
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent monomeric and dimeric Ru(II) acyclic carbene complexes as selective sensors for NH3/amine vapor and humidity†
Jingqi
Han
 ,
Shun-Cheung
Cheng
,
Shek-Man
Yiu
,
Man-Kit
Tse
and
Chi-Chiu
Ko
*
,
Shun-Cheung
Cheng
,
Shek-Man
Yiu
,
Man-Kit
Tse
and
Chi-Chiu
Ko
*
Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China. E-mail: vinccko@cityu.edu.hk
First published on 5th October 2021
Abstract
A new class of luminescent bis(bipyridyl) Ru(II) pyridyl acyclic carbene complexes with environmentally-sensitive dimerization equilibrium have been developed. Owing to the involvement of the orbitals of the diaminocarbene ligand in the emissive excited state, the phosphorescence properties of these complexes are strongly affected by H-bonding interactions with various H-bonding donor/acceptor molecules. With the remarkable differences in the emission properties of the monomer, dimer, and H-bonded amine adducts together with the change of the dimerization equilibrium, these complexes can be used as luminescent gas sensors for humidity, ammonia, and amine vapors. With the responses to amines and humidity and the corresponding change in the luminescence properties, a proof-of-principle for binary optical data storage with a reversible concealment process has been described.
Introduction
Smart materials that exhibit a significant change in their absorption and emission colors in response to external stimulation have attracted increasing research interest because of their potential applications in the fields of luminescent probes and sensors,1 light-emitting diodes,2 cell imaging,3 information security,4 and so on.Ammonia and volatile organic amine detections are essential because of their wide applications in food industries,5 environmental monitoring,6 and even disease diagnosis.7 As a result, there is a high demand for developing reliable sensors with high sensitivity and fast response to reduce food poisoning, ocular irritation, and respiratory infection. In this context, various optical amine sensors based on metal–organic frameworks and coordination polymers have been developed.8 On the other hand, moisture level or humidity is another important indicator for food quality assurance.9 Humidity sensors have also found applications in different areas,10 such as skin-humidity level monitoring,10a breath humidity detection,10b moisture detection in organic solvents,10c and sealed packaging humidity value visualization.10d
To date, commercial semiconductor-based sensors for humidity and amines have been developed. However, their applications as sensors in food product quality monitoring, consumer products, or rapid screening tools are limited due to the need of a continuous energy supply, an electronic device for reading the signal, high cost, or poor gas selectivity. For practical applications, luminescent sensors for amine and humidity detection, particularly those with considerable changes suitable for naked-eye detection, are highly desired but have been hardly developed.8,10d,11 Compared with fluorescent sensors, phosphorescent sensors have better emission characteristics, including large Stokes shift and longer-lived lifetimes that provide methods to distinguish sensing emission signals from background fluorescence.12 Moreover, phosphorescent transition metal complexes with environmental sensitive emission properties are ideal candidates for designing luminescent sensors.13
Based on the strongly dependent phosphorescent properties of the acyclic carbene complexes towards the conformational variations and the intermolecular interactions with hydrogen-bonding donor and acceptor molecules of the acyclic carbene ligands,14 herein, we report the design of a new class of bis(bipyridyl) Ru(II) complexes with pyridyl acyclic diaminocarbene ligands as amine and humidity chemosensor. It is hypothesized that the emission energy would be strongly affected by H-bonding interactions with various H-bonding donor and acceptor molecules due to the strong involvement of the orbitals of the diaminocarbene ligand. Interestingly, the N-deprotonated diaminocarbene ligand would dimerize upon coordination to electron-deficient Ru(II) system by forming strong intermolecular H-bonding. As a result, these complexes exhibit dual phosphorescent properties. The H-bonded dimerization equilibrium and its impacts on the photophysical properties have been studied. Based on the shift of the dimerization equilibrium with various types of H-bonding donor and acceptor molecules and their different H-bonding interactions, the dimeric and monomeric forms of the complex can serve as luminescent humidity and amine sensors. With reference to its responses to amines and humidity and the corresponding luminescence responses, a proof-of-principle for binary optical data storage15 has been described.
Using the synthetic methodology for pyridyl diaminocarbene ligands,14 a series of bis(bipyridyl) Ru(II) complexes with pyridyl diaminocarbene ligands (1–3) has been synthesized by the coordination of substituted 2-aminopyridine ligands to the isocyano bis(bipyridyl) Ru(II) precursors16 (Scheme 1). The structures of all these complexes have been determined by X-ray crystallography. The crystal data and selected bonding parameters are summarized in Tables S1–S4 in ESI.† It is interesting to note that the complex units dimerize with two strong intermolecular H-bonds as evidenced by a short N⋯N distances17 in the range of 2.97–2.99 Å and N⋯H distances of ca. 2.10–2.11 Å in the crystal structures of 1 (Fig. 1a) and 2 (Fig. S1, ESI†). Unlike other structurally-related N-deprotonated carbene ligands in the iridium(III) cyclometalates,14b the N-phenyl group of acyclic carbene ligands is cis to C–Ru in these H-bond dimers. For 3, only a tiny amount of single crystals with quality suitable for X-ray crystallography in the recrystallization can be obtained. The difference of these single crystals from the bulk solid of 3 is verified by the different powder XRD patterns of the bulk solid compared to the simulated diffraction patterns from the single-crystal data of 3 (Fig. S2a, ESI†). This contrasts with the close agreement of the simulated patterns and the powder XRD patterns of the bulk solid of 1 (Fig. S2b, ESI†). The crystal structure of these single crystals of 3 is in the monomer form with the N-phenyl group trans to C–Ru (Fig. 1b). Although the bonding parameters of these complexes are within the typical bonding parameter ranges,18 a close scrutiny of acyclic carbene ligands in these complexes reveals that Ru–C(carbene) of 3 is slightly shorter (2.01 Å for 3vs. 2.03–2.04 Å for 1 and 2) with a larger N–C–N angle (116.6° for 3vs. 111.2–111.5° for 1 and 2). Besides, the positions of N-deprotonation in the diaminocarbene ligands are different. In the structure of 3, the nitrogen attached to the pyridine of the diaminocarbene is deprotonated, whereas the nitrogen attached to the chlorophenyl ring is deprotonated in the structures of 1 and 2. This may be due to the difference between trans and cis conformation. Moreover, the presence of electron-withdrawing CF3 substituent on the pyridine of 3 also renders its amine substituent more acidic.
 | ||
| Scheme 1 Synthetic route to 1–3. | ||
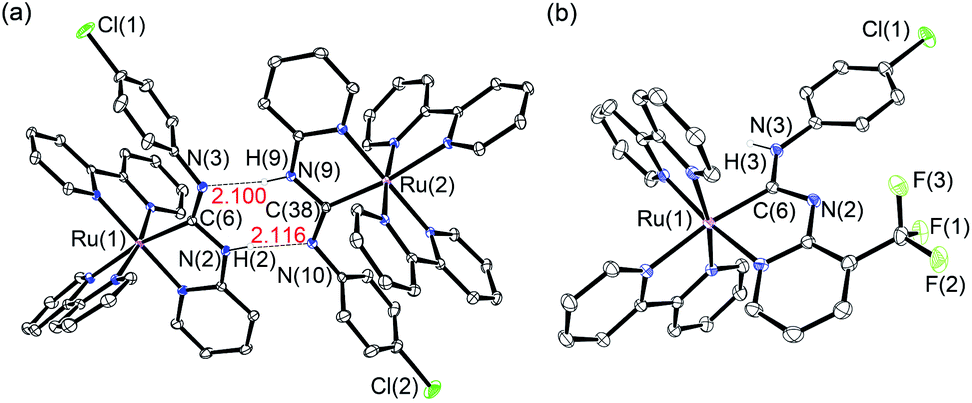 | ||
| Fig. 1 Perspective drawings of (a) H-bond dimer of 1 and (b) 3. | ||
Environment-dependent photophysical properties
The strong intermolecular H-bonding between the acyclic diaminocarbene leading to dimerization, as revealed from the X-ray crystallography, has also been observed in the solution. The absorption of 1 and 2, in particular the lowest-energy absorption band, in dichloromethane solution does not follow the Beer–Lambert law (Fig. 2a). Fitting the lowest-energy absorption data, where monomer has minimal absorption, to the dimerization equilibrium model,19 a linear correlation in the plot of [C]/A½vs. A½ (Fig. 2a: inset and Fig. S3a, ESI†) where [C] is the concentration of the complex and A is the absorbance. From the linear regression of the plots, the dimerization equilibrium constants (4.28 × 105 for 1 and 5.44 × 104 for 2), and thus the absorptivity of the H-bonded dimer and monomeric form (Table S5 in ESI, Fig. 2b, and Fig. S3b, ESI†) have been calculated. The dimerization equilibrium of 1 in dichloromethane solution is further supported by the decrease of intensity of the N–H stretch of the monomeric form at 3506 cm−1 relative to the N–H stretch of the dimeric form at 3437 cm−1 upon increasing the concentration (Fig. S4†). The extent of the shifting of the N–H stretching frequency from monomeric to dimeric form due to the strong N–H⋯N hydrogen bonding interactions is commonly observed in other systems.20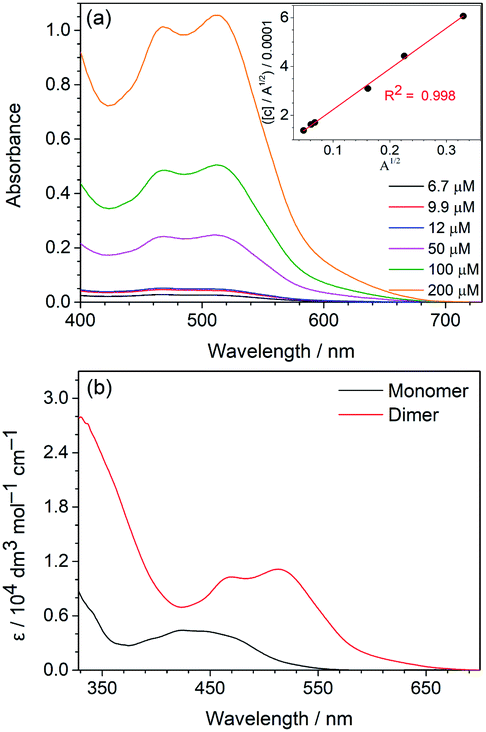 | ||
| Fig. 2 (a) Overlaid UV-vis absorption spectra of different concentrations of 1 in dichloromethane solutions. Inset: A plot of [C]/A½vs. A½ of 1 at 615 nm and the linear regression fit. (b) Calculated absorption spectra of the dimeric and monomeric forms of 1 in dichloromethane solution based on the dimerization equilibrium analysis. | ||
On the contrary, the absorption features in the spectra of 3 do not change with the concentration (Fig. S5a, ESI†). The close resemblance of the lowest-energy absorption band of 3 to the dimeric form of 1 and 2 suggests that the major form of 3 in the dichloromethane solution is the dimer form. This is corroborated by the observation of the characteristic dimeric emission of 3 (see below). The much higher dimerization affinity of 3 than 1 and 2 is likely due to the presence of electron-withdrawing CF3 substituent, which can strengthen the H-bonding interaction by enhancing the proton donating ability of the N–H group of the diaminocarbene ligand.
As similar to bis(bipyridyl) Ru(II) complexes, these complexes show intense ligand-centered ππ* transitions in the UV region and moderately intense metal-to-ligand charge transfer (MLCT) transitions in the visible region. Compared to [Ru(bpy)3]2+, the MLCT absorption bands/shoulders of these Ru(II) carbene complexes are broader and of much lower energy, which can be attributed to the mixing of two different MLCT transitions [dπ(Ru) → π*(bpy)] and [dπ(Ru) → π*(N^Ccarbene)]. The lowest-energy MLCT transition in these complexes is ascribed to MLCT[dπ(Ru) → π*(N^Ccarbene)]. This is further supported by the high sensitivity and the drastic red-shift of this band upon dimerizing the acyclic carbene. The red-shifted MLCT absorption in these complexes (450–470 nm) compared to [dπ(Ru) → π*(R-bpy)] transition in [Ru(R-bpy)3]2+ (410–430 nm),21 is due to the lower-lying π*(N^Ccarbene) orbital than that of the bipyridyl ligands (R-bpy).14e,18c
As with absorption properties, the dimerization equilibrium of 1 and 2 in dichloromethane solution would result in strongly concentration-dependent emission properties (Fig. 3a), comprising two emission components characterized by two different emission lifetimes in the range of 81–88 ns and 193–264 ns. The longer-lived high-energy and the shorter-lived low-energy emission components are ascribed to the emission of the monomeric and dimeric form, respectively. The evolution of a lower-energy emission band, corresponding to the emission of the dimeric form, at higher concentrations is attributed to the increase of the ratio of the dimeric form. With distinguishable emission lifetimes, the two emission bands can be well-resolved using time-resolved emission spectroscopy (Table S5 in ESI, Fig. 3b and S3c in ESI†). Concerning the assignment of the lowest-energy absorption band and the red-shift of the emission upon dimerization, these emissions are likely originated from the 3MLCT [dπ(Ru) → π*(N^Ccarbene)] excited state. Based on the ratios of absorptivity, emission intensity, and absorbance, the luminescence quantum yields of the monomeric and dimeric forms (Table S5, ESI†) have been determined. For 3 in dichloromethane solution, the emission does not change with its concentration with a lifetime of 160 ns and emission energy similar but slightly higher than those of the dimeric form of 1 and 2 (Fig. S5b, ESI†). Therefore, it is also ascribed to the MLCT [dπ(Ru) → π*(N^Ccarbene)] phosphorescence of the dimeric form.
 | ||
| Fig. 3 (a) Overlaid normalized emission spectra of different concentrations of 1 in dichloromethane solutions. Excitation wavelength: 436 nm. (b) Time-resolved emission spectra of short-lived and long-lived components of 1 recorded at time windows of 0–5 ns and 1–200 μs, respectively, after the excitation laser pulse. | ||
As intermolecular hydrogen bonding is the major driving force for dimerization, the extent of dimer formation would be strongly affected by the environments, particularly H-bonding donor/acceptor molecules. Given the drastic differences in the emission properties of the monomer and dimer, the environment-dependent dimerization is examined by the solvatochromic behavior of 1. The significant solvatochromism of 1, readily distinguishable by the naked eyes, is shown in Fig. S6, ESI.† As dimerization is concentration-dependent, the absorption and emission spectra were recorded for a fixed concentration of 23 μM in different solvent media. On top of solvatochromic shifts commonly observed in other charge transfer transitions,13,22 the intensity ratio of the monomeric and dimeric MLCT absorption of 1 in the visible region varies with the nature of the solvent (Fig. S6c, ESI†). In non-polar dichloromethane, the absorption intensity of the dimeric form (lower-energy band) relative to the monomer is much higher than that in polar or H-bonding solvents. The complex has the lowest absorption intensity of the dimeric MLCT transition in the strongest hydrogen-bonding organic solvent (methanol).
Consistent with the absorption properties, the intensity of the lower-energy emission band relative to the higher energy band also follow the order of dichloromethane > ethyl acetate > acetonitrile > ethanol ≫ methanol (Fig. S6d, ESI†). Except for methanol, the emission decays of 1 in all the studied solvents follow clear biexponential kinetics for the monomeric and dimeric forms described in the dichloromethane solution. In methanol solution, the emission decay is almost single exponential with a lifetime similar to the monomeric form observed in ethanol, suggesting that the complex is mainly the monomeric form in methanol (Table S6, ESI†). The shift of dimerization equilibrium to the monomeric form is due to the solvation stabilization by the H-bonding interaction of the acyclic carbene with methanol.
The change in the monomer/dimer ratio is also clearly observed from the emission properties in different types of low-temperature solid matrices. In 77 K EtOH/MeOH glass, 1 and 2 only exhibit the phosphorescence with maxima at 584–587 nm (Fig. S7a, ESI†), ascribed to the emission of the monomeric form. This is consistent with the results in the solvatochromic study of 1, in which only the emission from the monomeric form is observed in methanol. In contrast, 3 with the most stable dimeric form show a dual emission derived from the monomeric and dimeric forms peaking at 562 and 651 nm, respectively. Based on the relative emission intensities, the dimeric form of 3 is predominant in 77 K EtOH/MeOH glassy medium. In 77 K dichloromethane, the ratio of the monomeric and dimeric emission change considerably (Fig. S7b, ESI†). The higher-energy monomeric emission of 3 almost disappears compared with the dimeric lower-energy emission at 654 nm in 77 K dichloromethane. Similarly, the higher-energy monomeric emission of 1 becomes a minor component in the dual emission bands. For 2, the monomeric emission is still the major component (Fig. S7b, ESI†). This is consistent with the trend of dimerization affinity 3 ≫ 1 > 2.
In acetonitrile solution, 1 (100 μM) exhibits a dual phosphorescence with the dimeric emission as the major component. As with the solvatochromism induced by protic solvents, gradual addition of water to the acetonitrile solution of 1 (100 μM) would lead to a blue-shift of the MLCT absorption bands and the growth of the high-energy emission band (Fig. 4). This is due to the shift of the dimerization equilibrium to the monomeric form as a result of the H-bonding interaction of the acyclic carbene with water.
 | ||
| Fig. 4 Overlaid (a) UV-vis absorption and (b) emission spectra of 1 in MeCN–H2O mixed solvent with different water fractions. Concentration: 100 μM. Photos of the solution captured under daylight and UV illumination are shown in the upper panel. | ||
Luminescent sensor for humidity monitoring
Inspired by the water-sensitive dimer-monomer transformation together with the resulting color and emission responses, the application of this complex as a luminescent humidity sensor has been explored. Neat film of mainly the dimeric form of 1 on quartz plate can be fabricated by spin-coating the dichloromethane solution of 1 (200 μM). The initially formed neat film shows an emission band peaking at 714 nm, similar to solid-state emission of the single crystal sample of 1 (Fig. S8, ESI†). This confirms that the dimeric form is the major component in the spin-coated film. When the film is stored in an atmosphere with relative humidity below 43%, the emission of the film remains unchanged. With a gradual increase of the humidity of the environment to 84%, the emission gradually shifts to the blue (λem 688 nm at 76% and λem 660 nm at 84%) with similar emission intensity (Fig. 5a). Further increase of the humidity to 94%, the emission shifts to 641 nm. Moreover, the emission intensity increases significantly by ca. 27 folds (Fig. 5b). This change is due to the conversion of the dimeric form to the monomeric form. By monitoring the emission maximum, the maximum humidity of storing environment can be determined. This is particularly useful as an optical humidity sensor for packaged food, in which the water activity (relative humidity) of the storage environment is used as an indicator for food quality. For broken food packages, where food is exposed to high moisture levels that allow the growth of microorganisms, the sensing film becomes strongly emissive to indicate the food spoilage.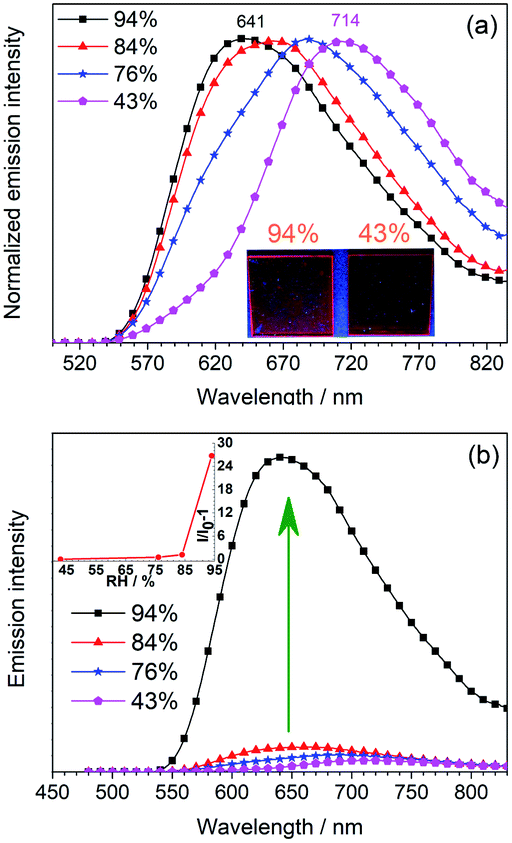 | ||
| Fig. 5 (a) Normalized emission spectra of spin-coated films of 1 in environments with relative humidity (RH) of 43%, 76%, 84% and 94%. Inset: photographs showing the emission of the films in the environments with RH of 43% and 94%. Excitation wavelength: 450 nm. (b) Emission spectra of the film in the environments with various RH. Inset: A plot of I/I0 − 1 versus RH. I0 = maximum emission intensity of the film in 43% RH. | ||
Luminescent sensor for amines
As discussed above, 1 would adopt a monomeric form in the presence of hydrogen-bonding donors. By loading a methanol solution of 1 on filter paper, the absorbed complex 1 on the filter paper (1Ms) is mainly the monomeric form. This is confirmed by its emission properties (λmax = 631 nm, Fig. 6) resembling that of the monomeric form in the solution state. With reference to the strong intermolecular hydrogen bonding interactions between the NH–C–N− moieties of the diaminocarbene ligands in the dimeric form, it is anticipated that the NH–C–N− moiety of the monomeric unit could also form strong hydrogen-bonding interactions with the (NH) group of amines. Upon exposure to ammonia and organic amines, the emission of 1 absorbed on filter paper (1Ms) changes significantly. The emission maximum of 1Ms rapidly shifts to the red from 631 nm to 723 nm with intensity decreases linearly with the concentration of ammonia of the environment from 1754 (2.7-fold) and to 3.96 × 105 ppm (48-fold) (Fig. 6a). | ||
| Fig. 6 (a) Emission spectra of the film upon exposure to different concentrations of NH3 vapor for 4 min. Inset: quenching efficiency Io/I of the film as a function of the vapor concentrations of NH3. (b) A plot of emission maximum changes (Δλmax) and (c) the changes of the emission intensities [(Iλmax-[Ru-A]/Ioλmax-[Ru-A])/(I631/Io631)] where Io and I are the emission intensities before and after exposure to the tested vapor, of 1Ms upon exposure to saturated ammonia and various organic amine vapors. | ||
The emission changes of 1Ms towards different types of saturated organic amines, butylamine (BUA), cyclohexylamine (CYA), isopropylamine (IPA), diethylamine (DEA), diisopropylamine (DIPA), and triethylamine (TEA), have been studied (Fig. S9, ESI†). Upon exposure to amine vapor for four minutes, the emission intensity decreases with the extent showing strong dependence on the nature and bulkiness of the amines. On the other hand, the emission is also red-shifted with Δλmax following the same order as the change in the emission intensity: BUA (103 nm) > CYA (102 nm) > IPA (98 nm) > DEA (83 nm) ≫ DIPA (19 nm) ≈ TEA (17 nm) (Fig. 6b and S9, ESI†). This trend does not correlate with the vapor pressure nor the basicity of the amines. In general, the extent of the change follows the order of primary amine > secondary amine > tertiary amine and reverse order of bulkiness. For all the studied primary amines, it follows the order of BUA > CYA > IPA. Thus, the least bulky butylamine induces the most drastic change of the emission intensity and maximum amongst all the studied organic amines.
As shown in the plot of [(Iλmax-[Ru-A]/Ioλmax-[Ru-A])/(I631/Io631)] (Fig. 6c), such changes are due to the decrease of emission band at 631 nm with a concomitant growth of new emission peaking at 713–734 nm. Because of the significant changes of the emission maximum induced by ammonia, primary and secondary amines, these changes are attributed to the formation of H-bonded carbene–amine adducts (Scheme 2). The sensitivity of the new emission band to the substituent on the amine further supports the adduct formation because the strength of the H-bonding is expected to be affected by the steric properties of the amine. The formation of H-bonded carbene–amine adduct is further verified by the isosbestic absorption in the UV-vis absorption spectral change in the titration of 1 (2 μM) with isopropylamine (IPA) (Fig. S10, ESI†). In the NMR study, the adduct is characterized by an upfield shift of NH proton of the diaminocarbene upon addition of isopropylamine (IPA) (Fig. S11, ESI†). In addition, the decrease of the diffusion coefficient in DOSY (Fig. S12, ESI†) after adding 75 mol equiv. of IPA is consistent with the decrease of molecular weight due to the shift of the dimerization equilibrium to the monomeric carbene–amine adduct. As these emission changes are due to the formation of the H-bonded carbene–amine adduct, the emission can be reverted when the amine vapor is removed (Fig. S13 and video, ESI†). Compared with other reported amine sensors,8 this amine sensor shows significantly improvement in its response time, stability, and reversibility, which can be reused many times and applied for real-time monitoring.
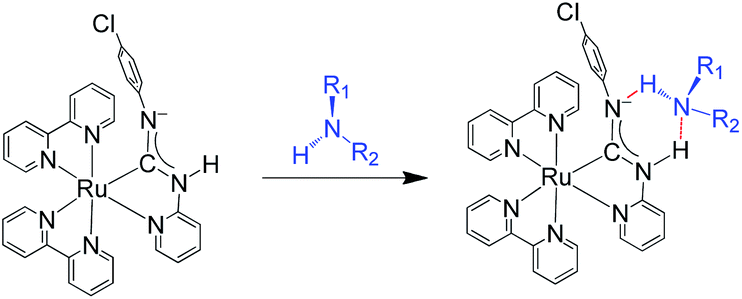 | ||
| Scheme 2 Formation of H-bonded carbene-amine adducts from 1Ms. | ||
In the case of very bulky secondary amine (DIPA) and tertiary amine (TEA), the carbene-amine adduct cannot be formed. At a result, the emission of 1Ms only shows minimal red-shift with much less decrease of the emission intensity after fuming with DIPA and TEA vapor (650 nm vs. 631 nm). As shown in Fig. 6c, the value of [(Iλmax-[Ru-A]/Ioλmax-[Ru-A])/(I631/Io631)] is close to 1 for DIPA and TEA, suggesting the quenching of the initial emission band.
Concealable optical data storage
Based on the readily distinguishable luminescence responses towards H-bonding donor and acceptor, a proof-of-principle for binary optical data storage15 has been proposed. A 5 × 8 dot matrix of 1 loaded on a filter paper (Fig. 7), as five 8-digit ASCII codes, is fabricated by dropping dichloromethane solution of 1 (5 mM). Unlike deposition by methanol solution, which displays strong orange phosphorescence of monomeric form, all the dots display weak dark red emission (Fig. 7). The emission of these dots could change from the dimeric form to orange phosphorescence of monomeric form by chemical encoding process with a drip of methanol from a glass capillary. The two different emission properties, weak dark red and bright orange emissions, can be used to store binary data of “0” and “1”, respectively. Since the methanol-induced conversion of the dimeric form to monomeric form involves the rearrangement of intermolecular packing, it is irreversible even after the removal of methanol. | ||
| Fig. 7 Photographs of the initial deposit 5 × 8 dots matrix (blank) and the methanol encoded, NH3 fumed, and methanol fumed matrix taken under UV irradiation showing the reversible concealment process of “CityU” in ASCII binary codes. | ||
As shown in Fig. 7, the initial deposit 5 × 8 dots translate into “00000”. The five ASCII codes for “CityU”, 01000011, 01101001, 01110100, 01111001, and 01010101, respectively, can be stored in the 5 × 8 dots by chemical encoding process with methanol. The data can be chemically concealed by fuming ammonia vapor for a few seconds, which converts all bright orange emissive dots into dark red emissive dots. This is due to the binding of the ammonia with the aminocarbene ligand of the emissive monomeric form (Scheme 2). After removing the NH3 vapor, the unbounded monomeric form can be regenerated, and thus the original bright orange emission can be restored to show the hidden ASCII codes. Such concealment–restoration processes are reversible for many cycles. The stored information can be cleared by fuming methanol vapor for 10 min, after which all dots show bright orange emission.
Conclusions
In conclusion, a new series of Ru(II) carbene complexes having environmental sensitive dimerization through intermolecular H-bonding have been successfully designed, synthesized, and characterized. Based on dimerization equilibrium analysis and time-resolved spectroscopy, the equilibrium, absorption and emission properties of the monomeric and dimeric forms have been elucidated. Using different coating/deposition techniques and conditions, thin films/depositions of the monomeric and dimeric forms can be fabricated. Importantly, the highly sensitive luminescent properties of these films towards various H-bonding donors and acceptors can serve as optical sensors for humidity monitoring and amine sensing, both of which are important in food safety and can be visualized by the naked eye. Taking the advantage of the multi-stimuli responsive properties of 1, a proof-of-principle for designing a binary optical data storage system with a reversible concealment process has been studied.Data availability
Experimental data including characterization data, NMR, absorption and emission spectra upon addition of IPA, dimerization analysis are in the figures or ESI.† Crystal data have been deposited at CCDC.Author contributions
J. Han performed most of the experiments, wrote the manuscript, and worked with S. C. Cheng on the photophysical study, vibrational spectroscopy, time-resolved spectroscopy, and data analysis. S. M. Yiu solved the X-ray crystal structures. M. K. Tse worked on the DOSY NMR study. C. C. Ko initiated, designed, and oversaw the project, discussed the results, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the General Research Fund (Project No. CityU 11306819 and CityU 11306820). J.-Q. Han acknowledges receipt of a University Postgraduate Studentship administrated by City University of Hong Kong.Notes and references
- (a) A. J. Metherell, C. Curty, A. Zaugg, S. T. Saad, G. H. Dennison and M. D. Ward, J. Mater. Chem. C, 2016, 4, 9664–9668 RSC; (b) B. Laramée-Milette, F. Nastasi, F. Puntoriero, S. Campagna and G. S. Hanan, Chem.–Eur. J., 2017, 23, 16497–16504 CrossRef PubMed; (c) M. Bar, S. Deb, A. Paul and S. Baitalik, Inorg. Chem., 2018, 57, 12010–12024 CrossRef CAS PubMed; (d) B. Chowdhury, S. Sinha and P. Ghosh, Chem.–Eur. J., 2016, 22, 18051–18059 CrossRef CAS PubMed; (e) D. R. Martir and E. Zysman-Colman, Chem. Commun., 2019, 55, 139–158 RSC; (f) J. S. Train, A. B. Wragg, A. J. Auty, A. J. Metherell, D. Chekulaev, C. G. P. Taylor, S. P. Argent, J. A. Weinstein and M. D. Ward, Inorg. Chem., 2019, 58, 2386–2396 CrossRef CAS PubMed; (g) Z. Zhang, G. J. Tizzard, J. A. G. William and S. M. Goldup, Chem. Sci., 2020, 11, 1839–1841 RSC.
- (a) D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed; (b) W. J. Finkenzeller, T. Hofbeck, M. E. Thompson and H. Yersin, Inorg. Chem., 2007, 46, 5076–5083 CrossRef CAS PubMed; (c) J. Lin, N.-Y. Chau, J.-L. Liao, W.-Y. Wong, C.-Y. Lu, Z.-T. Sie, C.-H. Chang, M. A. Fox, P. J. Low, G.-H. Lee and Y. Chi, Organometallics, 2016, 35, 1813–1824 CrossRef CAS; (d) E. Longhi, J. M. Fernandez-Hernandez, A. Lordache, R. Fröhlich, H.-P. Josel and L. de Cola, Inorg. Chem., 2020, 59, 7435–7443 CrossRef CAS PubMed; (e) V. W.-W. Yam, A. K.-W. Chan and E. Y.-H. Hong, Nat. Rev. Chem., 2020, 4, 528–541 CrossRef CAS.
- (a) R. Zhang, Z. Ye, Y. Yin, G. Wang, D. Jin, J. Yuan and J. A. Piper, Bioconjugate Chem., 2012, 23, 725–733 CrossRef CAS PubMed; (b) F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC; (c) F. Chen, F. Xiao, W. Zhang, C. Lin and Y. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 26964–26971 CrossRef CAS PubMed; (d) A. M.-H. Yip and K. K.-W. Lo, Coord. Chem. Rev., 2018, 361, 138–163 CrossRef CAS; (e) L. Wang, S. Monro, P. Cui, H. Yin, B. Liu, C. G. Cameron, W. Xu, M. Hetu, A. Fuller, S. Kilina, S. A. McFarland and W. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 3629–3644 CrossRef CAS PubMed; (f) K. L. Smitten, P. A. Scattergood, C. Kiker, J. A. Thomas and P. I. P. Elliott, Chem. Sci., 2020, 11, 8928–8935 RSC.
- (a) M. Zuo, W. Qian, T. Li, X.-Y. Hu, J. Jiang and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 39214–39221 CrossRef CAS PubMed; (b) L. Jia, B. Zhang, J. Xu, T. Zhu, R. Chen and F. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 19955–19964 CrossRef CAS PubMed; (c) X. Li, Y. Xie, B. Song, H.-L. Zhang, H. Chen, H. Cai, W. Liu and Y. Tang, Angew. Chem., Int. Ed., 2017, 56, 2689–2693 CrossRef CAS PubMed; (d) J. Han, J. Sun, Y. Li, Y. Duan and T. Han, J. Mater. Chem. C, 2016, 4, 9287–9293 RSC.
- (a) J. L. Pablos, S. Vallejos, A. Munoz, M. J. Rojo, F. Serna, F. C. Garcia and J. M. Garcia, Chem. – Eur. J., 2015, 21, 8733–8736 CrossRef CAS PubMed; (b) Y. Hu, X. Ma, Y. Zhang, Y. Che and J. Zhao, ACS Sens., 2016, 1, 22–25 CrossRef CAS; (c) M. Gao, S. Li, Y. Lin, Y. Geng, X. Ling, L. Wang, A. Qin and B. Z. Tang, ACS Sens., 2016, 1, 179–184 CrossRef CAS.
- Z. Wang, X. Yuan, S. Cong, Z. Chen, Q. Li, F. Geng and Z. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 15065–15072 CrossRef CAS PubMed.
- M.-J. Dai, Y.-L. Lin, H.-C. Lin, H.-W. Zan, K.-T. Chang, H.-F. Meng, J.-W. Liao, M.-J. Tsai and H. Cheng, Anal. Chem., 2013, 85, 3110–3117 CrossRef CAS PubMed.
- (a) A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420–1425 RSC; (b) X. Shen and B. Yan, J. Mater. Chem. C, 2015, 3, 7038–7044 RSC; (c) K. Vikrant, V. Kumar, K.-H. Kim and D. Kukkar, J. Mater. Chem. A, 2017, 5, 22877–22896 RSC; (d) J. Zhang, J. Ouyang, Y. Ye, Z. Li, Q. Lin, T. Chen, Z. Zhang and S. Xiang, ACS Appl. Mater. Interfaces, 2018, 10, 27465–27471 CrossRef CAS PubMed; (e) J. Ni, M.-Y. Li, Z. Liu, H. Zhao, J.-J. Zhang, S.-Q. Liu, J. Chen, C.-Y. Duan, L.-Y. Chen and X.-D. Song, ACS Appl. Mater. Interfaces, 2020, 12, 12043–12053 CrossRef CAS PubMed; (f) N. Marino, M. L. Calatayud, M. O. Arroyo, A. P. Álvarez, N. Moliner, M. Julve, F. Lloret, G. D. Munno, R. R. García and I. Castro, Magnetochemistry, 2021, 7, 65 CrossRef CAS.
- R. S. Singhal, P. R. Kulkarni and D. V. Rege, Handbook of Indices of Food Quality and Authenticity, Woodhead Pub., Cambridge, UK, 1997 Search PubMed.
- (a) W. Jeong, J. Song, J. Bae, K. R. Nandanapalli and S. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 44758–44763 CrossRef CAS PubMed; (b) Y. Kitamura, R. Ichikawa and H. Nakano, Mater. Chem. Front., 2018, 2, 90–95 RSC; (c) P. Kumar, R. Sakla, A. Ghosh and D. A. Jose, ACS Appl. Mater. Interfaces, 2017, 9, 25600–25605 CrossRef CAS PubMed; (d) Y. Cheng, J. Wang, Z. Qiu, X. Zheng, N. L. C. Leung, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2017, 29, 1703900 CrossRef PubMed.
- (a) S. Tunsrichon, C. Sukpattanacharoen, D. Escudero, N. Kungwan, S. Youngme and J. Boonmak, Inorg. Chem., 2020, 59, 6176–6186 CrossRef CAS PubMed; (b) Y. Yao, Y. Zhou, T. Zhu, T. Gao, H. Li and P. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 15338–15347 CrossRef CAS PubMed; (c) W. Xu, F. Li, Z. Cai, Y. Wang, F. Luob and X. Chen, J. Mater. Chem. C, 2016, 4, 9651–9655 RSC; (d) Y. Jiang, Y. Cheng, S. Liu, H. Zhang, X. Zheng, M. Chen, M. Khorloo, H. Xiang, B. Z. Tang and M. Zhu, Natl. Sci. Rev., 2021, 8, nwaa135 CrossRef CAS; (e) S. R. Halper and R. M. Villahermosa, ACS Appl. Mater. Interfaces, 2009, 1, 1041–1044 CrossRef CAS PubMed.
- (a) S. Abbas, I. Din, A. Raheel and A. T. Din, Appl. Organomet. Chem., 2020, 34, 5413 CrossRef; (b) D.-L. Ma, S. Lin, W. Wang, C. Yang and C.-H. Leung, Chem. Sci., 2017, 8, 878–889 RSC; (c) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- (a) M. D. Ward, Coord. Chem. Rev., 2006, 250, 3128–3141 CrossRef CAS and references therein; (b) S. G. Baca, H. Adams, C. S. Grange, A. P. Smith, I. Sazanovich and M. D. Ward, Inorg. Chem., 2007, 46, 9779–9789 CrossRef CAS PubMed; (c) T. L. Easun, W. Z. Alsindi, N. Deppermann, M. Towrie, K. L. Ronayne, X.-Z. Sun, M. D. Ward and M. W. George, Inorg. Chem., 2009, 48, 8759–8770 CrossRef CAS PubMed; (d) M. D. Ward, Dalton Trans., 2010, 39, 8851–8867 RSC; (e) H. Feng, K.-C. Chan, F. Zheng, C.-W. Lai, S.-M. Yiu and C.-C. Ko, Chem.–Eur. J., 2013, 19, 15190–15198 CrossRef CAS PubMed; (f) L. T.-L. Lo, S.-W. Lai, S.-M. Yiu and C.-C. Ko, Chem. Commun., 2013, 49, 2311–2313 RSC.
- (a) J. Han, Y.-K. Chun, S.-L. Chan, S.-C. Cheng, S.-M. Yiu and C.-C. Ko, CCS Chem., 2021, 3, 2345–2359 CrossRef; (b) J. Han, K.-M. Tang, S.-C. Cheng, C.-O. Ng, Y.-K. Chun, S.-L. Chan, S.-M. Yiu, M.-K. Tse, V. A. L. Roy and C.-C. Ko, Inorg. Chem. Front., 2020, 7, 786–794 RSC; (c) H. Na and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 6353–6360 CrossRef CAS PubMed; (d) H. Na, P.-N. Lai, L. M. Cañada and T. S. Teets, Organometallics, 2018, 37, 3269–3277 CrossRef CAS; (e) C.-O. Ng, S.-C. Cheng, W.-K. Chu, K.-M. Tang, S.-M. Yiu and C.-C. Ko, Inorg. Chem., 2016, 55, 7969–7979 CrossRef CAS PubMed.
- (a) H. B. Sun, S. J. Liu, W. P. Lin, K. Y. Zhang, W. Lv, X. Huang, F. W. Huo, H. R. Yang, G. Jenkins, Q. Zhang and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef PubMed; (b) L. Hu, Y. Duan, Z. Xu, J. Yuan, Y. Dong and T. Han, J. Mater. Chem. C, 2016, 4, 5334–5341 RSC; (c) T. Panda, D. K. Maiti and M. K. Panda, ACS Appl. Mater. Interfaces, 2018, 10, 29100–29106 CrossRef CAS PubMed; (d) J. Hai, H. Wang, P. Sun, T. Li, S. Lu, Y. Zhao and B. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 44664–44672 CrossRef CAS PubMed.
- J. M. Villegas, S. R. Stoyanov, W. Huang, L. L. Lockyear, J. H. Reibenspies and D. P. Rillema, Inorg. Chem., 2004, 43, 6383–6396 CrossRef CAS PubMed.
- (a) Hydrogen Bonding - New Insights, ed. S. J. Grabowski, Springer, 2006 Search PubMed; (b) W. C. Hamilton and J. A. Ibers, Hydrogen Bonding in Solids, Benjamin, New York, 1968 Search PubMed.
- (a) V. W.-W. Yam, C.-C. Ko, B. W.-K. Chu and N. Zhu, Dalton Trans., 2003, 3914–3921 RSC; (b) D. Schleicher, H. Leopold, H. Borrmann and T. Strassner, Inorg. Chem., 2017, 56, 7217–7229 CrossRef CAS PubMed; (c) J. Soellner, I. Císařová and T. Strassner, Organometallics, 2018, 37, 4619–4629 CrossRef CAS; (d) W. T. Kender and C. Turro, J. Phys. Chem. A, 2019, 123, 2650–2660 CrossRef CAS PubMed.
- For details of the dimerization equilibrium analysis, please refer to: (a) L. T.-L. Lo, C.-O. Ng, H. Feng and C.-C. Ko, Organometallics, 2009, 28, 3597–3600 CrossRef CAS; (b) L. T.-L. Lo, W.-K. Chu, C.-Y. Tam, S.-M. Yiu, C.-C. Ko and S.-K. Chiu, Organometallics, 2011, 30, 5873–5881 CrossRef CAS.
- K.-I. Sugawara, J. Miyawaki, T. Nakanaga, H. Takeo, G. Lembach, S. Djafari, H.-D. Barth and B. Brutschy, J. Phys. Chem., 1996, 100, 17145–17147 CrossRef CAS.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. zon Zelwewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS; (b) S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS.
- (a) J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 CrossRef CAS; (b) B. P. Sullivan, J. Phys. Chem., 1989, 93, 24–26 CrossRef CAS; (c) C.-C. Ko, A. W.-Y. Cheung, L. T.-L. Lo, J. W.-K. Siu, C.-O. Ng and S.-M. Yiu, Coord. Chem. Rev., 2012, 256, 1546–1555 CrossRef CAS; (d) C.-O. Ng, L. T.-L. Lo, S.-M. Ng, C.-C. Ko and N. Zhu, Inorg. Chem., 2008, 47, 7447–7449 CrossRef CAS PubMed; (e) C.-C. Ko, J. W.-K. Siu, A. W.-Y. Cheung and S.-M. Yiu, Organometallics, 2011, 30, 2701–2711 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details (synthetic procedures, characterization data, physical measurements and instrumentation, quantum yield determination), crystal data, powder XRD and simulated patterns of 1 and 3, selected bonding parameters for 1, 2 and 3, absorption spectra of the monomeric and dimeric form of 2, concentration-dependent absorption and emission spectra of 3, solid-state emission of the crystal sample and solvatochromic properties of 1, emission spectral changes of 1Ms upon addition of various amines, UV-vis absorption, 1H NMR, and DOSY NMR spectra of 1 after addition of IPA, and other supporting experimental data. CCDC 2076382–2076384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04074j |
| This journal is © The Royal Society of Chemistry 2021 |