as disproportionation, seem to stop although HCOOH is still present, experiments were carried out to assess possible product inhibition due to either water or methyl formate. The addition of 500 μmoles of H2O (Table 6, entry 8) slightly improved the methanol yield under otherwise identical standard conditions (Table 6, entry 10), thus excluding water as a possible catalyst poison.39 In contrast, when 487 μmoles of methyl formate were added to a reaction vessel or an autoclave (Table 6, entry 9), only small amounts of extra methyl formate were detected in the reactor. However, formic acid conversion remained unchanged (Table 6, entries 9 and 10, 84% vs. 85%): a result which can be explained by increased HCOOH dehydrogenation, as judged by the overall gas volume evolved (325 mL vs. 283 mL). For the reactions according to entries 8 and 9 in Table 6 an additional cooling trap was installed avoiding loss of methyl formate.
To improve selectivity in favour of MeOH, we explored the possibility of carrying out HCOOH disproportionation in a closed autoclave without and with added H2 (initial overpressure) to reduce and eventually suppress the competing HCOOH dehydrogenation. Experiments were performed under the standard conditions while varying the initial H2 pressure (0, 1 and 60 bars, Table 8). Performing the reaction in a closed autoclave using either 10 or 50 μmol of 5 (Table 8, entries 2 and 4, relative to reactions performed in open vessels entries 1 and 3) resulted in a pressure increase of 3.0 and 3.3 bars, respectively, due to HCOOH conversion. Notably, MeOH yield and selectivity improved in both cases when operating in a closed system. Experiments with initial hydrogen overpressure were performed using 10 μmoles of catalyst 5. While no change was observed with just 1 extra bar hydrogen (compare Table 8, entries 2 and 5), MeOH yield and selectivity were 19% and 37% respectively and the TON reached 69 (Table 8, entry 6) when 60 bars of hydrogen were initially admitted into the autoclave. Under these conditions, however, formation of MeOH by HCOOH hydrogenation is very likely, in line with observations by Laurenczy and Himeda when using iridium catalyst B (Scheme 1) under comparable conditions.22d However complex 5 is not able to catalyse the direct hydrogenation of CO2 to either MeOH or HCOOH as observed when a mixture of H2 and CO2 (35:15 bars) was heated at 90 °C in toluene (20 mL) for 24 h in the presence of 5 (10 μmol).40
| Entry | Gas (overpressureb, bar) | Gas volume [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | TON | Conversione (%) |
|---|---|---|---|---|---|---|---|---|
| a General conditions: all reactions, except those of entries 1 and 3, were performed in autoclaves; HCOOH 10 mmol, catalyst 5 10 μmol, toluene 20 mL, 90 °C, 6 h. b Initial pressure at room temperature. c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100. d See SI2 for calculation of selectivity. e Based on recovered HCOOH. f By the end of the reaction, the overall pressure had increased by 3.0 bar. g Catalyst 5 50 μmol, 4 h. h Catalyst 5 50 μmol, 4 h, by the end of the reaction, the overall pressure had increased by 3.3 bar. i By the end of the reaction, the overall pressure had increased by 2.6 bar. j By the end of the reaction, the overall pressure had increased by 6.4 bar. The experiments, except those in entries 2 and 4, were performed at least twice, standard deviations of MeOH yield and selectivity are 3 to 17% of the average, except for entry 6 (40% and 30%, respectively). Besides the listed products, traces of CO were detected (Table SI-11 and Fig. SI-19). | ||||||||
| 1 | Ar (0) | 272 | 23 | 267 | 8.6 | 9.5 | 29 | 94 |
| 2f | Ar (0) | 272 | 47 | 352 | 12 | 13 | 39 | 96 |
| 3g | Ar (0) | 285 | 5.0 | 503 | 15 | 17 | 10 | 95 |
| 4h | Ar (0) | 290 | 97 | 563 | 20 | 21 | 13 | 99 |
| 5i | H2 (1) | — | 20 | 365 | 11 | 13 | 38 | 91 |
| 6j | H 2 (60) | — | 44 | 647 | 19 | 37 | 69 | 62 |
In the system under investigation, two catalytic processes are taking place at the same time: HCOOH dehydrogenation and disproportionation. Experimental evidence and a perusal of literature on the topic suggests two steps common to both: (1) formation of a metal hydride formate species, arising from interaction of the catalyst precursor with HCOOH, followed by (2) formate decarboxylation through β-hydride transfer to the metal with generation of a metal dihydride species. Selectivity in favour of either process is determined at the next stage: the metal dihydride species may release a hydrogen molecule (dehydrogenation), either through dihydrogen reductive elimination or protonation by HCOOH, with regeneration of the metal hydride formate species. NH proton-assisted dehydrogenation, while not mandatory, may also be operative using catalyst 5, thus favoring dehydrogenation over disproportionation. Alternatively, a hydride may be transferred to an activated/protonated molecule of HCOOH which is so reduced to methanediol (disproportionation). The latter is then dehydrated to formaldehyde which is in turn reduced to methanol through the same hydride transfer mechanism. Depending on the catalytic system and the reaction conditions, either formate decarboxylation or the following step, hydrogen release for dehydrogenation or hydride transfer for disproportionation may be the rate determining step. Inspired by literature precedents,41–43 we carried out a series of experiments based on the results of which we propose a plausible general mechanism for the present system as illustrated in Scheme 4.
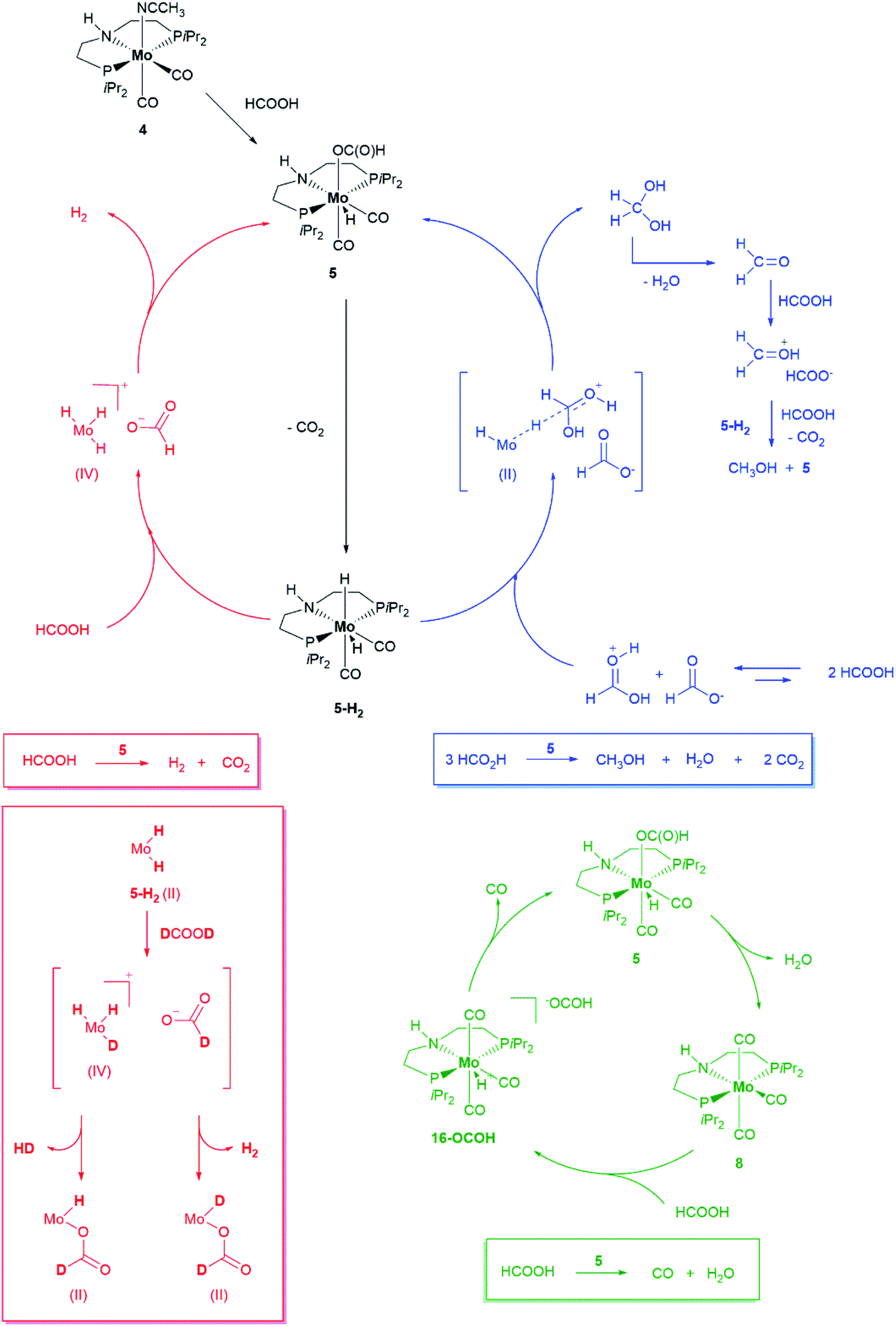 | ||
| Scheme 4 Proposed mechanisms for HCOOH dehydrogenation (red), disproportionation (blue) and decarbonylation (green) promoted by 5. Evidence for the formation of a Mo(IV) species is based on the detection by NMR of H2 and HD following addition of DCOOD to Mo(H)n species (see Fig. SI-31†). | ||
In the scheme, HCOOH decarboxylation is also accounted for, as this process accounts for one of the catalysts resting states (vide infra). In the presence of a large excess of HCOOH, catalyst precursor 4 is converted within minutes at room temperature to new hydride species as suggested by two triplets in the 1H NMR spectrum (toluene-d8) at δ = −4.16 ppm (JHP = 28.9 Hz) and δ = −5.16 ppm (JHP = 29.7 Hz) in a 2:1 ratio, correlated to two singlets in the same ratio in the 31P{1H} spectrum at δ = 67.1 ppm and δ = 66.8 ppm respectively, which suggests the equivalence of the phosphorus donors in both species (Fig. 2, a and SI8†).
 | ||
| Fig. 2 1H and 31P{1H} NMR spectra of a toluene-d8 solution of {Mo(CH3CN)(CO)2(HN[(CH2CH2P(CH(CH3)2)2]2} 4 in the presence of 100 equivalents of HCOOH ([Mo] 10−2 M, [HCOOH] 1 M), before (a) and after heating at 90 °C for 1 hour (b). Spectra were recorded at room temperature. Signals related to complex 5 are marked by red dots. | ||
One of the two hydrides (δ = −4.16 ppm (JHP = 28.9 Hz) could be unambiguously identified as the Mo-formate species 5. Regarding the other hydride at δ = −5.16 ppm (JHP = 29.7 Hz), we postulate either the formation of an isomer of 5 or a Mo(IV) dihydride arising from 5 following its further protonation made possible by the large excess of HCOOH. Interestingly, this species was no longer observable once all HCOOH had been consumed (Fig. 2b). Complex 5 was prepared by addition of 1.2 equivalents of HCOOH to 4 in toluene at room temperature. The reaction was very selective and only one species was observed, which could be easily isolated and purified for complete characterization. Its solid-state structure could be established by X-ray analysis. Protonation of 4 by HCOOH affords the 7-coordinated Mo(II)-complex 5 which adopts a pentagonal bipyramid coordination (Fig. 3).44 The PNP ligand is equatorial, one CO molecule and one hydride complete the set of ligands in the shared plane, although the hydride sits slightly above it. The second CO and the formate ligand occupy the axial positions, with the formate ligand cis to the hydrogen on the nitrogen. Notably, an intermolecular hydrogen bond between these groups is present in the solid state. 5 is relatively stable in the solid state: after exposure to air for two hours, the sample shows a slight change in colour but its solution 1H and 31P NMR spectra reveal no difference as to those of a sample kept under argon. Nonetheless, we suggest keeping 5 under argon for prolonged storage.
 | ||
| Fig. 3 Molecular structure of {Mo(H)(OCOH)(CO)2[HN(CH2CH2P(CH(CH3)2)2)2]} 5. Displacement ellipsoids correspond to 30% probability. Carbon-bound hydrogen atoms of the PNP ligand are omitted for clarity. | ||
Under standard reaction conditions, 5 afforded better catalyst performance compared to 4 which, being extremely air sensitive, must be handled in the glove box and is liable to degradation (which might explain the inferior performance as to 5). Therefore 5 is a convenient, easy to handle substitute for 4. 5 is also the predominant hydride species observed in toluene-d8 solution in the presence of excess HCOOH when the sample is heated to 90 °C directly in the NMR instrument probe and both H2 and MeOH are also detected (SI9, Fig. SI-25†). After HCOOH has been consumed, 5 is still present in solution suggesting it might be a catalyst resting state (Fig. 2b). The other species observed both during and after catalysis (31P{1H} NMR spectrum at δ = 75.9 ppm) could be identified as [MoPNP(CO)3] 8, by comparison of its spectroscopic features with an independently synthetized sample.26a This species arises from dehydration of 5, following formate decarbonylation,41 as could be demonstrated by thermal decomposition of [MoH(PNP)(CO)2O13COH] 13C-5, a sample of 5 isotopically labelled at the formyl group (SI9, SI10†). However, we did not observe the formation of water or that of a species formally arising from H2O oxidative addition to molybdenum although the formation of water is expected by stoichiometry.
As shown in Table 2, entry 3, also complex 8 is catalytically active. Its performance under standard reaction conditions in terms of hydrogen and MeOH production is in between that of 4 and 5, thus suggesting a common catalytically active species, regardless of the catalyst precursor used. As long as excess HCOOH is present in solution, 8 does not represent a dead end in catalysis (Scheme 4, green cycle): protonation affords 16-OCOH whose reduced electron density at the metal labilizes coordinated CO ligands.
Upon heating, one CO molecule is lost thus regenerating 5 and, from this, the catalytic active species (SI13†). Further evidence in support of the decisive role played by protonation stems from the observation that 8 is not able to promote catalytic hydrogenation of carbonyl compounds and amides.26 Because in the reaction with 8 an excess of HCOOH is necessary to completely shift the equilibrium towards 16-OCOH, the latter could not be obtained as an analytical pure sample. Further evidence in support of its identity though was obtained by comparing its spectroscopic properties with those of the analogous 16-BF4, obtained by protonation of 8 with just one equivalent of the strong acid HBF4 (Fig. SI-33 and SI-34†).42 The solid-state structure of 16-BF4 is shown in Fig. 4. If 13C-5 is heated up to 90 °C without any added HCOOH, the NMR spectra of the solution show the presence, of unconverted 13C-5 and exclusively labelled at CO 13C-8. Besides, new molybdenum hydride species emerge [Mo(PNP)(CO)x(H)n], the hydrides of which resonate in the range −4.0 and −6.0 ppm (SI10, Fig. SI-26, -28 and -29†) which all exhibit similar spectroscopic properties but the exact structure of which could not be determined. None appears to be an isomer of 13C-5 and therefore they are believed to arise from β-hydride transfer to the metal from the coordinated formate in 13C-5. Their hydride peaks all show a single well-defined triplet, the intensity of which cannot be compared to any other signal in the spectrum to establish whether it corresponds to one or two hydrogens.43 Low temperature spectra recorded down to −76 °C did not help clarify this point (Fig. SI-30†). No signal for dihydrogen at δ = 4.5 ppm could be observed suggesting no further evolution of such species by dihydrogen release. Yet, they quickly and quantitatively react with HCOOH at room temperature to regenerate 5 (SI12, Fig. SI-32†). Gas evolution is observed together with a tiny peak for hydrogen in the 1H spectrum. If DCOOD is used instead,45 both H2 and DH are detected,46 suggesting the formation of a transient Mo(IV) species (SI11†).47 Such species arises from protonation of the Mo-hydride species either at the metal or at the hydride: in the latter case, through the reversible isomerization of the initially formed non-classical [Mo(II)Hn(η-H2) to Mo(IV)Hn+2].48
 | ||
| Fig. 4 Molecular structure of {Mo(H)(CO)3[HN(CH2CH2P(CH(CH3)2)2)2]}BF416-BF4. Displacement ellipsoids correspond to 30% probability. Carbon-bound hydrogen atoms and the BF4 anion are omitted for clarity. | ||
According to our mechanistic proposal, the Mo hydride species generated from 5 decarboxylation are the key to HCOOH disproportionation (blue cycle) as well: they reduce the protonated HCOOH49 by hydride transfer to give methanediol which is subsequently dehydrated to formaldehyde. The latter is then further reduced to MeOH. If hydride transfer from the metal occurs to HCOOH2+, a protonated and thus activated molecule of formic acid, then it does not require coordination of the substrate to the metal and can be regarded as an example of catalytic ionic transfer hydrogenation.50 Indeed 5 is able to promote the stoichiometric and quantitative reduction of benzaldehyde to benzylalcohol at room temperature if a strong acid such as HBF4 is used (SI14†). The reaction requires heating if HCOOH is used instead as the proton source, (SI15†). The detection of 5 in the presence of excess HCOOH even at 90 °C suggests that β-hydride elimination to generate a reactive molybdenum hydride species, key to both dehydrogenation and disproportionation, is a slow process. We have carried out catalytic experiments under the standard reaction conditions using isotopomers of HCOOH. The observed KIE is a composite of the KIE of both dehydrogenation and disproportionation and is therefore difficult to disentangle. Yet it is evident, when comparing the gas evolution curves (Fig. 5), that the drop in activity brought about by using DCOOH in place of HCOOH is higher than that observed with HCOOD.
 | ||
| Fig. 5 Left: time course of generated gas volume through dehydrogenation and disproportionation of formic acid and its partially and fully deutero-substituted derivatives with 5 under the standard reaction conditions: HCOOH 10 mmol, catalyst 5 10 μmol, toluene 20 mL, 90 °C, 6 h. Gas volumes are not corrected by blank values. Right: amount of formed products, methanol, methyl formate, partially and fully deuterated counterparts. | ||
We could not establish the relative amounts of H2, DH and D2 in the gas phase and therefore the selectivity in the two processes. However, the highest MeOH yield is obtained using HCOOD, 10%, as compared to 8.6% with HCOOH (Table 9). The lowest one, 2.8%, is recorded with DCOOD. Himeda and Laurency observed the highest yield with HCOOH/D2O using the iridium catalyst B (Scheme 1) at atmospheric pressure: they could also establish a trend in MeOH selectivity which increased from 2.1% in H2O/H2SO4 to 4.0–4.5% in D2O/D2SO4. Their findings are explained with the slower generation of H2 (or HD/D2) in the latter case, as protonation of an iridium-hydride species is the slow step for their system.22c At variance to our system, they observed almost no influence on the reaction rate with deuteration of the formyl site H–COOH.
| Entry | HCOOH isotopomers | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | Conversione (%) |
|---|---|---|---|---|---|---|---|
| a General conditions: formic acid 10 mmol, catalyst 5 10 μmol, solvent 20 mL, 90 °C, 6 h. b Calculated after correcting volumes of evolved gases by blank volume (5.4 mL). c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100. d See SI2 for calculation of selectivity. e Based on recovered HCOOH isotopomers (Table SI-12). Because of technical limitations, it was not possible for HCOOD, DCOOH and DCOOD, to detect and quantify HD and D2, and therefore to establish the exact composition of the gas phase and the selectivity in MeOH for entries 2, 3 and 4. | |||||||
| 1 | HCOOH | 272 | 23 | 267 | 8.6 | 9.5 | 94 |
| 2 | HCOOD | 222 | 64 | 288 | 10 | — | 97 |
| 3 | DCOOH | 187 | 18 | 162 | 5.4 | — | 90 |
| 6 | DCOOD | 92 | n.d. | 95 | 2.8 | — | 63 |
Catalyst 5 is an 18-electron coordinatively saturated species. HCOOH dehydrogenation usually entails decarboxylation of the coordinated formato either through β-hydride elimination or, alternatively, through dissociation of the latter and subsequent hydride transfer from the free formate.51 In the former case, a vacant coordination site for the incoming hydride cis to the formato ligand is required. Labilization of a coordinated CO may ease this process and the formation of a reactive molybdenum hydride species. Therefore, we carried out a catalytic experiment using two equivalents Me3NO (to the catalyst), under otherwise standard conditions.
Me3NO-assisted decarbonylations are generally thought to occur via oxygen atom transfer to the carbonyl carbon atom which is then released from the metal coordination sphere as CO2.52 Following the addition of Me3NO, the yield and selectivity in MeOH improved from 8.6 to 11.0% and from 9.5 to 13%, respectively as to the standard conditions. We did not attempt to isolate the complex(es) arising from 5 decarbonylation but the altered reactivity in favour of disproportionation might be explained by a change in the Mo-hydride(s) hydricity with the changes in the metal coordination sphere (Table 10).53
| Entry | Additive (eq. to catalyst) | Gas volumeb [mL] | MeOH [μmol] | HCOOMe [μmol] | MeOH yieldc (%) | MeOH selectivityd (%) | Conversione (%) |
|---|---|---|---|---|---|---|---|
| a General conditions: HCOOH 10 mmol, catalyst 5 10 μmol, toluene 20 mL, 90 °C, 6 h. b Calculated after correcting volumes of evolved gases by blank volume (5.4 mL). c Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100. d See SI2 for calculation of selectivity. e Based on recovered HCOOH. Each experiment was performed at least twice, standard deviations of MeOH yield and selectivity are, respectively, 16% and 17% for entry 1 and 47% and 49% for entry 2. Besides the listed products, CO was detected (Table SI-13 and Fig. SI-20). | |||||||
| 1 | — | 272 | 23 | 267 | 8.6 | 9.5 | 94 |
| 2 | Me 3 NO (2) | 287 | 61 | 315 | 11 | 13 | 93 |
Finally, we tried to assess whether the PNP ligand supporting the Mo complex is directly involved in the elementary steps of the catalytic processes or not. Because of the presence of a protic NH ligand fragment, we wondered whether the catalyst might be operating through a bifunctional mechanism, in which the ligand nitrogen function as a H+ donor and the resulting amido bond N–Mo as a H+ acceptor.54 It is well-established that the possibility of such mechanism positively affects (de)hydrogenation and transfer (de)hydrogenation reactions in which hydrogen atoms are involved. In the present case, catalysis takes place under acidic conditions and ligand deprotonation at nitrogen does not constitute a prerequisite for catalyst turnover. The bifunctional mechanism has been recently revised showing that NH or NM bonds are not cleaved or formed during catalysis in several cases.55 Yet the presence of a hydrogen at nitrogen serves to stabilize the transition state of rate-determining steps through the formation of N–H⋯O hydrogen-bonding interactions between the ligand and the substrate, so that a metal ligand-assisted mechanism is operative (Dub-type mechanism55). Alkylation of the H–N group on the ligand backbone may negatively affect and ultimately shut down catalysis if such stabilization is operative and very effective. Under certain conditions, activity can even improve.56 In the present case, when the N-methylated Mo-complex 9 (Fig. 6) was applied for HCOOH conversion under the standard reaction conditions (Table 11, entry 2) activity was retained although with about 16% less total evolved gas volume as compared with 5 (Table 11, entry 1), thus proving that a hydrogen at the ligand nitrogen is not mandatory for catalysis. Noteworthy, the effect on dehydrogenation and disproportionation is quite different: methylation of the ligand nitrogen reduces the yield in H2 from 53% to 41%, but the yield, selectivity and TON for MeOH improve, from 8.6%, 9.5% and 29, respectively, with 5 (Table 11, entry 1) to 17%, 22% and 55, respectively, with 9 (Table 11, entry 2).
 | ||
| Fig. 6 Molecular structure of {Mo(H)(CO)2(CH3CN)[CH3N(CH2CH2P(CH(CH3)2)2)2]} 9. Displacement ellipsoids correspond to 30% probability. Hydrogen atoms are omitted for clarity. | ||
| Entry | [Mo] | Gas volb (mL) | H2c (mmol) | H2 yieldc (%) | MeOH [μmol] | HCOOMe [μmol] | MeOH Yieldd (%) | MeOH selectivitye (%) | TON | Conversionf (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a General conditions: HCOOH 10 mmol, catalyst 10 μmol, toluene 20 mL, 90 °C, 6 h. b Calculated after correcting volumes of evolved gases by blank volume (5.4 mL). c For calculation see SI2. d Yield is calculated as follows {[(mmol MeOH + mmol MF) × 3]/× mmol HCOOH} × 100. e See SI2 for calculation of selectivity. f Based on recovered HCOOH. g catalyst 20 μmol. h Experiment performed for 9 hours. Each experiment was performed at least twice, standard deviations of yields and selectivity are 15–24% of the average. Besides the listed products, CO was detected (Table SI-14 and Fig. SI-21). | ||||||||||
| 1 | 5 | 272 | 5.3 | 53 | 23 | 267 | 8.6 | 9.5 | 29 | 94 |
| 2 | 9 | 227 | 4.1 | 41 | n.d. | 575 | 17 | 22 | 55 | 83 |
| 3g | 9 | 229 | 4.2 | 42 | 6.8 | 520 | 16 | 20 | 26 | 82 |
| 4h | 9 | 253 | 4.5 | 45 | n.d. | 649 | 19 | 30 | 57 | 70 |
Thus, catalyst 9 combines a high MeOH yield and selectivity with the highest TON observed so far for a non-noble metal-based catalyst in HCOOH disproportionation to methanol.
It has been shown that the hydrogen at the nitrogen of the supporting ligand may help lower the kinetic barrier for H2 release from the metal catalyst by acting as a proton shuttle.57 Its replacement with a methyl as in 9 would then indirectly favour HCOOH disproportionation over dehydrogenation. On the other hand, ligand methylation may increase electron density at molybdenum and increase the hydride donor ability of 9-derived hydrides, thus promoting formic acid reduction.53b By doubling the catalyst amount from 10 to 20 μmol (Table 11, entry 3) almost the same results were obtained, while extending the reaction time to 9 hours slightly improved MeOH yield, selectivity and TON (Table 11, entry 4).
Conclusions
In the present investigation the catalytic disproportionation of formic acid in the presence of non-noble metal complexes has been realized for the first time. In general, this transformation allows for an alternative access of methyl formate and methanol from renewable resources. Specifically, molybdenum (0) complexes with aliphatic aminophosphine pincer ligands promote formic acid dehydrogenation with hydrogen yields up to 56% (catalyst 5). Parallel to formic acid dehydrogenation, formic acid disproportionation takes place with a MeOH selectivity up to 21%. This latter process is improved utilizing the N-methylated complex 9 (TON = 57; MeOHselectivity: 30%). Selectivity (37%) and catalyst performance (TON = 69) could be further enhanced, when the reaction was performed under hydrogen pressure (60 bars). When compared to most catalysts reported so far for formic acid disproportionation, e.g. iridium complexes, 5 appears to be more active and shows comparable activity compared to the state-of-the-art ruthenium complex C. Mechanistic investigations allowed to propose plausible catalytic cycles for formic acid dehydrogenation and disproportionation: molybdenum hydride species have been detected arising from decarboxylation of the formate complex 5, likely the rate determining step. Whether such dihydride species are protonated to evolve hydrogen or transfer a hydride to protonated and thus activated formic acid determines the selectivity of the process. Current limitations of the MeOH yield are explained because of product inhibition of 5. We believe that these insights will allow for a more rational development of advanced catalysts for FA disproportionation in the future.Data availability
We have added all the experimental data in the ESI.† If readers would like further explanation they can contact the authors directly.Author contributions
K. J., H. J., E. A. and M. B. conceived and coordinated the project. E. A. planned and performed together with W. B. the NMR investigation. A. K., E. A., R. S., J. S. and T. L. performed the catalytic reactions. T. L. and E. A. synthesized the respective Mo complexes. A. S. performed X-ray crystal structure analysis. E. A., H. J., M. B. and K. J. wrote the manuscript. All authors discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mrs Petra Bartels (LIKAT) is gratefully acknowledged for the synthesis of several batches of complex 5. Susann Buchholz and Susanne Schareina (both LIKAT) are acknowledged for performing GC and HPLC measurements.Notes and references
- J. Hietala, A. Vuori, P. Johnson, I. Pollari, W. Reutemann and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, 2016, pp. 1–22, DOI:10.1002/14356007.a12_013.pub3.
- R. van Putten, T. Wissink, T. Swinkels and E. A. Pidko, Fuelling the hydrogen economy: Scale-up of anintegrated formic acid-to-power system, Int. J. Hydrogen Energy, 2019, 44, 28533 CrossRef CAS.
- A. J. M. Miller, D. M. Heinekey, J. M. Mayer and K. I. Goldberg, Catalytic Disproportionation of Formic Acid to Generate Methanol, Angew. Chem., Int. Ed., 2013, 52, 3981 CrossRef CAS PubMed.
- D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
- C. Chauvier and T. Cantat, A Viewpoint on Chemical Reductions of Carbon−Oxygen Bonds in Renewable Feedstocks Including CO2 and Biomass, ACS Catal., 2017, 7, 2107 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment, Chem. Rev., 2018, 118, 434 CrossRef CAS PubMed.
- https://www.methanol.org/about-methanol/ .
- Selected references: (a) L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X.-D. Wen and D. Ma, Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts, Nature, 2017, 544, 80 CrossRef CAS PubMed; (b) M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide, Nature, 2013, 495, 85 CrossRef CAS PubMed; (c) R. D. Cortright, R. R. Davda and J. A. Dumesic, Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water, Nature, 2002, 418, 964 CrossRef CAS PubMed.
- S. S. Araya, V. Liso, X. Cui, N. Li, J. Zhu, S. L. Sahlin, S. H. Jensen, M. P. Nielsen and S. K. Kær, A Review of The Methanol Economy: The Fuel Cell Route, Energies, 2020, 13, 596 CrossRef.
- (a) S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions, Chem. Soc. Rev., 2021, 50, 4259 RSC; (b) X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis, Chem. Rev., 2020, 120, 7984 CrossRef CAS PubMed; (c) A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes, Chem. Rev., 2017, 117, 9804 CrossRef PubMed.
- (a) S. Xie, W. Zhang, X. Lan and H. Lin, CO2 Reduction to Methanol in the Liquid Phase: A Review, ChemSusChem, 2020, 13, 6141 CrossRef CAS PubMed; (b) M. F. Hertrich and M. Beller, Metal-Catalysed Hydrogenation of CO2 into Methanol, Top. Organomet. Chem., 2019, 63, 1 CAS , in Organometallics for Green Catalysis, ed. P. H. Dixneuf and J.-F. Soulé; ; (c) S. Kar, J. Kothandaraman, A. Goeppert and G. K. S. Prakash, Advances in catalytic homogeneous hydrogenation of carbon dioxide to methanol, J. CO2 Util., 2018, 23, 212 CrossRef CAS.
- (a) B. G. Schieweck, P. Jürling-Will and J. Klankermayer, Structurally Versatile Ligand System for the Ruthenium Catalyzed One-Pot Hydrogenation of CO2 to Methanol, ACS Catal., 2020, 10, 3890 CrossRef CAS; (b) J. D. Erickson, A. Z. Preston, J. C. Linehan and E. S. Wiedner, Enhanced Hydrogenation of Carbon Dioxide to Methanol by a Ruthenium Complex with a Charged Outer-Coordination Sphere, ACS Catal., 2020, 10, 7419 CrossRef CAS; (c) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. vom Stein, U. Englert, M. Hölscher, J. Klankermayer and W. Leitner, Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium–Triphos catalyst: from mechanistic investigations to multiphase catalysis, Chem. Sci., 2015, 6, 693 RSC; (d) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499 CrossRef CAS PubMed.
- (a) F. K. Scharnagl, M. F. Hertrich, G. Neitzel, R. Jackstell and M. Beller, Homogeneous Catalytic Hydrogenation of CO2 to Methanol – Improvements with Tailored Ligands, Adv. Synth. Catal., 2019, 361, 374 CAS; (b) J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst, Angew. Chem., Int. Ed., 2017, 56, 1890 CrossRef CAS PubMed.
- A. P. C. Ribeiro, L. M. D. R. S. Martins and A. J. L. Pombeiro, Carbon dioxide-to-methanol single-pot conversion using a C-scorpionate iron(II) catalyst, Green Chem., 2017, 19, 4811 RSC.
- Selected examples of “indirect” CO2 reduction to MeOH, for a more comprehensive list see ref. 6: (a) E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Efficient Hydrogenation of Organic Carbonates, Carbamates and Formates Indicates Alternative Routes to Methanol Based on CO2 and CO, Nat. Chem., 2011, 3, 609 CrossRef CAS PubMed; (b) E. Balaraman, Y. Ben-David and D. Milstein, Unprecedented Catalytic Hydrogenation of Urea Derivatives to Amines and Methanol, Angew. Chem., Int. Ed., 2011, 50, 11702 CrossRef CAS; (c) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, Direct Hydrogenation of Amides to Alcohols and Amines under Mild Conditions, J. Am. Chem. Soc., 2010, 132, 16756 CrossRef CAS; (d) E. M. Lane, Y. Zhang, N. Hazari and W. H. Bernskoetter, Sequential Hydrogenation of CO2 to Methanol Using a Pincer Iron Catalyst, Organometallics, 2019, 38, 3084 CrossRef CAS; (e) Z. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Catalytic Hydrogenation of Cyclic Carbonates: A Practical Approach from CO2 and Epoxides to Methanol and Diols, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS PubMed.
- (a) S. Kar, A. Goeppert and G. K. S. Prakash, Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed; (b) R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, Hydroxide Based Integrated CO2 Capture from Air and Conversion to Methanol, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed, and references therein; (c) S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, Mechanistic Insights into Ruthenium-Pincer-Catalyzed Amine-Assisted Homogeneous Hydrogenation of CO2 to Methanol, J. Am. Chem. Soc., 2019, 141, 3160 CrossRef CAS PubMed.
- (a) T. M. Rayder, A. T. Bensalah, B. Li, J. A. Byers and C.-K. Tsung, Engineering Second Sphere Interactions in a Host−Guest Multicomponent Catalyst System for the Hydrogenation of Carbon Dioxide to Methanol, J. Am. Chem. Soc., 2021, 143, 1630 CrossRef CAS PubMed; (b) T. M. Rayder, E. H. Adillon, J. A. Byers and C.-K. Tsung, A Bioinspired Multicomponent Catalytic System for Converting Carbon Dioxide into Methanol Autocatalytically, Chem, 2020, 6, 1742 CrossRef CAS; (c) W.-Y. Chu, Z. Culakova, B. T. Wang and K. I. Goldberg, Acid-Assisted Hydrogenation of CO2 to Methanol in a Homogeneous Catalytic Cascade System, ACS Catal., 2019, 9, 9317 CrossRef CAS; (d) C. A. Huff and M. S. Sanford, Cascade catalysis for the homogeneous hydrogenation of CO2 to methanol, J. Am. Chem. Soc., 2011, 133, 18122 CrossRef CAS PubMed.
- R. Kanega, N. Onishi, S. Tanaka, H. Kishimoto and Y. Himeda, Catalytic Hydrogenation of CO2 to Methanol Using Multinuclear Iridium Complexes in a Gas−Solid Phase Reaction, J. Am. Chem. Soc., 2021, 143, 1570 CrossRef CAS PubMed.
- (a) J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Biomass oxidation to formic acid in aqueous media using polyoxometalate catalysts – boosting FA selectivity by in situ extraction, Energy Environ. Sci., 2015, 8, 2985 RSC; (b) K. Li, L. Bai, P. N. Amaniampong, X. Jia, J.-M. Lee and Y. Yang, One-Pot Transformation of Cellobiose to Formic Acid and Levulinic Acid over ionic-Liquid-based Polyoxometalate Hybrids, ChemSusChem, 2014, 7, 2670 CrossRef CAS PubMed; (c) P. Zhang, Y.-J. Guo, J. Chen, Y.-R. Zhao, J. Chang, H. Junge, M. Beller and Y. Li, Streamlined hydrogen production from biomass, Nat. Catal., 2018, 1, 332 CrossRef CAS; (d) https://www.oxfa.eu/en/technology/ .
- (a) K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols, Chem. Rev., 2018, 118, 372 CrossRef CAS PubMed; (b) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction, Chem. Rev., 2015, 115, 12936 CrossRef CAS PubMed; (c) G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, Highly efficient reversible hydrogenation of carbon dioxide to formates using a ruthenium PNP-pincer catalyst, ChemCatChem, 2014, 6, 1526 CrossRef CAS; (d) R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Mechanistic Studies on the Reversible Hydrogenation of Carbon Dioxide Catalyzed by an Ir-PNP Complex, Organometallics, 2011, 30, 6742 CrossRef CAS; (e) T. Schaub and R. A. Paciello, A Process for the Synthesis of Formic Acid by CO2 Hydrogenation: Thermodynamic Aspects and the Role of CO, Angew. Chem., Int. Ed., 2011, 50, 7278 CrossRef CAS PubMed.
- P. Sabatier and A. Mailhe, C. R. Hebd. Seances Acad. Sci., 1911, 152, 1212 Search PubMed.
- (a) A. F. Sasayama, C. E. Moore and C. P. Kubiak, Electronic effects on the catalytic disproportionation of formic acid to methanol by[Cp*Ir(III)(R-bpy)Cl]Cl complexes, Dalton Trans., 2016, 45, 2436 RSC; (b) K. Sordakis, A. Tsurusaki, M. Iguchi, H. Kawanami, Y. Himeda and G. Laurenczy, Carbon Dioxide to Methanol: The Aqueous Catalytic Way at Room Temperature, Chem.–Eur. J., 2016, 22, 15605 CrossRef PubMed; (c) A. Tsurusaki, K. Murata, N. Onishi, K. Sordakis, G. Laurenczy and Y. Himeda, Investigation of Hydrogenation of Formic Acid to Methanol using H2 or Formic Acid as a Hydrogen Source, ACS Catal., 2017, 7, 1123 CrossRef CAS; (d) K. Sordakis, A. Tsurusaki, M. Iguchi, H. Kawanami, Y. Himeda and G. Laurenczy, Aqueous phase homogeneous formic acid disproportionation into methanol, Green Chem., 2017, 19, 2371 RSC; (e) X. Yan and X. Yang, Mechanistic Insights into Iridium Catalyzed Disproportionation of Formic Acid to CO2 and Methanol: A DFT Study, Organometallics, 2018, 37, 1519 CrossRef CAS; (f) C.-Z. Zhou, Y.-R. Zhao, Y.-J. Guo, P. Zhanga and Y. Li, Formic acid disproportionation into formaldehyde triggered by vanadium complexes with iridium catalysis under mild conditions in N-methylation, Green Chem., 2021, 23, 2918 RSC.
- (a) S. De, L. Gevers, A.-H. Emwas and J. Gascon, Conversion of Formic Acid into Methanol Using a Bipyridine-Functionalized Molecular Heterogeneous Catalyst, ACS Sustainable Chem. Eng., 2019, 7, 3933 CrossRef CAS; (b) S. Yamaguchi, Y. Maegawa, N. Onishi, R. Kanega, M. Waki, Y. Himeda and S. Inagaki, Catalytic Disproportionation of Formic Acid to Methanol by an Iridium Complex Immobilized on Bipyridine-Periodic Mesoporous Organosilica, ChemCatChem, 2019, 11, 4797 CrossRef CAS; (c) S. Yamaguchi and S. Hashimoto, Mechanism of Formic Acid Disproportionation Catalyzed by an Iridium Complex Immobilized on Bipyridine-Periodic Mesoporous Organosilica: A Case Study Based on Kinetics Analysis, Asian J. Org. Chem., 2020, 9, 99 CrossRef CAS.
- (a) S. Savourey, G. Lefèvre, J.-C. Berthet, P. Thuéry, C. Genre and T. Cantat, Efficient Disproportionation of Formic Acid to Methanol Using Molecular Ruthenium Catalysts, Angew. Chem., Int. Ed., 2014, 53, 10466 CrossRef CAS PubMed; (b) C. Chauvier, A. Imberdis, P. Thuéry and T. Cantat, Catalytic Disproportionation of Formic Acid to Methanol by using Recyclable Silylformates; Cantat also reported the metal-free disproportionation of HCOOH mediated by organoboranes, Angew. Chem., Int. Ed., 2020, 59, 14019 CrossRef CAS PubMedC. Chauvier, P. Thuéry and T. Cantat, Metal-free disproportionation of formic acid mediated by organoboranes, Chem. Sci., 2016, 7, 5680 RSC; (c) H. Fujita, S. Takemoto and H. Matsuzaka, Tin−Ruthenium Cooperative Catalyst for Disproportionation of Formic Acid to Methanol, ACS Catal., 2021, 11, 7460 CrossRef CAS.
- M. C. Neary and G. Parkin, Dehydrogenation, disproportionation and transfer hydrogenation reactions of formic acid catalyzed by molybdenum hydride compounds, Chem. Sci., 2015, 6, 1859 RSC.
- (a) T. Leischner, A. Spannenberg, K. Junge and M. Beller, Molecular Defined Molybdenum−Pincer Complexes and Their Application in Catalytic Hydrogenations, Organometallics, 2018, 37, 4402 CrossRef CAS; (b) T. Leischner, L. A. Suarez, A. Spannenberg, K. Junge, A. Nova and M. Beller, Highly selective hydrogenation of amides catalysed by a molybdenum pincer complex: scope and mechanism, Chem. Sci., 2019, 10, 10566 RSC; (c) T. Leischner, A. Spannenberg, K. Junge and M. Beller, Synthesis of Molybdenum Pincer Complexes and Their Application in the Catalytic Hydrogenation of Nitriles, ChemCatChem, 2020, 12, 4543 CrossRef CAS.
- (a) X. Li, A.-E. Surkus, J. Rabeah, M. Anwar, S. Dastigir, H. Junge, A. Brueckner and M. Beller, Cobalt Single-Atom Catalysts with High Stability for Selective Dehydrogenation of Formic Acid, Angew. Chem., Int. Ed., 2020, 59, 15849 CrossRef CAS PubMed; (b) A. Leval, H. Junge and M. Beller, Manganese(I) κ2-NN complex-catalyzed formic acid dehydrogenation, Catal. Sci. Technol., 2020, 10, 3931 RSC; (c) A. Leval, A. Agapova, C. Steinlechner, E. Alberico, H. Junge and M. Beller, Hydrogen production from formic acid catalyzed by a phosphine free manganese complex: investigation and mechanistic insights, Green Chem., 2020, 22, 913 RSC; (d) W. Zhou, Z. Wei, A. Spannenberg, H. Jiao, K. Junge, H. Junge and M. Beller, Cobalt-Catalyzed Aqueous Dehydrogenation of Formic Acid, Chem.–Eur. J., 2019, 25, 8459 CrossRef CAS PubMed; (e) M. Andérez-Fernández, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, A stable manganese pincer catalyst for the selective dehydrogenation of methanol, Angew. Chem., Int. Ed., 2017, 56, 559 CrossRef PubMed; (f) D. Mellmann, E. Barsch, M. Bauer, K. Grabow, A. Boddien, A. Kammer, P. Sponholz, U. Bentrup, R. Jackstell, H. Junge, G. Laurenczy, R. Ludwig and M. Beller, Base-Free Non-Noble-Metal-Catalyzed Hydrogen Generation from Formic Acid: Scope and Mechanistic Insights, Chem.–Eur. J., 2014, 20, 13589 CrossRef CAS; (g) A. Boddien, D. Mellmann, F. Gaertner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Matthias, Efficient Dehydrogenation of Formic Acid Using an Iron Catalyst, Science, 2011, 333, 1733 CrossRef CAS PubMed.
- (a) D. Niks and R. Hille, Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion, Protein Sci., 2019, 28, 111 CrossRef CAS PubMed; (b) J. Y. Yang, T. A. Kerr, X. S. Wang and J. M. Barlow, Reducing CO2 to HCO2− at Mild Potentials: Lessons from Formate Dehydrogenase, J. Am. Chem. Soc., 2020, 142, 19438 CrossRef CAS PubMed; (c) A. Bassegoda, C. Madden, D. W. Wakerley, E. Reisner and J. Hirst, Reversible Interconversion of CO2 and Formate by a Molybdenum-Containing Formate Dehydrogenase, J. Am. Chem. Soc., 2014, 136, 15473 CrossRef CAS PubMed; (d) A. Bassegoda, C. Madden, D. W. Wakerley, E. Reisner and J. Hirst, Correction to “Reversible Interconversion of CO2 and Formate by a Molybdenum-Containing Formate Dehydrogenase”, J. Am. Chem. Soc., 2015, 137, 4592 CrossRef CAS PubMed.
- S. Tanga and H. Zhao, Glymes as versatile solvents for chemical reactions and processes: from the laboratory to industry, RSC Adv., 2014, 4, 11251 RSC.
- L. Zhang, G. Raffa, D. H. Nguyen, Y. Swesi, L. Corbel-Demailly, F. Capet, X. Trivelli, S. Desset, S. Paul, J.-F. Paul, P. Fongarland, F. Dumeignil and R. M. Gauvin, Acceptorless dehydrogenative coupling of alcohols catalysed by ruthenium PNP complexes: Influence of catalyst structure and of hydrogen mass transfer, J. Catal., 2016, 340, 331 CrossRef CAS.
- Z. E. Clarke, P. T. Maragh, T. P. Dasgupta, D. G. Gusev, A. J. Lough and K. Abdur-Rashid, A Family of Active Iridium Catalysts for Transfer Hydrogenation of Ketones, Organometallics, 2006, 25, 4113 CrossRef CAS.
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, Manganese Pincer Complexes for the Base-Free, Acceptorless Dehydrogenative Coupling of Alcohols to Esters: Development, Scope, and Understanding, ACS Catal., 2017, 7, 2022 CrossRef CAS.
- (a) S. Elangovan, B. Wendt, C. Topf, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao, W. Baumann, K. Junge and M. Beller, Improved Second Generation Iron Pincer Complexes for Effective Ester Hydrogenation, Adv. Synth. Catal., 2016, 358, 820 CrossRef CAS; (b) S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, Iron-Based Catalysts for the Hydrogenation of Esters to Alcohols, J. Am. Chem. Soc., 2014, 136, 7869 CrossRef CAS PubMed; (c) E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst, J. Am. Chem. Soc., 2014, 136, 10234 CrossRef CAS PubMed; (d) N. E. Smith, W. H. Bernskoetter and N. Hazari, The Role of Proton Shuttles in the Reversible Activation of Hydrogen via Metal−Ligand Cooperation, J. Am. Chem. Soc., 2019, 141, 17350 CrossRef CAS PubMed; (e) J. B. Curley, W. H. Bernskoetter and N. Hazari, Additive-Free Formic Acid Dehydrogenation Using a PincerSupported Iron Catalyst, ChemCatChem, 2020, 12, 1934 CrossRef CAS.
- W. Zhao and J. Sun, Triflimide (HNTf2) in Organic Synthesis, Chem. Rev., 2018, 118, 10349 CrossRef CAS PubMed.
- W. H Bernskoetter and N. Hazari, Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts, Acc. Chem. Res., 2017, 50, 1049 CrossRef PubMed.
- (a) J. A. Widegren and R. G. Finke, A review of the problem of distinguishing true homogeneous catalysis from soluble or other metal-particle heterogeneous catalysis under reducing conditions, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS; (b) R. H. Crabtree, Resolving Heterogeneity Problems and Impurity Artifacts in Operationally Homogeneous Transition Metal Catalysts, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- This test was made, although no reference in the literature could be found concerning the possibility of Mo reacting with Hg to form an amalgama. Yet at least one other example has been reported in which Hg has been used to assess the homogeneous nature of a molybdenum catalyst: J. A. Buss, G. A. Edouard, C. Cheng, J. Shi and T. Agapie, Molybdenum Catalyzed Ammonia Borane Dehydrogenation: Oxidation State Specific Mechanisms, J. Am. Chem. Soc., 2014, 136, 11272 CrossRef CAS PubMed.
- (a) D. J. Tindall, M. Menche, M. Schelwies, R. A. Paciello, A. Schäfer, P. Comba, F. Rominger, A. S. K. Hashmi and T. Schaub, Ru(0) or Ru(II): A Study on Stabilizing the “Activated” Form of Ru-PNP Complexes with Additional Phosphine Ligands in Alcohol Dehydrogenation and Ester Hydrogenation, Inorg. Chem., 2020, 59, 5099 CrossRef CAS PubMed; (b) R. Konrath, K. Sekine, I. Jevtovikj, R. A. Paciello, A. S. K. Hashmi and T. Schaub, Performance enhancing additives for reusable ruthenium-triphos catalysts in the reduction of CO2 to dimethoxymethane, Green Chem., 2020, 22, 6464 RSC.
- The choice to add 500 μmoles of water to the standard reaction volume, 20 mL toluene, is based on the maximum solubility of water in toluene 0.0270 mol/L at 25°C as reported in J. Irchnerova and G. B. Cave, Can. J. Chem., 1976, 54, 3909 CrossRef.
- (a) Y. Zhang, P. G. Williard and W. H. Bernskoetter, Synthesis and Characterization of Pincer-Molybdenum Precatalysts for CO2 Hydrogenation, Organometallics, 2016, 35, 860 CrossRef CAS; (b) S. Chakraborty, O. Blacque and H. Berke, Ligand assisted carbon dioxide activation and hydrogenation using molybdenum and tungsten amides, Dalton Trans., 2015, 44, 6560 RSC; (c) R. Pal, T. L. Groy and R. J. Trovitch, Conversion of Carbon Dioxide to Methanol Using a C−H Activated Bis(imino)pyridine Molybdenum Hydroboration Catalyst, Inorg. Chem., 2015, 54, 7506 CrossRef CAS PubMed; (d) M. Minato, D.-Y. Zhou, K.-I. Sumiura, Y. Oshima, S. Mine and T. Ito, Reactivity Patterns of O2, CO2, Carboxylic Acids, and Triflic Acid with Molybdenum Silyl Hydrido Complexes Bearing Polydentate Phosphinoalkyl−Silyl Ligands: Pronounced Effects of Silyl Ligands on Reactions, Organometallics, 2012, 31, 4941 CrossRef CAS.
- (a) J. Halpern and A. L. W. Kemp, The Decarbonylation of Formic Acid by Ruthenium (II) Chloride, J. Am. Chem. Soc., 1966, 20, 5147 CrossRef; (b) W. C. Kaska, S. Nemeh, A. Shlrazi and S. Potuznik, Reduction of Carbon Dioxide by {2,6-Bis[(di-tert-butylphosphino)methyl]phenyldihydrido rhodium(III), Organometallics, 1988, 7, 13 CrossRef CAS; (c) S. Ogo, H. Nishida, H. Hayashi, Y. Murata and S. Fukuzumi, Aqueous Transformation of a Metal Diformate to a Metal Dihydride Carbonyl Complex Accompanied by H2 Evolution from the Formato Ligands, Organometallics, 2005, 24, 4816 CrossRef CAS; (d) C. W. Machan, M. D. Sampson and C. P. Kubiak, A Molecular Ruthenium Electrocatalyst for the Reduction of Carbon Dioxide to CO and Formate, J. Am. Chem. Soc., 2015, 137, 8564 CrossRef CAS PubMed; (e) M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, Reductive Cleavage of CO2 by Metal−Ligand-Cooperation Mediated by an Iridium Pincer Complex, J. Am. Chem. Soc., 2016, 138, 6445 CrossRef CAS PubMed; (f) A. Anaby, M. Feller, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, Bottom-Up Construction of a CO2-Based Cycle for the Photocarbonylation of Benzene, Promoted by a Rhodium(I) Pincer Complex, J. Am. Chem. Soc., 2016, 138, 9941 CrossRef CAS PubMed.
- (a) Ö. Öztopcu, C. Holzhacker, M. Puchberger, M. Weil, K. Mereiter, L. F. Veiros and K. Kirchner, Synthesis and Characterization of Hydrido Carbonyl Molybdenum and Tungsten PNP Pincer Complexes, Organometallics, 2013, 32, 3042 CrossRef PubMed; (b) R. Castro-Rodrigo, S. Chakraborty, L. Munjanja, W. W. Brennessel and W. D. Jones, Synthesis, Characterization, and Reactivities of Molybdenum and Tungsten PONOP Pincer Complexes, Organometallics, 2016, 35, 3124 CrossRef CAS.
- Selected examples of Mo(II)-dihydride complexes: (a) D. Lyons, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, Synthesis, X-Ray Crystal Structure, and Reactions of Dihydridopentakis(trimethyl phosphine)molybdenum(II) : Crystal Structure of the Carbon Dioxide Insertion Product, (Formato-O,O’) hydridotetrakis-(trimethyl phosphine) molybdenum (II), J. Chem. Soc., Dalton Trans., 1984,(4), 695 RSC; (b) G. J. Kubas, R. R. Ryan and C. J. Unkefer, Molecular Hydrogen Complexes. 5. Electronic Control of η2-H2 versus Dihydride Coordination. Dihydride Structure of MoH2(CO)(R2PC2H4PR2)2 for R = Et, t-Bu versus η2-H2 for R = Ph, J. Am. Chem. Soc., 1987, 109, 8113 CrossRef CAS; (c) T. Adrian George and R. C. Tisdale, Synthesis of Mono(dinitrogen) Complexes of Molybdenum, Inorg. Chem., 1988, 27, 2909 CrossRef. Formation of Ammonia and Hydrazine; (d) T.-Y. Cheng, D. J. Szalda, J. Zhang and R. Morris Bullock, Synthesis and Structure of CpMo(CO)(dppe)H and Its Oxidation by Ph3C+, Inorg. Chem., 2006, 45, 4712 CrossRef CAS PubMed; (e) Q. X. Dai, H. Seino and Y. Mizobe, Preparation and Reactions of Molybdenum and Tungsten Hydride Complexes Containing the Tetraphosphane Ligand meso-o-C6H4(PPhCH2CH2PPh2)2, Eur. J. Inorg. Chem., 2011, 141–149 CrossRef CAS; (f) S. Zhang and R. Morris Bullock, Molybdenum Hydride and Dihydride Complexes Bearing Diphosphine Ligands with a Pendant Amine: Formation of Complexes with Bound Amines, Inorg. Chem., 2015, 54, 6397 CrossRef CAS PubMed; (g) S. L. Apps, A. J. P. White, P. W. Miller and N. J. Long, Synthesis and reactivity of an N-triphos Mo(0) dinitrogen complex, Dalton Trans., 2018, 47, 11386 RSC.
- M. Kakeya, K. Hoshino, T. Asaeda, T. Nakada, K. Osakada, D. Casanova, P. Alemany, J. M. Bofill and S. Alvarez, Shape and Symmetry of Heptacoordinate Transition-Metal Complexes: Structural Trends, Chem.–Eur. J., 2003, 9, 1281 CrossRef PubMed.
- C. J. Jameson, Isotope Effects on Chemical Shifts and Coupling Constants, eMagRes, 2007, 1–17, DOI:10.1002/9780470034590.emrstm0251.
- J. Y.-C. Chen, A. A. Martı, N. J. Turro, K. Komatsu, Y. Murata and R. G. Lawler, Comparative NMR Properties of H2 and HD in Toluene-d8 and in H2/HD@C60, J. Phys. Chem. B, 2010, 114, 14689 CrossRef CAS PubMed.
- Selected examples of Mo(IV)-polyhydride species: see40a and (a) J. C. Fettinger, B. A. Pleune and R. Poli, First Structure of a Cyclopentadienyl Trihydride d2 System: A Pseudotrigonal Prism Rather Than the Expected Pseudooctahedron and Its Mechanism of Hydrogen Scrambling, J. Am. Chem. Soc., 1996, 118, 4906 CrossRef CAS; (b) P. Meakin, L. J. Guggenberger, W. G. Peet, E. L. Muetterties and J. P. Jesson, Stereochemically Nonrigid Eight-Coordinate Molybdenum and Tungsten Tetrahydrides, J. Am. Chem. Soc., 1973, 95, 1467 CrossRef CAS; (c) F. Abugideiri, J. C. Fettinger, B. Pleune, R. Poli, C. A. Bayse and M. B. Hall, Synthesis, Structure, and Hydride-Deuteride Exchange Studies of CpMoH3(PMe2Ph)2 and Theoretical Studies of the CpMoH3(PMe3)2 Model System, Organometallics, 1997, 16, 1179 CrossRef CAS.
- M. Besora, A. Lledós and F. Maseras, Protonation of transition-metal hydrides: a not so simple process, Chem. Soc. Rev., 2009, 38, 957 RSC.
- (a) S. Rondinini, P. Longhi, P. R. Mussini and T. Mussini, Autoprotolysis Constants in Nonaqueous Solvents and Aqueous Organic Solvent Mixtures, Pure Appl. Chem., 1987, 59, 1693 CAS; (b) E. S. Wiedner and J. C. Linehan, Making a Splash in Homogeneous CO2 Hydrogenation: Elucidating the Impact of Solvent on Catalytic Mechanisms, Chem.–Eur. J., 2018, 24, 16964 CrossRef CAS PubMed.
- (a) R. Morris Bullock, Catalytic Ionic Hydrogenations, Chem.–Eur. J., 2004, 10, 2366 CrossRef PubMed; (b) T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, Isolation and Crystal Structure of a Water-Soluble Iridium Hydride: A Robust and Highly Active Catalyst for Acid-Catalyzed Transfer Hydrogenations of Carbonyl Compounds in Acidic Media, J. Am. Chem. Soc., 2003, 125, 4149 CrossRef CAS PubMed.
- O. Blum and D. Milstein, Hydride elimination from an iridium(III) alkoxide complex: a case in which a vacant cis coordination site is not required, J. Organomet. Chem., 2000, 593−594, 479 CrossRef CAS.
- (a) T.-Y. Luh, Trimethylamine N-oxide - A Versatile Reagent for Organometallic Chemistry, Coord. Chem. Rev., 1984, 60, 255 CrossRef CAS; (b) Y.-C. Gao, Q.-Z. Shi, D. L. Kershner and F. Basolo, Oxygen Atom Transfer Reaction to Metal Carbonyls. Kinetics and Mechanism of CO-Substitution Reactions of Mo(CO)5L in the Presence of (CH3)3NO, Inorg. Chem., 1988, 27, 188 CrossRef CAS.
- (a) E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. Morris Bullock, A. J. M. Miller and A. M. Appel, Thermodynamic Hydricity of Transition Metal Hydrides, Chem. Rev., 2016, 116, 8655 CrossRef CAS PubMed; (b) T.-Y. Cheng, B. S. Brunschwig and R. M. Bullock, Hydride Transfer Reactions of Transition Metal Hydrides: Kinetic Hydricity of Metal Carbonyl Hydrides, J. Am. Chem. Soc., 1998, 120, 13121 CrossRef CAS.
- M. Yamakawa, H. Ito and R. Noyori, The Metal-Ligand Bifunctional Catalysis: A Theoretical Study on the Ruthenium(II)-Catalyzed Hydrogen Transfer between Alcohols and Carbonyl Compounds, J. Am. Chem. Soc., 2000, 122, 1466 CrossRef CAS.
- (a) P. A. Dub and J. C. Gordon, Metal−Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms, ACS Catal., 2017, 7, 6635 CrossRef CAS; (b) P. A. Dub and J. C. Gordon, The role of the metal- bound N–H functionality in Noyori-type molecular catalysts, Nat. Rev. Chem., 2018, 2, 396 CrossRef CAS.
- P. A. Dub, B. L. Scott and J. C. Gordon, Why Does Alkylation of the N-H Functionality within M/NH Bifunctional Noyori-Type Catalysts Lead to Turnover, J. Am. Chem. Soc., 2017, 139, 1245 CrossRef CAS PubMed.
- (a) A. Friedrich, M. Drees, J. Schmedt auf der Günne and S. Schneider, Highly Stereoselective Proton/Hydride Exchange: Assistance of Hydrogen Bonding for the Heterolytic Splitting of H2, J. Am. Chem. Soc., 2009, 131, 17552 CrossRef CAS PubMed; (b) N. E. Smith, W. H. Bernskoetter and N. Hazari, The Role of Proton Shuttles in the Reversible Activation of Hydrogen via Metal−Ligand Cooperation, J. Am. Chem. Soc., 2019, 141, 17350 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04181a |
| This journal is © The Royal Society of Chemistry 2021 |