
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitride protonation and NH3 binding versus N–H bond cleavage in uranium nitrides†
Megan
Keener
 ,
Rosario
Scopelliti
and
Marinella
Mazzanti
*
,
Rosario
Scopelliti
and
Marinella
Mazzanti
*
Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
First published on 18th August 2021
Abstract
The conversion of metal nitrides to NH3 is an essential step in dinitrogen fixation, but there is limited knowledge of the reactivity of nitrides with protons (H+). Herein, we report comparative studies for the reactions of H+ and NH3 with uranium nitrides, containing different types of ancillary ligands. We show that the differences in ancillary ligands, leads to dramatically different reactivity. The nitride group, in nitride-bridged cationic and anionic diuranium(IV) complexes supported by –N(SiMe3)2 ligands, is resistant toward protonation by weak acids, while stronger acids result in ligand loss by protonolysis. Moreover, the basic –N(SiMe3)2 ligands promote the N–H heterolytic bond cleavage of NH3, yielding a “naked” diuranium complex containing three bridging ligands, a nitride (N3−) and two NH2 ligands. Conversely, in the nitride-bridged diuranium(IV) complex supported by –OSi(OtBu)3 ligands, the nitride group is easily protonated to afford NH3, which binds the U(IV) ion strongly, resulting in a mononuclear U–NH3 complex, where NH3 can be displaced by addition of strong acids. Furthermore, the U–OSi(OtBu)3 bonds were found to be stable, even in the presence of stronger acids, such as NH4BPh4, therefore indicating that –OSi(OtBu)3 supporting ligands are well suited to be used when acidic conditions are required, such as in the H+/e− mediated catalytic conversion of N2 to NH3.
Introduction
Uranium nitrides were identified more than 100 years ago as active catalysts in the conversion of dinitrogen (N2) to ammonia (NH3),1 and very recently, the stoichiometric conversion of N2 into NH3 by molecular uranium complexes was reported.2 Notably, dinuclear uranium(III) complexes2a,3 or dinuclear U(IV) complexes combined with an external reducing agent,2b were capable of carrying out the four electron reduction of N2. Full cleavage of N2 to nitrides has also been reported,2c,4a,b where the resulting hydrazido and nitride ligands could be further reduced and protonated, yielding stoichiometric amounts of NH3.2,4bThe conversion of metal nitrides to NH3 is an essential step in dinitrogen fixation, where effective nitride protonation is crucial in building catalytic cycles.5 A key challenge in developing catalytic N2 reduction to NH3, is the simultaneous addition of reducing agent and acid (H+), while maintaining the structure of the catalyst, by avoiding ligand protonation and irreversible binding of NH3. However, studies addressing the reactivity of nitrides with protonating agents6 are limited to d-block metals, with few instances of imido (NH2−), amido (NH2−), and ammonia (NH3) bound products being isolated and characterized. Protonation of the ligand7 or the metal center,8 rather than the nitride, is common, and favoring protonation of the nitride can be challenging. The irreversible binding of NH3, or its reactivity with the metal complex, may also constitute an important disadvantage in catalyst design. Despite the inherent stability of the N–H bonds in NH3, activation under mild conditions can occur through ligand–metal cooperation,9 where the presence of basic ligands can result in the redox-neutral heterolysis of NH3 and subsequent ligand protonolysis. Facile N–H activation of NH3via metal–ligand cooperative addition was also reported in one instance for a uranium complex.10
Since the first examples of uranium nitrides were isolated in the gas phase11 and in solution,4a,12 uranium nitride complexes have been the subject of an increasing number of reports. Mononuclear, dinuclear, polynuclear, mono- and bis-nitride complexes that contain uranium in oxidation states ranging from (III) to (VI) have been synthesized.13 Several systems have revealed high reactivity towards small molecules, such as CO2, CO, H2, and N2,2a,b,3,13f,i,k,14a–q and demonstrated their ability to promote C–H bond activation.13m,15a–d Moreover, addition of excess strong acid to terminal or bridging uranium nitrides, derived from azide or N2, has been reported to yield variable amounts of ammonia (20–100%).2c,4b,13i,l,14o,q However, the isolation and characterization of partially protonated, intermediate species have not been reported thus far. Additionally, the site of first protonation, the parameters controlling the yield, or the fate of the uranium complex have not yet been elucidated.
We reasoned that investigating the protonation of uranium-nitrides, supported by different ancillary ligands, and their interaction with the resulting NH3, could provide insight into the parameters controlling the reactivity. This would allow us to harness significant elements for the design of complexes active in the H+/e− mediated catalytic conversion of N2 to NH3.
We have previously reported that –OSi(OtBu)3 are versatile ancillary ligands for the synthesis of bridging and terminal uranium nitrides in oxidation states ranging from (III) to (VI).13g,i,l,14q,16 Moreover, they have allowed the isolation of nitride and oxide bridged diuranium(III) complexes which promote the stoichiometric conversion of N2 to NH3.2a,3,4b We also found that using –OSi(OtBu)3 (complex A; Scheme 1)14h,i,lversus –N(SiMe3)2 (B and C)13k,m,17,18 as supporting ligands leads to significant differences in reactivity of analogous nitride and oxide bridged diuranium complexes with respect to reducing agents (KC8), electrophilic molecules such as CO2, CO, and H2 (Scheme 1), and N2 reduction.2a,18
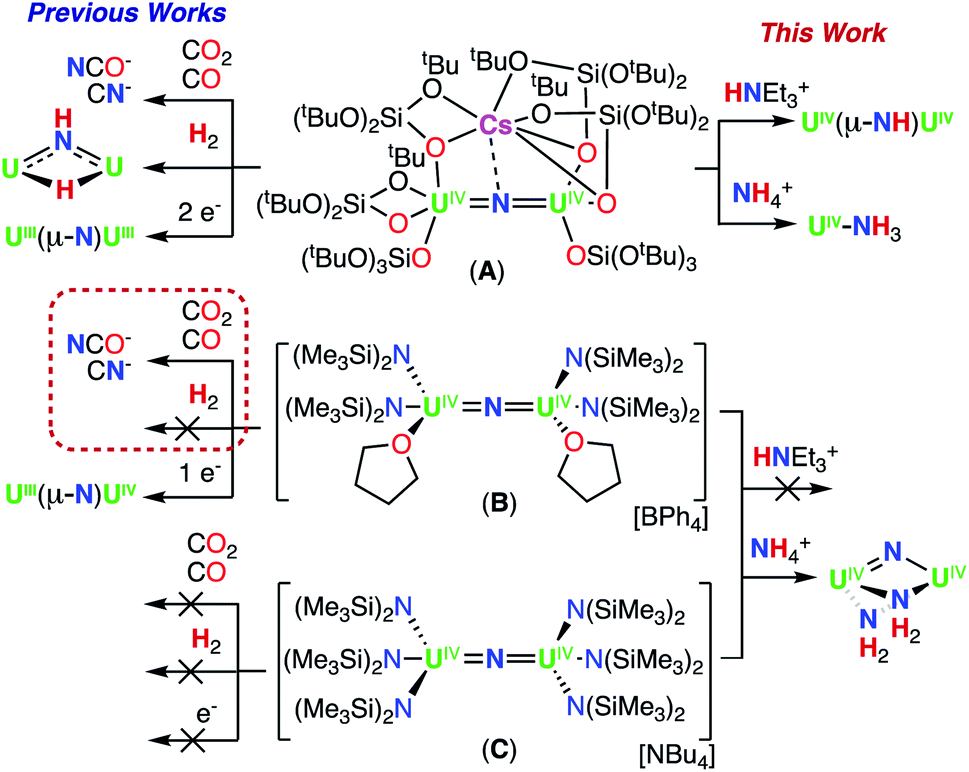 | ||
| Scheme 1 Previously reported dinuclear UIV-nitride complexes A, B, and C and their previous and currently described reactivity. | ||
Herein, we report the comparative studies for the reactions of H+ and NH3 with the diuranium(IV) bridging nitrides, A, B and C. We show that the different ancillary ligands lead to dramatically different reactivity, resulting in the isolation of a stable terminal NH3 complex (2) and a “naked” diuranium complex containing three bridging ligands, a nitride (N3−) and two NH2 ligands, providing important information on the species that can be formed during conversion of nitride to ammonia.
Results and discussion
Reactivity of OSi(OtBu)3-containing complexes
First, we were interested in probing the reactivity of the previously reported anionic bridging nitride (A), supported by –OSi(OtBu)3 ligands. The nitride link in this complex has been described to be more reactive towards electrophiles than in the analogous –N(SiMe3)2 complex C (Scheme 1) but its reactivity with acids was not investigated.13kAddition of 1.0 equiv. of NH4BPh4 to a solution of A in d8-THF at −40 °C resulted in the partial consumption of A and appearance of new resonances in the 1H NMR spectrum (Fig. S1†). While some resonances remain unidentified, two sets of resonances were assigned to the complexes [(UIV(OSi(OtBu)3)3(THF))2(μ-NH)] (1), which could be cleanly produced using the weaker acid, HNEt3BPh4 (Scheme 2 and the following section), and [UIV(OSi(OtBu)3)3(THF)2(NH3)][BPh4] (2; see below).
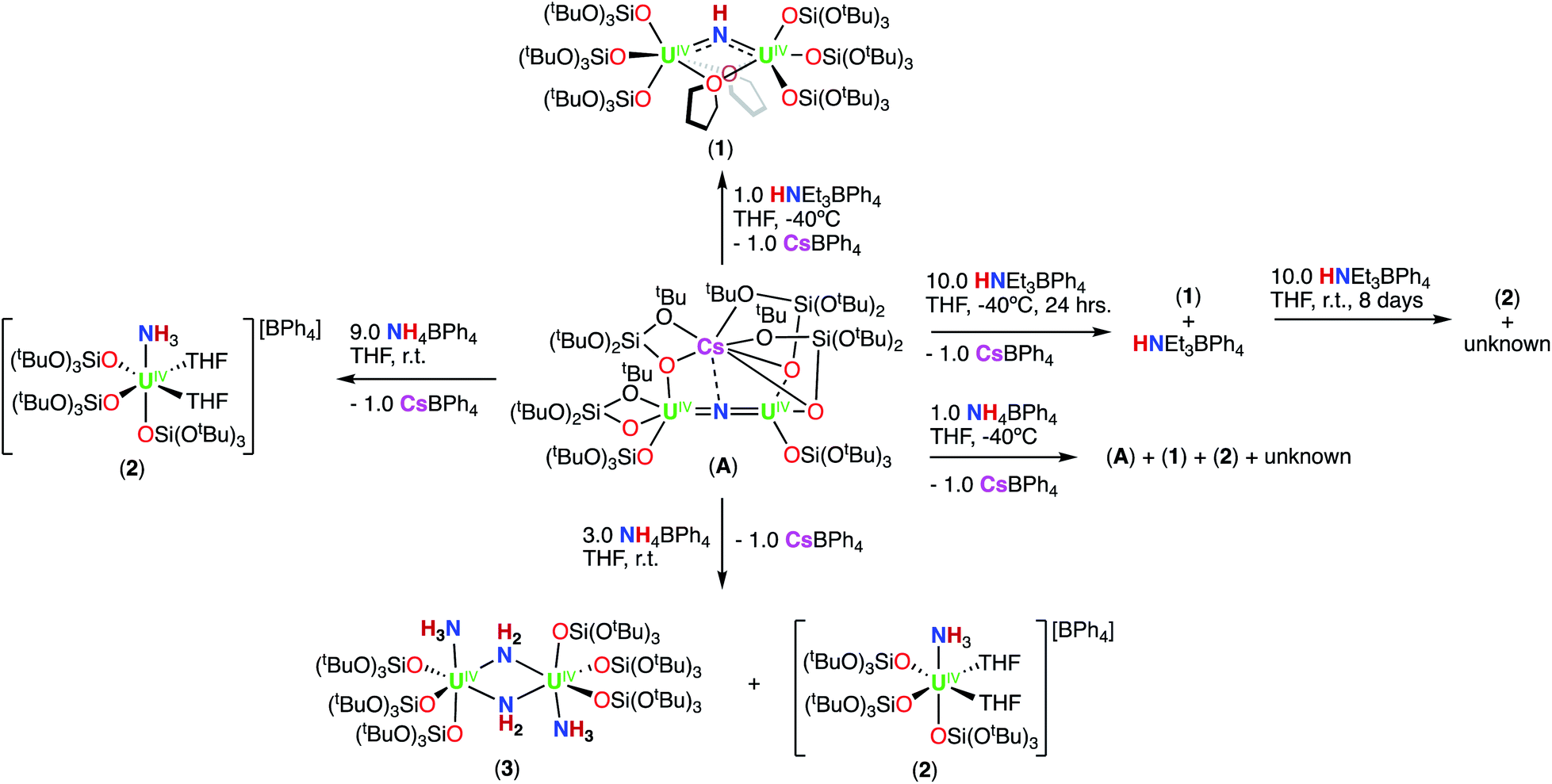 | ||
| Scheme 2 Reactivity of complex A toward various H+ (acid) sources. | ||
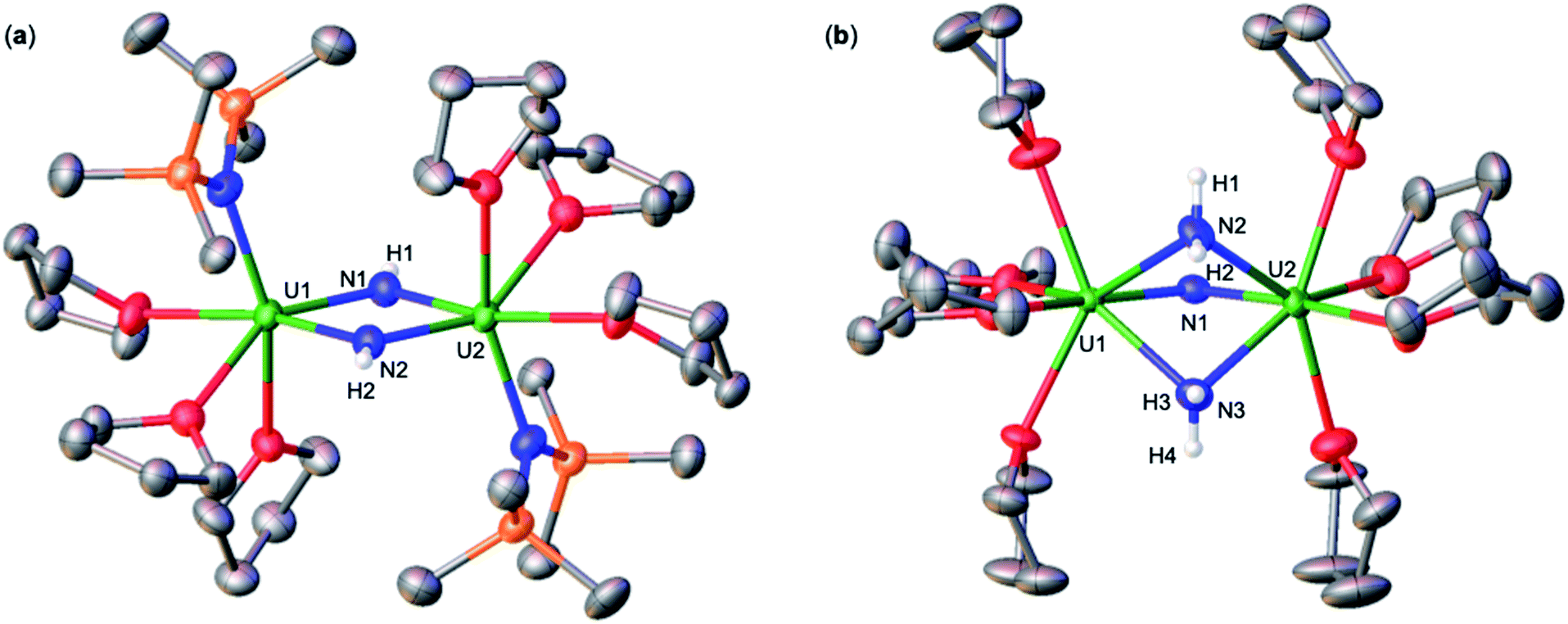
Addition of 3.0 equiv. of NH4BPh4 to a solution of A in d8-THF resulted in immediate formation of a teal solution and precipitation of a white solid. Crystals of two products were isolated at −40 °C from a concentrated Et2O solution of the reaction mixture and characterized by XRD analysis as the complexes [UIV(OSi(OtBu)3)3(THF)2(NH3)][BPh4] (2) (Fig. 1a), and [(UIV(OSi(OtBu)3)3(NH3))2(μ-NH2)2] (3) (Fig. 1b). Analysis by 1H NMR spectroscopy of the reaction mixture was consistent with the formation of complexes 2 and 3 in a 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. S2†). Few pure crystals of 3 could be obtained for 1H NMR spectroscopy, but attempts to isolate larger quantities of pure material remained unsuccessful, resulting in mixtures of both 2 and 3. The 1H NMR spectrum of complex 3 in d8-THF displays two signals at δ 5.1 and −5.6 ppm, corresponding to the –OSi(OtBu)3 ancillary ligands, and two resonances at −10.7 and −70.6 ppm assigned to the amido (NH21−) and NH3 resonances, respectively.
:1 ratio (Fig. S2†). Few pure crystals of 3 could be obtained for 1H NMR spectroscopy, but attempts to isolate larger quantities of pure material remained unsuccessful, resulting in mixtures of both 2 and 3. The 1H NMR spectrum of complex 3 in d8-THF displays two signals at δ 5.1 and −5.6 ppm, corresponding to the –OSi(OtBu)3 ancillary ligands, and two resonances at −10.7 and −70.6 ppm assigned to the amido (NH21−) and NH3 resonances, respectively.
 | ||
| Fig. 1 Molecular structures of (a) [UIV(OSi(OtBu)3)3(THF)2(NH3)][BPh4], 2, and (b) [(UIV(OSi(OtBu)3)3(NH3))2(μ-NH2)2], 3, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms on the –OSi(OtBu)3 ligands, methyl groups, and the BPh4 anion in (a) have been omitted for clarity. | ||
Addition of excess (9.0 equiv.) NH4BPh4 to a solution of A in d8-THF, resulted in a pale blue mixture. After 12 hours, the 1H NMR spectrum of this mixture shows only the presence of the signals assigned to complex 2 (Fig. S4†). Large plate, teal crystals suitable for XRD analysis of [UIV(OSi(OtBu)3)3(THF)2(NH3)][BPh4] (2), were obtained from a concentrated Et2O solution at −40 °C (Fig. 1b and Scheme 2), in 82% yield. The 1H NMR spectrum of complex 2 in d8-THF displays three aromatic BPh4 phenyl signals and one broad resonance at δ 9.6 ppm, corresponding to the –OSi(OtBu)3 ancillary ligands. The resonance at −160.1 ppm is assigned to the uranium bound NH3.
The clean formation of 2, from the reaction of A with excess NH4BPh4 and its isolation in 82% yield, cannot be explained only by the protonation of the nitride, but requires binding of NH3 released during protonation by NH4BPh4. In order to confirm that the NH3 ligand in 2, arises both from nitride protonation and from added NH4BPh4, we performed the reaction of A with isotopically enriched 15NH4BPh4 to yield 2-14/15N. Adding a solution of HCl in Et2O to 2-14/15N leads to the formation of both 15NH4Cl and 14NH4Cl (97% total yield), indicated by a doublet and triplet respectively in the 1H NMR spectrum. The ratio of 15NH4Cl:14NH4Cl is quite large (8:1 ratio), because of the large excess (9.0 equiv.) of 15NH4BPh4 used in the initial synthesis of A → 2-14/15N. The NH3 ligand in 2 binds quite strongly to the U(IV) center, as it could not be removed under dynamic vacuum for a few hours, but is removed upon addition of strong acids.
The isolation of complex 3, when 3.0 equiv. of NH4BPh4 are added to A, suggests that the formation of 2 proceeds through the bis-amido intermediate 3, which can be further protonated to yield the mononuclear terminal NH3 complex. We suggest that the formation of the bis-amido intermediate 3, involves a putative mono-amido complex, [(UIV(OSi(OtBu)3)3(NH3))2(μ-NH2)][BPh4] (3int) that undergoes ligand disproportionation to yield 2 and 3 (Scheme S1†).
In order to isolate intermediates with a lower degree of protonation, we investigated the reaction of A with HNEt3BPh4, a weaker acid compared to NH4BPh4. Addition of 1.0 equiv. of HNEt3BPh4 to a solution of A in d8-THF at −40 °C resulted in a green solution and precipitation of a white solid (CsBPh4). The 1H NMR spectrum of the reaction mixture reveals the complete consumption of A and the clean formation of a new species (Fig. S5†). Pale green crystals of [(UIV(OSi(OtBu)3)3)2(μ-THF)2(μ-NH)] (1) were obtained in 69% yield from a concentrated THF solution at −40 °C (Scheme 2 and Fig. 2). The 1H NMR spectrum of complex 1 in d8-THF displays a broad resonance at δ 0.72, corresponding the –OSi(OtBu)3 ancillary ligands, and another broad resonance at δ 150.3 ppm assigned to the imido (NH2−) group. A similar chemical shift (176.5 ppm) was observed for the imido group in the previously reported bis-imido bridged complex [K2{[U(OSi(OtBu)3)3]2(μ-NH)2}].13i
 | ||
| Fig. 2 Molecular structure of [(UIV(OSi(OtBu)3)3)2(μ-THF)2(μ-NH)][BPh4], 1, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms and methyl groups on the –OSi(OtBu)3 ligands have been omitted for clarity. | ||
We also investigated the addition of excess HNEt3BPh4 to A, in order to determine if protonation of the imido (NH2−) group in 1, would yield the NH3 complex, 2. Addition of excess (10 equiv.) HNEt3BPh4 to a solution of A in d8-THF at −40 °C resulted in the formation of a pale green solution with precipitation of a white solid (CsBPh4) (Scheme 2). The reaction mixture was brought to room temperature and analysis by 1H NMR spectroscopy revealed resonances consistent with the presence of 1 and unreacted HNEt3BPh4. Further stirring of the solution for an additional 2 days at room temperature resulted in a blue solution, with partial consumption of 1. After 8 days, 1H NMR spectroscopy indicated the complete consumption of 1 with appearance of the resonance at δ 9.6 ppm, assigned to the NH3 complex (2) in 40% yield, together with unidentified signals (Fig. S6a–d†). The absence of the resonance at δ −160.1 ppm, assigned to the uranium bound NH3, is attributed to the fast exchange of the NH3 ligand in the presence of excess HNEt3BPh4. We were able to confirm this by addition of 10.0 equiv. HNEt3BPh4 to a solution of 2 in d8-THF. Over 12 hours, we see that the signal corresponding to the uranium bound NH3 disappears (Fig. S7†). These results indicate that 1 is further protonated to the –NH3 containing species by excess HNEt3BPh4, but requires longer reaction times to yield 2. The formation of 2 (40% yield) in this reaction arises from cleavage of the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NU bridge, which requires the formation of other U-containing products that we were unable to identify.
NU bridge, which requires the formation of other U-containing products that we were unable to identify.
Structural characterization of OSi(OtBu)3-containing complexes
The solid-state molecular structures of complexes 1, 2, and 3 were determined by X-ray diffraction studies. The metrical parameters are presented in Table 1, including those previously reported for complex A.| Complex | A (ref. 14h) | 1 | 2 | 3 | B (ref. 13k) | 4 | 5 |
|---|---|---|---|---|---|---|---|
| U–Nnitride | 2.058(5) | — | — | — | 2.055(3) | — | 2.062(3); 2.018(3) |
| U–Nimido | — | NH: 2.243(7); 2.219(6) | — | — | — | NH: 2.179(5); 2.198(6) | — |
| U–Namido | — | — | NH3: 2.540(4) | NH3: 2.608(3) | — | NH2: 2.449(3); 2.455(3) | |
| NH2: 2.451(3); 2.463(2) | |||||||
| U–N–U | 170.2(3) | — | — | — | 168.97(14) | — | 107.41(12) |
| U–NH–U | — | 118.3(3) | — | — | — | 106.6(2) | — |
| U–NH2–U | — | — | — | 106.5(1) | — | — | 84.36(9) |
Complex 1 crystalizes in the space group C2/c with one molecule per asymmetric unit. The solid-state molecular structure of 1 shows the presence of a neutral diuranium(IV) complex where each uranium is bound by three –OSi(OtBu)3 ancillary ligands and are bridged by an imido (NH2−) ligand and two THF molecules (Fig. 2). The U–N–U bond angle changes dramatically from linear in A (U–N–U: 170.2(3)°) to bent in 1 (118.3(3)°). The U–N bond distances in 1 are elongated (U1–N1: 2.243(7), U2–N1: 2.219(6) Å) in comparison to A, consistent with a bridging imido (μ-NH).14l
Complex 2 crystalizes in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule per asymmetric unit. Its solid-state molecular structure shows the presence of an ion pair consisting of one BPh4− anion and the [UIV(OSi(OtBu)3)3(THF)2(NH3)] cation comprising of a terminal UIV–NH3 bound ligand (Fig. 1a). Only two examples of crystallographically characterized U–NH3 complexes have been reported so far: the bis(l,1,1,5,5,5-hexafluoropentane-2,4-dionato)NH3-uranyl(VI)19 and a series of trinuclear complexes of formula [(NH3)8U(μ-N)(NH3)3(X)2U(μ-N)U(NH3)8]Yn·ZNH3 (X = NH3, Br−, or Cl−; n = 6–8; Y = Cl− or Br−; Z = 26, 21, or 6).20 Where the U–NH3 bond distances in these examples, 2.48(6)19 and 2.605(3)20 respectively, are consistent with the U1–N1 (2.540(4) Å) bond distance in complex 2. Additionally, the U–NH3 bond distance in 2 is dramatically elongated in comparison to previously reported terminal amido (NH2) complexes, [U(1,2,4-(tBu)3C5H2)2(NH2)2] (U–Namido; 2.228(4) Å), [U(TrenTIPS)(NH2)] (U–Namido; 2.194(5) Å),21 and [U(COTTIPS2)(Cp*)(NH2)] (U–Namido; 2.217(4) Å),10 further supporting a terminal bound NH3 molecule. The protons of the NH3 ligand can be crystallographically identified, making their assignment unambiguous.
with one molecule per asymmetric unit. Its solid-state molecular structure shows the presence of an ion pair consisting of one BPh4− anion and the [UIV(OSi(OtBu)3)3(THF)2(NH3)] cation comprising of a terminal UIV–NH3 bound ligand (Fig. 1a). Only two examples of crystallographically characterized U–NH3 complexes have been reported so far: the bis(l,1,1,5,5,5-hexafluoropentane-2,4-dionato)NH3-uranyl(VI)19 and a series of trinuclear complexes of formula [(NH3)8U(μ-N)(NH3)3(X)2U(μ-N)U(NH3)8]Yn·ZNH3 (X = NH3, Br−, or Cl−; n = 6–8; Y = Cl− or Br−; Z = 26, 21, or 6).20 Where the U–NH3 bond distances in these examples, 2.48(6)19 and 2.605(3)20 respectively, are consistent with the U1–N1 (2.540(4) Å) bond distance in complex 2. Additionally, the U–NH3 bond distance in 2 is dramatically elongated in comparison to previously reported terminal amido (NH2) complexes, [U(1,2,4-(tBu)3C5H2)2(NH2)2] (U–Namido; 2.228(4) Å), [U(TrenTIPS)(NH2)] (U–Namido; 2.194(5) Å),21 and [U(COTTIPS2)(Cp*)(NH2)] (U–Namido; 2.217(4) Å),10 further supporting a terminal bound NH3 molecule. The protons of the NH3 ligand can be crystallographically identified, making their assignment unambiguous.
Complex 3 crystalizes in the space group P with 0.5 molecules per asymmetric unit. Its solid-state molecular structure can be described as a neutral complex consisting of a dinuclear UIV/UIV core, comprising of two [UIV(OSi(OtBu)3)3(NH3)] units bridged by two amido (NH21−) ligands (Fig. 1b). The U1–N1 and U2–N4 (2.608(3) Å) bond distances are consistent with a terminal NH3, as found in 2 (U1–N1: 2.540(4) Å). The U1–N2 = 2.451(3) Å and U1–N3 = 2.463(2) Å bond distances are consistent with two bridging amido (NH21−) ligands,14q with delocalized charge on the uranium ions as indicated by the nearly symmetrical U–Namido metrical parameters. The protons of the imido ligands can be crystallographically identified, making their assignment unambiguous.
Next, we investigated protonation of the previously reported dinuclear UIV-nitride complexes bearing more basic –N(SiMe3)2 ancillary ligands.
Reactivity of N(SiMe3)2-containing complexes
First, we probed the reactivity of complex, B with CO2, CO, and H2 to compare it with that of complexes A and C (Scheme 1). Previous work has shown that B has an increased reactivity toward reducing agents in comparison to the all –N(SiMe3)2, complex C, but the reactivity with CO2, CO, and H2 was not explored in the previous report.13m We found that complex B reacts with 2.0 equiv. of 13CO2 in d8-THF over 4 days, in which B is fully consumed and results in a complicated 1H NMR spectrum (Fig. S11†). Attempts to grow single crystals from the reaction mixture were unsuccessful. However, after the removal of volatiles and hydrolysis with D2O (pD = 12) of the reaction residue, N13CO− was identified by 13C NMR spectroscopy (Fig. S12†), similarly to what observed for complex A.Complex B reacts with 2.0 equiv. of 13CO in d8-THF over 6 days, in which B is fully consumed and results in a complicated 1H NMR spectrum (Fig. S13†). Attempts to grow crystals from the reaction mixture were unsuccessful. However, after quenching the reaction mixture with D2O (pD = 12), 13CN− was identified by 13C NMR spectroscopy (Fig. S14†). This indicates that, as previously reported for A, which reacts readily with CO to yield [Cs(U(OSi(OtBu)3)3)2(μ-CN)(μ-O)],14i the nucleophilic character of the bridging nitride promotes the cleavage and deoxygenation of CO to afford a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond and a bridging oxo. Conversely, the all –N(SiMe3)2, complex C, has been previously shown to be unreactive in the presence of stoichiometric and excess quantities of CO.13k These results suggest an increased nucleophilicity of the nitride in the cationic complex B, compared to the anionic complex C. However, complex B does not react with H2 similarly to what was found for complex A (Fig. S15†). The lack of reactivity with H2 indicates a decreased nucleophilicity of the nitride in B compared to A, which was found to activate H2 yielding the first example of a bridging imido-hydride complex, [Cs(U(OSi(OtBu)3)3)2(μ-NH)(μ-H)].14l Therefore, we became interested in how these differences in reactivity would extend to the reaction with acids (HNEt3BPh4 and NH4BPh4).
N triple bond and a bridging oxo. Conversely, the all –N(SiMe3)2, complex C, has been previously shown to be unreactive in the presence of stoichiometric and excess quantities of CO.13k These results suggest an increased nucleophilicity of the nitride in the cationic complex B, compared to the anionic complex C. However, complex B does not react with H2 similarly to what was found for complex A (Fig. S15†). The lack of reactivity with H2 indicates a decreased nucleophilicity of the nitride in B compared to A, which was found to activate H2 yielding the first example of a bridging imido-hydride complex, [Cs(U(OSi(OtBu)3)3)2(μ-NH)(μ-H)].14l Therefore, we became interested in how these differences in reactivity would extend to the reaction with acids (HNEt3BPh4 and NH4BPh4).
Next, we probed the reactivity of cationic bridging nitride (B), which was synthesized as reported by our group13m by successive protonolysis of one –N(SiMe3)2 ligand in the complex D, previously reported by Fortier and Hayton,13b and of the metal-amide methanide bond in E, using a total of 2.0 equiv. of HNEt3BPh4 (Scheme 3). Addition of excess (4.0 equiv.) HNEt3BPh4 to a solution of B in d8-THF, resulted in unreacted starting materials as seen by 1H NMR spectroscopy (Fig. S16†). Therefore, we pursued the protonation using the stronger acid, NH4BPh4, resulting in different reactivity compared to what was found for the –OSi(OtBu)3 complex, A.
 | ||
| Scheme 3 Reactivity of complexes B, C, D, and E toward various H+ (acid) sources. | ||
1H NMR studies showed that addition of 1.0 equiv. of NH4BPh to a solution of B in d8-THF resulted in the formation of a new species displaying a resonance at δ 12.6 ppm and unreacted B (Fig. S17†). Golden crystals suitable for XRD analysis were obtained from the reaction mixture at −40 °C after 12 hours. The solid-state molecular structure shows the presence of a dinuclear UIV/UIV bridging bis-imido (NH2−) complex, [(UIV(N(SiMe3)2)(THF)3)2(μ-NH)2][BPh4]2 (4) (Fig. 3a and Scheme 3). The product is insoluble in most common solvents including toluene, Et2O, THF, and n-hexanes, but is soluble in pyridine. The 1H NMR spectrum of the isolated crystals of 4 in d5-pyridine, displays a resonance at δ 13.2 ppm, three aromatic BPh4 phenyl signals, and THF resonances due to the displacement upon coordination of d5-pyridine, but were unable to identify resonances for the imido (NH2−) groups (Fig. S18†).
 | ||
| Fig. 3 Molecular structures of (a) [(UIV(N(SiMe3)2)(THF)3)2(μ-NH)2][BPh4]2, 4, and (b) [(UIV(THF)4)2(μ-N)(μ-NH2)2][BPh4]3, 5, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms and methyl groups on the ancillary ligands, and BPh4 counterions have been omitted for clarity. | ||
Attempts to isolate 4 analytically pure failed most likely due to the cocrystallization of B. Recrystallization of a pyridine solution containing 4, by slow diffusion of Et2O at −40 °C, resulted in single crystals suitable for XRD analysis, identifiable as the pyridine adduct of 4, [(UIV(N(SiMe3)2)(pyridine)3)2(μ-NH)2][BPh4]2 (4-pyr; Fig. S24†). All metrical parameters are analogous to the THF adduct of 4. Attempts to isolate larger quantities of 4-pyr proved to be unsuccessful.
1H NMR studies showed that addition of 2.0 equiv. NH4BPh4 to B in d8-THF after 20 minutes, results in the partial disappearance of the signals of B, and in the appearance of the resonance at δ 12.6 ppm assigned to 4. The 1H NMR spectrum of the reaction mixture measured after 12 hours showed complete disappearance of the signals corresponding to B and to 4, with concomitant appearance of the resonance assigned to HN(SiMe3)2 (Fig. S19a and b†). Furthermore, the reaction mixture, after standing 12 hours at −40 °C, afforded gold crystals of [(UIV(THF)4)2(μ-N)(μ-NH2)2][BPh4]3 (5) in 83% yield (Fig. 3b and Scheme 3). The product is insoluble in most common solvents including toluene, Et2O, THF, and n-hexanes, but is soluble in pyridine. The 1H NMR spectrum of complex 5 in d5-pyridine displays three aromatic BPh4 phenyl signals, THF resonances due to the displacement upon coordination of d5-pyridine, and two amido (NH21−) resonances at −468 ppm. Additionally, complex 5 can also be obtained in 89% yield through direct addition of 3.0 equiv. NH4BPh4 to complex E. Therefore, isolating complex B is not required for the synthesis of complex 5 (Scheme 3).
The presence of two amido (NH2−) bridging groups in 5, can be interpreted in terms of the binding and concomitant N–H cleavage of two NH3 molecules. This involves protonation of the UIV–N(SiMe3)2 bonds, resulting in the release of four molecules of HN(SiMe3)2. To date, there is only one other example of the facile N–H activation of NH3 by an uranium complex. The UIV “tucked-in” mixed-sandwich complex, [U(η-COTTIPS2)(η5:κ1-C5Me4CH2)], was reported to promote the ligand assisted NH3 activation yielding the terminal UIV–NH2 complex [U(COTTIPS2)Cp*(NH2)].22 Uranium complexes of primary amides remain rare,21 and complex 5 provides the second example of a bridging amide.13a
In order to confirm that the two bridging amido (NH21−) moieties in 5, are derived from NH3, we prepared the 15N labeled analogue 5-14/15N by reacting the isotopically enriched 15NH4BPh4 with B. Adding a solution of HCl in Et2O to 5-14/15N leads to the formation of both 15NH4Cl and 14NH4Cl (95% yield), indicated by a doublet and triplet respectively in the 1H NMR spectrum (2:1 ratio, 15NH4Cl:14NH4Cl).
These results indicate that formation of the bridging nitride bis-amido complex (5) occurs via protonation of two –N(SiMe3)2 ligands, binding of the resulting NH3, and subsequent N–H heterolysis of NH3 assisted by the remaining two basic –N(SiMe3)2 ligands. We suggest that the reaction proceeds first through the imido (NH2−) intermediate (4), which arises from protonation of one UIV–N(SiMe3)2 bond and N–H heterolysis of one NH3 molecule with concomitant elimination of two HN(SiMe3)2 ligands. This is likely to occur via a nitride-amido bridged complex, [(UIV(N(SiMe3)2)(THF))2(μ-NH2)(μ-N)][BPh4]2 (4int) but the higher nucleophilicity of the nitride, compared to B, results in a proton redistribution, yielding complex 4 (Scheme S2†). Further protonation of a UIV–N(SiMe3)2 bond in 4, followed by NH3 binding and N–H heterolysis, should afford a bis-imido, mono-amido complex, [(UIV(THF)4)2(μ-NH)2(μ-NH2)][BPh4]3 (5int), but subsequent rearrangement of the protons instead yields complex 5. Such redistribution of protons can be explained in terms of an increased stability of the nitride compared to the imide when two N(SiMe3)2 ligands are replaced by two THF and one amido ligands. Previous studies demonstrated the important effect of ancillary ligands on the stability and reactivity of bridging uranium nitrides.13k
Next, we probed the protonation and reactivity of the neutral nitride complex, C. Complex C is analogous to B in that it contains –N(SiMe3)3 ancillary ligands, but has two additional ligands within the complex. Previous studies from our group showed that C is less reactive towards small molecule activation in comparison to complexes B and A (Scheme 1). This was interpreted, based on DFT studies, in terms of a decreased nucleophilicity of the nitride moiety, due to an increased bond order of the bridging nitride in the UIV–N(SiMe3)3 complex C compared to the UIV–OSi(OtBu)3 complex A.13k Therefore, we investigated if protonation of the amide ligands and NH3 activation could also occur in complex C.
Similar to the reactivity of B with HNEt3BPh4, addition of 4.0 equiv. of HNEt3BPh4 to a solution of C in d8-THF resulted in no reaction (Fig. S20†). Alternatively, treatment with 3.0 equiv. NH4BPh4 resulted in an immediate color change from brown to golden yellow. Within 5 minutes, golden crystals of 5 suitable for XRD analysis were obtained in 72% yield. Analysis by 1H NMR spectroscopy and elemental analysis indicated 5 is the only product isolated, showing similar reactivity to complex B. This suggests that the previously determined unreactive nature of C can be circumvented by use of a strong acid, promoting a series of protonation/protonolysis reactions with subsequent N–H heterolysis of NH3 to 5.
Structural characterization of N(SiMe3)2-containing complexes
The solid-state molecular structures of complexes 4 and 5 were determined by X-ray diffraction studies. The metrical parameters are presented in Table 1, including those previously reported for complex B.Complex 4 crystalizes in the space group P with 0.5 molecules per asymmetric unit. Its solid-state structure can be described as an ion pair consisting of two BPh4− anions and a UIV/UIV mixed dication comprising of two [(UIV(N(SiMe3)2)(THF)3)2] units bridged by two imido (NH2−) ligands. The U1–N1 = 2.198(6) Å and U1–N2 = 2.179(5) Å bond distances are similar and fully consistent with bridging imido (NH2−) ligands (Fig. 3a).13i All distances are consistent with delocalized charge on the uranium ions as indicated by the nearly symmetrical U–Nimido metrical parameters. The protons of the amido ligands can be crystallographically identified, making their assignment unambiguous.
Complex 5 crystalizes in the space group P21/c with one molecule per asymmetric unit. The solid-state molecular structure of 5 shows the presence of an ion pair consisting of three BPh4− anions and a UIV/UIV trication comprising of two [(UIV(THF)4)] units bridged by one nitrido (N3−) and two amido (NH21−) ligands, forming a face-sharing geometry (Fig. 3b). The U1–N1 and U2–N1 (2.062(3); 2.018(3) Å) bond distances are consistent with the presence of a bridging nitride (μ-N).13b The U–N–U bond angle changes dramatically from linear in B (U–N–U: 168.97(14)°) to highly bent in the diamond core geometry of 5 (107.41(12)°), and is analogous to the previously reported complex D (Scheme 3).13b,m The U1–N2 = 2.455(3) Å and U1–N3 = 2.449(3) Å bond distances are consistent with two bridging amido (NH21−) ligands, with nearly symmetrical U–Namido metrical parameters. The protons of the NH2 ligands can be crystallographically identified, making their assignment unambiguous. The coordination around the U(IV) ions is unprecedented for molecular nitride complexes as it contains only solvent molecules and bridging amido ligands, as all ancillary –N(SiMe3)3 ligands have been removed upon reaction with NH4BPh4.
Conclusion
In summary, we have investigated the reactions of H+ and NH3 with a series of nitride bridged diuranium(IV) complexes differing in the type and number of supporting ligands.We found that the nitride ligand is easily protonated by 1.0 equiv. of weak acid (HNEt3BPh4) in the –OSi(OtBu)3 supported complex A, yielding an imido bridged complex 1. Further protonation of the imido moiety to a terminal NH3 complex, 2, can be achieved by using a large excess of weak acid and longer reaction time, or by means of a stronger acid. These results indicate that the uranium-nitride bond is easily protonated to afford NH3. Ammonia binds the U(IV) ion strongly in the resulting mononuclear U–NH3 complex 3, but can be displaced by addition of strong acid. Furthermore, the U–OSi(OtBu)3 bonds were found to be stable, even in the presence of stronger acids, such as NH4BPh4, therefore indicating that –OSi(OtBu)3 supporting ligands are well suited for use when acidic conditions are required, such as in the H+/e− mediated catalytic conversion of N2 to NH3.5j,23
Conversely, a very different reactivity is observed for both the cationic and anionic nitride bridged complexes, B and C, supported by –N(SiMe3)2 ligands. Opposed to complex A, the nitride in both B and C are more resistant toward protonation by acids. For example, when a weak acid, such as HNEt3BPh4, is employed, the nitride in these complexes is unreactive. This is consistent with the lesser nucleophilic character of the bridging nitride in N(SiMe3)2-containing complexes compared to the analogous –OSi(OtBu)3 systems, which was also supported by their reactivity toward small molecules (CO, CO2 or H2). Alternatively, addition of the stronger acid, NH4BPh4, resulted in the complete loss of –N(SiMe3)2 supporting ligands, while the bridging nitride remains intact. Moreover, the basic –N(SiMe3)2 ligands promote the N–H heterolytic cleavage of NH3, yielding a stable bis-NH2, mono-nitride bridged complex (5), where only ancillary solvent molecules support the metal center. These results demonstrate that basic supporting ligands, such as –N(SiMe3)2, present several disadvantages compared to the –OSi(OtBu)3 ligands for usage in the development of catalysts for N2 conversion to NH3. Utilizing OSi(OtBu)3-containing complexes is not only advantageous for their resistance toward acids, but also for the high reactivity of bound nitrides to yield NH3. In contrast, N(SiMe3)2-supported complexes may be of interest for studies pertaining to the heterolytic bond activation of NH3.
Author contributions
M. K. carried out the synthetic experiments and analysed the experimental data. R. S. carried out the X-ray single crystal structure analyses. M. M. originated the central idea, coordinated the work, and analysed the experimental data. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 200021_178793 and the École Polytechnique Fédérale de Lausanne (EPFL). We thank F. Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses.Notes and references
- (a) F. Haber, Ammonia, German patent DE 229126, 1909; (b) F. Haber, Angew. Chem., 1914, 27, 473–477 CrossRef CAS.
- (a) M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS; (b) P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS PubMed; (c) X. Q. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed.
- M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS.
- (a) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433–3436 CrossRef CAS; (b) N. Jori, L. Barluzzi, I. Douair, L. Maron, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2021, 143(29), 11225–11234 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed; (b) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed; (c) B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS; (d) Y. Nishibayashi, Nat. Chem., 2011, 3, 502–504 CrossRef CAS PubMed; (e) C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC; (f) K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 5666–5669 CrossRef CAS PubMed; (g) N. B. Thompson, M. T. Green and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 15312–15315 CrossRef CAS PubMed; (h) Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS; (i) S. J. K. Forrest, B. Schluschass, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed; (j) F. Masero, M. A. Perrin, S. Dey and V. Mougel, Chem.–Eur. J., 2021, 27, 3892–3928 CrossRef CAS.
- (a) J. J. Scepaniak, J. A. Young, R. P. Bontchev and J. M. Smith, Angew. Chem., Int. Ed., 2009, 48, 3158–3160 CrossRef CAS PubMed; (b) K. C. MacLeod, S. F. McWilliams, B. Q. Mercado and P. L. Holland, Chem. Sci., 2016, 7, 5736–5746 RSC; (c) B. M. Lindley, Q. J. Bruch, P. S. White, F. Hasanayn and A. J. M. Miller, J. Am. Chem. Soc., 2017, 139, 5305–5308 CrossRef CAS PubMed; (d) A. K. Hickey, L. A. Wickramasinghe, R. R. Schrock, C. Tsay and P. Muller, Inorg. Chem., 2019, 58, 3724–3731 CrossRef CAS PubMed.
- T. J. Hebden, R. R. Schrock, M. K. Takase and P. Muller, Chem. Commun., 2012, 48, 1851–1853 RSC.
- F. S. Schendzielorz, M. Finger, C. Volkmann, C. Wurtele and S. Schneider, Angew. Chem., Int. Ed., 2016, 55, 11417–11420 CrossRef CAS.
- (a) E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS PubMed; (b) T. Shima and Z. M. Hou, J. Chem. Soc., Dalton Trans., 2010, 39, 6858–6863 RSC; (c) Y. H. Chang, Y. Nakajima, H. Tanaka, K. Yoshizawa and F. Ozawa, J. Am. Chem. Soc., 2013, 135, 11791–11794 CrossRef CAS PubMed; (d) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS; (e) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS; (f) M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef CAS; (g) L. Nurdin, Y. Yang, P. G. N. Neate, W. E. Piers, L. Maron, M. L. Neidig, J. B. Lin and B. S. Gelfand, Chem. Sci., 2021, 12, 2231–2241 RSC.
- J. A. H. Frey, G. N. Cloke and S. M. Roe, Organometallics, 2015, 34, 2102–2105 CrossRef.
- (a) D. W. Green and G. T. Reedy, J. Chem. Phys., 1976, 65, 2921–2922 CrossRef CAS; (b) R. D. Hunt, J. T. Yustein and L. Andrews, J. Chem. Phys., 1993, 98, 6070–6074 CrossRef CAS; (c) L. Andrews, X. Wang, R. Lindh, B. O. Roos and C. J. Marsden, Angew. Chem., Int. Ed., 2008, 47, 5366–5370 CrossRef CAS.
- (a) W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835–1838 CrossRef CAS PubMed; (b) G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2008, 47, 3040–3042 CrossRef CAS PubMed.
- (a) L. Barluzzi, F. C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC; (b) S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed; (c) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS; (d) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS; (e) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed; (f) D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266, 2–15 CrossRef; (g) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS; (h) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed; (i) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC; (j) J. Du, D. M. King, L. Chatelain, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC; (k) C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC; (l) L. Barluzzi, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 19047–19051 CrossRef CAS PubMed; (m) C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed; (n) M. D. Straub, L. M. Moreau, Y. S. Qiao, E. T. Ouellette, M. A. Boreen, T. D. Lohrey, N. S. Settineri, S. Hohloch, C. H. Booth, S. G. Minasian and J. Arnold, Inorg. Chem., 2021, 60, 6672–66790 CrossRef CAS PubMed; (o) L. Maria, I. C. Santos, V. R. Sousa and J. Marcalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed.
- (a) A. L. Odom, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 1998, 120, 5836–5837 CrossRef CAS; (b) P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS; (c) F. Geoffrey, N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef; (d) W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS PubMed; (e) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS; (f) S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS; (g) P. D. Dau, P. B. Armentrout, M. C. Michelini and J. K. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 7334–7340 RSC; (h) M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 4074–4078 CrossRef CAS PubMed; (i) M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290–12294 CrossRef CAS; (j) M. D. Walter, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef CAS; (k) P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem.–Eur. J., 2017, 23, 2950–2959 CrossRef CAS; (l) M. Falcone, L. N. Poon, F. F. Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS; (m) E. Lu, B. E. Atkinson, A. J. Wooles, J. T. Boronski, L. R. Doyle, F. Tuna, J. D. Cryer, P. J. Cobb, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, N. Kaltsoyannis and S. T. Liddle, Nat. Chem., 2019, 11, 806–811 CrossRef CAS; (n) M. A. Boreen, G. D. Rao, D. G. Villarreal, F. A. Watt, R. D. Britt, S. Hohloch and J. Arnold, Chem. Commun., 2020, 56, 4535–4538 RSC; (o) L. Chatelain, E. Louyriac, I. Douair, E. L. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 337 CrossRef CAS; (p) D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS; (q) L. Barluzzi, F. C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096–8104 RSC.
- (a) R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS; (b) P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kuhn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS; (c) K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M. H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS; (d) M. Yadav, A. J. Metta-Magana and S. Fortier, Chem. Sci., 2020, 11, 2381–2387 RSC.
- C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS.
- D. K. Modder, C. T. Palumbo, I. Douair, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2021, 12, 6153–6158 RSC.
- D. K. Modder, C. T. Palumbo, I. Douair, F. Fadaei-Tirani, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2021, 60, 3737–3744 CrossRef CAS.
- D. A. Johnson, J. C. Taylor and A. B. Waugh, J. Inorg. Nucl. Chem., 1979, 41, 827–831 CrossRef CAS.
- S. S. Rudel, H. L. Deubner, M. Muller, A. J. Karttunen and F. Kraus, Nat. Chem., 2020, 12, 962–967 CrossRef CAS PubMed.
- D. M. King, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619–5622 CrossRef CAS.
- J. A. H. Frey, G. N. Cloke and S. M. Roe, Organometallics, 2015, 34, 2102–2105 CrossRef.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, NMR spectra, crystallographic data. CCDC 2097071, 2097072 and 2097366–2097369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03957a |
| This journal is © The Royal Society of Chemistry 2021 |