
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect air capture of CO2via crystal engineering
Radu
Custelcean
 *
*
Chemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: custelceanr@ornl.gov
First published on 13th September 2021
Abstract
This article presents a perspective view of the topic of direct air capture (DAC) of carbon dioxide and its role in mitigating climate change, focusing on a promising approach to DAC involving crystal engineering of metal–organic and hydrogen-bonded frameworks. The structures of these crystalline materials can be easily elucidated using X-ray and neutron diffraction methods, thereby allowing for systematic structure–property relationships studies, and precise tuning of their DAC performance.
 Radu Custelcean | Radu Custelcean is a senior research scientist at Oak Ridge National Laboratory, where for the past 18 years has been involved in basic and applied research in molecular recognition, crystal engineering, separation science and technology, and carbon capture. Following a serendipitous discovery of atmospheric CO2 capture by crystallization of a simple guanidine compound in 2016, direct air capture (DAC) became his favorite research topic. His ultimate goal is to develop energy-efficient, cost-effective DAC technologies that will help address climate change. |
Introduction
Our continuing reliance on fossil fuels as a major source of energy created the impetus for effective emissions reductions through large-scale deployment of carbon capture and storage (CCS) technologies for mitigating climate change. While CCS technologies have traditionally been implemented at point sources of CO2 emissions, such as coal- or gas-fired power plants, recent integrated assessment models have increasingly emphasized the need for negative emissions technologies (NETs), that is, technologies that remove CO2 out of the atmosphere, to limit global warming below 2 °C by 2100.1,2Direct air capture (DAC) refers to NETs that capture CO2 from ambient air using engineered chemical processes.3–7 DAC technologies have the potential to remove past emissions and restore the atmospheric CO2 concentration to an optimal level below 350 ppm, and lower the global temperature back within the optimum range of the pre-industrial Holocene period.8 DAC can also compensate for the CO2 emissions from the economy sectors that are difficult to decarbonize, such as steel and cement industries, long-distance transportation and shipping, and on-demand reliable electricity, which together account for about 27% of the global CO2 emissions.9 Furthermore, DAC offers the potential for high capacity (Gt-CO2 scale), the flexibility of placement near the storage sites, and the possibility to generate high-purity CO2 streams that can be converted into value-added products.1,2
To achieve these ambitious goals, DAC technologies must overcome a number of technical and economic challenges. First, the very low atmospheric CO2 concentration (currently about 415 ppm) dictates that DAC sorbents bind CO2 strongly and selectively against other components in the air. Indeed, simple thermodynamic considerations define the lower end of the CO2 binding constant (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) to 3.7, corresponding to a free energy of binding of at least −21 kJ mol−1.10 Furthermore, considering the entropy of binding is typically negative, the enthalpy of CO2 binding needs to be significantly exothermic (at least −50 kJ mol−1). With regard to selectivity, DAC sorbents need to be selective for CO2 against water, which is 10–100 times more abundant than CO2 in the atmosphere and tends to bind relatively strongly to most sorbents. Second, considering the enormous magnitude of DAC technologies deployment required to make an impact on the climate – up to 10 Gt CO2 need to be captured annually by 20501,2 DAC sorbents will have to be manufactured on a massive scale (Mt per year), need to be chemically robust to survive tens of thousands of loading/unloading cycles, and cost less than $10 per kg.11 As a result of these stringent requirements, there are currently few viable CO2 sorbents for DAC, mostly based on aqueous alkaline bases (e.g., NaOH, KOH),12 solid-supported amines,13,14 or moisture-swing anion-exchange resins.15
K) to 3.7, corresponding to a free energy of binding of at least −21 kJ mol−1.10 Furthermore, considering the entropy of binding is typically negative, the enthalpy of CO2 binding needs to be significantly exothermic (at least −50 kJ mol−1). With regard to selectivity, DAC sorbents need to be selective for CO2 against water, which is 10–100 times more abundant than CO2 in the atmosphere and tends to bind relatively strongly to most sorbents. Second, considering the enormous magnitude of DAC technologies deployment required to make an impact on the climate – up to 10 Gt CO2 need to be captured annually by 20501,2 DAC sorbents will have to be manufactured on a massive scale (Mt per year), need to be chemically robust to survive tens of thousands of loading/unloading cycles, and cost less than $10 per kg.11 As a result of these stringent requirements, there are currently few viable CO2 sorbents for DAC, mostly based on aqueous alkaline bases (e.g., NaOH, KOH),12 solid-supported amines,13,14 or moisture-swing anion-exchange resins.15
With their ordered, well-defined structures, crystalline organic materials (e.g., metal–organic frameworks, hydrogen-bonded frameworks) offer a unique and promising approach to DAC. Their crystalline structures can be easily elucidated using X-ray and neutron diffraction techniques, leading to a precise understanding of the relationship between their structures and functions. Moreover, their DAC performance could in principle be tuned and systematically improved by employing crystal engineering principles. Indeed, crystal engineering, whose primary objective has been the design and synthesis of functional organic and metal–organic crystals, has seen significant progress over the last decades, evolving from analysis of crystal structures and intermolecular interactions, to design of crystals with predetermined topologies, to property design and optimization. Thus, crystal engineering, which for many years has been limited to crystal structure engineering, is now transforming into crystal property engineering.16 The crystal property of interest here, which will be discussed in detail in this perspective article, is atmospheric CO2 capture, towards development of effective and energy-efficient DAC technologies.
DAC with metal–organic frameworks
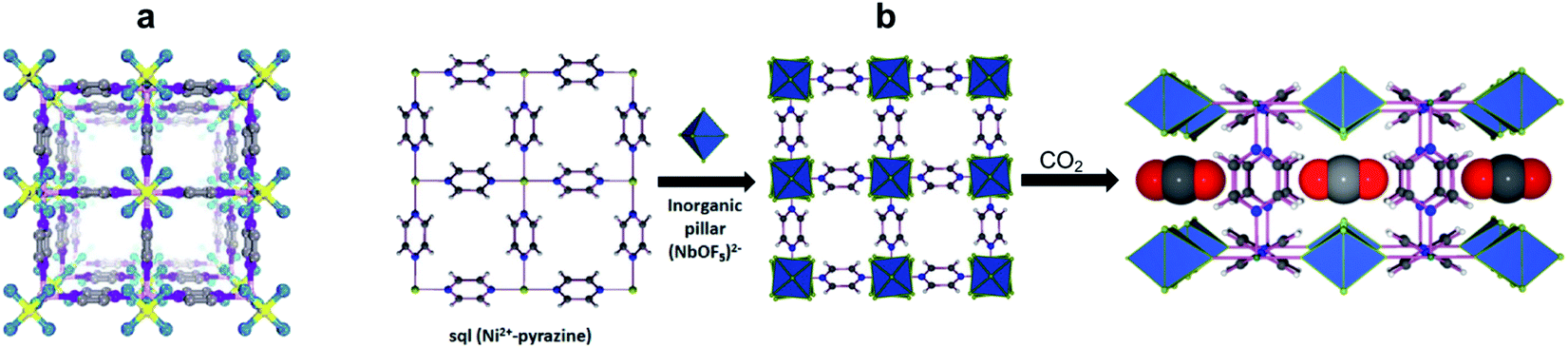 | ||
| Fig. 1 MOFs/HUMs for DAC by physisorption. (a) Crystal structure of SIFSIX-3-Ni. (b) Crystal structure of NbOFFIVE-1-Ni and binding of CO2 in its pores. Adapted with permission from ref. 20. Copyright (2016) American Chemical Society. | ||
In an initial study, Zaworotko et al. performed screening of five porous sorbents for their ability to capture CO2 under DAC conditions: SIFSIX-3-Ni, HKUST-1, Mg-MOF-74/Mg-dobdc, zeolite-13X, and TEPA-SBA-15.17 These sorbents represent four classes of porous materials: HUMs (SIFSIX-3-Ni), MOFs (HKUST-1, Mg-MOF-74/Mg-dobdc), zeolites, and amine-modified mesoporous silicate (TEPA-SBA-15) as a representative chemisorbent for comparison with the physisorbents. Temperature-programmed desorption (TPD) measurements under DAC conditions found that, while the SIFSIX-3-Ni performed best among the MOFs, the CO2/H2O selectivity was poor for all physisorbents. In contrast, the chemisorbent TEPA-SBA-15 showed far superior DAC performance and CO2/H2O selectivity. Nevertheless, the promising performance of the SIFSIX-3-Ni HUM compared to the other MOF sorbents indicated that tuning pore size and functionality could lead to improved DAC by this class of materials.
Fine tuning the pores of the HUMs can be easily done by substituting the SiF62− anionic pillars with TiF62−, which leads to the isostructural TIFSIX-3-Ni sorbent with tighter binding sites for CO2, and as a result about three times higher uptake of CO2 under DAC conditions.19 The measured enthalpy of CO2 adsorption for TIFSIX-3-Ni, of −50 kJ mol−1, is slightly higher than for the SIFSIX-3-Ni analogue (Qst − 45 kJ mol−1), and significantly higher than the corresponding values for other MOF materials (e.g., Mg-MOF-74, −42 kJ mol−1; UiO-66-NH2, −38 kJ mol−1; HKUST-1 −23 kJ mol−1). Accordingly, the CO2 capacity (0.41 mmol g−1) and CO2/H2O selectivity (12.8) measured under DAC conditions for TIFSIX-3-Ni, was significantly higher compared to other physisorbents. A related crystalline material, with anionic pillars consisting of NbOF5− (NbOFFIVE-1-Ni) was described by Eddaoudi et al. (Fig. 1b).20 This sorbent has slightly higher gravimetric uptake (1.3 mmol g−1) and enthalpy of CO2 adsorption (−54 kJ mol−1) than TIFSIX-3-Ni. Its crystal structure obtained in the presence of carbon dioxide revealed the CO2 is tightly bound within the square-shaped channels, surrounded by four electronegative F atoms from the NbOF5− anions interacting with the electropositive C atom of CO2 (Fig. 1b). These strong and complementary interactions provide an optimal CO2 binding site, consistent with the relatively high enthalpy of adsorption compared with other physisorbents. Unfortunately, for both TIFSIX-3-Ni and NbOFFIVE-1-Ni, the binding sites also strongly bind water, which competes with the CO2 adsorption.21 Nevertheless, the regeneration of these materials can be done at relatively low temperatures (90–100 °C), under purging with an inert gas or vacuum.
The superior DAC performance of TIFSIX-3-Ni and NbOFFIVE-1-Ni compared to SIFSIX-3-Ni highlights how subtle changes in pore size and chemistry can have a dramatic effect on the CO2 sorption performance of a series of isostructural materials. However, competition with water vapor remains a significant challenge, especially for DAC applications. Even though the incorporation of strong electrostatics from the inorganic pillars combined with ultramicropores offers improved CO2 capture performance from moist air, it is not enough to compete with chemisorbents.21 Furthermore, like zeolites that have high affinity to water, water adsorption in the pores of MOFs or HUMs competes with the CO2 adsorption, leading to longer regeneration times, higher regeneration energies, and reduced working capacity for the DAC process.
Functionalization of magnesium dioxybenzenedicarboxylate (Mg/dobdc, or Mg-MOF-74) with ethylenediamine (ed) led to an early example of MOF capable of multiple cycles of CO2 adsorption/release via chemisorption under practical DAC conditions using simulated dry air (400 ppm CO2).22 Ethylenediamine was grafted to the pores of this MOF through coordination of the open metal sites with one of the amine groups, which left the second NH2 group pendent inside the pore and available to react with CO2 (Fig. 2a). The measured cyclic capacity of ed–Mg/dobdc was about 1.50 mmol g−1, sustained over 4 consecutive cycles. For comparison, under the same conditions, the measured cyclic capacity of Mg/dobdc was 1.35 mmol g−1 in the first cycle, and dropped to 1.06 mmol g−1 after 4 cycles.
 | ||
| Fig. 2 Amine-functionalized MOFs for DAC by chemisorption. (a) Crystal structure of ed–Mg/dobdc. Reproduced with permission from ref. 22. Copyright (2012) American Chemical Society. (b) Crystal structure of mmen–Mg2(dobpdc) showing the CO2 binding by pendent amine groups. Reproduced with permission from ref. 23. Copyright (2012) American Chemical Society. (c) Crystal structure of ed–Mg2(dobpdc). Reproduced with permission from ref. 25. Copyright (2014) Royal Society of Chemistry. (d) Crystal structure of H2NNH2–Mg/dobdc. Reproduced with permission from ref. 26. Copyright (2016) Royal Society of Chemistry. | ||
An expanded analogue of Mg/dobdc, magnesium 4,4′-dioxido-3,3′-biphenyldicarboxylate [Mg2(dobpdc)], was functionalized with N,N′-dimethylethylenediamine (mmen) to afford mmen–Mg2(dobpdc) (Fig. 2b).23 After pore activation, its CO2 adsorption capacity at 25 °C and 0.39 mbar, conditions simulating ambient air, was 2.0 mmol g−1, 15 times higher than measured for Mg2(dobpdc) under the same conditions. The isosteric heat of adsorption for mmen–Mg2(dobpdc) is −71 kJ mol−1, consistent with a chemisorption mechanism involving amine reaction with CO2 to form carbamate, which was confirmed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The CO2 cyclic capacity measured using dry air was 1 mmol g−1, which was maintained after 10 consecutive adsorption/desorption cycles. Regeneration of this MOF sorbent involved heating at 150 °C under nitrogen for 30 min.
This MOF adsorbent exhibits step-shaped isotherms, allowing for a relatively large working capacity with only a small increase in temperature. In direct contrast, classical adsorbents, including most amine-functionalized solids, exhibit Langmuir-type isotherms requiring high desorption temperatures to achieve large working capacities. Spectroscopic and diffraction measurements indicated a mechanism of CO2 uptake involving cooperative insertion of CO2 into the metal–amine bonds, resulting in a carbamate group coordinated to the metal center, and further stabilized by hydrogen bonding to an ammonium group formed by proton transfer from the carbamic acid to the free pendent amine group from an adjacent mmen.24 This cooperative mechanism leading to the formation of one-dimensional ammonium carbamate chains explains the sudden uptake of large amounts of CO2 and the unusual step-shaped isotherms. The ammonium-carbamate hydrogen bonding may also facilitate the proton transfer during the regeneration of these sorbents, which only require about 2.2–2.3 MJ kg−1 CO2, as measured by differential scanning calorimetry (DSC).
The ethylenediamine-functionalized analogue of Mg2(dobpdc), ed–Mg2(dobpdc) (Fig. 2c), showed a significant CO2 uptake of 2.83 mmol g−1 at 0.39 mbar and 25 °C.25 A chemisorption mechanism consisting of carbamic acid formation by reaction of the free amine groups with CO2 was proposed based on in situ IR measurements, which is consistent with the estimated stoichiometry of 0.83 moles CO2 per moles of ED, and the relatively low isosteric heat of adsorption of about −50 kJ mol−1. CO2 desorption required heating at 150 °C for 2 h under air purge.
Further increase in the CO2 capacity was possible by functionalizing Mg/dobdc with the shortest possible diamine, hydrazine (H2NNH2) (Fig. 2d).26 The ultrahigh concentration of amine groups in this sorbent, coupled with their ability to form carbamic acid by reaction with CO2, led to the unprecedented CO2 capacity of 3.89 mmol g−1 at the atmospheric CO2 concentration of 0.4 mbar and 25 °C. The formation of carbamic acid was confirmed by in situ IR and 13C NMR spectroscopies, and the measured isosteric heat of adsorption of −90 kJ mol−1 confirmed the chemisorption of CO2. The regeneration energy determined by DSC is 3.02 MJ kg−1 CO2. Regeneration of the sorbent was done by heating at 130 °C under nitrogen flow. The DAC performance of this sorbent was minimally impacted by the presence of humidity.
Despite their favorable structural and thermodynamic features, amine-functionalized MOFs with stepped isotherms like ed–Mg2(dobpdc) and mmen–Mg2(dobpdc) were found to have some kinetic limitations under DAC conditions. Specifically, the kinetics of CO2 adsorption at concentrations of 400 ppm were measured from breakthrough experiments and characterized through an Avrami model, with the mass transfer rate limited by the cooperative insertion of CO2. Notably, the shape of the isotherm and the kinetic factors severely diminish the CO2 capture fraction at low concentrations, reducing the viability of these MOF sorbents for practical DAC applications.27 Nevertheless, the MOFs' structural tunability allows for optimization of CO2 adsorption under DAC conditions. For example, small modifications of the appended diamine in Mg2(dobpdc) led to shifts in the threshold pressure for CO2 adsorption by over 4 orders of magnitude.28 Furthermore, it was demonstrated that the adsorption kinetics can also be improved through selection of appropriate diamines that lead to faster CO2 adsorption under DAC conditions.29 Replacing the appended diamines with tetraamines led to more stable MOFs that can cooperatively capture CO2 from humid air, and can be regenerated with steam.30
An alternative approach to MOF functionalization with amines is to decorate the pores with Zn–OH groups that mimic the active site of carbonic anhydrase. This approach has been recently demonstrated with a Zn–benzotriazolate MOF postsynthetically modified to generate nucleophilic Zn–OH binding sites for CO2 (Fig. 3).31 After activation by heating at 100 °C under high vacuum, the resulting MOF sorbent exhibited remarkably steep CO2 uptake at low pressures, with a CO2 capacity of 2.20 mmol g−1 at 0.4 mbar, and an isosteric heat of adsorption of −71 kJ mol−1. The enthalpy of binding is consistent with a chemisorption mechanism, which was confirmed by DRIFTS experiments that revealed the conversion of Zn–OH to Zn–O2COH groups. DFT calculations revealed the formation of hydrogen-bonded bicarbonate dimers that crosslink the Zn clusters in the framework. Notably, the CO2 cyclic capacity measured over multiple consecutive cycles with simulated air mixtures was somewhat lower (i.e., 5.8 wt%) than the corresponding value measured from the single-component isotherm. Furthermore, this material performed poorly in breakthrough experiments conducted in the presence of humidity. Postsynthetic exchange of Zn–OAc groups in the framework with Ni–OH resulted in an heterobimetallic analogue with even steeper CO2 adsorption isotherm, higher CO2 capacity of 2.7 mmol g−1 at 0.4 mbar, and more exothermic heat of adsorption of −84 kJ mol−1.32 DRIFTS experiments and DFT calculations revealed that, unlike in the Zn–OH system, the Ni–O2COH species do not engage in intercluster hydrogen bonding, which could explain its intrinsically higher affinity for CO2. Breakthrough cycling experiments with simulated air yielded a slightly lower CO2 cyclic capacity of 1.93 mmol g−1, though its structure appeared to be stable to repeated cycling with humid gas streams. However, the Ni–OH MOF adsorbed more water (2.95 mmol H2O per g) compared to the Zn–OH analogue (1.07 mmol H2O per g), resulting in an overall decreased DAC performance under humid conditions.
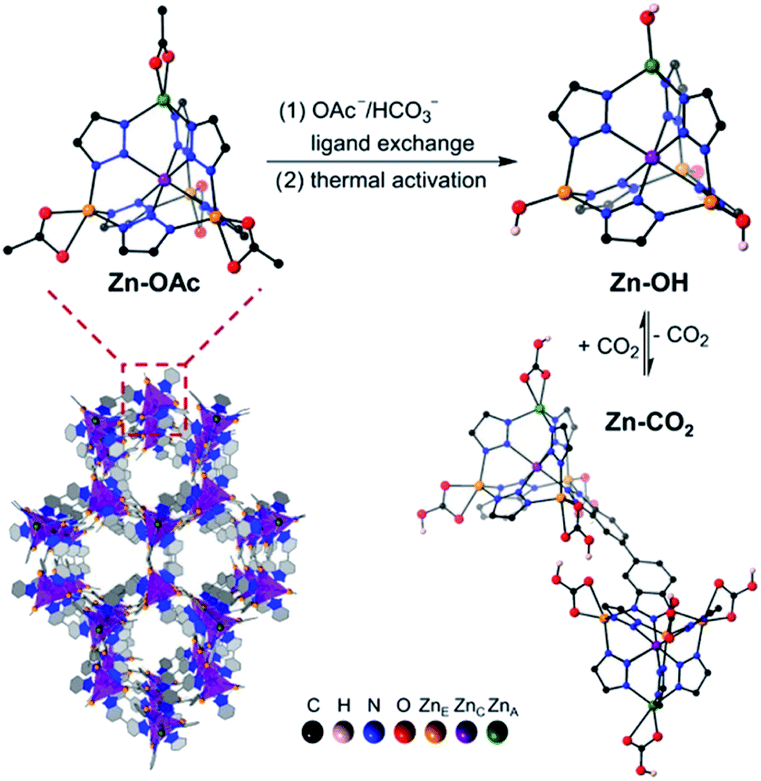 | ||
| Fig. 3 Crystal structure of a Zn–benzotriazolate MOF functionalized with Zn–OH binding sites and reaction with CO2. Reproduced with permission from ref. 32. Copyright (2020) American Chemical Society. | ||
DAC by crystallization of hydrogen-bonded frameworks
The allure of MOFs comes from their aesthetic porous structures that can be precisely designed and engineered for optimal binding of gas molecules like CO2. Porosity, though, is a high-maintenance functionality. Pore activation is often an elaborate and tedious process. Maintaining an open structure over multiple adsorption/desorption cycles takes effort and energy, as nature abhors vacuum and will do anything to fill those pores with whatever is available in the air. Particularly, water condensation in the pores is a problem for DAC applications, as the energy required to desorb water may be substantial, sometimes exceeding the energy of CO2 desorption.33Hydrogen-bonded frameworks (HBFs) offer a viable alternative to MOFs. Like MOFs, the structures of HBFs can be precisely designed and tuned using the principles of crystal engineering. While permanently porous HBFs have recently become a reality,34 HBF porosity is not actually a prerequisite for DAC. As an alternative approach, the CO2 capture can be done in aqueous solutions, as long as the absorption process leads to the formation of stable and insoluble HBF crystals that incorporate CO2 in their structures (typically as carbamic acid, carbamate, or carbonate anions). In such cases, the HBF–CO2 crystals can be simply removed from solution by filtration, followed by CO2 desorption from the isolated solids, and recycling of the water-soluble HBFs. Ideally, this strategy combines the benefits of aqueous solvents, such as fast kinetics of CO2 absorption, easy scalability, and low cost, with the advantages of crystalline sorbents, such as structural tunability and lower regeneration energies.
A proof of concept for DAC by crystallization of HBFs came from a simple amine system, m-xylylenediamine (MXDA).35 An aqueous solution of MXDA left open to air absorbed atmospheric CO2 and formed insoluble crystals of MXDA·CO2 (Fig. 4a). X-ray structural analysis revealed a carbamic acid product linked into extensive chains via NH⋯O hydrogen bonds. Additional COOH⋯NH2 hydrogen bonds crosslink the chains into two-dimensional networks. Finally, the Ph groups pack into hydrophobic layers that sandwich the hydrophilic hydrogen-bonded layers in a reverse lipid bilayer-like structure (Fig. 4b). It was proposed that this crystal packing prevents the water from penetrating into the hydrophilic layers, thereby resulting in low aqueous solubility of MXDA·CO2. Heating the crystals at 103–120 °C releases the CO2 and regenerates MXDA, which can be recycled. The anhydrous nature of this HBF presents a great advantage for a DAC process, in that it avoids the energy-intensive water evaporation step typically associated with aqueous solvents or hygroscopic sorbents.
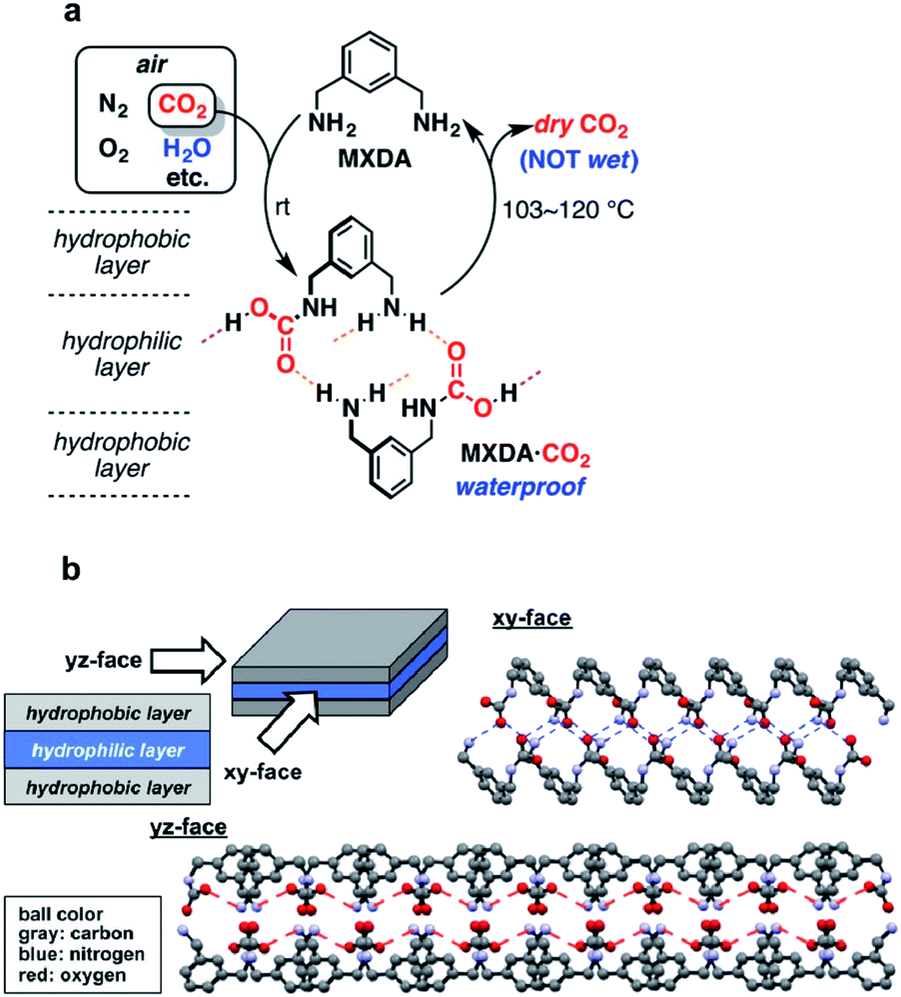 | ||
| Fig. 4 DAC by crystallization of MXDA. (a) DAC cycle involving atmospheric CO2 absorption by an aqueous solution of MXDA. (b) Crystal structure of the carbamic acid formed from the reaction of MXDA with CO2. Reproduced with permission from ref. 35. Copyright (2017) American Chemical Society. | ||
The success of the above DAC strategy hinges on the premise that the resulting HBF·CO2 complex has sufficiently low aqueous solubility to drive the CO2 absorption and crystallization equilibria in spite of the very low concentration of CO2 in the air. However, most amines form products with CO2 that are relatively soluble in water; MXDA is more the exception than the rule. Thus, there remains the need to identify design principles for engineering amines or related organic bases that form insoluble carbamic acids or carbamate/(bi)carbonate crystals upon reaction with atmospheric CO2, if this DAC approach is to become more general.
Research at Oak Ridge National Laboratory led recently to development of a new, energy-efficient approach to DAC based on crystallization of hydrogen-bonded bis-iminoguanidinium (BIG) (bi)carbonate salts.36 BIGs are a new class of anion receptors readily synthesized by imine condensation of aldehydes or ketones with aminoguanidinium salts (Fig. 5).37–39 BIGs form with oxyanions (e.g., sulfate, carbonate) HBFs of very low aqueous solubilities (on par with BaSO4 or CaCO3), a property that can be exploited for selective separation of these anions by crystallization. The low aqueous solubilities of these salts can be attributed to a number of structural factors: favorable π-stacking of the planar BIG cations, strong charge-assisted hydrogen bonding of the oxyanions by the iminoguanidinium groups, and reduced dehydration penalty of oxyanions, as they crystallize as hydrated clusters.
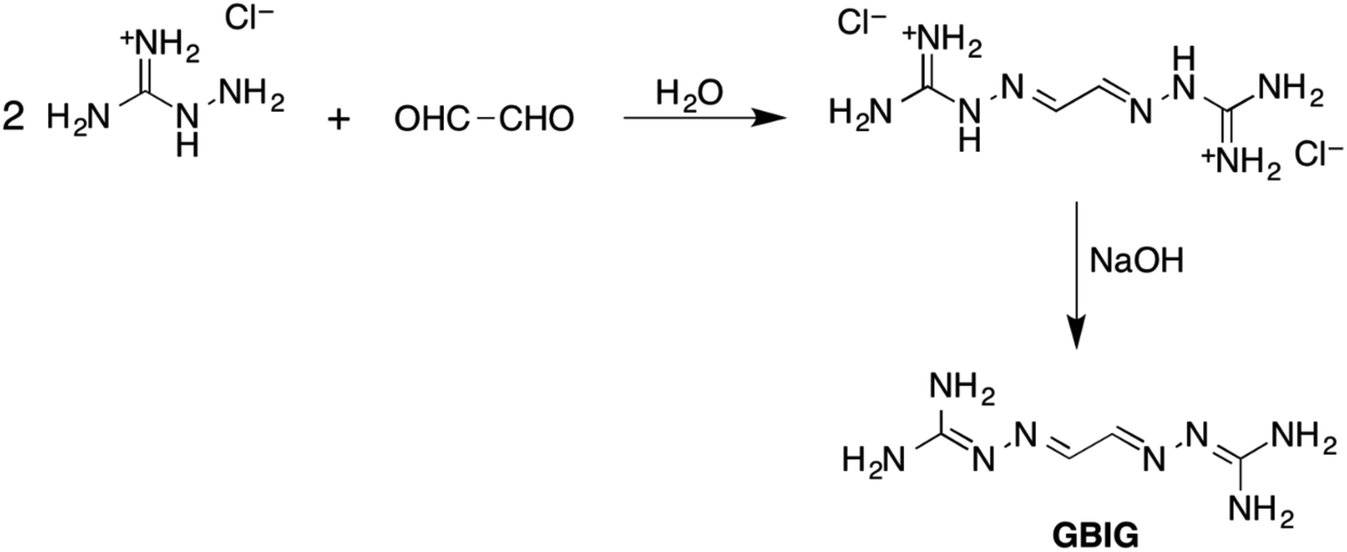 | ||
| Fig. 5 Synthesis of a BIG exemplified by glyoxal-bis-iminoguanidine (GBIG). Reproduced with permission from ref. 39. Copyright (2020) Royal Society of Chemistry. | ||
The prototype glyoxal-bis-iminoguanidine (GBIG), first reported in the chemical literature in 1898 by Johannes Thiele (Fig. 5),40 provided the proof-of-concept for CO2 capture by BIG crystallization. An aqueous solution of GBIG left overnight under a CO2 atmosphere led to crystallization of a hydrated GBIG bicarbonate salt. Single-crystal X-ray diffraction analysis (Fig. 6) revealed the formation of centrosymmetric (HCO3−)2 dimers with H⋯O contact distances of 1.71 Å (Fig. 6a). Each bicarbonate anion additionally accepts three guanidinium and two water hydrogen bonds (Fig. 6b). The bicarbonate dimers are linked by the water molecules into one-dimensional ladder-shaped clusters (Fig. 6c). On the other hand, the essentially planar GBIGH22+ cations stack along the same direction, with a mean interplanar distance of 3.25 Å (Fig. 5d). Finally, the cationic stacks flank the anionic clusters in a close-packed arrangement (Fig. 5e).41
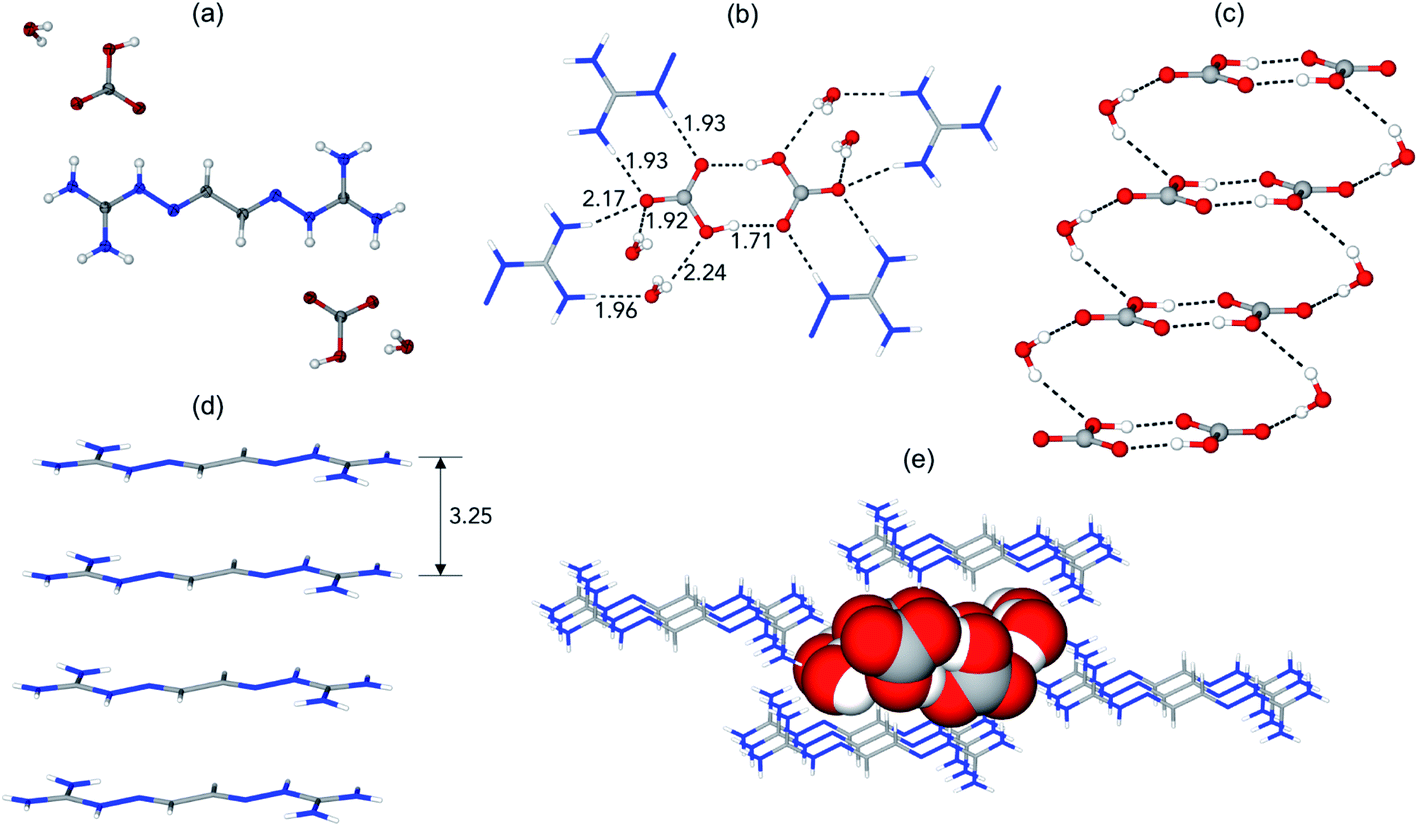 | ||
| Fig. 6 X-ray crystal structure of GBIG–HCO3. (a) ORTEP representation, with 50% ellipsoids. (b) Hydrogen bonding of bicarbonate dimers, with contact distances shown in Å. (c) Ladder-shaped hydrogen-bonded [(HCO3−)2(H2O)2]n anionic cluster. (d) Stack of GBIGH22+ cations, with the mean distance between adjacent cations shown in Å. (e) Crystal packing viewed along the crystallographic a axis, with the cationic stacks (shown as stick models) flanking the anionic cluster (shown as space-filling model). Reproduced with permission from ref. 41. Copyright (2019) Elsevier. | ||
The CO2 absorption by GBIG consists of the following elementary steps: protonation of GBIG by water to generate GBIGH22+ cations and HO− anions, CO2 transport from air into the aqueous solution, reaction of CO2 with HO− to generate HCO3−, and crystallization of GBIGH22+ and HCO3− ions into GBIGH2(HCO3)2(H2O)2. The reaction enthalpy for the overall CO2 capture process is −61 kJ mol−1 CO2, close to the lower end of the 60–80 kJ mol−1 range of absorption enthalpies for amine-based sorbents. Heating the crystals at 120 °C under a flow of air in the oven releases the CO2 and regenerates GBIG quantitatively, thereby closing the CO2-separation cycle. Experimental and computational investigations provided evidence for a CO2 release mechanism consisting of surface-initiated low-barrier proton transfer from the GBIG cations to the bicarbonate anions with the formation of carbonic acid dimers, followed by CO2 and H2O release in the rate-limiting step, with a measured activation energy of 102 ± 12 kJ mol−1.41
The CO2 capture cycle with GBIG was tested with a flue gas simulant (Fig. 7).41 A CO2 crystallization yield of 89% and quantitative GBIG regeneration were sustained for ten consecutive cycles. The CO2 capacity of the isolated carbonate solid, based on its molecular formula, is 6.1 mmol g−1. However, this is only a theoretical number that represents the upper limit achievable for the cyclic capacity. The actual working capacity will depend on the exact composition of the initial GBIG solution and other variables selected in the process design. The regeneration energy of GBIG is 151.5 kJ mol−1 CO2, 24% lower than the regeneration energy of monoethanolamine (MEA), a benchmark industrial CO2 solvent for which excessive energy is consumed in heating and vaporization of water.
 | ||
| Fig. 7 CO2 separation via GBIG bicarbonate crystallization. A flue gas simulant (12.8% CO2, EPA standard) is bubbled through an aqueous GBIG solution, leading to crystallization of GBIG bicarbonate. The solid is collected by filtration, then heated at 120 °C to release the CO2 and regenerate the GBIG sorbent. The filtrate containing the residual GBIG, and the regenerated GBIG solid are recycled. | ||
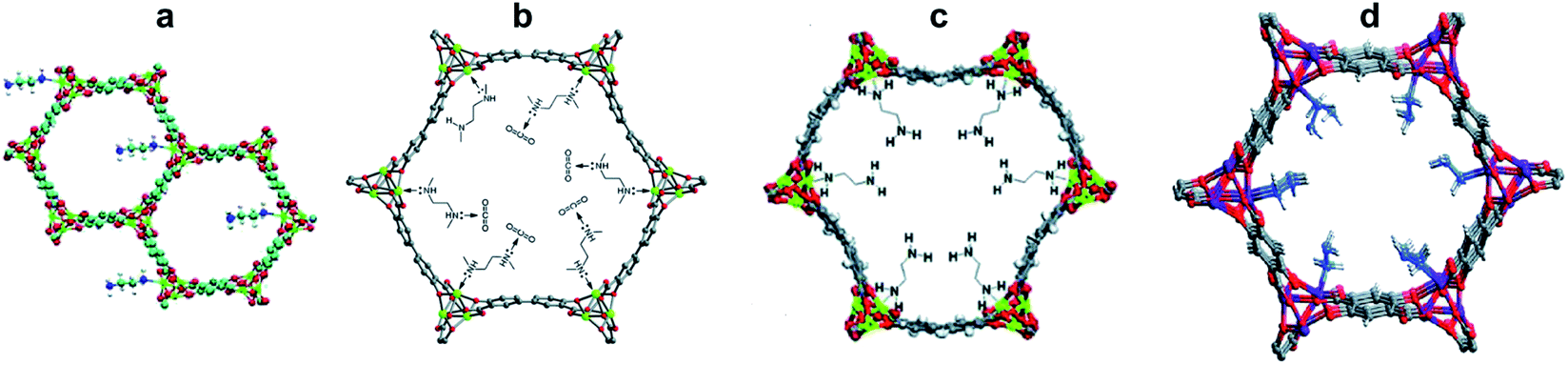
While GBIG was found to capture CO2 from flue gas energy efficiently, it proved inadequate for DAC. Exposure of an aqueous GBIG solution to air simply led to crystallization of the hydrated free ligand. In direct contrast, 2,6-pyridine-bis(iminoguanidine) (PyBIG), a different analogue from the BIG family, proved effective for DAC.42 An aqueous solution of PyBIG open to air reacted with atmospheric CO2 and crystallized as a hydrated carbonate salt with the formula PyBIGH2(CO3)(H2O)4. This corresponds to a theoretical CO2 capacity of 2.6 mmol g−1. The crystal-structure analysis by X-ray and neutron diffraction revealed extended one-dimensional carbonate–water clusters and stacked PyBIGH22+ cations (Fig. 8). It appears that the very low aqueous solubility of the PyBIG carbonate salt (Ksp = 1.0 × 10−9), on a par with CaCO3, drives the overall DAC equilibrium despite the very low concentration of CO2 in the air. Another favorable factor is the alkalinity of the PyBIG solution (pH ≈ 10), which reacts with the CO2 from air and generates carbonate anions in sufficient concentration to achieve supersaturation of the PyBIG carbonate salt. A likely structural factor contributing to the low solubility of the PyBIG carbonate salt is the strong hydrogen bonding of the carbonate anions by the guanidinium groups and the water molecules included in the crystals, as found by single-crystal neutron diffraction combined with electron density measurements from high-resolution X-ray diffraction.43 Heating of the carbonate crystals at 120 °C under a flow of air in the oven releases the CO2 and regenerates the PyBIG ligand quantitatively, thereby closing the CO2-separation cycle. The measured enthalpy of regeneration is 223 kJ mol−1, of which 148 kJ mol−1 (66%) is wasted for water desorption.44 Thus, crystal engineering of anhydrous BIG carbonate crystals would greatly improve the efficiency of the DAC process. Another component of the heat of regeneration is the sensible heat of 65 kJ mol−1 necessary to heat the crystals from ambient temperature to the 120 °C temperature of regeneration. This brings the overall heat of regeneration to 288 kJ mol−1.45
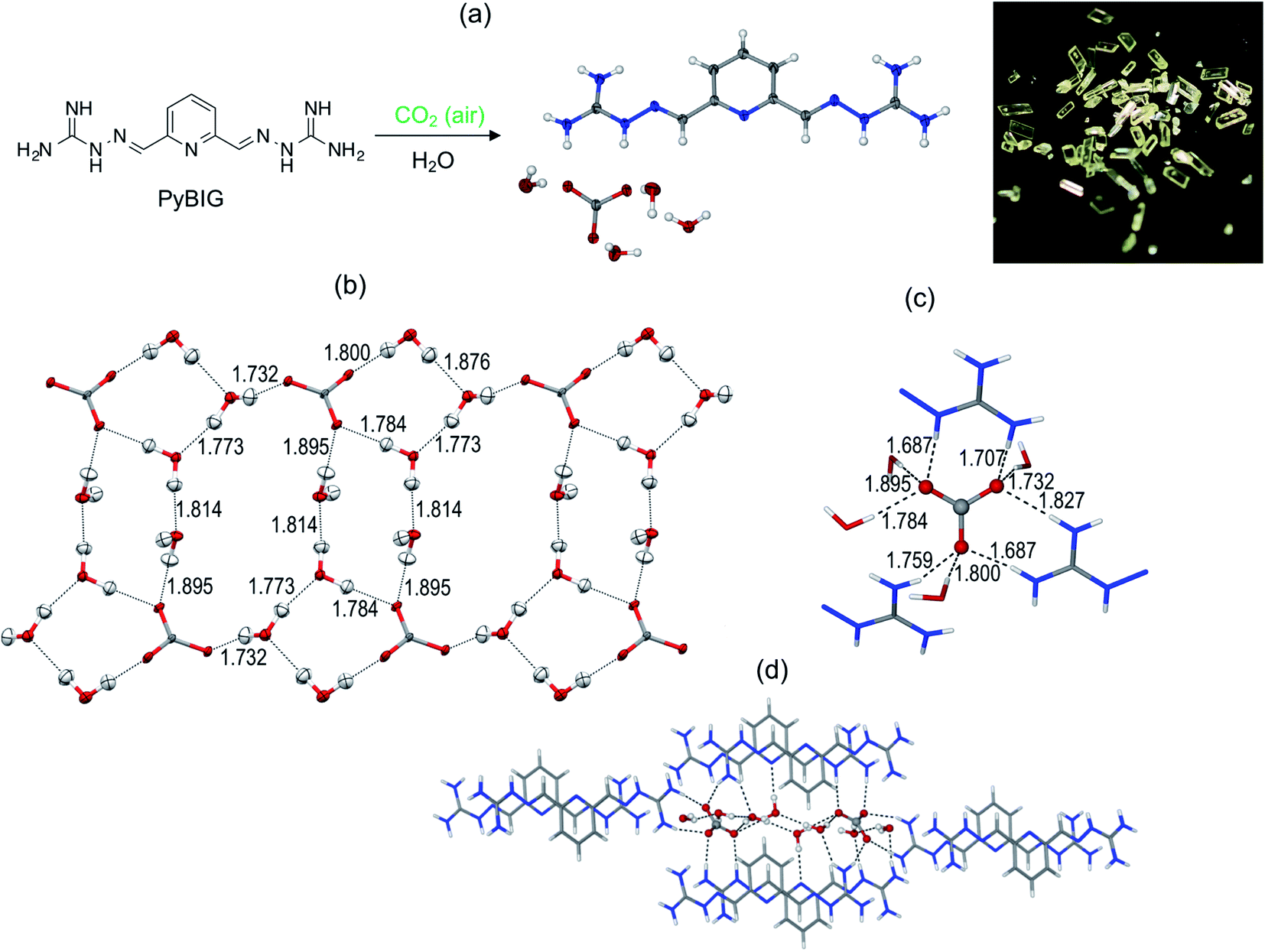 | ||
| Fig. 8 DAC with PyBIG. (a) Crystallization of hydrated PyBIG carbonate from aqueous PyBIG and atmospheric CO2. (b) Extended carbonate–water clusters from the single-crystal neutron diffraction structure, with the hydrogen bond contacts in Å. (c) Carbonate binding site in the crystal, showing the hydrogen bond contacts (Å) from the guanidinium groups and water molecules. (d) Crystal packing consisting of cationic PyBIGH22+ stacks that flank the anionic [CO3(H2O)42−]n clusters. | ||
In order to understand the structural factors determining the DAC chemistry of BIGs, we initiated a systematic structure–properties relationship study, focusing initially on the very basic series of BIG structures resulting from glyoxal (GBIG) and its simple analogs methylglyoxal (MGBIG) and diacetyl (DABIG) (Fig. 9).10 To a large extent, DAC by BIG crystallization is driven by the solubility difference between the neutral BIG ligand and the corresponding BIG carbonate salt. For GBIG, the solubility ratio between the free ligand and its bicarbonate salt is only 1.6, too small to drive DAC. In principle, the equilibrium DAC reactions could be shifted towards the carbonate solids by engineering BIG crystals with higher solubility and BIG carbonate crystals with lower solubilities. Our study found that relatively minor modifications in the molecular structure of GBIG, i.e., substituting one or two hydrogen atoms with methyl groups, led to dramatic differences in the crystal structures of the corresponding MGBIG and DABIG. This, in turn, led to large increases in their aqueous solubilities, by factors of 96 and 14 relative to GBIG, respectively. One notable difference between the crystal structures of GBIG and those of MGBIG and DABIG is the presence of π-stacking in the former, and the absence of it in the latter, apparently due to increased conformational flexibility and steric hindrance caused by the methyl groups. On the other hand, the crystal structures of the corresponding carbonate salts of MGBIG and DABIG revealed relatively planar BIG cations that are closely stacked in the crystals. Another structural feature that enhances the stability of these carbonate crystals is the inclusion of at least two water molecules per carbonate anion, and the formation of extended carbonate–water clusters. As a result, the aqueous solubilities of the carbonate salts of DABIG and MGBIG are lower than the corresponding solubilities of the neutral ligands by factors of 38 and 72, respectively, thereby providing the thermodynamic driving force required for the DAC reactions.
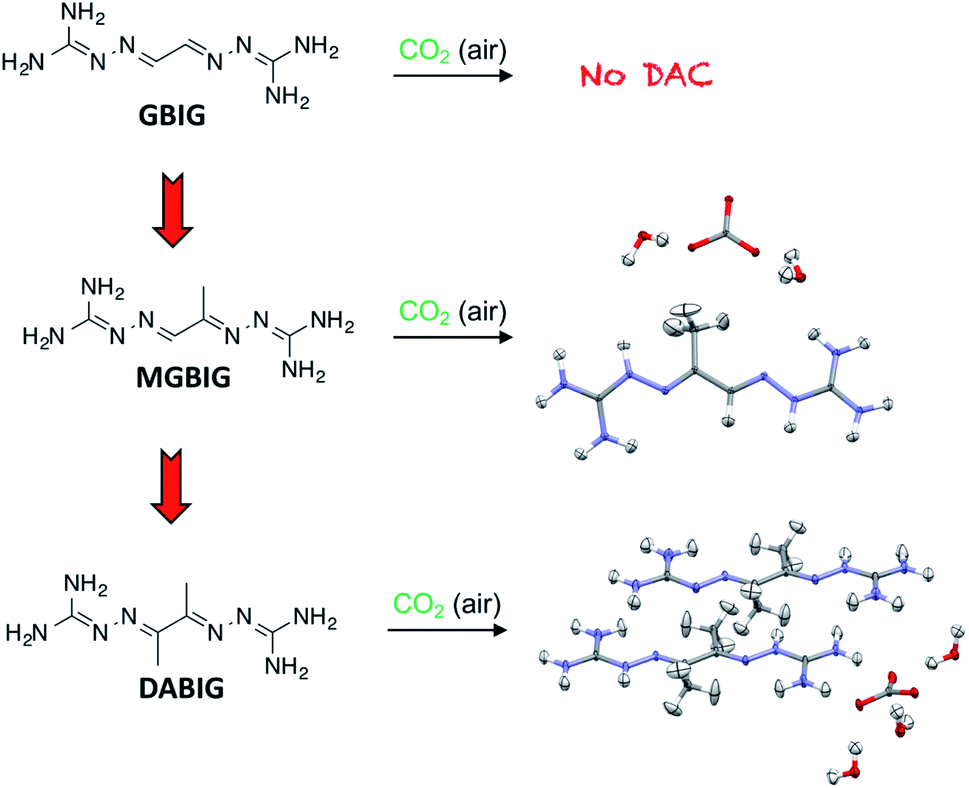 | ||
| Fig. 9 Dialing in DAC by crystal engineering of BIGs. An aqueous solution of GBIG left open to air led to crystallization of the free ligand, whereas the corresponding solutions of the methylated analogs MGBIG and DABIG led to DAC. | ||
Preliminary experiments confirmed both MGBIG and DABIG are effective at DAC. An aqueous solution of DABIG left open to air reacted with CO2 and yielded a hydrated crystalline carbonate salt with molecular formula (DABIGH+)(DABIGH22+)0.5(CO32−)(H2O)3 (Fig. 9). This corresponds to a theoretical CO2 capacity of 2.4 mmol g−1. The CO2 and water are released by heating the crystals at 80–120 °C (under nitrogen purging) and the free DABIG ligand is regenerated quantitatively. The total regeneration energy, consisting of reaction enthalpy and sensible heat is 422 kJ mol−1. On the other hand, an aqueous solution of MGBIG left open to air reacted with CO2 and yielded two main crystalline carbonate phases with the formulas (MGBIGH+)2(CO32−)(H2O)2 and (MGBIGH22+)(CO32−)(H2O)2. The corresponding theoretical CO2 capacities are 2.1 and 3.5 mmol g−1, respectively. The measured regeneration energies of the two phases are 358 and 323 kJ mol−1, respectively, more than twice as large compared to the corresponding value for GBIG (151.5 kJ mol−1). Nevertheless, the temperatures of regeneration are very similar for GBIG, MGBIG, and DABIG, in the range of 120 to 160 °C (under a flow of air).10
Outstanding challenges and future opportunities
With its structural precision and the prospect for tuning the CO2 binding, transport, and release in crystalline frameworks, crystal engineering of MOFs and HBFs offers unique opportunities for DAC. Table 1 summarizes the key properties for some of these materials, relevant to their DAC performance.| Sorbent | CO2 capacity mmol g−1 | Heat of regeneration kJ mol−1 | T of regeneration °C | Reference |
|---|---|---|---|---|
| a Based solely on the Qst value. b Measured by DSC – includes desorption enthalpy and sensible heat. c Regenerated under inert gas purging. d Regenerated under air flow. e Regenerated under vacuum. f Capacity calculated from the molecular formula of the crystallized solid. | ||||
| MOFs | ||||
| TIFSIX-3-Ni | 0.41 | 50a | 90c | 19 |
| NbOFFIVE-1-Ni | 1.3 | 54a | 105c | 20 |
| ed–Mg(dobdc) | 1.5 | NA | 120c | 22 |
| mmen–Mg2(dobpdc) | 1 | 103b | 150c | 23 |
| ed–Mg2(dobpdc) | 2.83 | 50a | 150d | 25 |
| NH2NH2–Mg(dobdc) | 3.89 | 133b | 130c | 26 |
| MOF-Zn–OH | 2.2 | 71a | 100e | 31 |
| MOF-Ni–OH | 2.7 | 84a | 100e | 32 |
|
||||
| HBFs | ||||
| MXDA | 5.6f | NA | 103–120c | 35 |
| GBIG | 6.1f | 151.5b | 120d | 41 and 46 |
| PyBIG | 2.6f | 288b | 120d | 42 and 44 |
| DABIG | 2.4f | 422b | 80–120c | 10 |
| MGBIG-1 | 2.1f | 358b | 80–120c | 10 |
| MGBIG-2 | 3.5f | 323b | 80–120c | 10 |
Atmospheric CO2 capture with MOFs can be done via either physisorption or chemisorption mechanisms. The former offers the prospect for lower regeneration energies, though that may come at the expense of reduced working capacities and lower CO2/H2O selectivities. These limitations can be addressed by functionalizing the MOF pores with amine or M–OH groups that react with CO2 and convert it into carbamic acid, carbamate, or bicarbonate anions. However, water condensation in the pores can lead to higher regeneration energies and sometimes chemical stability issues. While MOFs tend to have superior working capacities compared to other porous material that are less crystalline, very few MOFs have been tested under realistic DAC conditions, with their reported performances often based solely on single-component CO2 sorption isotherms. The reported regeneration energies are also typically based on measurement of isosteric heat of reactions from sorption isotherms at low CO2 loading, without taking into account the sensible heat or the heat required to remove the co-adsorbed water. MOF regeneration is often done by purging with an inert gas, which dilutes the output of CO2, thereby requiring additional separation steps before the carbon dioxide is ready to be compressed for storage or utilization.
Crystallization of HBFs presents an alternative approach to DAC that obviates the need for sorbent porosity. The challenge, in this case, is to identify organic bases, such as amines or guanidines, that form crystalline HBFs of low aqueous solubilities with CO2. Unfortunately, crystal engineering is still not sophisticated enough to predict solubilities of molecular crystals or organic salts. While advances in computational methods may one day allow for ab initio design of crystal structures, aqueous solubilities, and free energies of reactions involved in DAC, structure–properties relationship studies remain for the moment the most reliable approach to developing new crystalline DAC systems. Along this line, one of the priorities is to identify design principles leading to exclusion of water from the BIG carbonate crystals, as water desorption comes with a high enthalpic cost, which can significantly increase the regeneration energy of the DAC process. Alternatively, developing improved desorption processes that avoid water evaporation or recover the energy stored in the water vapor during condensation could make the overall DAC process more energy sustainable. As in the case of MOFs, the CO2 desorption step has typically been done under inert gas or air, though vacuum- or steam-assisted desorption may be feasible for some HBFs and need to be explored further.
In addition to favorable thermodynamics of CO2 capture, crystallization, and desorption, fast kinetics for these steps are also critical for a viable DAC system. For instance, crystallization of BIGs with atmospheric CO2 is thermodynamically driven by the very low aqueous solubility of the BIG–CO3 salts, but the rates of CO2 uptake by the aqueous BIGs are generally too slow, with loading times on the order of days. The rates of CO2 uptake can be greatly accelerated by combining BIG–CO3 crystallization with CO2 absorption by aqueous amino acids or peptides, which work in synergy with BIGs to achieve effective and energy-efficient DAC.44–46
As illustrated in Fig. 10, the DAC cycle with amino acids/BIGs comprises the following steps: (i) fast CO2 absorption by the aqueous amino acid (glycine is shown as a representative example), yielding the corresponding bicarbonate salt (via a carbamate intermediate); (ii) crystallization of the carbonate anions with BIG, which regenerates the amino acid; (iii) CO2 release from the BIG carbonate solid and BIG regeneration.
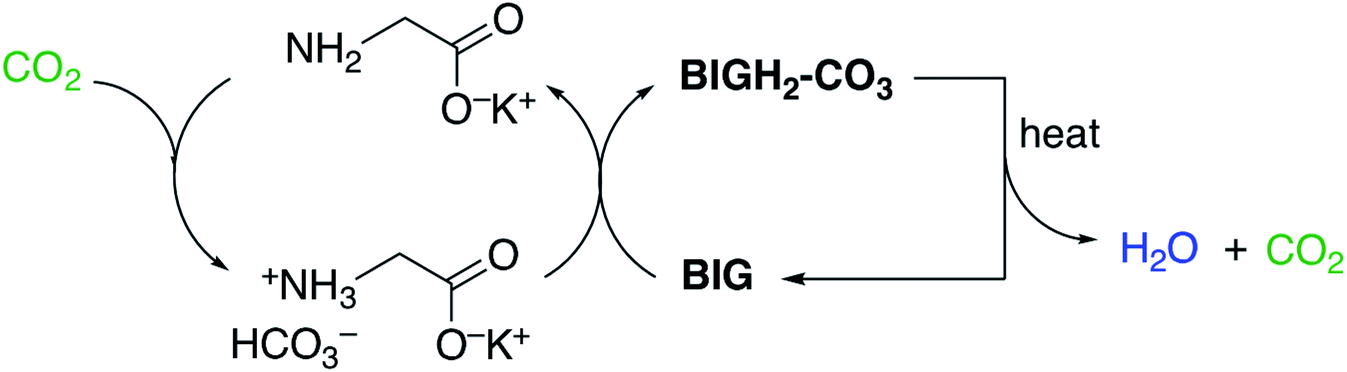 | ||
| Fig. 10 DAC by CO2 absorption with aqueous amino acids and carbonate crystallization with BIGs. Reproduced with permission from ref. 45. Copyright (2019) American Chemical Society. | ||
While crystal engineering may be critical in the initial design of novel crystalline DAC systems, developing a practical and efficient DAC technology also involves equally important chemical engineering aspects, such as process design and optimization, scale up, design, manufacturing, and testing of efficient air–solid or air–liquid contactors, crystallizers, and solid–liquid separators. Optimization of the CO2 desorption to minimize the required temperature and maximize the purity of the CO2 is another important aspect. Along this line, purging with inert gas or air should be avoided as much as possible if high purity for the CO2 product is needed. Instead, vacuum- or steam-aided desorption are preferred, which not only result in high-purity CO2, but also lower the regeneration temperatures. Once all components and operations are in place and working synergistically, the economic and environmental feasibility, and the carbon footprint of the ensuing DAC technology needs to be evaluated through rigorous technoeconomic and lifecycle analyses.
Finally, crystalline organic materials are not ready yet for large-scale deployment in DAC applications, as both MOFs and HBFs need to be manufactured at a much larger scale (Mt per year) at a reasonable cost (<$10 per kg) and tested over tens of thousands of loading/unloading cycles under realistic DAC conditions. Nevertheless, understanding and improving their fundamental DAC chemistry is a critical first step in developing viable DAC technologies, and it is the author's hope that this article will stimulate further research along this direction.
Author contributions
Conceptualization, funding acquisition, writing – original draft, writing – review & editing, R. C.Conflicts of interest
The author is an inventor on a couple of patents on CO2 capture by crystallization of BIGs47,48 and seeks to license and commercialize a DAC technology based on this approach.Acknowledgements
This research was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division.Notes and references
- National Academies of Sciences, Engineering, and Medicine, Negative emissions technologies and reliable sequestration: A research agenda, The National Academies Press, Washington, DC, 2019, DOI:10.17226/25259.
- The role of negative emission technologies in addressing our climate goals, ed. J. Wilcox, P. Renforth and F. Kraxner, Frontiers Media, Lausanne, 2020 Search PubMed.
- N. McQueen, K. V. Gomes, C. McCormick, K. Blumanthal, M. Pisciotta and J. Wilcox, Progress in Energy, 2021, 3, 032001 CrossRef.
- D. Kramer, Phys. Today, 2020, 73, 44–51 CrossRef CAS.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- A. Gambhir and M. Tavoni, One Earth, 2019, 1, 405–409 CrossRef.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- J. Hansen, M. Sato, P. Kharecha, K. von Schuckmann, D. J. Beerling, J. Cao, S. Marcott, V. Masson-Delmote, M. J. Prather, E. J. Rohling, J. Shakun, P. Smith, A. Lacis, G. Russell and R. Ruedy, Earth Syst. Dyn., 2017, 8, 577–616 CrossRef.
- S. J. Davis, et al. , Science, 2018, 360, eaas9793 CrossRef PubMed.
- R. Custelcean, N. J. Williams, X. Wang, K. A. Garrabrant, H. J. Martin, M. K. Kidder, A. S. Ivanov and V. S. Bryantsev, ChemSusChem, 2020, 13, 6381–6390 CAS.
- H. Azarabadi and K. S. Lackner, Appl. Energy, 2019, 250, 959–975 CrossRef.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- C. Gebald, J. A. Wurzbacher, P. Tingaut, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2011, 45, 9101–9108 CrossRef CAS PubMed.
- X. Shi, H. Xiao, K. S. Lackner and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4026–4029 CrossRef CAS PubMed.
- G. R. Desiraju, IUCrJ, 2017, 4, 710–711 CrossRef CAS PubMed.
- A. Kumar, D. G. Madden, M. Lusi, K.-J. Chen, E. A. Daniels, T. Curtin, J. J. Perry IV and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- A. Kumar, C. Hua, D. G. madden, D. O'Nolan, K.-J. Chen, L.-A. J. Keane, J. J. Perry IV and M. J. Zaworotko, Chem. Commun., 2017, 53, 5946–5949 RSC.
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS PubMed.
- D. G. Madden, H. S. Scott, A. Kumar, K.-J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry IV and M. J. Zaworotko, Philos. Trans. R. Soc., A, 2017, 375, 20160025 CrossRef PubMed.
- S. Choi, T. Watanabe, T.-H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- T. M. McDonald, et al., Cooperative insertion of CO2 in diamine-appended metal-organic frameworks, Nature, 2015, 519, 303 CrossRef CAS PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 RSC.
- P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- L. A. Darunte, T. Sen, C. Bhawanani, K. S. Walton, D. S. Sholl, M. J. Realff and C. W. Jones, Ind. Eng. Chem. Res., 2019, 58, 366–377 CrossRef CAS.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed.
- J. D. Martell, P. J. Milner, R. L. Siegelman and J. R. Long, Chem. Sci., 2020, 11, 6457–6471 RSC.
- E. J. Kim, R. L. Siegelman, H. Z. H. Jiang, A. C. Forse, J.-H. Lee, J. D. Martell, P. J. Milner, J. M. Falkowski, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, Science, 2020, 369, 392–396 CrossRef CAS PubMed.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. Winston Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 489–497 CrossRef CAS.
- J. A. Wurzbacher, C. Gebald, N. Piatkowski and A. Steinfeld, Environ. Sci. Technol., 2012, 46, 9191–9198 CrossRef CAS PubMed.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS PubMed.
- R. Custelcean, Reducing Atmospheric Carbon Dioxide through Direct Air Capture, Scientia, 2021, 138, 20–23, DOI:10.33548/SCIENTIA613.
- R. Custelcean, N. J. Williams and C. A. Seipp, Angew. Chem., Int. Ed., 2015, 54, 10525 CrossRef CAS PubMed.
- R. Custelcean, N. J. Williams, C. A. Seipp, A. S. Ivanov and V. S. Bryantsev, Chem.–Eur. J., 2016, 22, 1997 CrossRef CAS PubMed.
- R. Custelcean, Chem. Commun., 2020, 56, 10272 RSC.
- J. Thiele and E. Dralle, Justus Liebigs Ann. Chem., 1898, 302, 275 CrossRef CAS.
- N. J. Williams, C. A. Seipp, F. M. Brethomé, Y.-Z. Ma, A. S. Ivanov, V. S. Bryantsev, M. K. Kidder, H. J. Martin, E. Holguin, K. A. Garrabrant and R. Custelcean, Chem, 2019, 5, 719–730 CAS.
- C. A. Seipp, N. J. Williams, M. K. Kidder and R. Custelcean, Angew. Chem., Int. Ed., 2017, 56, 1042–1045 CrossRef CAS PubMed.
- C. G. Gianopoulos, Z. Chua, V. V. Zhurov, C. A. Seipp, X. Wang, R. Custelcean and A. A. Pinkerton, IUCrJ, 2019, 6, 56–65 CrossRef CAS PubMed.
- F. M. Brethomé, N. J. Williams, C. A. Seipp, M. K. Kidder and R. Custelcean, Nat. Energy, 2018, 3, 553–559 CrossRef.
- R. Custelcean, N. J. Williams, K. A. Garrabrant, P. Agullo, F. M. Brethomé, H. J. Martin and M. K. Kidder, Ind. Eng. Chem. Res., 2019, 58, 23338–23346 CrossRef CAS.
- R. Custelcean, K. A. Garrabrant, P. Agullo and N. J. Williams, Cell Rep. Phys. Sci., 2021, 2, 100385 CrossRef CAS.
- R. Custelcean, C. A. Seipp and N. J. Williams, US Pat., 10583387, 2020 Search PubMed.
- R. Custelcean, N. J. Williams and C. A. Seipp, US Pat., 10633332, 2020 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |